
Abstract
Significance:
Tetrahydrobiopterin (BH4) is most well known as a required cofactor for enzymes regulating cellular redox homeostasis, aromatic amino acid metabolism, and neurotransmitter synthesis. Less well known are the effects dependent on the cofactor's availability, factors governing its synthesis and recycling, redox implications of the cofactor itself, and protein–protein interactions that underlie cell death. This review provides an understanding of the recent advances implicating BH4 in the mechanisms of cell death and suggestions of possible therapeutic interventions.
Recent Advances:
The levels of BH4 often reflect the sum of synthetic and recycling enzyme activities. Enhanced expression of GTP cyclohydrolase, the rate-limiting enzyme in biosynthesis, increases BH4, leading to improved cell function and survival. Pharmacologically increasing BH4 levels has similar beneficial effects, leading to enhanced production of neurotransmitters and nitric oxide or reducing oxidant levels. The GTP cyclohydrolase-BH4 pairing has been implicated in a type of cell death, ferroptosis. At the cellular level, BH4 counteracts anticancer therapies directed to enhance ferroptosis via glutathione peroxidase 4 (GPX4) activity inhibition.
Critical Issues:
Because of the multitude of intertwined mechanisms, a clear relationship between BH4 and cell death is not well understood yet. The possibility that the cofactor directly influences cell viability has not been excluded in previous studies when modulating BH4-producing enzymes.
Future Directions:
The importance of cellular BH4 variations and BH4 biosynthetic enzymes to cell function and viability makes it essential to better characterize temporal changes, cofactor activity, and the influence on redox status, which in turn would help develop novel therapies. Antioxid. Redox Signal. 37, 171–183.
Introduction
This review focuses on the involvement of tetrahydrobiopterin (BH4) in cell function and death. One of the ironies of life is that life depends on cell death (36) for several aspects governing normal organ development, function, and repair in disease (7, 35, 109). The existing knowledge of what role BH4 plays in the decision between life and death has been inadequate, especially on the BH4 role in many types of cell death (38).
This understanding is important because the triggers and mediators of cell death are critical to discovering new interventions. Cell death can be seen from the perspective of physiological and pathological processes; examples of physiological cell death processes would be apoptosis and senescent death and pathological processes would be necrosis and stress-induced cell death.
BH4 is linked to reactive oxygen species (ROS), especially those that cause cell dysfunction and cell death. ROS constitute a group of redox-active compounds that can react with proteins, lipids, and other biomolecules. Superoxide radical anion (O2 •−), the one-electron reduction product of oxygen, is the main product released by specialized enzymes such as NADPH oxidase (NOX, EC 1.6.3.1) and as a by-product of the oxidation of xanthine by xanthine oxidase (XO, EC 1.17.3.2) (10, 78).
Under certain circumstances, O2 •− is also released from mitochondria, nitric oxide synthase (NOS, EC 1.14.13.39) uncoupling, and the redox cycling of certain drugs. Superoxide produces hydrogen peroxide (H2O2) through a spontaneous or superoxide dismutase (SOD, EC 1.15.1.1)-catalyzed dismutation. Both iron and copper ions can mediate the decomposition of H2O2 via the Fenton reaction, generating highly reactive hydroxyl radical species (•OH) that participate in nonregulated oxidative mechanisms as •OH reacts at diffusion-limiting rates (72).
Since the Fenton reaction is slow, the relative significance of •OH in cellular oxidative processes is controversial (98). H2O2 is also the substrate of several iron heme-containing peroxidases, including myeloperoxidase (MPO, EC 1.11.1.7), a critical antimicrobial enzymatic defense generating hypohalous acids such as hypochlorous acid (HOCl), an oxidant considered the most relevant in vivo (21).
Another critical reaction of O2 •− is the radical combination reaction with nitric oxide (•NO or NO), mainly released from NOS (40) and through reduction of nitrite (NO2 −) (24) to produce peroxynitrite anion (ONOO−), a biological oxidant with similar potency to •OH (5). The significance of O2 •− and O2 •−-derived ROS in disease mechanisms rests on the idea that their production occurs at fluxes high enough to overwhelm antioxidant enzymatic defenses, such as SOD, peroxiredoxins (PRDX, EC 1.11.1.15), glutathione peroxidase (GPX, EC 1.11.1.9), and catalase (EC 1.11.1.6), and low-molecular-weight antioxidants, such as ascorbate, vitamin E, and glutathione.
Another factor is the specificity of ROS signaling or the specific targeting of vital cellular components that can trigger maladaptive cell responses. There is evidence that H2O2 signaling specificity is mediated by preferential reaction with peroxiredoxin that can serve as a redox relay of target/effector proteins (88). In addition, nitration of specific proteins or cellular components by ONOO− (25, 52) can lead to signaling dysfunction and even cell death.
BH4 is a required cofactor for NO production by NOS (83). Under conditions of deficient supply of the cofactor, NOS shifts activity from an NO to O2 •−-generating enzyme due to the uncoupled NADPH to L-arginine oxidation activity (92, 93). The O2 •− release and increase in H2O2 and possibly ONOO− can contribute to oxidant stress-triggered cell death in conditions where the supply of BH4 is below optimal levels (100) (Fig. 1).

Since exogenous treatment with BH4 generally leads to improved cell functions (1) and cell survival, the role of BH4 in cell death mechanisms has gained attention as a possible therapeutical target. To develop a viable therapy, one has to understand how BH4 is created, how deficiency of BH4 causes many disorders, and how BH4 interjects itself in many fundamental pathogenetic streams.
Regulation of Cellular BH4 Levels
The importance of BH4 to several physiological processes can be obtained from the manifestations of genetic mutations causing BH4 deficiency. Deficient cellular BH4 is linked to motor impairments (37), intellectual disabilities (32), hypertension, endothelial dysfunction, and cancer (111), while increased BH4 levels are commonly associated with inflammation (41).
The BH4 steady-state concentrations vary depending on the tissue and cell type, but are generally found in the low nano- to micromolar range. The maintenance of BH4 in a range hinges on the expression level of synthetic and recycling enzymes, the expression of partners' proteins, and possibly other factors, as recent discoveries indicate. The loss of synthetic capacity, however, is a driver of BH4 deficiency.
Many enzymes contribute to production and maintenance of BH4 levels (Fig. 2). The first enzyme in the biosynthetic pathway of BH4 is GTP cyclohydrolase I (GTPCH-I, GTPCH, or GCHI, EC 3.5.4.16). The transcriptional regulation of this enzyme is a central mechanism affecting cellular BH4 levels. GTPCH initiates the biosynthesis of BH4 by catalyzing the hydrolysis of GTP, which is the rate-limiting step in the synthetic pathway.

The expression of GTPCH-I and BH4 synthesis is enhanced severalfold in various cell types, including inflammatory epithelial and vascular cells (47), upon stimulation with interferon-alpha and gamma (IFN-α and IFN-γ), interleukin-1 beta (IL1-β), tumor necrosis factor (TNF)-alpha, and lipopolysaccharide (LPS). Gene expression is mediated by the nuclear factor-kappa B (NFkB) transcription factor, which in combination with Janus tyrosine kinase-2 (JAK2, EC 2.7.10.2)–signal transducer and activator of transcription 1 (STAT1), potentiates transcription (47).
Induction of GTPCH-I expression by H2O2 via activation of the JAK2-STAT1 pathway has also been described (46, 77). Nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor and master regulator of numerous antioxidants and antiapoptotic proteins, stimulates GTPCH expression, as was shown in cells transfected with Nrf2; the converse was observed in Nrf2 knockout (KO) mice. Nrf2 KO mice also show increased BH4 oxidation (65). The interaction between the GCH promoter region and Nfr2 was established by chromatin immunoprecipitation assay and protein expression by Western blots (104).
GTPCH mRNA is detected in human neuroblastoma cell lines, and cAMP increases levels of BH4 by rapidly stimulating GCH gene transcription (45). GCH transcription by these various mechanisms is associated with improved cell function, survival, and even differentiation. Thus, it is almost certain that GTPCH/BH4 exerts pleiotropic effects in several cellular functions beyond regulation of inflammation, as initially proposed.
As described above, proinflammatory conditions increase the activity of GTPCH-I and BH4 production. Proinflammatory cytokines stimulate the activation of the kynurenine pathway. Binding screening assays of kynurenine bioactive compounds to sepiapterin reductase (SPR, EC 1.1.1.153), an NADPH-dependent reductase in the BH4 synthetic pathway, identified xanthurenic acid, coming from the kynurenine pathway, as a potent inhibitor of SPR (43).
The inhibition of SPR has been proposed to explain some adverse effects characteristic of BH4 deficiency associated with IFN-α in the treatment of cancer and viral infections (84, 85). Moreover, since both kynurenine and BH4 are stimulated in chronic pain (81), kynurenine metabolites may represent a new approach to alleviate debilitating pain in some clinical conditions.
The activity of constitutive GTPCH levels is also susceptible to regulation by protein–protein interaction with the GTPCH feedback regulatory protein (GFRP) (42, 102). GTPCH-I forms a 1:2 complex with the GFRP, which mediates allosteric regulation of GTPCH-I activity, with phenylalanine as a positive and BH4 as a negative effector, respectively (31, 42). Formation of the GTPCH: GFRP complex is mediated by both BH4 and 7,8-dihydrobiopterin (7,8-BH2) with similar efficiency (106).
However, it is unclear whether the effects on GTPCH activity are comparable. Whether the expression of GFRP attains similar levels to GTPCH is also not known. Conflicting data on the role of GFRP regulating BH4 levels are found in the literature (64, 82) as recently, GFRP overexpression was considered to have no significant effect on endothelial BH4 levels (82). In contrast, we and others described that enhanced GFRP expression in endothelial cells after H2O2 exposure was followed by a decrease in BH4 (50, 51).
This finding would indicate that increased oxidation of the cofactor under oxidant stress conditions is not the only explanation for depletion of BH4 (73). The regulation of GFRP may be a common mechanism to increase BH4. Early reports indicated that LPS-treated myelomonocytic (THP-1) and human umbilical vein endothelial cells enhanced GTPCH activity while downregulating GFRP levels (99). These findings expand the idea that the GTPCH:GFRP complex is a metabolic sensor that maintains BH4 and aromatic amino acid homeostasis in inflammatory and oxidant signaling.
This idea is further supported by phosphorylation of GTPCH-I on serine 81 in anticluster of differentiation 3 (CD3)-stimulated T cells, which likely weakens the interaction with GFRP, attenuating GFRP inhibition. In addition, casein kinase II (EC 2.7.11.1) inhibition diminished GTPCH-I phosphorylation and BH4 production (57). The GTPCH-I phosphomimetic mutant (S81D) further corroborated that there is a GTPCH-I (S81D) escape from GFRP negative feedback (57). This evidence indicates that the GTPCH:GFRP complex is likely amenable to modulation by pharmacological effectors to increase or decrease GTPCH-I activity, representing a way of altering endogenous BH4 synthesis to treat disease conditions depending on the effector molecule.
BH4 Cofactor as an Antioxidant
It is well known that five key enzymes require BH4 for optimal enzyme activity. These are phenylalanine hydroxylase (PAH, EC 1.14.16.1), tyrosine 3-monooxygenase (or tyrosine hydroxylase [TH, EC 1.14.16.2]), tryptophan 5-monooxygenase (TPH, EC 1.14.16.4), alkylglycerol monooxygenase (EC 1.14.16.5), and NOS (Fig. 3). Most of the cellular functions of BH4 are associated with the levels of the enzyme products, namely tyrosine, L-DOPA, 5-hydroxytryptophan, glycerol+aldehyde, and NO+citrulline, respectively.
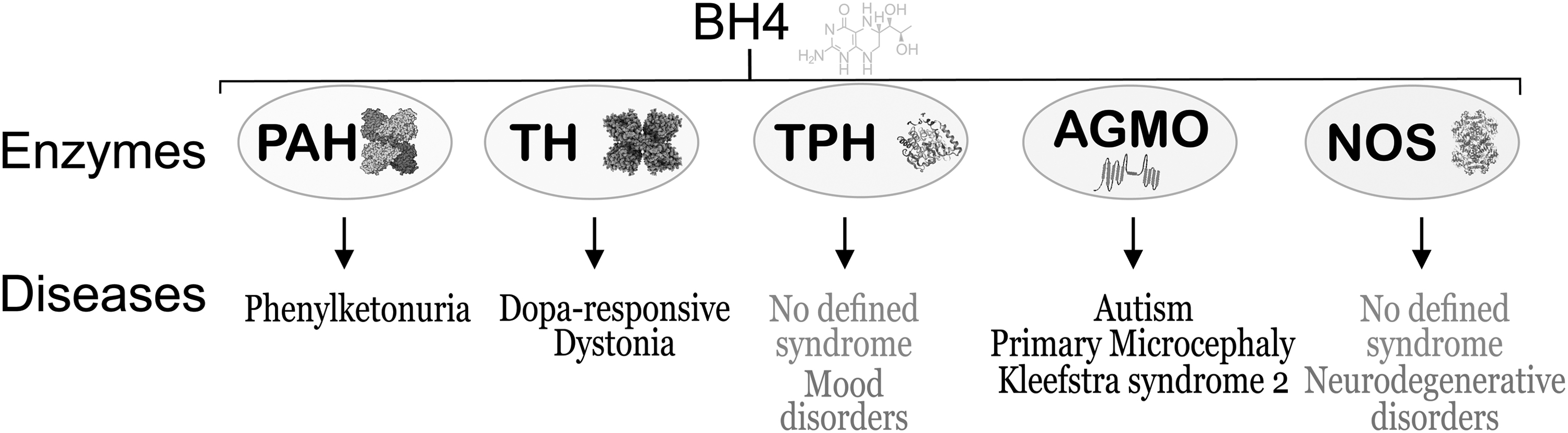
These enzymes vary in their requirement for BH4. The Km value of BH4 for TH is about 10–30 μM, for PAH is about 2–3 μM, and for NOS is about 0.02–0.03 μM (49, 89). Interestingly, if found to be deficient in activity, three of the enzymes are associated with well-defined clinical syndromes and the other two have an indirect impact on clinical disorders (Fig. 3). The implications of BH4 deficiency, being varied and complex, are discussed herewith.
The BH4 activity as a reducing compound may explain its biological activity independent of its cofactor properties. Kinetic and cellular studies have established the BH4 reactivity toward several oxidizing species, including oxygen (23), O2 •− (97), ONOO− (63), H2O2/horseradish peroxidase (HRP, EC 1.11.1.7) (3), and iron in several forms, including iron-containing proteins and iron porphyrins such as cytochrome c (15). Through these reactions, the cellular BH4 supply can become limiting and it may explain its relatively rapid turnover of 3–5 h, as shown in endothelial cells upon inhibition of GTPCH (75).
Moreover, in the case of accumulating 7,8-BH2 as the BH4 oxidation product, this reaction can further disrupt the normal functioning of NOS (94), inhibiting NO production and increasing O2 •− release. Although BH4 shows a relatively favorable redox potential, ∼0.27 V (39), for the reaction with several oxidizing species compared with ascorbate with a redox potential of 0.33 V (12), BH4 efficiency as a low-molecular-antioxidant hinges on local cellular concentrations. This concept of the local milieu is important to understand as the final biological effect of an oxidant–antioxidant reaction would depend upon the redox reaction potential and the reaction rate of the oxidant with neighboring molecules (Fig. 4).
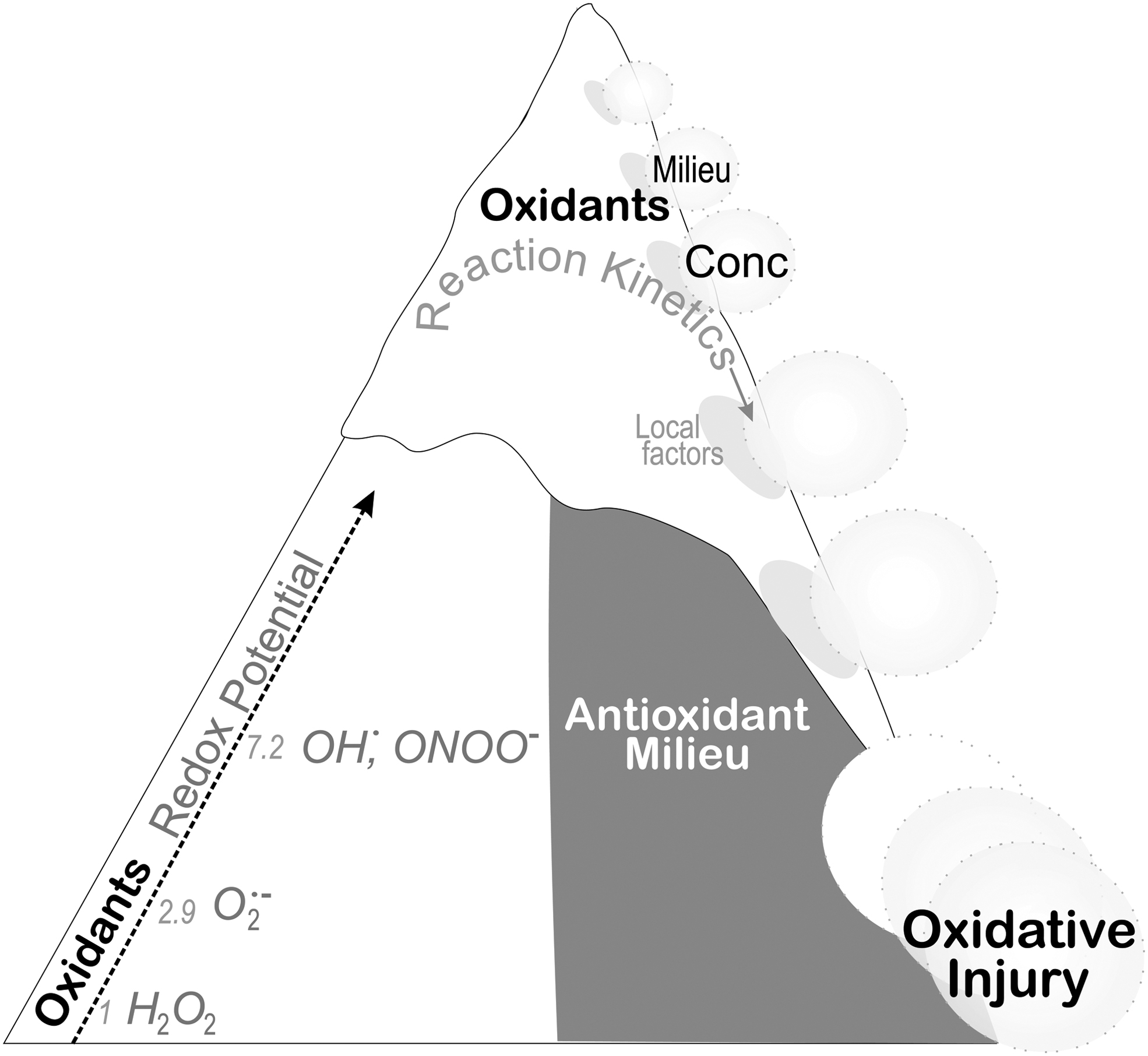
Ascorbate and glutathione reach concentrations severalfold higher than BH4, indicating that their reactions with radical species and oxidants occur at a faster rate than BH4. From this viewpoint, ascorbate and glutathione protect BH4 from oxidation by acting as a first-line defense. In fact, the stabilizing activity of ascorbate treatment supporting an increase in endothelial BH4 occurs independently of increased cofactor synthesis. However, ascorbate treatment decreased 7,8-BH2 accumulated in the cells, suggesting that ascorbate, by shielding BH4 from oxidation, effectively increases its cellular concentration (20, 44).
The direct reaction of BH4 with oxidants may still represent an antioxidant pathway. The one-electron oxidation reaction mediated by free radical species will produce a BH4-free-radical that could further react with another oxidant to generate the two-electron oxidation product, quinonoid dihydrobiopterin (qBH2). qBH2 is reduced back to BH4 via NADH/dihydropteridine reductase (DHPR, EC 1.5.1.34) (100) or qBH2 can rearrange to a more stable 7,8-BH2, which is reduced back to BH4 via NADPH/dihydrofolate reductase (DHFR, EC 1.5.1.3).
In the case that the BH4 radical does not react fast enough with another oxidant to generate qBH2 or 7,8-BH2, there is the possibility that it reacts with oxygen to generate the 4a-peroxytetrahydropterin (4a-peroxyBH4) radical analogous to the intermediate generated in the amino acid hydroxylase (TH and PAH) catalytic cycle (22). The 4a-peroxyBH4 radical will most likely propagate oxidative reactions through several mechanisms. The biochemistry of these reactions can be exceedingly complicated and more studies are needed to understand their biological relevance.
BH4 and Cell Functions
Recent developments in the literature indicate that enzymes in the BH4 pathway regulate cell functions through mechanisms loosely connected with BH4 synthesis. One aspect is the possible influence of the BH4 pathway in regulation of folate-dependent reactions. It has been assumed that DHFR represents the center, controlling both dihydrobiopterin (BH2)/BH4 and methyltetrahydrofolate/homocysteine.
Although in vitro data have not firmly established these interactions, a new DHPR KO mouse model showed that DHPR activity increases liver BH2 levels severalfold. This result indicates that DHFR may not be the most important enzyme in the BH4 salvage pathway. In addition, unexpectedly, DHPR KO showed altered folate metabolites, such as low tetrahydrofolate and 5-methyltehydrofolate levels (103). The DHPR KO model provided an important biochemical connection between BH4 and the folate pathway.
DHPR is confirmed to maintain cellular tetrahydrofolate levels, which may explain the fact that low tetrahydrofolate levels are characteristic in DHPR-deficient subjects (112). There is also cross talk between the folate metabolic pathway and the BH4 metabolic pathway (26, 69), initially suspected from the relationship between plasma and CSF levels of both substances in various diseases (11, 34), and manipulations of folate levels by supplementation (16).
Another interesting study by Lange et al. described the interaction of SPR with ornithine decarboxylase (ODC, EC 4.1.1.17), a regulatory enzyme in polyamine synthesis, in MYCN2 (MYCN-2 Tet-on) neuroblastoma cells (55). Impaired cell proliferation was demonstrated in SPR knockdown using SPR siRNA to a similar extent as ODC knockdown, suggesting that SPR has a regulatory effect on ODC activity.
The lack of evidence for downregulation of ODC by siRNA knockdown of SPR, and similarly of SPR by siRNA knockdown of ODC, further supports the protein–protein interaction in the regulatory mechanism. Tumor sample analysis demonstrated a good correlation between low SPR gene expression and patient survival prognosis. Unfortunately, this study did not provide information on BH4 or oxidant changes in SPR deficiency, which could be a factor enhancing ODC inhibition or potentiating inhibition of cancer cell proliferation.
Supporting evidence for pharmacological inhibition of SPR as an anticancer strategy is still very limited. In studies with hepatocellular carcinoma (HCC), Wu et al. examined the effects of SPR depletion by gene editing and pharmacological SPR activity inhibition in cancer progression (101). In HCC tumors, the SPR expression levels were higher than those in healthy tissue. Treatment of HCC cells with SPR inhibitors did not affect cell proliferation; however, SPR protein depletion induced mitochondrial-dependent apoptosis that was not reversed by BH4 treatment.
SPR activity does not affect cell proliferation as cell proliferation in HCC expressing the inactive SPRD275G mutant was unchanged. A systematic RNA seq analysis of cells treated with SPR siRNA led to identification of an apoptosis-associated pathway with Fox3a-mediated Bim expression as a potential effector of SPR depletion. Thus, this study provides additional support to the idea that SPR, through interactions with partner proteins, regulates the protein activity to regulate cell survival.
New data from studies with recombinant SPR revealed that the reductase activity of SPR may influence the redox mechanism in cellular functions. These studies established that SPR catalyzes the redox cycling of several redox-active compounds, including menadione and 9, 10-phenanthrenequinone, to increase O2 •− production (Fig. 5). The substrate sepiapterin did not affect redox cycling activity; however, redox-cycling quinones inhibited the reduction of sepiapterin (105).
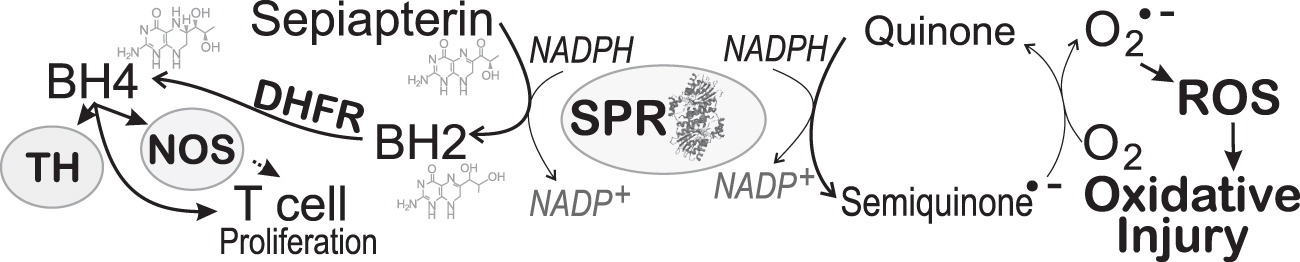
Notably, inhibitors of sepiapterin reduction such as N-acetylserotonin and indomethacin did not affect SPR redox cycling activity. Moreover, the mutation, D257H, in the C-terminal sepiapterin binding region of SPR minimally affected redox cycling activity, but eliminated sepiapterin reduction (105). With the lack of effect of the mutant SPRD257G on cell proliferation, one cannot entirely reject a role of SPR in this process as increased ROS may be involved. These novel functions of multifaceted SPR provide further nuance to the role of BH4 in physiological and pathological conditions.
The impact of the BH4 pathway in T cell biology is not well defined. A recent study examined in vivo effects using genetic mutants with T cell-specific GTPCH KO and with inhibitors of SPR. Repressed T cell-mediated autoimmunity and allergic responses were associated with low BH4 (19). Although this study did not find defective T cell development in GTPCH KO, adult BH4-deficient T cells showed reduced proliferation upon stimulation. The role of the BH4 pathway in effective T cell proliferation and activation was further verified in experiments using pharmacological inhibition of SPR.
Deficient expression of GTPCH in T cells did not affect the biogenic amine or amino acid content. However, loss of GTPCH was found to increase the expression of proteins involved in iron regulation, including mitoferrin, ferritin (FTMT, EC 1.16.3.1), heme oxygenase-1 (HO1, EC 1.14.14.18), and frataxin (FRDA, EC 1.16.3.1). The mechanisms by which BH4 deficiency enhances iron regulatory protein expression, however, are unknown. Overexpression of GTPCH indicated that T cell development and distribution of CD4+ and CD8+ T cells were unaffected.
However, upon stimulation of CD4+, T cells displayed enhanced T cell proliferation. It has not been established that enhanced T cell proliferation was due to regularization of iron regulatory protein expression. These data could have shed light on the BH4 regulatory mechanisms of T cell growth (Fig. 5). Notably, in increasing T cells, BH4 appears to confer not only enhanced proliferation but also anticancer immunity. This complex study hints at the unrealized role of BH4 controlling iron regulatory gene expression and the fine-tunning of adult T cell proliferation with essential consequences in inflammation and cancer progression.
It is recognized that BH4 is an essential cofactor in monoamine neurotransmitter synthesis and that variations in BH4 may represent a physiological regulatory mechanism of transmitter levels. These activities are due to the BH4 effect on both TH-Fe(III) reduction in the initial steps of catalysis and inactivation and aggregation of the enzyme when present at low levels (90).
Cook et al. (18) recently demonstrated the novel role of BH4 in sulfonation of neurotransmitters as a mechanism controlling their activity. They showed that BH4 binds with high affinity and selectivity to the sulfotransferase 1A3 (SULT1A3, EC 2.8.2.1) catechin site to inhibit enzyme activity (Ki 23 nM). The suggested mechanism favors a classical feedback inhibition mechanism in which BH4 concentrations result in allosteric inhibition of the enzyme and augmentation of neurotransmitter availability.
The dual role of BH4 in regulating neurotransmitter production and inactivation via the sulfonation pathway adds another factor to consider in the treatment with BH4, as minor variations of BH4 in the brain may, in fact, effectively help neurotransmitter regulation.
BH4 in Cell Survival and Death
Selective neuronal cell death in specific brain regions is characteristic of progressive neurological diseases. In Parkinson's disease, the selective loss of dopaminergic neurons in the substantia nigra causes significant loss of dopaminergic transmission in the striatum (27). Spinal neuron death is a substantial consequence of spinal cord injury (76). Increased neuronal cell death leading to decreased functions and overall brain mass is characteristic of Alzheimer's disease (9, 14). Neuronal cell death is likely also involved in death and developmental disabilities consequent of neonatal hypoxic-ischemic encephalopathy (70).
The exact underlying cause of neuronal cell loss in neurological diseases is not yet well established, although one of the culprits is oxidants that cause cell dysfunction and cell death. Environmental toxicants and redox-active substances combined with genetic factors are proposed to activate endogenous mechanisms such as inflammation in the cascade of events leading to accumulation of oxidative or nitrative modifications and cell dysfunction. Inhibition of adenosine triphosphate synthesis is considered a critical initiating event in cellular dysfunction, generally culminating in mitochondrial-dependent cell death.
Apoptotic cell death by the intrinsic pathway occurs after mitochondrial membrane permeabilization by the B cell lymphoma 2 family proteins, which several stressors, including ROS, can stimulate. Resident macrophages and other professional phagocytes recognize apoptotic cells, initiating efferocytosis, the process of engulfing dying cells and digesting cell contents (Fig. 6). This process decreases the possibility of cells undergoing gasdermin E– or deafness, autosomal dominant 5-mediated secondary necrosis (71).
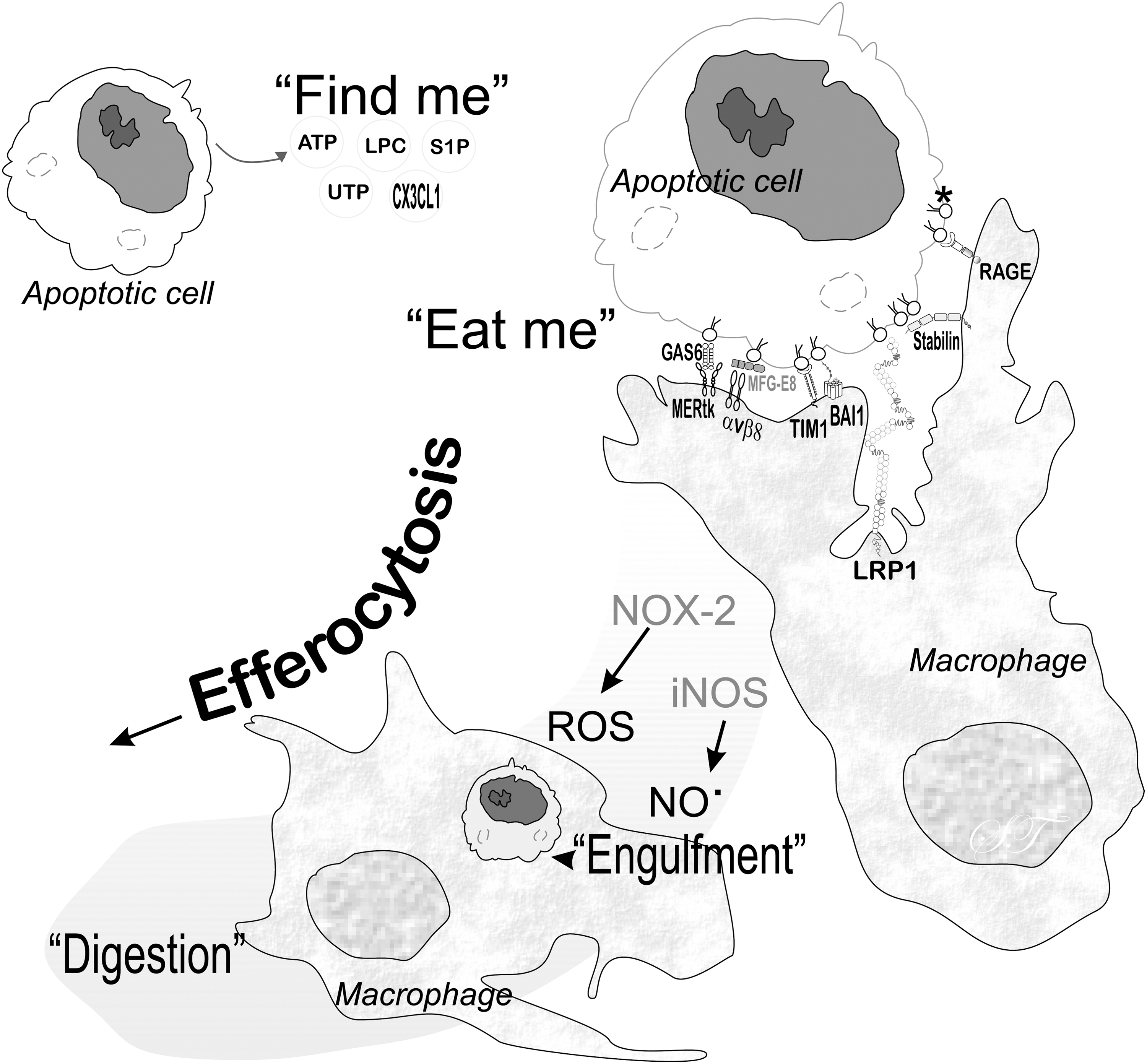
Efferocytosis prevents dying cells from releasing damage-associated molecular patterns (67) that could trigger an inflammatory response. In the process of efferocytosis, macrophages generate ROS from NOX-2, which needs to be controlled for favorable outcomes (56). NO from the inducible isoform of NOS is involved in efferocytosis associated with inflammation (110).
Acute oxidant injury is believed to be involved in necrotic cell death with cell rupture and release of intracellular contents. This type of cell death triggers an inflammatory response, enhancing cytokine signaling. Necroptosis is a type of cell death initiated by TNF signaling. It involves activation of receptor-interacting serine/threonine-protein kinase (RIPK, EC 2.7.11.1) and RIPK1, activation of the kinase activity of RIPK3, and oligomerization of phosphorylated mixed lineage kinase domain-like pseudokinase (MLKL) (8). Since this cell death mechanism requires the activation of various proteins, it is considered a regulated necrotic cell death process (Fig. 7). The use of RIPK1 inhibitors represents a possible strategy to inhibit necroptosis, reducing inflammation and brain injury (59, 61).
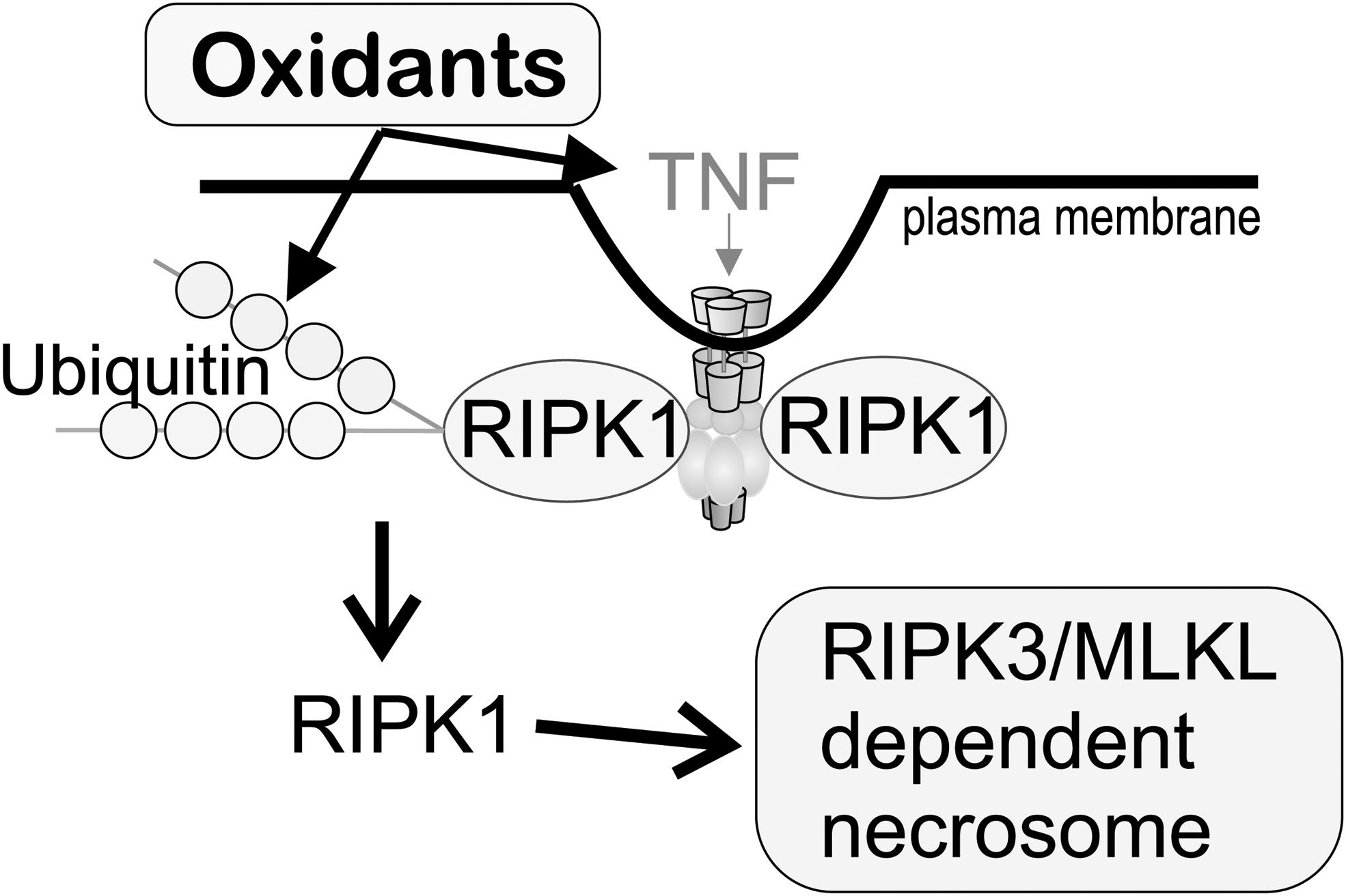
Necrostatin-1 (Nec-1) is not only a necroptosis inhibitor that targets RIPK1 activity (Ki 494 nM) but Nec-1 also shows an inhibitory effect on indoleamine 2, 3-dioxygenase (IDO, EC 1.11.1.7), a potent immunosuppressive molecule (62, 86). Nec-1 has shown neuroprotective effects in neonatal ischemia–reperfusion injury (17), spinal cord injury, and traumatic brain injury (107). Nec-1 has recently been found to have off-target effects on ferroptosis (33), and other studies link both together (66).
Nec-1s is an Nec-1 analog, with superior selectivity and metabolic stability in blocking RIP1 and without off-target inhibition of indoleamine 2, 3-deoxygenase in contrast to Nec-1 (87). Using nonspecific markers, Nec-1s has been shown to decrease oxidative stress in neurons derived from neuroprogenitor cells (48). Although Lannielli et al. showed Nec-1s to be neuroprotective in dopaminergic neurons in an MPTP mouse model of Parkinson's disease (48), another study showed no difference with treatment (28).
In newborn brains, there is a continuum of cell loss from fetal life to the time of delivery. Cell loss is part of normal nervous system development and shaping. Neuronal cells die via a regulated cell death process (53). It is well established that neuronal and preoligodendrocytes are susceptible to hypoxia–ischemia (HI)–reperfusion injury with increased ROS production (13, 30). MRI imaging findings in antenatal HI support the notion of an acute injury phase followed by a delayed phase guided by activation of inhibition of metabolic and signaling pathways that could trigger an inflammatory response.
However, biomarkers of specific acute brain injury in vivo are elusive, which complicates a clear identification of injury mechanisms. Recent studies on necroptosis in HI injury identified RIPK3, not RIPK1, as the effector protein in the pathway leading to oligodendrocyte necroptosis in the developing brain (68). Our group has identified BH4 as a developmental factor determining the brain's vulnerability to HI and development of motor impairments and death (96).
The typical low antenatal brain BH4 levels were associated with an increased risk of uncoupled NOS activity with increased ROS-mediated signaling and cell injury. This mechanism is supported by studies showing that maternal BH4 treatment augmented fetal brain BH4 levels, which reduced both the severity of motor deficits (95, 96) and HI-induced death (108).
The role of BH4 in cell survival and growth has been recently linked to the regulation of mTORC1, a nutrient, energy, and redox sensor pathway. Reduced BH4 production due to genetic mutations of biosynthetic enzymes can inhibit phenylalanine hydroxylase (PAH) activity (60, 100). Tyrosine is the product of hydroxylation of phenylalanine, which serves in several other pathways, including dopamine and melanin synthesis.
Mouse models of PAH and SPR deficiency show notorious growth retardation, which is corrected by tyrosine-rich diets. It was found that increased autophagy in BH4-deficient mice was linked to mammalian target of rapamycin (mTOR, EC 2.7.11.1) inactivation (54). Since mental retardation is a significant consequence of inadequate production of tyrosine and BH4, the role of autophagy in developmental brain disorders associated with deficient BH4 levels warrants investigation.
The effect of BH4 variations in the inflammatory response is not well characterized. Recent studies with specific inhibition of macrophage GTPCH showed that BH4-deficient macrophages increase IL-1β upon stimulation with LPS/IFNγ. The increased cytokine production was not dependent on NO inhibition, but was likely mediated by metabolic remodeling (4). Since similar metabolic remodeling was observed with iNOS KO models, which still produced BH4, the main effector appears to be NO.
Others have shown that the GTPCH inhibitor (DAHP) combined with triptolide in rat brain cerebral ischemia results in a significant neuroprotective effect. This response was associated with iNOS inhibition and reduction in GFAP-positive cells, causing apoptosis and PI3K/Akt/mTOR activation (58). In addition, increased iNOS expression in spinal cord injury is associated with poor outcomes. Microribonucleic acid (miR)-124 is highly expressed in the brain, regulating neurogenesis and postnatal neural differentiation.
There is an association of miR-124 with GTPCH-1-inhibited spinal neuronal apoptosis and inflammatory response to LPS-induced BH4 and iNOS activation. Although these data support a role for BH4 in inflammation, they do not clarify if BH4 reduction alters the course of cytokine-stimulated processes, including GTPCH and iNOS expression.
Genetic defects of cytosolic SOD (SOD1) and glutathione peroxidase 4 (GPX4, EC 1.11.1.12) have detrimental consequences on neuronal survival (6, 74). Enhanced SOD1 aggregation induces selective loss of motor neurons in amyotrophic lateral sclerosis (ALS), a progressive paralytic and ultimately fatal disease. Mutations in the gene encoding SOD1 in familial ALS increase toxicity due to protein misfolding and the likely ability of aggregates to spread from neuron to neuron in a prion-like manner.
In contrast, GPX4 is essential for early embryonic development, and GPX4 KO embryos die in utero. Studies with conditional KO models showed poor survival, neuronal cell death, and enhanced astrogliosis. GPX4 is the only known enzyme that reduces phospholipid hydroperoxide (P-LOOH) to phospholipid alcohol (P-LOH) using reduced glutathione as the reducing power. Lipoxygenases (LOXs) produce P-LOOH as a reaction intermediate, with arachidonate 5-lipoxygenase (ALOX5, EC 1.13.11.34) and ALOX12/ALOX15 (EC 1.13.11.31) as essential mediators of neuroinflammation and neurodegeneration.
In addition, iron can have a role in the initiation of lipid peroxidation by generating potent oxidant species such as hydroxyl radical or perferryl-oxo species. It follows that cell death triggered by either depletion of glutathione, a low-molecular-weight antioxidant, or inhibition of GPX4, a phospholipid hydroperoxidase, has been identified as another mechanism of cell death, ferroptosis. Ferroptosis is described as a form of cell death intimately linked with ROS, sulfhydryl defense, lipid peroxidation, and iron-mediated oxidation (Fig. 8) (2, 29, 33, 79, 91). Low-molecular-weight antioxidants such as α-tocopherol, iron chelators, and BH4 appear to counteract this form of cell death.

The increased expression of GTPCH-I was recently demonstrated in a CRISPR-mediated activation screen upon RSL3-mediated GPX4 inhibition in cancer cells. It was shown that increased BH4 protected cells from death, which indicated a direct role in inhibiting lipid peroxidation and, as also shown, augmenting reduced CoQ levels.
Additional studies have indicated that BH4 efficiency in scavenging of the lipid peroxide radical is enhanced by recycling of BH2 by the DHFR/NADPH system (80). Since these studies use GPX4 inhibition, it would be important to establish whether BH4 responses are specific to GPX4 inhibition or if increased iron-mediated lipid peroxidation induces similar responses (Fig. 9). In the brain, BH4 cofactor activity of NOS also influences these reactions by inhibiting ROS and increasing NO, a powerful inhibitor of lipid peroxidation.

The role of BH4 in ferroptosis is intriguing, but in pathological conditions, this involvement needs to be confirmed.
Conclusions
In summary, an understanding of the role of BH4 in cell death has evolved beyond the cofactor role. Factors that determine its availability, from variables that affect synthesis and recycling to the redox implications of BH4 itself, added to the interaction with other redox pathways, which is dependent on the levels of BH4 available in the local milieu, along with many protein–protein interactions, may ultimately determine the fate of a cell.
Future studies characterizing how BH4 variations affect cell turnover and maintenance will be vital to understand its therapeutic profile.
Footnotes
Authors' Contributions
J.V-V. contributed to the conceptualization, research, drafting of the manuscript, review of graphics, and the intellectual and technical content of the study. Z.S. participated in the conceptualization, writing, research, and revising its intellectual and technical content. S.T. participated in the conceptualization, writing, research, graphics, and revising its intellectual and technical content. All authors assume responsibility and accountability for the results.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by funding from National Institutes of Health, National Institute of Neurological Disorders and Stroke, NS114972 (S.T.), NS117146 (J.V-V. and S.T.), and NS081936 (J.V-V. and S.T.).