
Abstract
Significance:
As the global prevalence of diabetes rises, diabetic complications are also increasing at an alarming rate. Peripheral neuropathy (PN) is the most prevalent complication of diabetes and prediabetes, and is characterized by progressive sensory loss resulting from nerve damage. While hyperglycemia is the major risk factor for PN in type 1 diabetes (T1D), the metabolic syndrome (MetS) underlies the onset and progression of PN in type 2 diabetes (T2D) and prediabetes.
Recent Advances:
Recent reports show that dyslipidemia, a MetS component, is strongly associated with PN in T2D and prediabetes. Dyslipidemia is characterized by an abnormal plasma lipid profile with uncontrolled lipid levels, and both clinical and preclinical studies implicate a role for dietary fatty acids (FAs) in PN pathogenesis. Molecular studies further show that saturated and unsaturated FAs differentially regulate the nerve lipid profile and nerve function.
Critical Issues:
We first review the properties of FAs and the neuroanatomy of the peripheral nervous system (PNS). Second, we discuss clinical and preclinical studies that implicate the involvement of FAs in PN. Third, we summarize the potential effects of FAs on nerve function and lipid metabolism within the peripheral nerves, sensory neurons, and Schwann cells.
Future Directions:
Future directions will focus on identifying molecular pathways in T2D and prediabetes that are modulated by FAs in PN. Determining pathophysiological mechanisms that underlie the injurious effects of saturated FAs and beneficial properties of unsaturated FAs will provide mechanistic targets for developing new targeted therapies to treat PN associated with T2D and prediabetes. Antioxid. Redox Signal. 37, 560–577.
Introduction
Diabetes is one of the fastest growing epidemics of the 21st century. Over 463 million adults worldwide currently have diabetes, and the number of diabetes cases is projected to reach 700 million by 2045 (66). This massive increase in diabetes cases over the next two decades will impact one tenth of the world's population, and result in a global rise in diabetes-related deaths and diabetic complications (132). Type 2 diabetes (T2D) is the predominant type of diabetes, accounting for 90%–95% of global diabetes cases, and is characterized by impaired insulin secretion and progressive insulin resistance (5, 66).
Before developing T2D, almost all individuals have prediabetes, a condition that precedes T2D and is a major risk factor for the development of T2D (41). In the United States alone, 1 in 3 adults have prediabetes placing them at risk of developing T2D. Although lifestyle interventions such as diet or exercise can slow the progression from prediabetes to T2D, 3%–11% of prediabetes cases progress to T2D each year (47, 100).
The global rise in both diabetes and prediabetes cases has led to a parallel increase in diabetic complications. Peripheral neuropathy (PN), the most prevalent complication of diabetes and prediabetes, occurs in ∼50% of T2D as well as 30% of prediabetes patients (14, 49, 105). PN is characterized by progressive peripheral nerve damage that leads to tingling, numbness, and/or pain (35). The distal-to-proximal loss of normal sensation arises first in the feet and progresses proximally in a stocking-and-glove distribution.
The progressive loss of sensory function in PN causes severe morbidity and is a leading cause for nontraumatic amputations (88). In addition, the financial expenditure related to PN health care costs in the United States is >US$10.9 billion annually (49). Therefore, there is a pressing need to identify the factors that lead to PN in T2D and prediabetes to develop new and targeted therapies for PN; however, the molecular mechanisms that drive PN are incompletely understood.
Dyslipidemia is increasingly recognized as an important risk factor for PN in T2D and prediabetes (50, 75, 137). Broadly defined as an abnormal plasma lipid profile, dyslipidemia commonly develops in patients with T2D and prediabetes, is a component of the metabolic syndrome (MetS) (2), and is thought to play a major role in tissue-specific complications associated with metabolic diseases.
The plasma lipid profile is influenced by diet, in particular the dietary fatty acid (FA) level and composition (39). Consuming an unhealthy diet, such as the western diet, composed of foods rich in saturated FAs (SFAs), is associated with increased plasma lipid levels, elevated low-density lipoprotein cholesterol, and the development of dyslipidemia in both human subjects and preclinical animal models. The American Diabetes Association advises patients with T2D and prediabetes to reduce the dietary intake of saturated and trans-FAs, and supplement their diet with sources of monounsaturated FAs (MUFAs) and polyunsaturated FAs (PUFAs) (77).
Dietary FAs have emerged as both potential mediators and treatments for PN in T2D and prediabetes (25, 114, 144). The molecular mechanisms that underlie the injurious or beneficial effect of FAs on nerve function are an active area of study. Herein, we first review the clinical and preclinical studies that delineate the importance of dietary FAs in modulating peripheral nerve function in PN associated with T2D and prediabetes. Second, we discuss the metabolic effects of dietary FAs on the peripheral nervous system (PNS) in T2D and prediabetes. Finally, we detail the molecular effects of FAs on complex lipid synthesis, lipid metabolism, and mitochondrial function in the PNS.
Dietary FAs
FAs are essential molecules for the nervous system, and are critical for maintaining neuronal health and function (130). FAs play a number of important cellular functions ranging from substrates for mitochondrial FA β-oxidation and mediators of intracellular signaling to components of complex lipid species in plasma membranes and organelles (30). Circulating FAs are the major source of FAs for the nervous system, and can be obtained from the diet or synthesized through de novo lipogenesis FA synthesis. The physiological destination and functional role of FAs are defined by the structure of the FA hydrocarbon chain.
FAs are composed of an aliphatic hydrocarbon chain terminating in a carboxylic acid functional group. Although FAs tend to be hydrophobic in nature due to the hydrophobicity of the hydrocarbon chain, the carboxylic acid is a polar functional group that participates in FA β-oxidation (30). During β-oxidation, two carbons are removed from the carboxyl end of the FA to generate acetyl-CoA, which enters the tricarboxylic acid cycle (TCA) to produce cellular energy.
FAs are classified by hydrocarbon chain length and degree of saturation, which correspond to the number of carbons and double bonds in the hydrocarbon chain, respectively (Fig. 1). Short-chain FAs contain 2–4 carbons, medium-chain FAs are composed of 6–12 carbons, long-chain FAs consist of 14–18 carbons, and very long-chain (VLC) FAs contain 20–24 carbons. FAs also have varying degrees of saturation. SFAs lack double bonds and have fully saturated hydrocarbon chains, while unsaturated FAs have one or more double bonds (Fig. 1A–C). MUFAs contain one double bond typically between the 9th and 10th carbons in the hydrocarbon chain, which causes a kink in the FA structure (Fig. 1B). Among other types, PUFAs constitute omega-3 or omega-6, which indicates the location of the first double bond closest to the methyl group within the hydrocarbon chain (Fig. 1C).
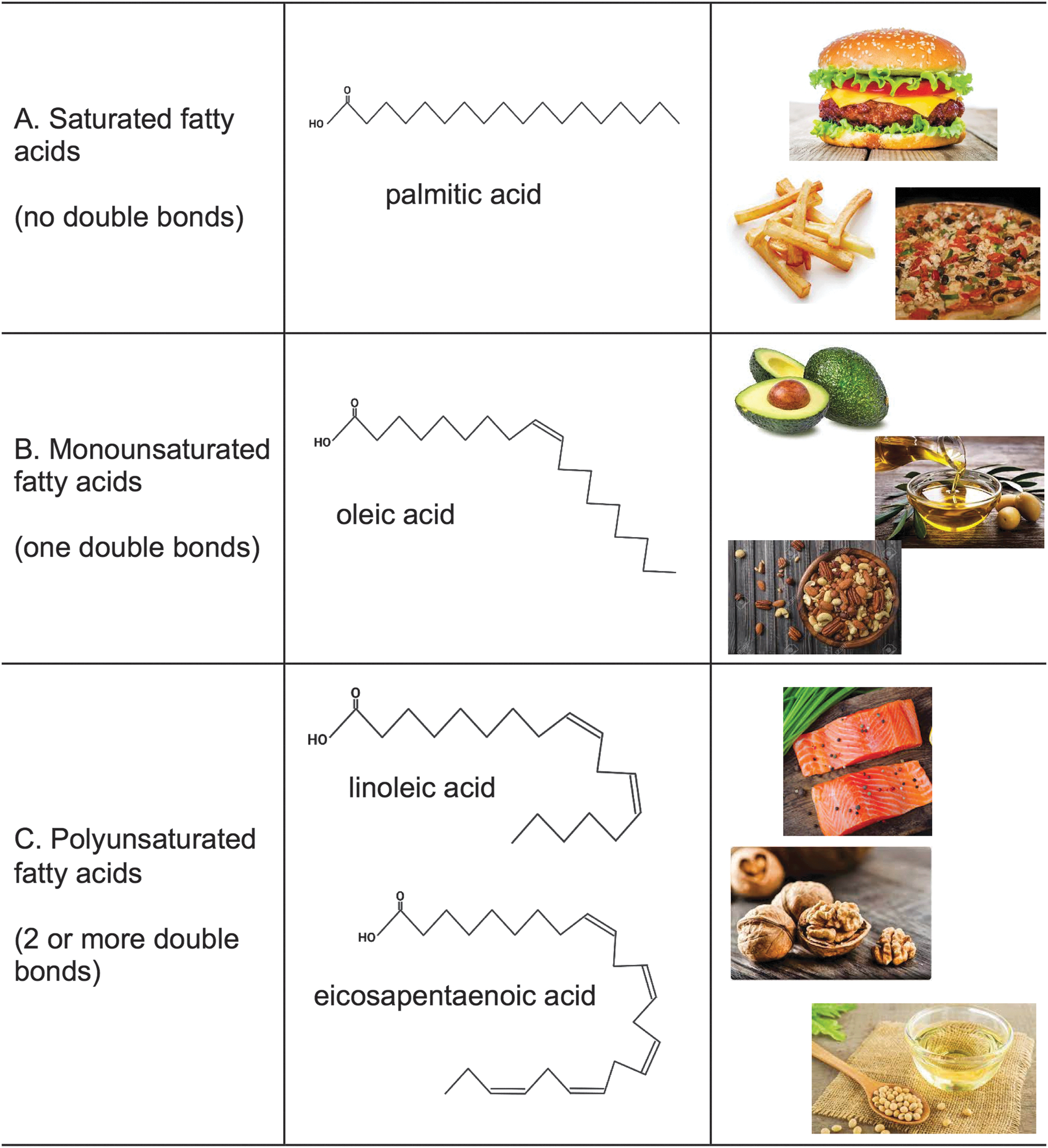
The most common omega-3 FAs are eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Bends in the hydrocarbon chain that result from a single double bond in MUFAs or multiple cis-double bonds in PUFAs play an important role in cellular membranes by preventing tight packing and improving membrane fluidity and curvature. The length and degree of saturation of FA acyl chains dictate the FA intracellular localization and function (1, 130). The hydrocarbon chain length of FAs dictates the intracellular localization and downstream metabolic and signaling pathways. The degree of saturation of a FA plays a key role in membrane fluidity and organelle dynamics.
There are two categories of FAs, including essential FAs, which must be obtained from the diet, and nonessential FAs, which may be synthesized by cells and tissues within the body. Essential FAs, including linoleic acid and linolenic acids, cannot be synthesized endogenously and must be obtained from dietary sources (28). These FAs play important roles in membrane fluidity, regulation of enzymatic reactions, and receptor signaling in the nervous system (130). Nonessential FAs can be either obtained from the diet or synthesized intracellularly through de novo lipogenesis pathways. Nonessential FAs include saturated, monounsaturated, and some polyunsaturated FAs.
Different types of foods contain distinct FA profiles (Fig. 1). Red meats, and processed and fried foods contain high levels of long-chain saturated FAs, whereas dairy products, such as milk and cheese, are rich in medium-chain saturated FAs (97). These foods are commonly found in the western diet, and have been associated with the development of both prediabetes and T2D.
Nuts, seeds, avocados, and plant-based oils are rich in MUFAs, such as oleic acid and palmitoleic acid (69). These foods are commonly consumed at high levels in the Mediterranean diet, and improve metabolic function in patients with T2D and prediabetes (108, 139). Omega-3 FAs are commonly found in foods such as fish, flaxseed, and plant-based oils. In T2D and prediabetes, the western diet plays a major role in the development of dyslipidemia, and a high-fat diet (HFD) rich in SFAs underlies the onset and progression of PN in preclinical models of prediabetes (57, 94).
Aberrant dietary FA intake promotes dyslipidemia in patients with T2D and prediabetes. In healthy individuals, insulin release after a meal suppresses FA release from adipose depots in the body, and FA metabolism is under tight regulation. FAs are stored as energy in triglycerides in adipose tissue, and serve as fuel for the heart and muscles. In dyslipidemia, however, high plasma FA levels result from both the diet and FA mobilization from adipose tissue (32). This elevation in plasma FAs contributes to insulin resistance and also increases FA levels, promoting metabolic dysfunction in peripheral tissues, such as the PNS.
The PNS
The PNS consists of the somatic nervous system and the autonomic nervous system. Although both the somatic nervous system and autonomic nervous system are impacted by T2D and prediabetes, this review will focus on the somatic nervous system. The somatic nervous system, which consists of both motor and sensory nerves, is responsible for transmitting sensory information from the periphery to the central nervous system (CNS) (Fig. 2). Sensory neurons are afferent neurons that transmit sensory information from receptors in the sensory nerve terminals to the CNS, whereas motor neurons are efferent neurons that carry information from the CNS to muscles and organs within the body.

Axons from peripheral neurons are myelinated or unmyelinated. Myelination in the PNS is carried out by resident Schwann cells (SCs), which produce lipids and proteins to insulate the axons and facilitate efficient nerve conduction (109) (Fig. 2). SCs myelinate a single segment of an axon, called the internode, which is separated from other internodes by unmyelinated regions called Nodes of Ranvier. The myelin sheath is a lipid-rich multilamellar structure composed of closely packed lipid bilayers (104, 121). This lipid-rich myelin sheath is composed of 70%–85% lipids, and is attuned to changes in FAs and metabolic flux (40).
Myelinated and unmyelinated axons are bundled into nerve fibers surrounded by a layer of connective tissue called the endoneurium. Sensory nerve fibers, including Aα, Aβ, Aδ, and C fiber types, are each responsible for relaying specific types of sensory signals. The large, myelinated Aα fibers receive signals from the muscle spindle and Golgi tendon, and are responsible for proprioception required for spatial position and movement.
The Aβ fibers include large and mid-diameter myelinated fibers, which carry touch and pressure sensations as well as mechanoreceptor-mediated signals. Small afferent Aδ fibers are thinly myelinated fibers, and transmit mechanoreceptor-triggered cold, touch, and pressure sensations. Finally, thin unmyelinated C fibers are <1 μm in length and are surrounded by nonmyelinating SCs that occur in groups, known as Remak bundles. These fibers serve as PNS nociceptors, and transmit heat, pain, and nociception sensory signals (37).
PN associated with diabetes and prediabetes tends to present as a loss of sensory function with only a small effect on motor function (38). The different anatomical locations of motor and sensory neuron cell bodies may contribute to the susceptibility of sensory neurons to diabetic metabolic flux (38). Motor neurons localized in the ventral horn inside the spinal cord are protected by the blood–brain barrier, whereas dorsal root ganglion (DRG) sensory neurons are located outside of the spinal cord, and lack either a blood–brain or blood–nerve barrier.
Consequently, unlike motor neurons, DRG sensory neurons are exposed to circulating metabolites, including FAs, glucose, and other metabolites, making them vulnerable to metabolic stressors and axonal injury in diabetes and prediabetes (38). Unmyelinated C nerve fibers are also particularly sensitive to injurious metabolites, such as FAs, likely secondary to the absence of a myelin sheath (Fig. 2) (38). Thus, the lack of protective barriers makes both DRG sensory neurons and C fibers more susceptible to metabolic injury.
The morphology of DRG sensory neurons also makes them more susceptible to injurious circulating metabolites. DRG neurons extend axons, which are among the longest axons in the body, extending up to 1 m in length from the cell body to the axon terminal. DRG neurons have a pseudounipolar morphology where the stem axon bifurcates into a peripheral axon branch and a central axon branch (91).
This unique morphology is key for transmitting sensory information from the peripheral axon branch to the bifurcation, and then to the CNS via the central axon branch (91). After a stimulus, action potentials are generated in the peripheral nerve branch and conducted toward the CNS. However, the extremely large axon to cell body ratio presents unique bioenergetics challenges for DRG neurons and axon health and function. To maintain energy homeostasis distally in the axon, DRG neurons employ mitochondrial axonal transport mechanisms to provide mitochondria throughout the entire length of the axon.
Clinical Data on the Impact of FAs on Neuropathy
Nerve injury from hyperglycemia was previously considered the principal molecular driver of PN, although the pathophysiology of type 1 diabetes (T1D) and T2D is distinct. Over the last decade, however, the multifactorial nature of T2D prompted clinical studies to investigate unique metabolic processes associated with PN in T1D and T2D. The concept that hyperglycemia is not the sole metabolic factor underlying PN in T2D was addressed in a Cochrane review evaluating the efficacy of glucose control for regulating PN in T1D compared with T2D subjects separately (13).
This systematic review compared 17 randomized, controlled studies consisting of subjects with T1D, T2D, or both. It found that glucose control significantly reduces PN development in T1D subjects, as identified by a decrease in abnormal nerve conduction studies and vibration thresholds. However, in subjects with T2D, glucose control had no significant impact on nerve conduction and vibration thresholds. These clinical studies suggest that the mechanisms underlying PN differ between T1D and T2D (12, 13).
In an effort to distinguish T2D risk factors that correlate with PN, the impact of the MetS on PN was evaluated (8). The MetS is a group of metabolic disorders that is highly prevalent in T2D subjects, ∼73%–83% prevalence, and is a major risk factor for T2D (119, 142). To assess the effect of MetS components on PN, a cohort of 2382 subjects with PN assessments from the Health, Aging, and Body Composition (Health ABC) study were evaluated for prevalence of PN, after stratification for glycemic status and MetS components. Interestingly, although PN was more common in subjects with diabetes, MetS components, including prediabetes and obesity, were linked to secondary measures of PN (16).
In addition, PN was more prevalent in subjects with a higher number of MetS components independent of glycemic status (16). Another cross-sectional, population-based study consisting of 4002 subjects from China sought to identify individual MetS components associated with PN. In agreement with previous studies, diabetes and obesity were the major metabolic drivers of PN (11). The three components of MetS that correlated with PN in T2D were diabetes, prediabetes, and obesity, which all intersect with components of dyslipidemia (16, 17, 79).
The association of dyslipidemia with PN was evaluated in a longitudinal study, the Danish arm of Anglo-Danish-Dutch study of Intensive Treatment of Diabetes in Primary Care (ADDITION-Denmark). This study examined incident PN in T2D subjects at a 13-year follow-up appointment after enrollment in the ADDITION-Denmark study (6). The cohort of 1,445 subjects showed that obesity along with reduced levels of high-density lipoprotein cholesterol, a MetS component, is a substantial risk factor for PN in T2D. In parallel, a separate study on 5249 T2D subjects from the Danish Centre for Strategic Research in Type 2 Diabetes (DD2) Cohort found that PN is associated with MetS components, including elevated triglyceride levels, hypertension, and obesity (23).
To determine whether obesity is an independent risk factor for PN in nondiabetic individuals, another study evaluated the 138 obese subjects and 46 lean controls and the prevalence of PN (15). This study discovered that obesity in normoglycemic individuals associates with PN. Altogether, these studies indicate that MetS is a major risk factor for PN. Since dyslipidemia is commonly associated with MetS in T2D and prediabetes, we contend that dietary FA intake may contribute to PN in T2D.
Based on the emerging role of dyslipidemia in PN pathogenesis, another study determined whether lipid lowering statins would reduce the prevalence of PN in T2D subjects (124). Despite numerous studies showing an association of PN with dyslipidemia, the incidence of PN was similar in both T2D subjects with and without statin therapy (72). Therefore, the reduction of triglyceride and cholesterol levels by statins had little effect on PN. These results suggest that overall dyslipidemia is not responsible for PN in T2D subjects, but rather that specific lipid species underlie neuropathy progression.
To identify specific lipid metabolites that associate with PN in T2D, we evaluated global plasma metabolomics in a cohort of T2D subjects with and without PN compared with lean control subjects from ADDITION-Denmark (111). We found that individuals with T2D and PN had changes in plasma metabolites related to lipid and energy metabolism compared with T2D subjects without PN.
This prompted us to evaluate alterations in plasma lipid metabolites identified by the global metabolomics analysis. The abundance, chain length, and saturation of both plasma FAs and complex lipids were appreciably altered in T2D subjects compared with lean controls (111). T2D subjects shifted from beneficial VLC unsaturated FAs in the plasma to toxic long-chain saturated FAs. In addition, plasma complex lipids showed an increase in diacylglycerol and phosphatidylethanolamine species, and a decrease in phosphatidylcholine, sphingomyelin, ceramide, and acylcarnitine species (111).
A separate study comparing obese T2D subjects with and without PN showed that plasma serine and 1-deoxyhydroceramide levels are inversely correlated with quantitative C fiber assessment as a measure of PN, suggesting that 1-deoxyhydroceramides are potential mediators of PN in T2D and obesity (44). Collectively, these studies suggest that PN is associated with changes in plasma lipid species and metabolites of lipid metabolism in T2D and prediabetes.
Dietary supplementation studies with MUFAs or PUFAs in T2D and prediabetic subjects with PN show significant beneficial effects on metabolic parameters and nerve function. A European Prospective Investigation into Cancer and Nutrition (EPIC)-InterAct study showed that plant-based omega-3 and omega-6 PUFAs inversely correlate with T2D, suggesting that PUFAs improve metabolic function in T2D subjects (43). In this study, T2D was inversely correlated with plant-based omega-3 α-linoleic acid and omega-6 PUFA linoleic acid, while EPA and DHA had no association. These results highlight the need to evaluate the effect of individual FAs, not overall FA levels.
To evaluate the connection between dietary PUFA consumption and PN, a National Health and Nutrition Examination Survey study found that dietary PUFA intake is associated with lower incident neuropathy in diabetic subjects (129). In addition, individual FAs γ-linolenic acid (67, 70) and EPA (98) confer significant improvements in PN associated with T2D. A similar dietary supplementation paradigm in subjects with T1D, however, had no effect on nerve conduction or sensory nerve function, further supporting the idea that metabolic factors contributing to PN in T1D and T2D are unique (76).
In addition, a recent cross-sectional study on 147 T2D subjects showed that high dietary iron intake and a high iron:PUFA ratio correlate with PN, suggesting that the iron level determines the level of benefit conferred by dietary PUFAs (71). The effect of different types of FAs on nerve function is likely the result of altered lipid metabolism within the nerve. However, the molecular mechanisms that are differentially regulated by saturated and unsaturated FAs in the nerve are still an active area of research.
Preclinical Data on the Impact of FAs on Neuropathy
Similar to humans, obese and prediabetic mice develop PN associated with dyslipidemia and metabolic dysfunction (57, 94, 136, 137). Preclinical animal models are a valuable tool for identifying molecular mechanisms and lipid changes that associate with PN in prediabetes and T2D. Mice fed a HFD have impaired glucose tolerance, increased body weight, and a higher percentage body fat compared with their standard diet-fed counterparts (11, 57, 94).
The HFD mice also develop PN characterized by a reduction in sural and sciatic nerve conduction velocities, an increase in hind paw latency, and a decrease in C fibers, as measured by the intraepidermal nerve fiber density (IENFDs) in the hind paw (57, 94). Genetic models of T2D also consistently develop the same PN phenotypes, along with overt T2D characterized by hyperglycemia, dyslipidemia, and PN (96). Three common murine models of T2D are (i) ob/ob mice with leptin deficiency, (ii) db/db mice that lack the leptin receptor, and (iii) mice fed a HFD and treated with one low dose of streptozotocin (STZ).
Both prediabetic and T2D animal models are dyslipidemic, and align with the metabolic phenotypes in prediabetic or T2D humans. Using drugs to reduce free FAs in rodents improves nerve function. For example, pioglitazone treatment in db/db mice improved small fiber function as measured by sural nerve conduction velocity and IENFD (58, 62) and acipimox treatment of male Zucker fa/fa rats, a robust model of T2D when fed a HFD, restored nerve conduction velocities (81). Although these studies indicate that dyslipidemia and FAs contribute to PN development and progression in murine models, the molecular changes that underlie these beneficial effects on PN are not completely understood.
As a first step to identify dysregulated pathways that underlie PN in prediabetes and T2D, we performed lipidomics (94, 122) and transcriptomics (94) studies on peripheral nerves from HFD murine models of prediabetes and T2D. We integrated lipidomic profiles from one sciatic nerve and transcriptomics from the other sciatic nerve for each mouse. This analysis showed dysregulation of lipid pathways in both the HFD-fed prediabetic mice and the HFD-STZ T2D mice (94).
Specifically, we found increases in triglycerides containing SFAs as well as increases in diacylglycerol acyltransferase 2 (DGAT2), the enzyme that carries out the committed step in triglyceride synthesis. Lipid metabolism was also altered in the sciatic nerve from db/db mice (122). These studies indicate that high levels of SFAs in the HFD are incorporated into the nerve lipidome altering the sciatic nerve lipid profile, which may contribute to PN in prediabetes and T2D.
To determine whether the FA composition of the diet contributes to PN in the HFD murine models, we and others compared the effects of dietary SFAs and unsaturated FAs on metabolic and PN phenotypes in prediabetic and T2D mice (114). Mice fed a SFA-rich HFD from 6 to 16 weeks of age developed PN and metabolic dysfunction. However, mice switched from the SFA-rich HFD to a MUFA-rich HFD exhibited a significant improvement in nerve function, including increased sensory and motor nerve conduction velocities and restoration of C fibers as measured by increased IENFDs.
Similarly, the Yorek laboratory discovered the beneficial effects of Menhaden oil supplementation for improving nerve function in murine models of PN. Menhaden oil is composed of elevated levels of omega-3 PUFAs EPA and DHA. Supplementation of the HFD with Menhaden oil led to significant improvements in sensory and motor nerve conduction velocities, thermal nociception, and IENFDs in HFD- and low-dose STZ-treated Sprague Dawley rats and C57BL6/J mice (24, 29, 127).
Altogether, these results show that dietary FAs modulate nerve function in rodent models of PN associated with prediabetes and T2D. Dietary SFAs underlie PN in rodent models of prediabetes and T2D, while dietary MUFAs and PUFAs improve nerve function. The molecular mechanisms that underlie the differential regulation of nerve function by SFAs and MUFAs are not completely understood. One possibility is that SFAs impair lipid metabolism within the nerve resulting in an altered nerve lipid profile that drives PN development and progression. We will next detail the mechanisms by which SFAs and MUFAs/PUFAs might differentially regulate lipid metabolic pathways in the nerve.
FAs and Nerve Lipid Metabolism
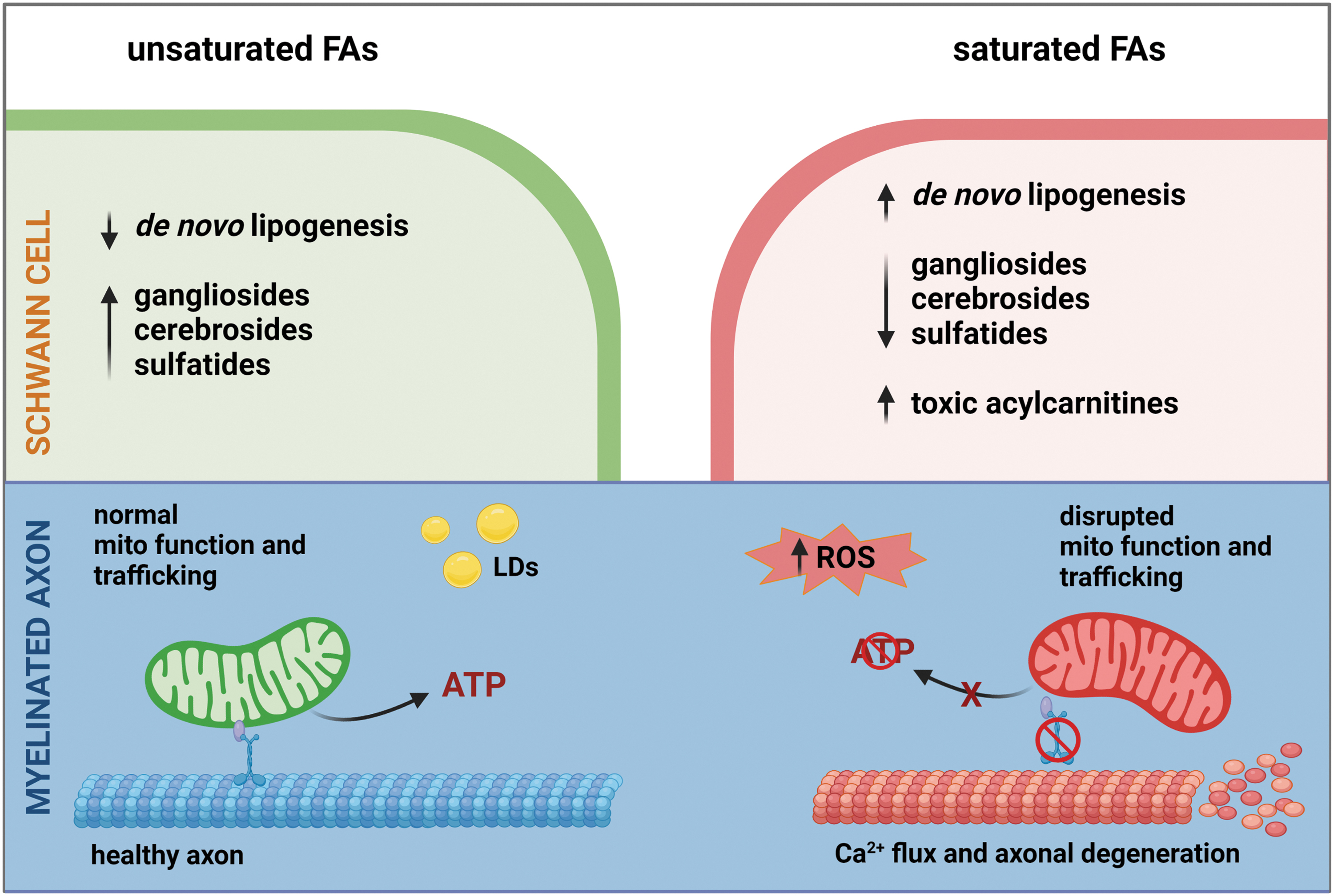
Under homeostatic conditions, neuronal lipid metabolism is centered on a balance of lipid uptake, de novo lipogenesis, and FA β-oxidation to maintain neuronal health and function (104, 130). However, dyslipidemia leads to an accumulation of neurotoxic lipids, aberrant lipid lipogenesis, and impaired mitochondrial energy production. Dietary FAs are fundamental players in the development of dyslipidemia and drastically impact these lipid metabolism processes.
Adipose and liver regulate circulating FA levels through uptake, storage, and mobilization of triglycerides and FAs (Fig. 3). After meal consumption, dietary FAs are esterified into triglycerides and incorporated into chylomicrons in the intestine (53). The majority of these chylomicrons are taken up by tissues with high lipoprotein lipase activity, such as adipose tissue and muscle, whereas chylomicron fragments are taken up by the liver. These FAs are converted into triglycerides in very-low-density lipoproteins (VLDLs) or remain nonesterified FAs, which are then packaged into lipoprotein particles and transported through the bloodstream to peripheral tissues, such as the peripheral nerves (3).

In obesity, excessive intake of the foods rich in SFAs is associated with weight gain, impaired glucose tolerance, and increased body fat mass. This elevation in adiposity is often associated with adipose tissue dysfunction characterized by inflammation, adipocyte expansion, impaired insulin signaling, dysfunctional triglyceride storage, and increased FA mobilization (18). The liver also becomes dysfunctional in obesity with an overload of intrahepatic lipid and decreased production of VLDL triglycerides (82). Therefore, adipose tissue and liver dysfunction dictate changes in circulating FAs, and are closely associated with the development of dyslipidemia and obesity (95).
FA Uptake
Circulating FAs taken up by neurons and SCs are oxidized to generate cellular energy or converted into complex lipids to support the nerve. Circulating lipids, such as triglycerides, in lipoprotein particles are hydrolyzed into free nonesterified FAs by lipoprotein lipase localized in the peripheral nerves on the surface of cells within the endoneurium, the connective tissue that surrounds a myelinated nerve (33, 61). The free FAs are then transported into neurons and SCs, and the mode of transport is determined by the FA chain length (51). Short- or medium-chain FAs are passively diffused into cells by a membrane flip-flop mechanism. Conversely, long-chain FAs and VLC FAs must be transported into the neurons or SCs by FA transporters.
Two major FA transporters in peripheral nerves are FA translocase (CD36) and FA transport protein (FATP) (Fig. 4). Both CD36 and FATP have a high affinity for long-chain saturated and unsaturated FAs, and contribute to obesity and metabolic dysfunction (102). CD36 is localized on the cell membrane of neurons and SCs, and facilitates FA transport through a central FA transport tunnel (102). FATPs consist of a single transmembrane domain, an extracellular FA binding site that allows for FA transport, and an intracellular ATP binding site with acyl-CoA synthetase activity that converts incoming FAs into acyl-CoAs (85).
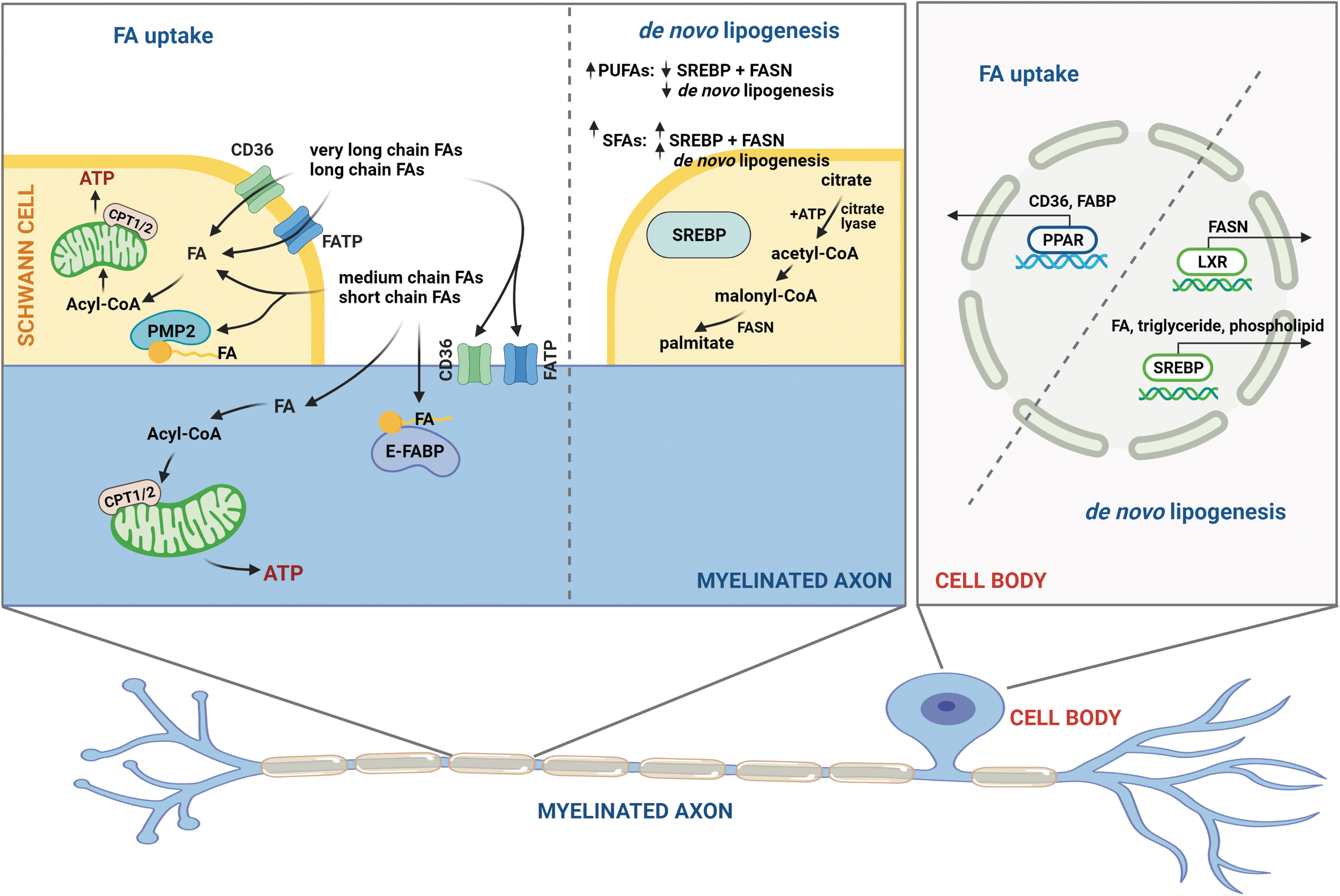
Once inside the cell, FA binding proteins (FABPs) facilitate intracellular FA distribution by acting as lipid chaperones to transport FAs to organelles (89, 90, 107). Only four of the six FABP isoforms are expressed in the nervous system, of which the peripheral myelin protein 2 (PMP2/M-FABP) isoform is found only in myelin in the peripheral nerves and the E-FABP isoform is mainly localized to neurons (133). The affinity of FABPs for FAs is proportional to FA hydrocarbon chain length and degree of saturation with the highest affinity for VLC FAs and fewer double bonds.
FA transporters in the dyslipidemic sciatic nerve are dramatically affected by circulating dietary FAs. Using a transcriptomics and lipidomics analysis, we recently found that the expression level of both lipoprotein lipase, Lpl, and CD36 in the sciatic nerve is modulated by a HFD, indicating that the higher level of FAs in both prediabetes (94) and T2D (63, 101) alters FA uptake in PN. Although both neurons and SCs express CD36 and FATP, the known role of CD36 and FATP in dyslipidemic sensory neurons is limited. In SCs, CD36 and FATP mediate uptake of circulating FAs, which profoundly affects the myelin lipid composition by incorporating FAs into the myelin sheath (104, 143). In fact, ablation of CD36 perturbs nerve remyelination pathways after injury, suggesting that CD36 is essential for the proper maintenance of the myelin sheath (36, 104).
FABPs play an important role in PN associated with Charcot Marie Tooth disease (CMT) (59), but recent evidence indicates that FABPs also contribute to PN in T2D and prediabetes (56). The FABP PMP2 is expressed in myelinating SCs and facilitates structural stability in myelinated large axons by anchoring myelin lipid bilayers (116, 126, 131, 145). Mutant PMP2 causes severe demyelinating PN characterized by decreased nerve conduction velocities in CMT type 1A, emphasizing the importance of this FABP for nerve function (59). In addition to providing a scaffold for myelin lipid bilayers, PMP2 has a strong affinity for both cholesterol and FAs, and is likely to play a major role in FA cellular transport and metabolism in SCs (104, 110, 145).
Indeed, we identified a downregulation of PMP2 gene expression in the sciatic nerve of db/db mice, indicating that elevated levels of circulating lipids in diabetic murine models impair nerve lipid homeostasis and potentially myelin structure (56). FABP5 is the major isoform expressed in DRG neurons and is also an important regulator of nerve function. Small nucleotide polymorphisms in FABP5 are associated with T2D, indicating that a loss of FABP function contributes to metabolic dysfunction (9). Indeed, omega-3 PUFAs increase FABP5 expression that provides neuroprotection after spinal cord injury. Increasing FABP5 levels in neurons also prevents palmitate-induced lipotoxicity (78).
FA transporters and FABPs are critical regulators of energy homeostasis in neurons, and contribute to the pathogenesis of diabetes, obesity, and their complications (74, 83). These studies suggest that dysregulation of FA transport by CD36, FATP, and FABPs may be a critical molecular target in PN associated with prediabetes and T2D.
FAs and Mitochondrial Function, Trafficking, and Dynamics in the PNS
After transport into the cell, FAs may be oxidized by mitochondrial β-oxidation to generate cellular energy in the form of ATP under homeostatic conditions. Synthesis of cellular ATP from FAs occurs in the mitochondria by sequential reactions including FA β-oxidation, followed by the TCA, and finally oxidative phosphorylation. A detailed description of mitochondrial bioenergetics and dynamics in PN has been previously published (115). DRG neurons and SCs rely on mitochondrial ATP production to maintain nerve health and function (130). In addition to ATP production, DRG neurons require mitochondrial trafficking mechanisms to transport mitochondria throughout the axon and provide energy at distal regions of the axon. Mitochondrial fusion/fission events are also required to maintain energy balance (115).
We recently discovered that the effect of FAs on DRG neuron mitochondrial function is dependent on FA hydrocarbon chain length and degree of saturation (112 –114). We observed a significant decrease in mitochondrial membrane potential in DRG neurons treated with diabetic concentrations of long-chain SFAs palmitate (C16:0) and stearate (C18:0), whereas shorter chain SFAs laurate (C12:0) and myristate (C14:0) did not affect mitochondrial depolarization (112, 113). The loss of mitochondrial membrane potential in long-chain SFA-treated DRG neurons was associated with loss of ATP in immortalized DRG neurons (113, 114).
In addition, long-chain SFA palmitate treatments profoundly affected DRG neuron mitochondrial bioenergetics marked by a significant and dose-dependent decrease in spare respiratory capacity, as well as an increase in mitochondrial uncoupling and proton leak (112). Interestingly, mitochondrial depolarization and ATP loss due to long-chain SFAs were completely abrogated with the addition of exogenous long-chain MUFA oleate (C18:1), potentially due to the formation of axonal lipid droplets (114).
These alterations in mitochondrial function were accompanied by impairments in axonal mitochondrial trafficking. Cell culture models of dyslipidemia show significant impairment in axonal mitochondrial trafficking in DRG axons (Fig. 5). DRG axons treated with long-chain SFAs palmitate (C16:0) and stearate (C18:0) had significant and dose-dependent decreases in the percentage of motile mitochondria (Fig. 5A, D, E) (113). Shorter chain SFAs laurate (C12:0) and myristate (C14:0) had no effect on the level of mitochondrial trafficking in DRG axons (Fig. 5A–C) (113).
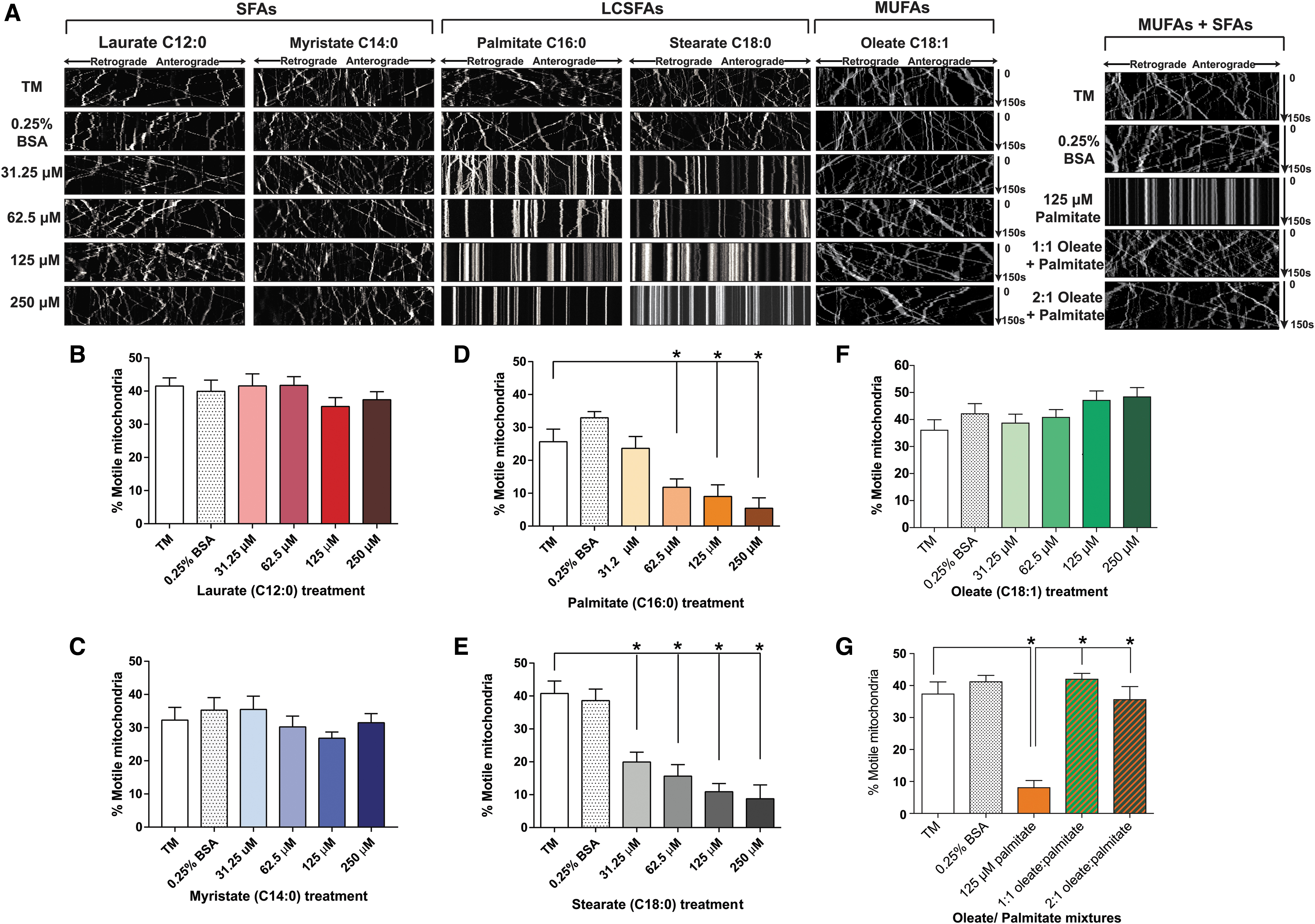
Long-chain MUFA oleate had no effect on mitochondrial trafficking, and supplementation of oleate into the palmitate treatments completely prevented the inhibition of mitochondrial trafficking caused by palmitate alone (Fig. 5A, F, G) (114). This suggests that long-chain SFAs with hydrocarbon chains >16 carbons in length have a detrimental effect on axonal mitochondrial trafficking and function, whereas shorter SFAs and MUFAs have no effect. Furthermore, the impairment in mitochondrial trafficking and function can be prevented by MUFA supplementation.
Mitochondrial fusion and fission dynamics are also profoundly impacted by complex lipids derived from FAs. Mitochondrial fission and fusion dynamics play an important role in regulating energy balance (115). Lipids including cardiolipin, phosphatidylethanolamine, phosphatidic acid, and diacylglycerols play a major role in mitochondrial fission/fusion, and the level of these lipids is modulated by FAs.
Cardiolipin is a mitochondria-specific phospholipid enriched with the essential FA linoleic acid that regulates mitochondrial fusion, fission, and function. The availability of linoleic acid modulates the level of cardiolipin in the mitochondria, which directly impacts mitochondrial fusion and fission dynamics and mitochondrial function (21, 45). Interestingly, despite elevated levels of linoleic acid in the HFD, there is a steep decrease in cardiolipin in the sciatic nerve of HFD-fed murine models of prediabetes (94), suggesting that increased linoleic acid levels in the HFD are not sufficient to maintain mitochondrial cardiolipin levels and prevent mitochondrial dysfunction caused by long-chain SFAs in the HFD.
Phosphatidylethanolamine, a phospholipid that is rich in the outer mitochondrial membrane and inner mitochondrial membrane, plays an important role in mitochondrial fusion. Phosphatidic acid stimulates mitochondrial fusion, whereas diacylglycerols promote fission. The interplay between these two lipid species tightly regulates fusion and fission dynamics (45). Although the effect of dietary FAs on mitochondrial fusion and fission remains incompletely characterized, mice fed a 60% HFD rich in SFAs exhibited significant alterations in the level of these complex lipids, indicating that changes in dietary FA composition may alter mitochondrial fusion and fission (94).
Much like DRG neurons, SCs can use mitochondrial β-oxidation of FAs to produce energy, but elevated levels of long-chain FAs result in SC mitochondrial dysfunction in PN associated with T2D and prediabetes. Elevated levels of long-chain FAs cause a loss of SC mitochondrial coupling efficiency and mitochondrial dysfunction (55). However, overexpression of long-chain acyl-CoA synthase 1 (Acsl1), which metabolically activates long-chain FAs for mitochondrial oxidation, is sufficient to rescue mitochondrial function. In the presence of elevated long-chain FAs, overexpression of Acsl1 in SCs alleviates oxidative stress, mitochondrial dysfunction, incomplete β-oxidation, and SC injury by improving metabolic activation of long-chain FAs (55).
Similarly, a murine model with SC-specific KO of the mitochondrial transcription factor A (TFAM) gene (Tfam), a transcription factor that is critical for mitochondrial biogenesis and function (73), developed severe PN (134). These TFAM SC knockout mice displayed mitochondrial dysfunction marked by a shift from lipid synthesis to FA β-oxidation (135). The loss of FA synthesis pathways significantly reduced crucial lipids in the myelin and instead triggered an elevation in acylcarnitine levels. These acylcarnitines were then excreted from the SCs triggering DRG neuron intracellular calcium flux and axonal degeneration. Therefore, functional SC mitochondria are critical for supporting peripheral neurons (134).
In conjunction with TFAM, two transcription factors, peroxisome proliferator-activated receptor coactivator 1 alpha (PGC-1α) and sirtuin 1 (SIRT1), play an important role in mitochondrial biogenesis. TFAM, PGC-1α, and SIRT1 respond to metabolic cues to preserve mitochondrial copy number and maintain efficient mitochondrial β-oxidation for ATP production. However, this pathway is compromised in PN associated with T2D (19, 34) and prediabetes (120). Conversely, caloric restriction enhances the mitochondrial biogenesis and mitochondrial oxidative capacity through the TFAM, PGC-1α, and SIRT1 pathway (34). These studies suggest that dysregulation of lipid uptake, β-oxidation, and mitochondrial biogenesis occurs in PN through the TFAM, PGC-1α, and SIRT1 pathway (19).
Oxidative Stress
Both hyperglycemia and dyslipidemia contribute to oxidative stress in diabetic PN (137, 138). Refer to Dr. Paul Fernyhough's Forum article for a detailed review about oxidative stress in T1D. Also, refer to Dr. Stephanie Eid's Forum article for an extensive review on oxidative stress in the PNS.
During β-oxidation, long-chain nonesterified FAs can also generate reactive oxygen species (ROS) in PN (125, 137). Basal levels of nonesterified FAs in both neurons and SCs are utilized for ATP generation, but excessive levels of FAs trigger the production of superoxide and other ROS species due to leaky electrons from the electron transport chain (130). The generation of ROS within the nerve causes nerve injury by triggering apoptosis in DRG neurons (117, 118, 138). Dyslipidemic Zucker fatty (fa/fa) rats with increased circulating nonesterified FAs had lower sensory nerve conduction velocities, tactile allodynia, and thermal and mechanical hypoalgesia associated with oxidative–nitrosative stress within the nerve (81).
Reducing oxidative–nitrosative stress within the peripheral nerves significantly improved nerve function (81). Fortunately, unsaturated FAs may provide therapeutic benefits by reducing oxidative stress in murine models of diabetic PN (144). Omega-3 PUFAs in Menhaden oil significantly lower neuronal oxidative stress by scavenging free radicals (48), which stabilizes mitochondrial function (7). This also prevents the generation of proinflammatory modulators (48). These results suggest that modulating the redox state may be a feasible approach for future PN therapeutics.
De Novo Lipogenesis
Although cells and tissues mainly obtain FAs from dietary sources (26, 87), nonessential FAs can be synthesized intracellularly through de novo lipogenesis. The canonical purpose of de novo lipogenesis is to generate FAs for triglyceride synthesis and energy storage (4).
In the first step of lipogenesis, ATP-citrate lyase converts citrate to acetyl-CoA (Fig. 4). Acetyl-CoA is then carboxylated to malonyl-CoA by the rate-limiting enzyme acetyl-CoA carboxylase, which is activated by insulin during the fed state and impaired by glucagon during the fasting state (128, 140). Finally, FA synthase (FASN) converts malonyl-CoA to palmitate, which can be incorporated into triglycerides. These de novo lipogenesis enzymes are under the tight regulation of two transcription factors: sterol regulatory element-binding protein (SREBP1) and peroxisome proliferator-activated receptor gamma (PPARγ), both of which are highly regulated by FAs (Fig. 4).
Regulation of FASN expression by SREBP1 and PPARγ, and therefore de novo lipogenesis, is sensitive to changes in FA levels (Fig. 4). Unsaturated, but not SFAs decrease the nuclear level of SREBP1 in a length-dependent and saturation-dependent manner (52). PUFAs decrease the expression of SREBP1 and FASN, which results in a decrease in lipogenesis (68) (Fig. 6). PPARγ activity is also regulated by numerous FAs that activate PPARγ to stimulate lipid uptake and maintain lipid homeostasis (42, 106). In addition, PPARγ cooperates with another nuclear receptor, liver X receptor (LXR), to regulate FA metabolism (64), a cross-talk that is promoted by unsaturated FAs (141) (Fig. 4). In murine models of prediabetes, LXR agonists protect DRG neurons from the SFA-induced endoplasmic reticulum stress response and reduced allodynia (46).
De novo lipogenesis is an essential process for myelinating SCs (87). SCs surround large-caliber axons, and produce myelin to insulate the axons and sustain saltatory conduction. Since myelination is an energetically expensive metabolic process, it requires careful coordination of RNA, protein, and lipid synthesis for membrane production (54, 86, 92, 103). FASN is critical for maintaining the lipid composition and myelination of the peripheral nerves (87).
In fact, SC-specific FASN depletion impairs PPARγ transcriptional regulation. Interestingly, increased FA uptake via rosiglitazone treatment compensated for insufficient FA synthesis in FASN-deficient mice. In line with these results, we have identified changes in PPARγ in prediabetic mice as well as an activation of PPARγ in the nerves of db/db mice receiving a rosiglitazone isomer, pioglitazone (58, 94). Therefore, altered de novo lipogenesis by insulin resistance and altered FA levels may contribute to PN in T2D and prediabetes.
Lipid Composition of DRG Neurons and Myelin
Careful coordination of FA uptake, β-oxidation, complex lipid synthesis, and de novo lipogenesis sustains the health and function of neurons and SCs within the peripheral nerves. Under homeostatic conditions, these lipid metabolic processes maintain the optimal proportion of lipids in the myelin and peripheral nerves to facilitate nerve conduction. Nerve tissue in the PNS contains free FAs, glycerolipids, sphingolipids, glycerophospholipids, cholesterol, and sterol lipids, which are uniquely distributed across each cell type (SC vs. neuron) and morphological localization (cell body vs. axon) (60, 93, 104).
These lipids are incorporated into membranes, lipid rafts, organelles, cytosolic lipid species, and myelin, and are critical for nerve function. To understand the pathogenesis of PN associated with T2D and prediabetes, it is critical to determine the effect of dietary FAs on peripheral nerve lipid composition. The FA properties of myelin can alter the speed and strength of sciatic motor nerve conduction (31), indicating that FAs associated with dyslipidemia may alter the nerve lipid composition and cause nerve injury. We contend that dyslipidemia alters the nerve lipidome, triggering injurious changes that affect myelination, sensory neuron function, and in turn, contribute to PN in T2D and prediabetes (94).
The myelin sheath is composed of 70%–85% lipids (104), and the major lipid classes in the myelin are 40% cholesterol, 40% phospholipids, and 20% glycolipids. The cholesterol and glycolipid levels are significantly higher in the myelin than in other tissues. Myelin phospholipid species include plasmalogen, phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, and sphingomyelin, while the glycolipids are mainly galactosylceramides.
Ethanolamine plasmalogen is the most abundant myelin phospholipid, but its role in the peripheral nerves is not fully understood. Several studies suggest that ethanolamine plasmalogen stabilizes the compact lipid structure in myelin membranes (80), prevents peroxidation of unsaturated myelin lipids (80), and mediates proper myelination within the peripheral nerves (27). This unique lipid profile is essential for maintaining myelin structure. Interestingly, many of these myelin lipids are altered in studies on the lipidomic alterations in whole nerves from HFD-fed mice (94).
Complex lipids within the myelin also contain a high level of VLC FAs compared with other tissues, which are important for maintaining the myelin integrity (22, 104, 123). Murine models deficient in ceramide synthase 2 (CerS2), the enzyme responsible for VLC sphingolipids synthesis, show that VLC sphingolipid deficiency is associated with severe multifocal detachment of myelin from the axon (65). Therefore, myelin integrity requires SC-mediated maintenance of VLC lipid levels within the myelin sheath. This study suggests that elevated levels of SFAs in dyslipidemia may impair CerS2 activity, thereby altering the level of VLC sphingolipids. Indeed, VLC glycosphingolipids including gangliosides (84), sulfatides (99), and galactosylceramides (99) were significantly depleted in murine models of T2D and prediabetes (Fig. 6).
Compared with myelin, the lipid content of DRG neurons is lower but plays an important role in sensory neuron function (10). The lipid composition of DRG neuron cell bodies and axons is distinct. Approximately 37% of the dry weight of the DRG neuron soma is lipid, consisting of 15.4% cholesterol, 4.8% galactolipid, and 57.1% phospholipids (10, 60). Fifteen percent of the dry weight of DRG axons is composed of lipids, which consists of 22.1% cholesterol, 7.7% galactolipid, and 56.4% phospholipid.
In both the DRG cell body and axons, the molar ratio of galactolipids was 2:1 with cerebrosides two times higher than that of sulfatides. Disruption of the DRG neuron lipidome in ApoE knockout mice is associated with severe peripheral sensory nerve defects and impaired electrophysiology (20), emphasizing the importance of the lipid composition for DRG neuron function.
Although the effect of dyslipidemia on complex lipid levels in DRG neurons is not completely understood, it is likely that dietary FAs also alter the complex lipid profile of DRG neurons in dyslipidemia. This contention is supported by evidence that SFAs and MUFAs profoundly and differentially effect sensory neurons both in vivo (120) and in vitro (112 –114), and that IENFD is also modulated by the degree of FA saturation in HFD-fed mice (94). Specific sulfatide, cerebroside, and phospholipid species were also significantly reduced in DRG from diabetic mice. These studies suggest that elevated dietary FAs associated with dyslipidemia contribute to changes in complex lipid species within the nerve, and may contribute to PN in T2D and prediabetes.
Conclusions
Clinical studies have identified an association between PN and dyslipidemia in T2D and prediabetes, suggesting that FAs contribute to the development and progression of PN. Indeed, preclinical studies show that diets rich in injurious SFAs cause PN, while beneficial unsaturated FAs significantly improve nerve function in murine models of prediabetes and T2D. Peripheral nerve function is dependent on maintaining a specific lipid profile, which is regulated by dietary FA uptake, mitochondrial FA β-oxidation, oxidative stress, and de novo lipogenesis in DRG neurons and SCs (130). Molecular studies that determine the effect of SFAs and unsaturated FAs on these metabolic pathways and alterations in both neurons and SCs will greatly improve our understanding of the pathophysiological changes that underlie T2D and prediabetic PN.
Determining how these pathways alter the nerve lipidome in relation to PN in preclinical rodent models will provide insight into the lipid changes that underlie PN. In addition, axoglial interactions between DRG axons and SCs may regulate nerve function through the transfer of lipids or metabolites between the two cell types. Therefore, the biology of FAs is likely to play an important role in the molecular mechanisms that underlie PN in T2D and prediabetes.
Authors' Contribution
A.E.R. and E.L.F. planned, conceptualized, and wrote the article. A.E.R., B.K., and E.L.F. reviewed and edited the article. B.K. created the figures and contributed to writing the article. Each author reviewed, revised, and approved the submitted article.
Footnotes
Author Disclosure Statement
All authors declare no competing interests.
Funding Information
This work was supported by the National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) grant numbers 1R24DK082841 and 1R21NS102924 (to E.L.F.) and 1F32DK112642 and K99/R00 DK119366 (to A.E.R.); the American Diabetes Association [grant number 7-12-BS-045] (to E.L.F.); the NeuroNetwork for Emerging Therapies; and the A. Alfred Taubman Medical Research Institute.