
Abstract
Significance:
It is estimated that close to 50 million cases of sepsis result in over 11 million annual fatalities worldwide. The pathognomonic feature of sepsis is a dysregulated inflammatory response arising from viral, bacterial, or fungal infections. Immune recognition of pathogen-associated molecular patterns is a hallmark of the host immune defense to combat microbes and to prevent the progression to sepsis. Mitochondrial antiviral signaling protein (MAVS) is a ubiquitous adaptor protein located at the outer mitochondrial membrane, which is activated by the cytosolic pattern recognition receptors, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5), following binding of viral RNA agonists.
Recent Advances:
Substantial progress has been made in deciphering the activation of the MAVS pathway with its interacting proteins, downstream signaling events (interferon [IFN] regulatory factors, nuclear factor kappa B), and context-dependent type I/III IFN response.
Critical Issues:
In the evolutionary race between pathogens and the host, viruses have developed immune evasion strategies for cleavage, degradation, or blockade of proteins in the MAVS pathway. For example, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) M protein and ORF9b protein antagonize MAVS signaling and a protective type I IFN response.
Future Directions:
The role of MAVS as a sensor for nonviral pathogens, host cell injury, and metabolic perturbations awaits better characterization in the future. New technical advances in multidimensional single-cell analysis and single-molecule methods will accelerate the rate of new discoveries. The ultimate goal is to manipulate MAVS activities in the form of immune-modulatory therapies to combat infections and sepsis. Antioxid. Redox Signal. 35, 1376–1392.
Introduction
The host defense against pathogens is classified as innate and adaptive immunity. Innate immunity provides the first line of defense against microbes by host mechanisms involving plasmatic protein cascades and cellular programs. The latter requires the activation of tissue-resident cells and recruitment of professional immune cells, which produce cytokines, such as interferons (IFNs), and other inflammatory mediators (44, 58, 94).
The initiation of innate immunity requires the sensing of pathogen-associated molecular patterns (PAMPs) by a plethora of nonclonal, germ line-encoded pattern-recognition receptors (PRRs) (53). The subcellular localization of these PRR families is either membrane-bound or cytosolic. Toll-like receptors (TLRs), C-type lectin receptors (CLRs), and formyl peptide receptors (FPRs) are membrane spanning receptors. Nod-like receptors (NLRs) and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are localized in the cytosol (139). These conserved receptors detect nonself-molecular patterns that are exclusively present in microbes, in particular nucleic acids (RNA, DNA), bacterial peptides and proteins (N-formyl methionine, flagellin), lipoproteins, bacterial carbohydrates (lipopolysaccharide, mannose), peptidoglycans, fungal β-glucans or chitin from parasites.
The presence of promiscuity of PRRs in regard to their ligand chemical structures is commonly observed and may help to sense a wide array of PAMPs (2, 81). To forestall pathogen evasion, the network of PRRs is considerably redundant, so infection with a given pathogen that contains many PAMPs typically activates several PRR pathways. In some cases, the same PAMP ligand can even activate more than one PRR pathway (e.g., long dsDNA binds to cyclic GMP-AMP synthase [cGAS] and absent in melanoma 2 [AIM2]) (42). On the other hand, the evolutionary processes of natural mutations and selection have shaped versatile strategies of fast replicating pathogens to escape immune recognition (70). Of note, the replication machinery of some viruses (e.g., hepatitis C virus [HCV], severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]) seems particularly error prone, which may serve the deliberate purpose of a sufficiently high mutation rate for better immune evasion. Zoonotic pathogens (e.g., causing mosquito-borne infections) have evolved evasion strategies to escape immune recognition in vastly diverse hosts (e.g., mosquitoes and mammalian hosts), which is often achieved by a metamorphic life cycle of the pathogen.
The immune recognition of viral nucleic acids is based on nucleotide sequences (A-rich or AU-rich) (28), abnormal subcellular localization (DNA in the cytosol), and various structural motifs of the RNA/DNA backbone (5′-cap, CpG motifs) (Table 1). Long double-stranded RNA (dsRNA), single-stranded DNA, and cytosolic RNA:DNA intermediates can occur during the replication of certain viruses, but are typically absent in noninfected mammalian cells. Thus, these nucleic acid species are recognized as PAMPs. The TLRs involved in nucleic acid immunity are TLR3 (dsRNA), TLR7 (single-stranded RNA [ssRNA]), TLR8 (ssRNA, adenosine-rich), and TLR9 (unmethylated CpG DNA) (42, 61, 132). The cGAS-STING (stimulator of interferon genes) cytosolic dsDNA sensing pathway is attached to the endoplasmic reticulum and is required for the defense against DNA viruses by way of an STAT6-dependent type I IFN response (14, 93).
Different Types of Viruses and Their Ligand Recognition by Cytoplasmic RIG-I-like Receptors
This table was modified from Lee et al. (70).
dsRNA, double-stranded RNA; DDX58, DExD/H-Box helicase 58; EBOV, Ebola virus; HCV, hepatitis C virus; IAV, influenza A virus; IFIH1, interferon induced with helicase C domain 1; LGP2, laboratory of genetics and physiology 2; MDA5, melanoma differentiation-associated gene 5; RIG-I, retinoic acid-inducible gene I; RSV, respiratory syncytial virus; SARS-CoV, severe acute respiratory syndrome coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SeV, Sendai virus; VSV, vesicular stomatitis virus; WNV, West Nile virus.
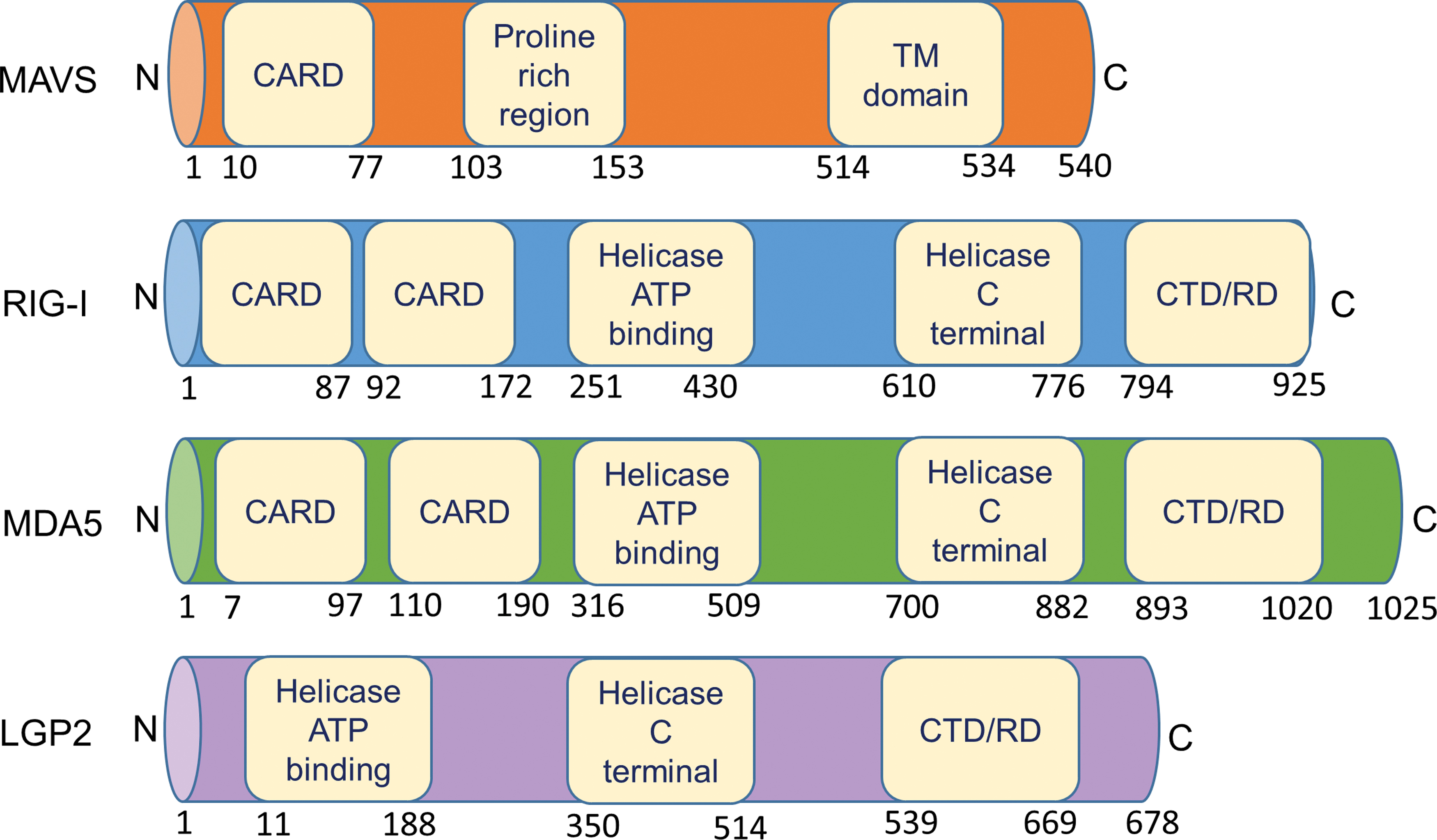
The RLR helicase family contains three members: RIG-I, melanoma differentiation associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) (109). RIG-I and MDA5 primarily detect dsRNA and are constitutively expressed in all tissues, although their functional predominance compared with other PRRs is variable in different cell types (57). RIG-I shows a preference to recognize viral dsRNA with 5′-triphosphate and 5′-diphosphate blunt ends (18–19 bp minimum size) missing the endogenous mammalian 2′-O-methylation of the first nucleotide (N-1 methylation) (37, 46, 123). RIG-I was also reported to detect ssRNA with 5′-phosphates and poly(dA-dT) DNA when converted into 5′-triphosphorylated RNA by RNA polymerase III (15, 101). The ssRNA may need to contain intramolecular base pairing patterns (e.g., stem-loop) to trigger recognition by RIG-I (83). While RIG-I recognizes the ends of dsRNA, MDA5 is a sensor for higher order nonterminal RNA structures including synthetic analogs including polyinosinic:polycytidylic acid (Poly(I:C)) (42). No MDA5-specific ligand seems to exist. The lower affinity of MDA5 to dsRNA, compared with RIG-I, is overcome by filamentous MDA5 oligomerization (42). LGP2 lacks a signaling domain, but can still facilitate the functions of MDA5 through cooperative binding, while inhibiting RIG-I-dependent immunity (42, 109, 113).
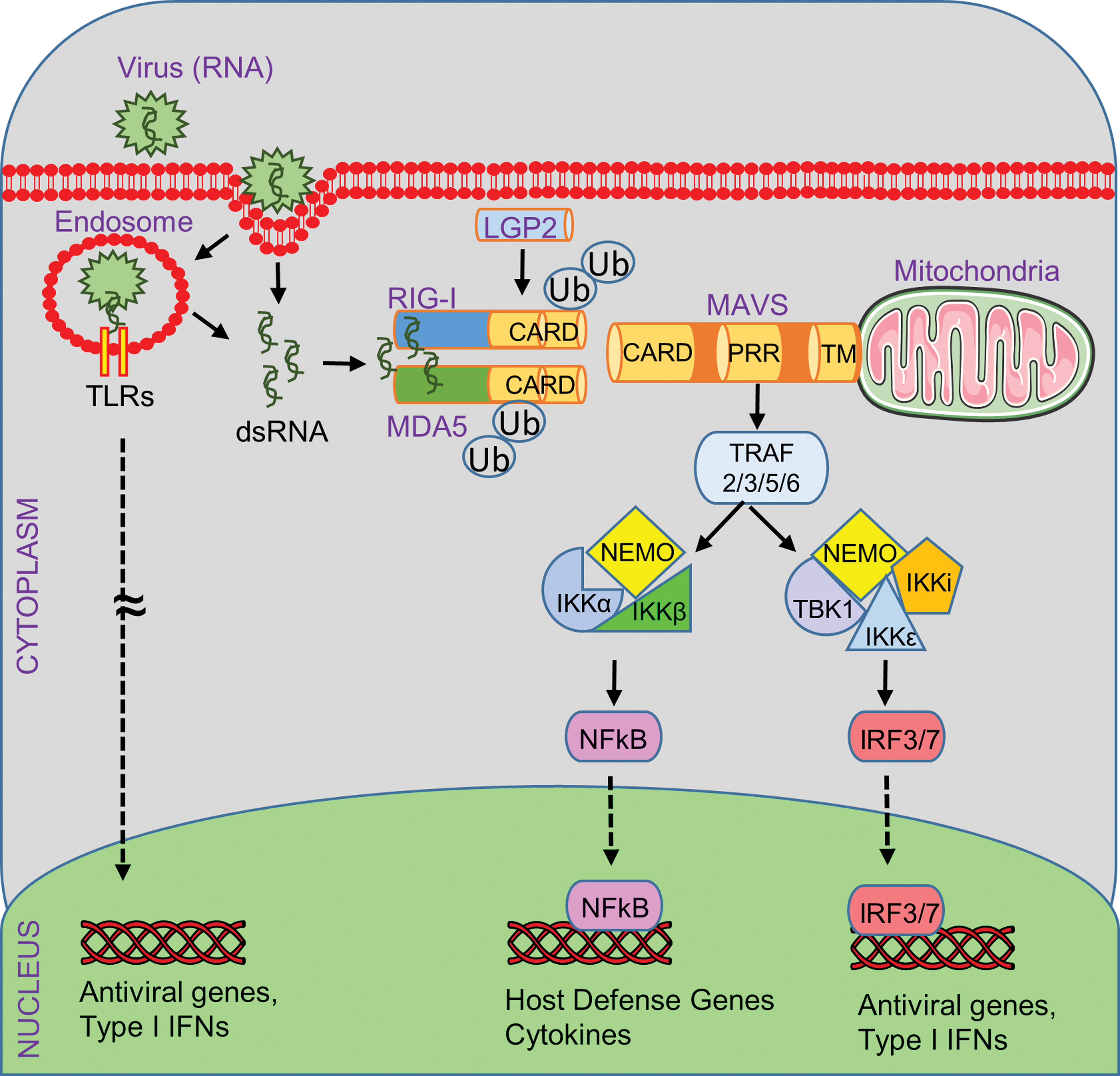
RIG-I and MDA5 activate the mitochondrial antiviral signaling protein (MAVS) (124). Alternative names for MAVS are IFN-β promoter stimulator 1 (IPS-1), virus induced signaling adaptor (VISA), and caspase activation recruitment domain adaptor inducing IFNβ (CARDIF) as it was independently discovered by four different laboratory teams in 2005 (60, 90, 124, 160). MAVS is a tail-anchored membrane protein located on peroxisomes, the mitochondrion-associated membrane (a subdomain of the endoplasmic reticulum), and the outer membrane of mitochondria (147). MAVS acts as an adaptor for activated RIG-I and/or MDA5 helicases to accomplish signal amplification and downstream activation of transcription factors (interferon regulatory factors [IRFs] such as IRF3, IRF7, IRF9; or nuclear factor kappa B [NFκB], etc.) for the induction of a robust type I/III IFN response (155). In addition, RIG-I mediates caspase-1 inflammasome activation and interleukin-1β (IL-1β) release independent of MAVS (62, 105, 106).
A rapid, effective, and selective immune recognition of viruses by MAVS and other PRR pathways is required to curtail virus replication and accomplish pathogen clearance. A failure of efficient nucleic acid sensing increases the risk for spread of infection with the consequences of dysbalanced inflammation, tissue injury, and sepsis (7, 73). Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection (129). Sepsis is a feared complication of systemic infection from viruses, bacteria, or fungi (7, 114, 127). It is estimated that almost 50 million of patients worldwide are diagnosed with sepsis each year with mortality rates over 20% (116). Almost all lethal cases of COVID-19 are diagnosed with sepsis (170). Here, we summarize the current knowledge of the MAVS nucleic acid sensing pathway concentrating on its roles in viral sepsis and its link to the respiratory burst. The activation of RLRs by mislocated or misprocessed host RNA and the strong association of gain-of-function mutations in MDA5 with autoimmune diseases, while important, are not a major focus of this review (32, 71, 112).
Expression, Structure, and Function of MAVS
The MAVS gene is a 540 amino acid protein localized on chromosome 20 in humans and chromosome 2 in mice. Human MAVS shares 51.8% amino acid identity with mouse MAVS (48). It is expressed in a constitutive and ubiquitous manner across many tissues and cell types (Fig. 1). MAVS molecules are localized on the outer mitochondrial membrane, peroxisomes, and endoplasmic reticulum of cells (19, 45, 147).
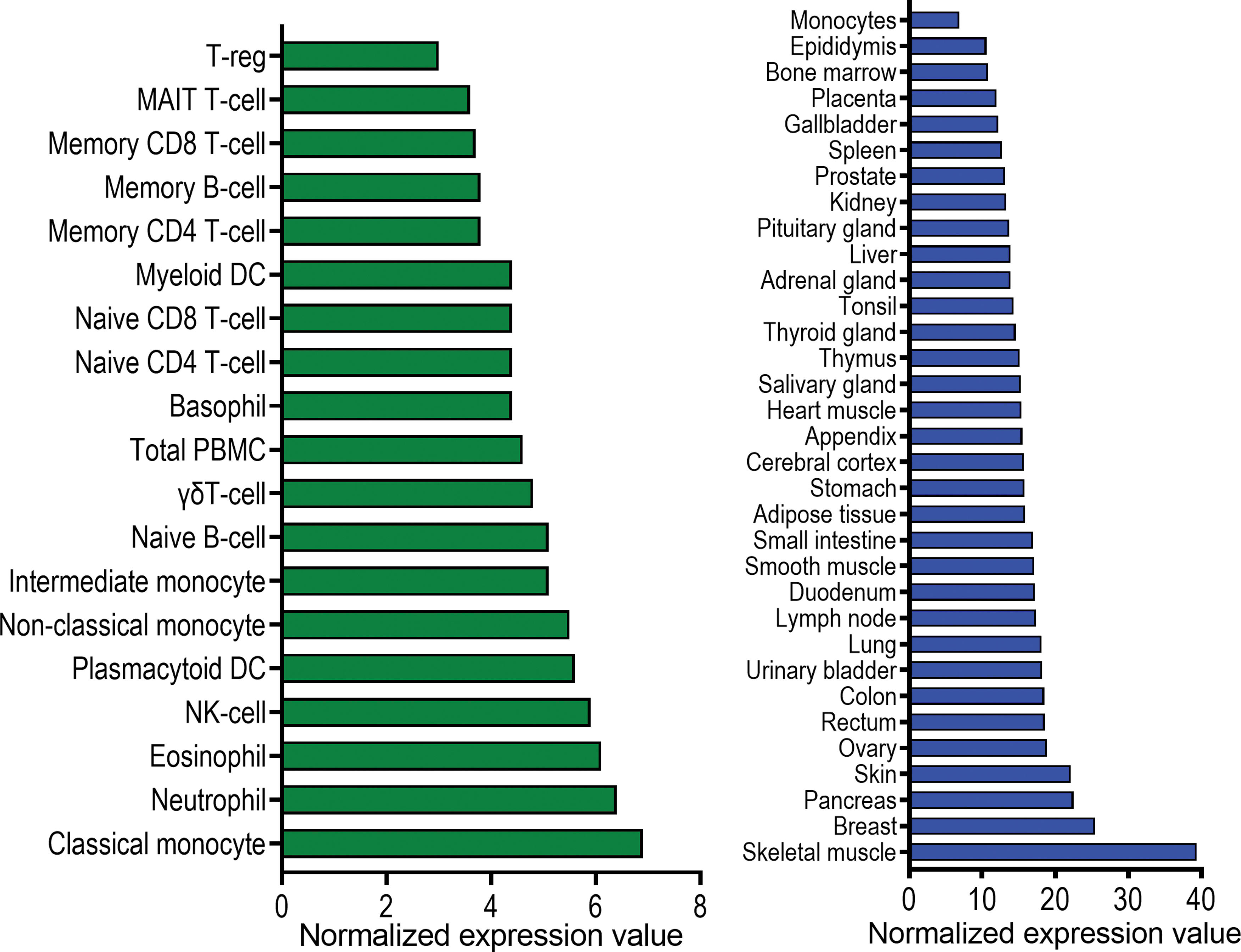
Genomic mapping of MAVS has revealed that it consists of three functional domains: an N-terminal caspase activation recruitment domain (CARD), a central proline-rich domain and a C-terminal transmembrane (TM) domain (Fig. 2) (124). The MAVS-CARD interacts with the CARD present in RIG-I and MDA5, while the C-terminal TM domain allows it to localize at the outer mitochondrial membrane (84). Activation of MAVS and the downstream cascades of signaling molecules aim to produce cytokines, which subsequently induce a protective response against the pathogens, thus resulting in viral clearance (19, 26, 45, 51).
There are several transcriptional regulators of MAVS, in particular a negative feedback loop by reactive oxygen species (ROS) molecules generated during the antiviral response, splice variants encoded by the gene, as well as IRF3, an antiviral gene transcription factor (11, 66, 68, 147). Translation of the polycistronic mRNA can be initiated in several sites, which can result in multiple shorter MAVS isoforms that lack the CARD (8, 107). Post-translational elements, such as E3 ubiquitin ligases, catalyze MAVS ubiquitination and degradation by the proteasome, while protein kinases bound to certain regions of MAVS can inhibit downstream signaling (11, 51).
Recent work regarding the molecular interactions of MAVS has revealed that during viral infection, RIG-I promotes the prion-like polymerization of MAVS on the outer mitochondrial membrane, which subsequently induces TNF receptor associated factor (TRAF)-dependent activation of inhibitor of nuclear factor kappa B (IκB) kinase (IKK) and TANK-binding kinase 1 (TBK1) (47). In addition, the phosphorylated MAVS recruits IRF3 via its positively charged phospho-binding domain, which ultimately results in further phosphorylation of IRF3 by TBK1 (85). In this context, the aggregation of the scaffold protein Fas associated factor-1 (FAF1) acts as a safety switch, which inhibits MAVS accumulation under normal conditions. However, FAF1 is phosphorylated by IκB kinase-ɛ (IKKɛ) during viral infection, which then allows MAVS to escalate an antiviral response (16).
The deletion of the CARD eliminates the signaling function of MAVS with the hallmark feature of defective IFN induction (124). Surprisingly, the C-terminal TM domain was also shown to be crucial for signal transduction, which highlights the importance of mitochondria in immunity as well as the potential of these organelles as therapeutic targets (51, 124, 134). Furthermore, overexpression of MAVS or RIG-I can trigger spontaneous activation of several transcription factors, NFκB, IRF3, and IRF7, which initiates the induction of IFN pathways for antagonizing viral replication (124).
At least 12 single-nucleotide polymorphisms (SNPs) were identified in the coding region of human MAVS. These SNPs can result in impaired function of MAVS due to altered intracellular distribution, inhibited signaling, and ineffective protein binding (159). The prevalence of SNPs in human populations seems to be very rare (159). In addition, natural MAVS isoforms without the CARD prevent a spontaneous aggregation of full-length MAVS for avoiding accidental activation and autoinflammation (107).
Structure and Function of RIG-I and MDA5 As MAVS Activators
All RLRs are characterized by a central DEAD box helicase/adenosine triphosphate binding domain and a C-terminal regulatory domain, necessary for RNA binding and to prevent constitutive activation (Fig. 2) (141). RIG-I and MDA5 possess two N-terminal CARDs that mediate their oligomerization following RNA binding. LGP2 is dissimilar to both RIG-I and MDA5 as it lacks the CARD signaling domains, implying that its biological functions arise from interactions with viral dsRNA ligands or other RLRs (9, 146). The precise role of LGP2 in innate immunity remains to be further investigated, but LGP2 seems to act as a specific regulator of RLR pathways (97, 120).
The binding of viral nucleic acids induces a conformational change in RIG-I and MDA5 to allow the formation of a tandem CARD tetramer in synchrony with translocation to the mitochondria (Fig. 3) (100). E3 ubiquitin ligases equip the CARDs of RIG-I and MDA5 with K63-polyubiquitin chains (100). This K63 ubiquitination-mediated signal activation mechanism is required for efficient RIG-I and MDA5 binding to MAVS, which will rapidly form prion-like MAVS aggregates using the mitochondrial outer membrane as scaffold (47). The CARD-like domain of MAVS is responsible for interacting with RIG-I, and the TM domain-containing region of MAVS mediates oligomerization and interaction with other adaptor proteins, in particular members of the tumor necrosis factor receptor-associated factor family (TRAF2, TRAF3, TRAF5, and TRAF6) (25). The recruitment of TRAFs is followed by the activation of the TBK1 and IKK complexes (IKKi, IKKɛ, NEMO) (25). The TBK1 complex promotes the phosphorylation and dimerization of IRF3 and IRF7, which subsequently translocate to the nucleus for binding to IFN-stimulated response elements of type I IFN-regulated genes (12, 54, 94). In addition, the activation of the IKK complex induces the nuclear translocation of NFκB for the induction of host defense genes, including proinflammatory cytokines (Fig. 3) (59, 111).
MAVS and Reactive Oxygen Species
Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) exert a plethora of functions in inflammation, cellular messaging, tissue injury, and sepsis (31). ROS are produced mainly by mitochondria and several oxidases such as NADPH oxidases (NOX1, NOX2) (17). Mitochondria spatially combine essential functions for antiviral immunity and ROS production (91, 153). Viral infections can initiate the production of ROS, which in turn activate a cellular antiviral host response program (63). Treatment of host cells with antioxidants before infection results in a weaker antiviral response and higher viral loads (63).
A prevailing viewpoint is that the activation and oligomerization of MAVS are redox sensitive. This concept is supported by experimental findings suggesting that ROS amplify RIG-I signaling, as well as that NOX2 enhances the MAVS signaling cascade (35, 130, 136, 144). ROS can induce MAVS oligomerization even in the absence of viral infection (10). Further evidence suggest that ROS can play a dual role in promoting RLR signaling, since decreased NOX2 presence results in reduced MAVS mRNA expression (65, 130, 140).
In addition to NOX2, several other proteins link ROS to MAVS-mediated nucleic acid sensing. For example, cytochrome C oxidase 5B (COX5B, Complex IV), is an enzyme that catalyzes the final step in the mitochondrial electron transport chain. In fact, overexpression of COX5B through interaction with autophagy-related-5 (ATG5) inhibits RLR-dependent MAVS signaling and suppresses ROS production, while no such effects were observed by TLR signaling (168). Thioredoxin-2, a small multifunctional redox-active protein in mitochondria, represses the production of ROS and thereby prevents MAVS activation in response to Poly(I:C) or vesicular stomatitis virus (VSV) (74).
The rate of ROS formation is influenced by the mitochondrial membrane potential (137). Interestingly, MAVS can induce a collapse in mitochondrial membrane potential in the context of apoptosis (72). ROS also oxidize and thereby damage organelles. In fact, MAVS was suggested to function as an autophagy receptor that mediates the removal of damaged mitochondria (135). More specifically, MAVS possesses an LC3-interaction region (LIR), which is typical for autophagy receptors and allows MAVS to interact with LC3-II. This MAVS-LC3-II complex is an important recognition site for autophagosomes (135).
MAVS interacts with and regulates the NOD-, LRR-, and pyrin domain containing protein 3 (NLRP3) inflammasome (131). In detail, MAVS facilitates NLRP3 oligomerization by recruiting it to close proximity with mitochondrial ROS, which is critical for NLRP3 activation (99). Moreover, sensing of dsRNA by RIG-I and MDA5 induces an MAVS-dependent K+-efflux, which is essential for NLRP3 inflammasome activation (29). The K+ currents can arise from membrane damage that is caused by ROS. N-acetylcysteine, a free radical scavenger, inhibits this process by preventing the efflux of K+ ions, thus preventing MAVS-dependent NLRP3 activation in response to cytosolic nucleic acids (29).
The Protective Roles of MAVS Against Viral Infections and Sepsis
While the annual number of sepsis cases is almost 50 million worldwide, the prevalence of viral sepsis is difficult to estimate. The origin of sepsis is less likely to be attributed to a viral pathogen; however, estimates place viral causes in about 30% of all sepsis cases (82). The most susceptible patient populations for these viral pathogens are neonates, young infants, pregnant women, elderly humans, and immunosuppressed individuals. Sepsis and septic shock are accompanied by multiple organ dysfunction and end-organ damage of vital organs, for instance, the lungs, kidneys, liver, heart, and brain (1).
MAVS signaling is required for immune recognition and host defense, which confer protection against viruses (Table 2). An established marker for 30-day sepsis mortality is the concentration of lactate in blood at the time of admission to the intensive care unit. Interestingly, lactate acts as a glycolysis-mediated RIG-I/MAVS pathway inhibitor by directly interacting with the TM domain of MAVS (166). In addition, reduction of lactate through inactivation of lactate dehydrogenase A increases type I IFN production and protects mice against viral infection (166). Thus, the inhibition of MAVS signaling by lactate and intracellular acidosis could contribute to the dysregulated host response and immunosuppression of viral sepsis, which increase the vulnerability of patients to secondary infections.
Studies on the Importance of MAVS in the Immunopathology of Infections
CHIKV, chikungunya virus; CVB, Coxsackie B virus; IFN, interferon; IL-1β, interleukin-1β; IκB, inhibitor of nuclear factor kappa B; IKK, IκB kinase; IRF, interferon regulatory factor; MAVS, mitochondrial antiviral signaling protein; NFκB, nuclear factor kappa B; NIK, NF-κB-inducing kinase; RLRs, RIG-I-like receptors; RRV, Ross River virus; UTR, untranslated region.
The genetic disruption of MAVS signaling in mice can dramatically increase their susceptibility for viral sepsis-associated mortality (134, 148). The replication cycle of ssRNA viruses generates double-stranded RNA intermediates as structural motifs for nucleic acid sensing by RIG-I and MDA5, although pathogenic viruses have often evolved escape strategies.
Chikungunya virus
Chikungunya virus (CHIKV) is a mosquito-transmitted, positive-sense, ssRNA virus, which preferentially targets myoblasts, fibroblasts, and some mononuclear phagocyte populations. CHIKV infections are usually mild-moderate, although some patients develop fatal disease due to severe sepsis and septic shock (115). CHIKV infection activates IRF3 via MAVS in human fibroblasts (154). Treatment of primary human monocytes and monocyte-derived dendritic cells (DCs) with RIG-I agonist (5′ triphosphate double-stranded RNA [5′pppRNA]) prevents infection through the RIG-I/MAVS/TBK1/IRF axis, but surprisingly this is largely independent of type I IFNs (96).
Coxsackie B virus
Coxsackie B virus (CVB) is a group of +ssRNA viruses that can cause myocarditis, aseptic meningitis, and sudden death. During CVB infection, the absence of MAVS or its adaptor molecule MDA5 in mice results in a stunted IFN response and thus early mortality. While all MAVS-deficient mice died within 5 days after infection, half of the heterozygous and wild-type animals survived until the end of the observation period (148).
Dengue virus
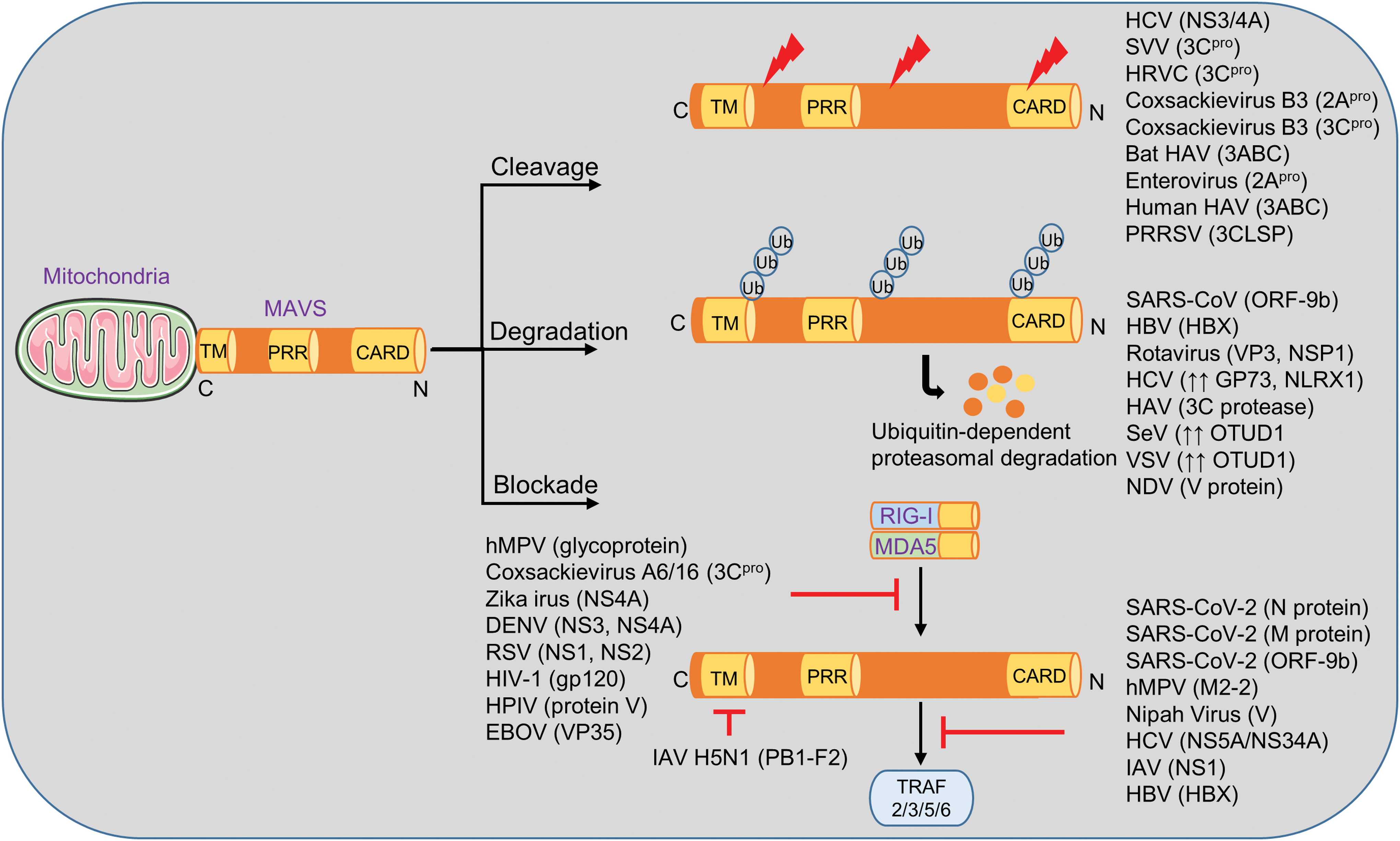
Dengue virus (DENV) is a mosquito-transmitted, +ssRNA virus. It was estimated that 390 million annual DENV infections occur in tropical regions (4). The majority of cases (∼75%) are without clinical symptoms, although DENV can cause sepsis in susceptible individuals. The mammalian 14-3-3ɛ protein binds phosphorylated serine and threonine residues of other proteins. 14-3-3ɛ is important for the translocation of RIG-I to the mitochondrial membrane which harbors MAVS. The nucleostemin 3 (NS3) protein of DENV binds to 14-3-3ɛ for blocking the RIG-I/MAVS pathway and subverting innate and adaptive immunity (Fig. 4) (13).
Ebola virus
Ebola virus (EBOV) carries a −ssRNA genome and causes hemorrhagic fever in humans that often progresses to multiorgan failure, sepsis, and death. MAVS deletion enables greater replication of EBOV because of an inadequate type I IFN response with insufficient gene expression of IFNα and other ISGs. Infection with a mouse-adapted EBOV resulted in death of all mice with either constitutive MAVS ablation or myeloid cell-specific MAVS deletion, while all MAVS-competent wild-type mice survived the infection (20). The relevance of the MAVS pathway as a host defense mechanism against Ebola disease is underscored by the fact that EBOV encodes the viral protein 35 (VP35) immune evasion protein. VP35 inhibits dsRNA recognition by RIG-I via disrupting RIG-I interactions with the endogenous dsRNA-binding helper protein activator of the interferon induced protein kinase (PACT), thus preventing subsequent IRF3, IRF7 activation and type I IFN production (86, 89). In addition, viral protein 24 (VP24) of EBOV inhibits IFNAR signaling by antagonizing nuclear localization of phosphorylated STAT1 (110).
Influenza A virus
RIG-I signaling is required for protection against influenza A virus (IAV) infection and reduces virus titers in the lungs. The activation of RIG-I is essential for preventing IAV infection of lung epithelial cells and hematopoietic cells. RIG-I-deficient mice display defective patterns in migratory DC activation, viral antigen presentation, and priming of CD8+ and CD4+ T cell responses during IAV infection. RIG-I-deficient mice were found more susceptible to IAV (PR8 virus strain) compared with wild-type mice in one study that showed survival rates of ∼10% compared with ∼70% (55, 80). In contrast, no differences in survival of RIG-I-deficient and MAVS-deficient mice after infection with IAV PR8 were observed by another group (157). It remains unclear how to reconcile these variable findings.
Hepatitis C virus
HCV is a +ssRNA virus causing high rates of chronic infections, liver cirrhosis, and an increased risk for hepatocellular carcinoma. The type I IFN response determines the complex pathophysiology of HCV infection, and recombinant IFNα was the standard therapy before introduction of direct-acting antivirals. One of the many factors involved in the pathogenesis of HCV-positive hepatitis is MAVS signaling (78). The nonstructural protease NS3–4A encoded by HCV cleaves MAVS (cleavage site: cysteine 508) and thereby inactivates it (Fig. 4) (77), which results in an insufficient production of IFNβ alongside reduced Janus kinase (JAK)/signal transducer and activator of transcriptions (STATs) and lower expression of ISGs, for instance, viperin and interferon alpha inducible protein 27 (IFI27) (3, 90, 117, 152). This immune evasion mechanism is counter-blocked by pharmacologic NS3–4A inhibitors (43, 78).
Nipah virus
Nipah virus (NiV), a highly pathogenic bat-borne +ssRNA member of the paramyxovirus family, also interferes with the MAVS pathway. NiV infection can present as the clinical picture of lethal encephalitis (21). A major viral protein, V, suppresses the RIG-I/MDA5 pathway by stabilizing the UBX domain containing protein 1 (UBXN1), a ubiquitin binding protein that disrupts MAVS oligomerization by interfering with the MAVS/TRAF signaling cascade (145). Protein V increases the binding affinity of MAVS with UBXN1 in addition to direct inhibition of MDA5, thus preventing protective immune signaling (145).
Respiratory syncytial virus
Respiratory syncytial virus (RSV) encodes a −ssRNA genome. RSV accounts for a large percentage of upper and lower respiratory tract viral infections in infants and young children. MAVS is required for the induction of type I IFNs and other proinflammatory cytokines in cultured mouse lung fibroblasts, macrophages, and conventional DCs (5). MAVS activities in response to RSV are a concerted program of synergizing cell types that include both nonimmune lung-resident cells and immune cells (18). Among these different cell types, alveolar macrophages are major producers of MAVS-dependent IFNs during RSV infection, which are required for the recruitment of inflammatory monocytes to the lungs (36). While MAVS-deficient mice are still capable of clearing RSV infection from lungs for recovery, they experience higher viral titers and broad defects in the early inflammatory response as analyzed by microarray profiling. CD103+ DCs in infected lungs appear to partially compensate MAVS deficiency, possibly through TLR3 and TLR7 signaling (18). In terms of adaptive immunity, MAVS deficiency is associated with defective RSV-specific antibody production, whereas the CD8+ responses to RSV remain normal (5).
Vesicular stomatitis virus
VSV encodes a −ssRNA genome and causes a natural self-limiting disease in hoofed animals or flu-like symptoms in humans. VSV-associated mortality in laboratory animal studies depends on the infectious dose and intact immune defenses. In experimental VSV infection, the genetic deletion of MAVS abolishes viral induction of IFNs and prevents the activation of NFκB and IRF3. MAVS-deficient mice are extremely susceptible to VSV (Indiana strain) when compared with wild-type mice (134). In fact, all heterozygous and homozygous mice died within 4 days after VSV infection, while survival rates in the wild-type cohort were around 70% in one study (134). In human bronchial epithelial cells, VSV infection initiates an IRF3-dependent expression of miR-576-3p as a feedback loop to curtail IFNs and antiviral host defense programs to avoid excessive inflammation and tissue injury (163). VSV infection reduces the expression of the RIG-I inhibitor, NLRP12 (163). The NEMO-like kinase (NLK) phosphorylates MAVS to initiate its degradation (75). Depletion of NLK increases antiviral effects and survival of VSV infection (75).
Ross River virus
Ross River virus (RRV) is a mosquito-borne, +ssRNA virus responsible for outbreaks of polyarthritis and rash in the Southwest Pacific region and Australia. RLR signaling has a protective function during RRV infection (40). MAVS-dependent production of type I IFNs in monocytes is a key factor in controlling and preventing acute RRV infection. Monocytes can elicit an effective immune response to contain the infection and reduce musculoskeletal damage under assistance by plasmacytoid DCs, which act independently of MAVS (40). In vitro production of type I IFNs by monocytes in response to RRV is dependent on MAVS, while RRV-infected mice with a functional MAVS gene experience lower viral loads (39).
Sendai virus
Sendai virus (SeV) is a −ssRNA virus that infects rodents, but not humans. Similar to other viruses, MAVS-mediated activation of IRF3 and NFκB is observed in macrophages responding to SeV infection. In fact, the gene expression of IFNα, IFNβ, and IL-6 is completely abolished in the absence of MAVS (134).
The West Nile virus
The West Nile virus (WNV) is a mosquito-born, +ssRNA flavivirus, which causes severe neurological infections (meningitis, encephalitis) in 1% of patients. Innate immune responses, critical for controlling WNV infection, are highly dependent on RIG-I, MDA5, and MAVS (24). Lack of both RLRs in engineered RIG-I/MDA5 double knockout mice results in a profound failure to sense 5′-triphosphate dsRNA intermediates during replication of WNV in host cells (24). MDA5, RIG-I, MDA5/RIG-I, or MAVS deficiencies are all associated with a significantly higher mortality to murine WNV infection (24). MAVS-dependent IFNAR signaling in myeloid cells protects against the progression of WNV infection into sepsis with massive complement activation (103). Defective MAVS increases the infiltration of myeloid cells and virus-specific T cells in the brain as a sign of insufficient control of viral replication and spread. In the absence of MAVS expression, a residual degree of type I IFNs and other proinflammatory mediators is likely produced via MAVS-independent pathways, such as TLRs, myeloid differentiation primary response 88 (MyD88), and possibly STING signaling (122, 138). MAVS activity in hematopoietic cells facilitates rapid WNV clearance and the early resolution of a dysregulated or pathogenic immune response. In one study, all mice with hematopoietic cell-specific MAVS deletion died by day 14 after WNV infection, while infection was lethal for only 10%–30% of wild-type mice in the control group (167).
Zika virus
Lastly, the recent emergence of +ssRNA Zika virus (ZIKV) has drawn additional attention toward the need to further understand the molecular basis of viral infections to combat global threats by infectious pathogens (102). Type I IFNs exert a protective role against infection by ZIKV, and consequently, the virus has developed several mechanisms to counteract IFNs (52, 143, 158). The ZIKV protein nonstructural protein 4A (NS4A) has been shown to directly bind to MAVS for interference of the domains, CARD and TM, which bridge RIG-I and MAVS, thus blocking the signal transduction and allowing sustained viral infection (Fig. 4). The consequence is an inadequate host response in ZIKV-positive patients along with attenuated release of type I IFNs (48). Noteworthy, MAVS-deficient mice are not extremely susceptible to ZIKV infection, which is in contrast to mice deficient for IFNAR or IRF3/5/7 triple knockout mice, which have completely disrupted type I IFN circuits (69). Thus, MAVS-independent IFN production seems important in preventing ZIKV disease and especially the feared central nervous system manifestations.
Emerging Evidence for MAVS Signaling in COVID-19
Severe acute respiratory syndrome coronavirus 2
SARS-CoV-2 encodes a large (∼30 kB) +ssRNA genome. SARS-CoV-2 causes 15%–30% of inapparent cases, although more severe COVID-19 symptoms and lethality occur in the elderly and immunocompromised patients. Almost all lethal COVID-19 cases are diagnosed with acute respiratory distress syndrome and sepsis (49, 170).
In general, the RIG-I/MDA5 pathway and the TLR family of receptors play important roles in the detection of coronaviruses (98, 125). Moreover, it has been shown that SARS and MERS interfere with downstream molecules of both the MAVS and TRAF3/6 pathways, by ubiquitination and degrading adaptor molecules, thus preventing IRF3 translocation into the nucleus and subsequent histone modifications (64).
It was recently reported that the RIG-I helicase domain binds the 3′ untranslated region of SARS-CoV-2 RNA in infected lung epithelial cells (161). It was suggested that this RIG-I binding directly inhibits the viral RNA-dependent RNA polymerase and confines SARS-CoV-2 replication in an MAVS- and IFN (type I and III)-independent manner (161). A stem-loop RNA that mimics physiological dsRNA and acts as a minimal RIG-I agonist conferred protection against SARS-CoV-2 infection in mice (88).
Deletion of RIG-I, MDA5, and MAVS in human epithelial cells facilitates increased SARS-CoV-2 replication, in conjunction with abolished type I/III IFN responses (119, 162). On the contrary, SARS-CoV-2 inhibits MAVS/RIG-I signaling by disrupting the molecular foundation of this signaling cascade. Transfection of isolated SARS-CoV-2 RNA elicits stronger RLR responses than infectious viral particles, suggesting immune escape mediated by viral proteins (67). More specifically, the dimerization domain of SARS-CoV-2 nucleocapsid protein (N) negatively regulates the K63-linked polyubiquitination and aggregation of MAVS, which results in reduced TBK1/IRF3 activation, IFNβ signaling, and inadequate antiviral immunity (149). The N protein itself can be targeted by host acetyltransferases or engineered peptides to reduce its activity (149). Likewise, ORF9b viral protein disrupts the type I/III IFN antiviral response by interfering with the K63-linked ubiquitination of NEMO, decreasing TBK1 phosphorylation, and suppressing phosphorylation of IRF3 during infection of human alveolar epithelial cells (41, 156). Interestingly, ORF9b from the homologous SARS-CoV also targets MAVS by triggering K48-linked ubiquitination and proteasomal degradation (128). NSP5 proteins of SARS-CoV and SARS-CoV2 antagonize RIG-I/MAVS-induced nuclear translocation of phosphorylated IRF3 and IFNβ expression (33). Furthermore, SARS-CoV-2 membrane (M) protein interferes with type I and III IFNs (30, 133, 169). HEK293T cells manipulated to express M protein showed a significantly lower expression of several inflammatory markers, including IFNβ, CXCL10, and other ISGs after challenge with SeV and Poly(I:C) (169). The M protein was identified by coimmunoprecipitations to interact with the RIG-I, MDA5, and TBK1 proteins, but not IRF3 (169). SARS-CoV-2 NSP12 also inhibits the RIG-I/MDA5/MAVS IFN response in HEK293T cells (150). Lactoferrin displays antiviral activities against SARS-CoV-2 by inducing the expression of several members of the IFN, TLR, and IRF families, as well as the MAVS gene (118).
A Role for MAVS in Nonviral Host/Microbe Interactions
RLRs can detect both bacterial and viral RNA molecules to initiate the production of IFNs through the MAVS adapter protein (76). Bacterial RNA contains 5′-triphosphate as well as other similar secondary structures, which are recognized by the RLRs (6, 76, 121). However, RNA, originating from certain Escherichia coli strains, does not induce traditional RLR-associated IFN transcription factors, but rather leads to the production of other cytokines such as TNF-α and IL-1β (22). In addition, MAVS has been suggested to play an important role in mitochondrial homeostasis and autophagy (135). MAVS protects against colitis, while MAVS deficiency can result in dysregulation of host/microbe interactions and the symbiotic relationship with the gut microbiota (50, 104). This, in turn, can create larger problems, namely an increased intestinal permeability and higher risk for infections. Furthermore, the downstream type I IFN production has a defensive nature, preventing extensive damage to the gut and reducing the multiplication of pathogens (27). Interestingly, MAVS-deficient mice are more sensitive to radiation- and chemotherapy-induced damage, a finding that highlights the pleiotropic roles of the RLR family (50, 126). Studies regarding the antibacterial role of the RIG-I/MDA5 pathway during Legionella pneumophila infection have revealed that bacterial RNA intermediates trigger an IFN-dependent response, and MAVS-deficient mice are partly defective in their production of IFNβ in infected lungs (92, 95).
MDA5/MAVS have also been observed to play a role in antifungal immunity. The leukocyte-mediated host resistance against Aspergillus fumigatus requires “vitality sensing” of fungal dsRNA by MDA5/MAVS (151). MAVS-deficient mice challenged with A. fumigatus displayed stunted production of type III IFNs (and to a lesser extent type I IFNs) combined with lower chemokine levels (CXCL9 and CXCL10) (151). Finally, the role of RIG-I/MDA5/MAVS encompasses the sensing of plasmodium RNA and recognition of small-self RNA generated by RNAse L for amplifying antiviral immunity (79, 87).
Conclusions and Future Directions
The MDA5/RIG-I/MAVS immune sensor pathway was discovered two decades ago, following the discovery of TLRs (124, 164). This pathway plays a critical role for the detection of distinct structural motifs of RNAs in the cytosol. Failure of immune recognition through MAVS results in an inadequate IFN response and facilitates the progression of localized viral infection into systemic inflammation and sepsis. While it is widely acknowledged that IFNs are important for the host defense to intracellular pathogens, type I/III IFNs often have a context-dependent function with a fine line between protective effects and detrimental outcomes (142). The intricate balance of the MAVS/IFN axis in human disease will need further studies. Emerging evidence suggests that MAVS signaling may also be activated by bacterial RNA and mammalian host RNA (38). It is therefore not completely surprising that inadequate overactivation of MDA5 in the absence of infection is strongly associated with certain autoimmune diseases (32, 56). In the future, we need to further define the molecular interactors that regulate the activity of MAVS. The ongoing progress in multidimensional, single-cell transcriptomics (23), advanced proteomics, proteogenomics, and single-molecule imaging will provide a powerful toolbox to better investigate disease processes as related to their cellular heterogeneity, protein/protein interactions, immune-metabolism, host/virus coevolution, and other aspects. So far, RIG-I agonists are being explored as vaccine adjuvants (108, 165). The ultimate goal would be to manipulate the MAVS pathways in the form of an immunomodulatory therapy to fight infections, rebalance tissue-injuring ROS production, and to reduce the global burden of infection-associated inflammation, sepsis, and septic shock.
Footnotes
Author Contributions
A.S. wrote the first version of the article and prepared the figures. K.K. and M.B. revised and edited the article. M.B. supervised and funded the work.
Acknowledgments
We thank Deepthi Sree Vamaraju for assisting with the figure preparation and article editing. We thank Kara Farquharson for reading and editing the final draft of the article. We thank the Evans Center for Interdisciplinary Biomedical Research at Boston University School of Medicine for their support of the Affinity Research Collaborative on “Respiratory Viruses: A Focus on COVID-19.” Animated figures were created with the elements from Servier Medical Art by Servier under a creative commons attribution 3.0 Unported License.
Author Disclosure Statement
The authors have no financial conflicts of interest and all the authors are responsible for the contents of this publication. All requests for materials and correspondence should be addressed to M.B.
Funding Information
This work was supported (to M.B.) by the National Institutes of Health (1R01HL141513, 1R01HL139641, 1R01AI153613, 1UL1TR001430) and the Deutsche Forschungsgemeinschaft (BO3482/3-3, BO3482/4-1).