
Abstract
Significance:
Thioredoxin 1 (Trx1) is a ubiquitous protein that is found in organisms ranging from prokaryotes to eukaryotes. Trx1 acts as reductases in redox regulation and protects proteins from oxidative aggregation and inactivation. Trx1 helps the cells to cope with various environmental stresses and inhibits programmed cell death. It is beneficial to neuroregeneration and resistance against oxidative stress-associated neuron damage. Trx1 also plays important roles in suppressing neurodegenerative disorders.
Recent Advances:
Trx1 is a redox regulating protein involved in neuronal protection. According to a previous study, Trx1 expression is increased by nerve growth factor (NGF) and necessary for neurite outgrowth of PC12 cells. Trx1 has been shown to promote the growth of neurons. Trx1 knockout or knockdown has the worse impact on cell viability and survival.
Critical Issues:
Trx1 has functions in central nervous system. Trx1 plays the defensive roles against oxidative stress in neurodegenerative diseases.
Future Directions:
In this review, we focus on the structure of Trx1 and basic functions of Trx1. Trx1 plays a neuroprotective role by suppressing endoplasmic reticulum stress in Parkinson's disease. Neurodegenerative diseases have no cure and carry a high cost to the health care system and patient's families. Trx1 may be taken as a new target for neurodegenerative disorder therapy. Further studies of the Trx1 roles and mechanisms on neurodegenerative diseases are needed. Antioxid. Redox Signal. 36, 1023–1036.
Introduction
For cell survival in the intracellular or extracellular environment, preserving a proper disulfide state is necessary. The key protein systems in the cytoplasm that control the thiol/disulfide redox balance, the intracellular disulfide reduction agents, are the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent thioredoxin reductase (TR)/thioredoxin (Trx) system (103) and the glutathione reductase/glutathione/glutaredoxin (Grx) pathway (61, 97, 133). In all three domains of life, the TR/Trx system is widely expressed in many microorganisms and some anaerobic bacteria (100, 122, 130). Trx1 is a redox-regulating protein (12 kDa) with a preserved CxxC catalytic site with reversible oxidation/reduction in both cysteine residues, whereas TR is a pyridine nucleotide disulfide oxidoreductase that acts in a homodimeric form (136).
The universality of Trx system is demonstrated by decreased function of Trx in critical biological activities, such as converting nucleotides to deoxynucleotides in bacteria or regulating transcription factor levels in eukaryotes (61, 130). In addition, the TR/Trx system provides the defense against oxidative stress, restores protein activity weakened by oxidative stress, modulates the activity of a redox stressor, and acts as hydrogen donors to detoxify enzymes that are essential for the response to oxidative stress (130). Three distinct members are identified in human cells. Thioredoxin 1 (Trx1) is identified and found in the cytoplasm and nucleus, thioredoxin 2 (Trx2) is documented as a mitochondrial type, and the third type is highly expressed in the sperms (3), which perform multivalent roles in cells. They act as reductions in redox control, guard against inactivation and oxidative accumulation (123, 135), assist the cells in coping with diverse environmental pressures, such as peroxynitrite, arsenate, and reactive oxygen species (ROS) (79, 91), and regulation of programmed death in cells (112) through denitrosylation (13). Few Trxs are also capable of functioning as a growth factor (110), also regulate inflammatory response (96), stimulate folding of protein (73), and play vital roles in the life cycles of phages and viruses (61).
Trx1 was identified as an enzyme involved in DNA synthesis and is capable of reducing ribonucleotide reductase (RNR) (80). In both eukaryotic and prokaryotic organisms, Trx1 was found to modulate the functions of numerous proteins (Fig. 1). Likewise, Trx1 stimulates methionine sulfoxide reductase (MSr), an enzyme functioning in aging and protein repair (83), and reduces a family of enzymes that convert H2O2 to water, peroxiredoxins (Prxs) (25, 137). Trx1 found in bacteria is important for the catalytic process of sulfonucleotide reductase (SR), which is found in the reduction of sulfur assimilation involved in the noncovalent interaction between Trx1 and its target proteins (24, 120) (Fig. 1).
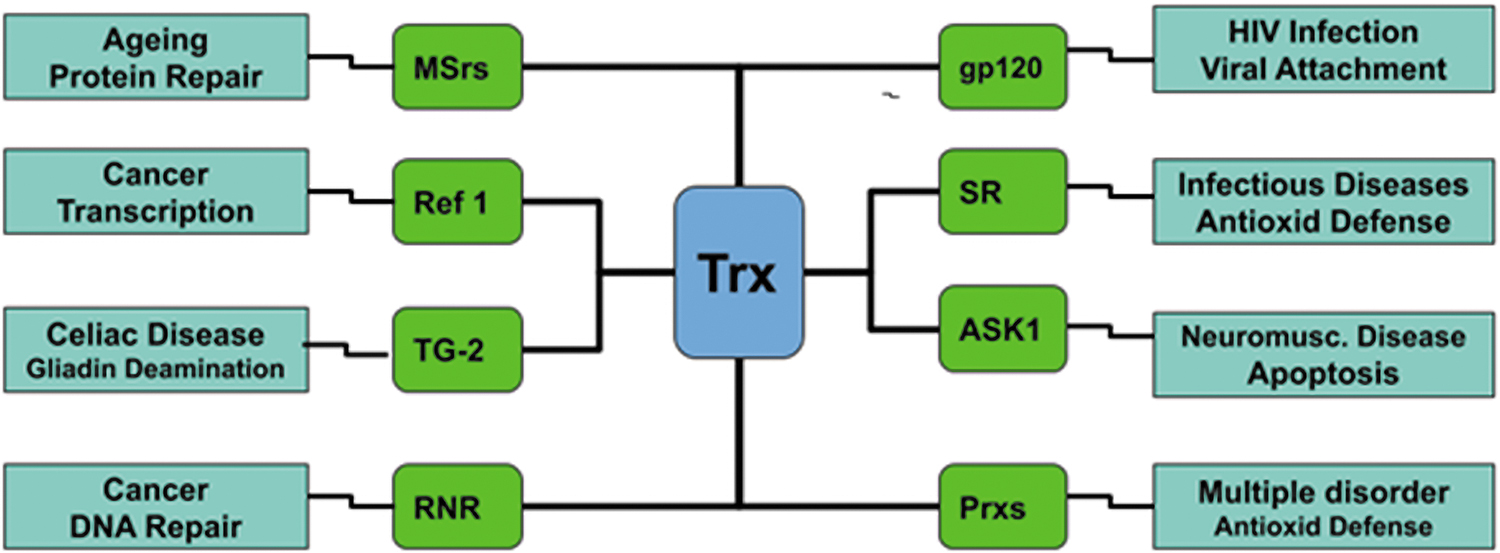
There are enormous studies on Trx1 (18). In this review, we first introduce the structural properties of this protein and the basic functions of this protein. Finally, we concentrate on the roles of Trx1 in the central nervous system (CNS), such as in neurodegenerative disorders.
Trx1 Structure
Trx1 contains a highly conserved catalytic sequence of the canonical CGPC. Trx1 uses mainly the residues of the cysteine motif CGPC to break disulfide bonds in oxidized substratum proteins (91). These two residues of cysteine get oxidized and form a disulfide until the catalytic cycle is over. Trx1 reductase can be converted back to a reduced state by the cost of a reduced form of NADPH (Fig. 2) (36).
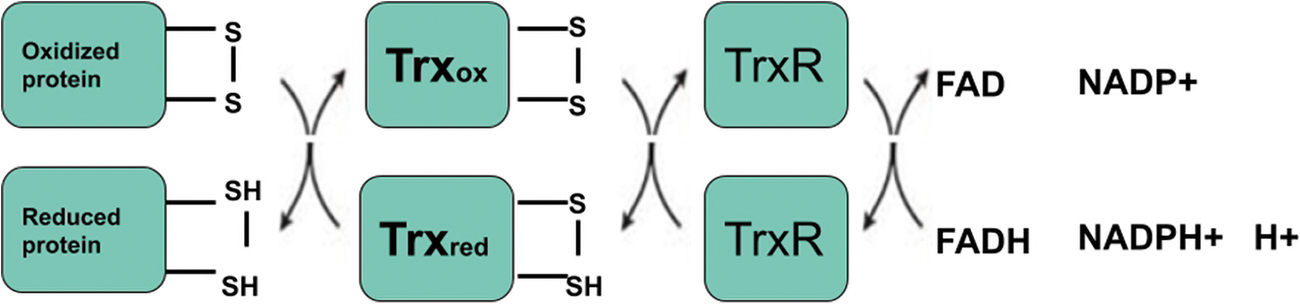
In 1975, Holmgren et al. first described the Trx1 protein structural architecture (62). Today, many oxidized and reduced Trxs crystal and solution structures are reported. The Trx1-fold consists of five β-strands with four α-helices (Fig. 3) (36). The α-helices and β-sheets are divided into the N-terminal motif β1α1β2α2β3 and the α3-helix-connected C-terminal motif β4β5α5α4. The N-terminal motif β strands run parallel, and the C terminal motif β strands run in an antiparallel manner. The α2 and α4 helices are located on one side of the central β sheet, whereas the α3 helix is located on the opposite side. The α3 helix has a perpendicular path toward the helices α2 and α4. The catalytic CGPC motif is situated on the protein surface inside a short section at the amino-end of the α2 helix. A minimal variant of this fold is called the fold Trx1. The β1 strand and the α1 helix are lacking (57). Most of the Trx fold proteins have a retained CXXC catalytic motif.

Trx1 Functions
Trx1 and Grx systems have been formed to maintain thiol redox homeostasis in cells. The fundamental function of these two systems is to remove disulfide bonds and thereby save cells from damage in the oxidizing environment. Together with selenoenzyme TR, the Trx network contains cytosolic isoforms (Trx1) and mitochondrial isoforms (Trx2) (102). One of the key pathways controlling the redox balance of cells and the fate of cells is the Trx1-dependent system. Trx1 plays various roles in the regulation of apoptosis and development, as well as chronic inflammation (65). It is also secreted as a cytokine and a chemokine (3). In response to stress and redox signals, Trx1 is also a regulator of cell activity that modulates different signal pathways, such as immune responses and transcription factor activities (97). Trx1 is an effective cell redox balance modulator and has been involved in neurodegenerative and cancer diseases. During embryo formation and adult physiology, the importance of the Trx1 was confirmed by the loss of function strategies of an individual member of this group. Extreme intracellular mass proliferation defects and blastocyst-level embryonic death are associated with targeted cytosolic Trx1 inactivation (87). There were pronounced apoptosis, anterior neural tube defect, and embryonic lethality in mice with mitochondrial Trx2 deficiency at E10.55 (102). In comparison, by controlling numerous stresses, the overexpression of Trx1 has contributed to neuronal survival. For instance, when presented with focal brain ischemia, Trx1 overexpression has a neuroprotective impact on neuronal cells in mice (124, 145) or damage occurred by chronic excitotoxicity (123).
Trx1 in CNS
In comparison with other tissues, the brain is more vulnerable to oxidative injury. Many variables contribute to the formation of reactive species, including high oxygen consumption, high iron content, and high fat content of unsaturated fatty acid, as well as low levels and inadequate activity of antioxidant enzymes such superoxide dismutase (SOD), catalase, and glutathione peroxidase (Gpx) (43). Trx1 and TR extensively exist in various tissues, including brain tissues (5).
Trx1 overexpression was shown to reduce damage to brain reperfusion injury (124) and successfully mitigates focal ischemic brain injury. The amount of damage caused by focal brain ischemia was minimized by systemic recombinant human Trx1 administration (56). Trx1 overexpression in transgenic (Tg) mice increases the life span of mice (93). Trx1 translocates from the cytosol to the nucleus, which is activated by phorbol ester, ultraviolet light, or other stimuli (97). Trx1 interacts with redox factor-1 to modulate transcription activity by changing the activity of transcription factors such protein-1 activator (AP-1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and hypoxia-inducible factor-1 (HIF-1) (60). Trx1 has a neurotrophic function and is recognized in cholinergic neurons as a primary neurotrophic factor (44). Trx1 is induced by nerve growth factor (NGF) and necessary for the outgrowth of neurites in PC12 cells (8). Trx1 is induced in glia by ischemia in the brain (63) and by nerve injury in motor neurons to exert a cytoprotective function against oxidizing stress and neuroprotective action.
Trx1 is involved in neuronal protection. Trx1 from activated macrophages is a key cholinergic neuron trophic factor in vitro (44). Trx1 has been shown to increase neuron growth and deletion or knockdown of Trx1 is crucial to cell viability and survival (31, 87, 93). Viability, longevity, and synaptic protein levels after traumatic brain injury (TBI) are affected by brain-derived neurotrophic factor (BDNF) (29). According to our findings, BDNF promotes cell survival and induces Trx1 expression in SH-SY5Y cells. SHSY5Y cell survival and proliferation may thus be connected to Trx1 expression driven by BDNF (10). BDNF binds and activates tropomyosin-related kinase B (TrkB), which subsequently activates phosphatidylinositol 3-kinase (PI3-K), which improves neuronal survival and proliferation (11, 67). TrkB inhibitors, such as K252a or PI3-K inhibitors, have been demonstrated to reduce BDNF-induced cell survival and Trx1 expression (Fig. 4). Thus, BDNF's effects on cell survival and Trx1 expression could be mediated by the TrkB/PI3-K pathway (10).

Neurodegeneration and Oxidative Stress
Neurodegenerative disorders are expected to rise as the second leading cause of death worldwide by 2040, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD). In both developing and developed nations, AD, PD, frontotemporal dementia, and other neurodegenerative disorders are the important health concerns (2). Therefore, it is important to understand the molecular pathways driving neurodegeneration and to establish appropriate therapeutic strategies (33, 50, 94). Protein misfolding disorders are often thought to be neurodegenerative diseases, and share common traits, such as misfolded protein accumulation, which leads to neuronal dysfunction and apoptosis. Endoplasmic reticulum (ER) production of misfolded aggregated proteins induces ER stress and triggers mitochondrial dysfunction, crucial in the development of neurodegenerative diseases (49, 118). Neurodegenerative diseases, in particular, are characterized by progressive neuronal dysfunction, which initially involves selected classes of neurons. The brain uses a high content of oxygen, including oxidized redox-active metals and polyunsaturated fatty acids, and is highly susceptible to oxidant harm. A major cause of neurodegenerative diseases is known to be the rise in oxidative stress with age (132). Existing research suggests that redox homeostasis and elevated oxidative stress are linked to the pathophysiology of multiple sclerosis, and alter brain stress response (108).
Impaired redox cell balance is attributed to age-related ailments and neurodegenerative diseases such as AD and PD (2). Oxidative stress has been reported to be a common reason of AD, PD, and ALS, and results in the destruction and death of neuronal cells that lead to the pathogenesis of these disorders (2). During the pathogenesis of neurodegenerative diseases, the principal cause of ROS is mitochondria (39). Inhibition of TrxR and mitochondrial dysfunction of 6-hydroxydopamine and paraquat result in elevated levels of H2O2 and cell death (84). Although Trx1 is a redox controlling protein, it also plays various roles in the defense of neurons and acts as a cofactor for NGFs (8). Trx1 is also known as an ER-stress modulator (9, 43).
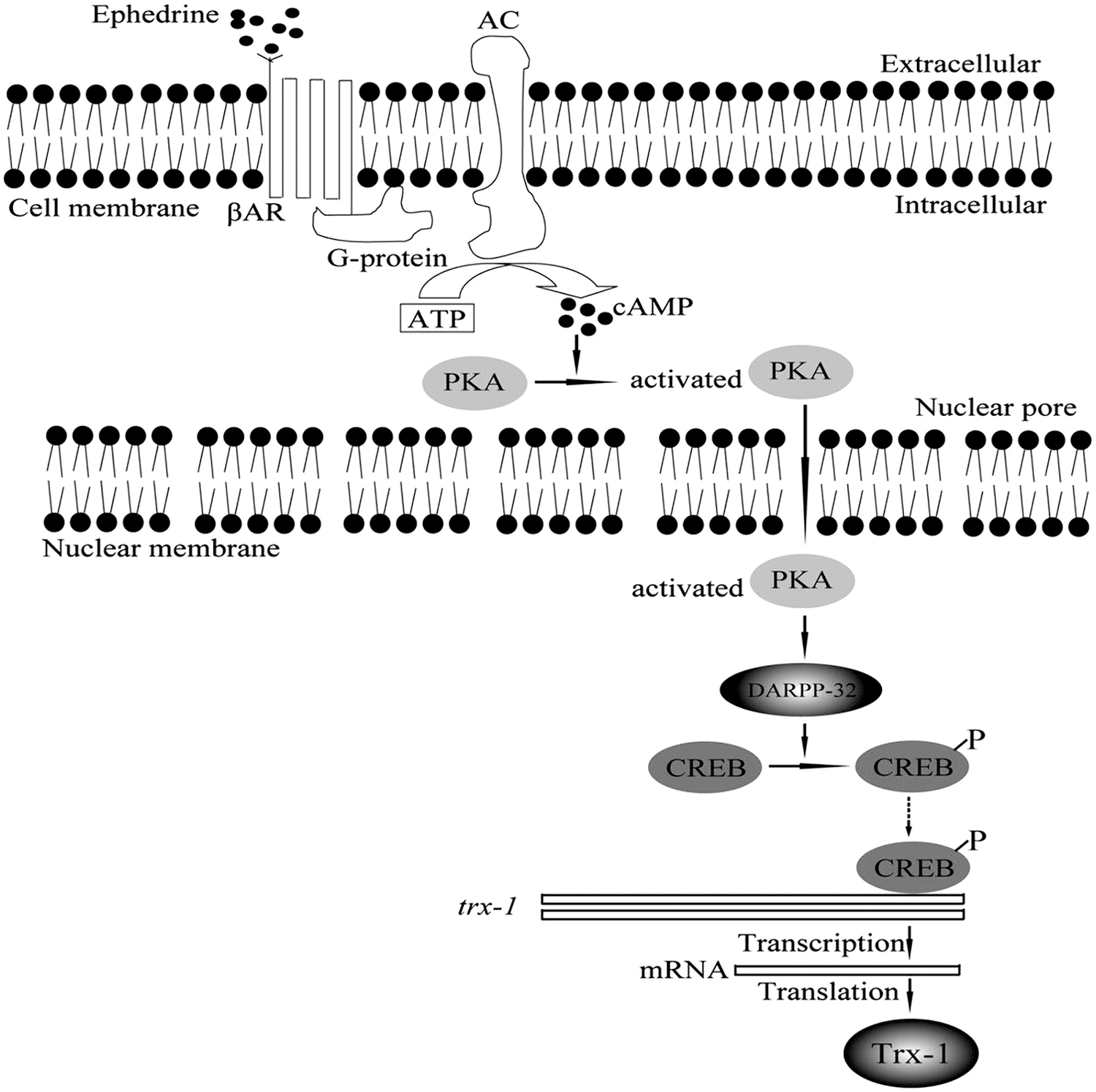
Trx1 expression was also induced by ephedrine (Eph). After PC12 cell exposure to Eph, Trx1 expression increases in a dose- and time-dependent manner (70). Dopamine and cyclic adenosine monophosphate (cAMP)-regulated neuronal phosphoprotein (DARPP-32) is involved in signaling transduction pathways regulated by a range of neurotransmitters, neuromodulators, and neuropeptides in dopaminergic neurons in the neostriatum and likely in other brain regions (95). Similar to Trx1, Eph increased DARPP-32 expression in a dose- and time-dependent manner. Eph-induced DARPP-32 expression was faster than Trx1. In addition, Trx1 small interfering RNA (siRNA) showed no effect on DARPP-32. On the contrary, DARPP-32 knockdown reduced Trx1 expression. These findings revealed that Trx1 was a DARPP-32 downstream. Thus, in PC12 cells, Eph regulates Trx1 expression through the DARPP-32 signaling pathway (Fig. 5) (70). It has been reported that DARPP-32 is activated by β-adrenergic receptors and phosphorylated by protein kinase A (PKA) in response to an increase in cellular cAMP level (59). Haloperidol enhances phosphorylation of ribosomal protein S6 (rpS6) at Ser235/236, which is independent of extracellular signal-regulated kinase (ERK)- and multicomponent mechanistic target of rapamycin complex 1 (mTORC1)-mediated activation of ribosomal protein S6 kinase 1 (S6K1), but needs intact PKA/DARPP-32 signaling (12, 131). Thus, DARPP-32 is an integrator of neural signal transmission and is the central molecular trigger that underlies the neurobiological alterations (45, 81).
Alzheimer's Disease
With more than 50% of all types of dementia, AD is the most common neurodegenerative disorder (109). AD is characterized by the development of neurofibrillary tangles (NFT) known as intraneuronal filamentous attachments and extracellular senile plaques (SPs) (117). There is strong evidence to support the theory of increased oxidative stress and circuit dysfunction in AD (19, 72). The evidence usually indicates that free radical contributes significantly to the pathogenesis of neuronal death in AD and that antioxidant systems can play an important role in preventing and regulating AD (117).
Trx1 is a highly preserved protein with many functions, from oxygen reactive foraging to chemokine activity (3). The role of Trx1 in AD seems important, since the brain tissue of AD patients shows a low level of Trx1 (88) and pronounced buildup of beta-amyloid peptide (117). In both the frontal cortex and the cornu ammonis (CA1) hippocampus regions of patients suffering from AD, the output of Trx1 is decreased. In addition, the treatment of beta-amyloid peptides in human SH-SY5Y neuroblastoma cells caused high, intermittent, and early oxidation of both Grx1 and Trx1, while overexpression of Trx1 entirely shields cells against the toxicity of beta-amyloid peptides, as shown by the viability of cellular expression (1).
Trx1 can be cleaved, creating a long peptide of 80 amino acids called thioredoxin 80 (Trx80). Trx80 was originally identified as a peripheral proinflammatory cytokine that was secreted by immune cells (38, 107). Interestingly, compared with normal brain tissue, Trx80 levels in AD were substantially diminished, even in areas with severe inflammatory alterations, suggesting that Trx80 dysfunction may be a basic feature of the disorder. In SH-SY5Y cells, Trx80 is able to suppress both β-amyloid accumulation and β-amyloid toxicity in vitro, and thus, improving Trx1 or Trx80 may be helpful in reacting to the toxic effects of β-amyloid cells (53). β-amyloid results from sequential cleavage of amyloid precursor protein (APP) by β- and γ-secretases. Normally, a nonamyloidogenic pathway, involving a-secretases, is predominant in brain. Two α-secretases, a disintegrin and metalloproteinase domain-containing protein (ADAM) 10 and 17, are responsible for the Trx1 proteolytic cleavage to Trx80 (53). The Trx80 levels of brain homogenates and cerebrospinal fluid in AD patients are also lowered relative to the equivalent controls (53). Trx80 is also greatly decreased in the cortex in AD. Trx80 inhibits and defends against amyloid beta peptide (Aβ) development by reducing the sensitivity of neurons to its toxicity (52). These results suggest that Trx80 plays a defensive function against the toxicity of amyloid beta-42 (Aβ42) and subsequently against the pathology of AD (52). The effects of the Trx80 and its underlying mechanism must be identified.
The expression Trx1 is increased in white matter astrocytes in AD brains (Table 1) (5, 119), while certain AD brain regions have been shown to decrease relative to controls (4). Postmortem examined brains were marked by decreased rates of Trx1 in the hippocampus and amnestic moderate cognitive impairment cerebellum (AMCI), a transitional phase between natural aging and adult T cell leukemia-derived factor (ADF)/human Trx1 (42, 119). Rats were prone to chronic, intermittent hypoxia, a reversible cause of cognitive decline in AD patients (71, 105), and memory loss and spatial learning are negatively associated with the level of Trx1 protein and hippocampal RNA (140). It has been seen that TrxR activity is increased in the AD brain cerebellum and amygdala, which indicates that TrxR activity increase is a countervailing resource in the face of oxidative stress and related with progressive neurodegeneration in AD (85). The reduced form of Trx1 is a negative regulator of apoptosis through binding apoptosis signal-regulating kinase 1 (ASK1) (121). Previous studies also indicate that ASK1 is involved in the cell death of neurons caused by Aβ (78). The overexpression of Trx1 also prevents SH-SY5Y cells from Aβ toxicity (1). The treatments of TrxR and Trx also help protect primary hippocampal cultures from the toxicity of Aβ, serving as a basic scavenger that obstructs neuronal lesions.
Thioredoxin System and Neurodegenerative Disease
Detailed view of involvement of Trx system in CNS disorders.
ADF, adult T cell leukemia-derived factor; AMCI, amnestic moderate cognitive impairment cerebellum; CSF, cerebrospinal fluid; CNS, central nervous system; FALS, family amyotrophic lateral sclerosis; HD, Huntington's disease; MPP+, 1-methyl-4-phenylpyridinium ion; mRNA, messenger RNA; SOD, superoxide dismutase; TR, thioredoxin reductase; Trx, thioredoxin.
Methionine sulfoxide is a reversible outcome of methionine oxidation that can be reduced by MSrs dependent on TrxR, but irreversible oxidation by methionine sulfone is infrequent and only happens in the presence of powerful oxidants. MSrs declined in the brain in AD (48) and modulate Aβ-induced neurotoxic effects in vivo (92). Aβ is also linked to many other diseases, such as muscle degeneration and glaucoma in mice, due to impaired Trx system pathogenesis (127).
Changes in Aβ rely on the system of Trx and the reduced amounts of this protein system render the cell more prone to neurotoxic Aß (78). The level of this peptide, Trx80, is dramatically decreased in the AD brain (52). Trx80 also inhibits Aβ accumulation and toxicity in vitro (53). Using models of Drosophila melanogaster, study showed that the expression of human Aβ42 in the fly brain results in gradual age-dependent accumulation of Aβ42 and reduces the life span, and reduced locomotive function, which were attenuated by Trx80 (58, 77). Soluble Aβ42 is strongly expressed in young flies, while age leads to Aβ42's gradual insoluble/fibrillary accumulation. In coexpressing flies Aβ42 and Trx80, Aβ42 aggregation was dramatically decreased and phenotype-dependent Aβ42 was reversed in early morbidity and locomotive operation (52).
Trx1 is also connected with the plasma membrane and is trafficked through the leaderless secretory pathway with a small amount of cytosolic proteins (23). Trx1 secretion is altered under oxidative stress. Trx1 and Grx1 are also liberated in the cerebrospinal fluid. Trx1 and Grx1 levels are correlated with the identified biomarkers, tau and phospho-tau, in 120 patients with AD at the early stages of mild cognitive impairment disease (4). The study suggests that Trx1 is involved in the pathogenesis of AD. A substantial reduction in Trx1 protein expression was shown in the cortices collected from aged AD patients by immunoblotting assay. This is well corroborated by evidence indicating a substantial decline in Trx1 levels in the amygdala and hippocampus/parahippocampal gyrus in earlier AD (85). Thus, Trx1 in the cerebrospinal fluid is an important biomarker for the progression of AD (4).
Caspase-6 (CASP6) activity in the Trx system has been linked to neurodegeneration, and the possible link between the two systems is yet to be investigated. In SH-SY5Y cells, the Trx1 redox state primarily mediates CASP6 proteolytic activity, whereas the availability of decreased Trx1 promotes CASP6 activation. The decreased Trx1 reduction capability of the cell was proven to be the operative factor in CASP6 activation using cell lysates, cell-free nuclear-enriched preparations, and purified enzymatic tests. This can set off a chain of responses that lead to the incapacitation of the cellular defense system and the promotion of cell death by depleting DJ-1. Detection of hippocampus tissue from a mouse model of AD showed that this finding in an in vitro model can be applied to the etiology of neurodegeneration. The role of cellular reducing capacity in CASP6 stimulation was primarily mediated by Trx1, as cellular glutathione depletion did not result in CASP6 activation or lamin-B1 cleavage in these experiments (66). Synaptosomal Grx1 and Trx1 levels were significantly lower in 1-month-old double transgenic mice expressing a chimeric mouse/human amyloid precursor protein (APP/PS1) mice, and the drop was persisted until 9 months of age, when obvious pathological hallmarks appeared, demonstrating that synaptosomal Grx1 and Trx1 levels are reduced throughout the APP/PS1 mice's life span. Reduced Grx1 levels result in a decreased capacity to reduce glutathionylated proteins to thiols, which may be severely cooperated in the synapse of APP/PS1 (76).
Parkinson's Disease
PD is the second highly common neurodegenerative disease that affects 0.3% of the general population and 1%–3% of the age group older than 65 (37, 40). Tremor, bradykinesia, and poor posture clinically describe idiopathic PD (51). In the midbrain substantia nigra pars compacta (SNpc), the presence of intracytoplasmic filamentous aggregates called as Lewy body in neurons and axons is defined as a pathological manifestation (68). The abnormal accumulation and aggregation of alpha synuclein protein (α-Syn), a major protein component of Lewy bodies, are a neuropathological hallmark of Parkinson's disease (139). Pathological aggregation of α-Syn is a well-known characteristic of several neurodegenerative illnesses, including PD, dementia with Lewy bodies, and multiple system atrophy, all of which are referred to as synucleinopathies (54). α-Syn is a protein with exceptional construction plasticity, as it can adopt a wide range of structural conformations, including oligomers, protofibrils, and fibrils (41, 89). In terms of neurotoxicity, stability, seeding, and propagation ability, each α-Syn conformation is unique. It has been suggested that the occurrence of structurally different α-Syn assemblies or “strains” may contribute to clinicopathological variation across synucleinopathies and aid in the development of strain-specific therapies (22, 90, 106). Nonmotor symptoms in PD are linked to the sequential development of Lewy bodies in various brain regions, including the olfactory bulb, dorsal motor nucleus of the vagal nerve, locus coeruleus, raphe nucleus, basal nucleus of Meynert, and pedunculopontine nucleus, in addition to deficits in both dopaminergic and nondopaminergic neurotransmission systems (15, 16, 116).
Several genetic variations and environmental factors have been identified to cause PD (125). There are several animal models for dopaminergic neuronal failure including 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) and 6-hydroxydopamine, as well as Tg mice models with null mutations in tensin homologue deleted on chromosome ten (PTEN)-induced putative kinase 1 (PINK1), Parkin, and DJ-1, or point mutations in genes located in different PARK loci. Models with mostly inherited gain-of-function mutations were developed, such as a-Syn and leucine-rich repeat kinase 2 (LRRK2), in which additional gene copies were inserted into the genome of the mouse (125). Each model represents different PD characteristics, but none of these models expresses a detailed understanding of PD pathologies (6).
PD is identified in the SNpc with eosinophilic and intracytoplasmic protein-containing inclusions in surviving neurons called Lewy bodies and Lewy dystrophic neurites together with gradual, chronic, and large depletion of neuromelanin-containing dopaminergic neurons (46). The substantia nigra cells use the dopamine neurotransmitter to interact with striatum neurons. Hence, a fall in the amount of nigral dopamine contributes to a reduction in striatal dopamine, and causes PD symptoms. The oxidative stress leads to the sequence of events, which result in neuronal dopaminergic degeneration in PD (68, 69). In addition, the monoamine oxidase (MAO) catalytic enzyme that catalyzes the oxidative deamination of dopamine and other transmitters of monoamine is identified to coincide with a selective loss of dopamine neurons of the substantia nigra (35, 98, 99). In addition, MAO-B converts MPTP to 1-methyl-4-phenylpyridinium ion (MPP+), which causes a serious and permanent PD-like syndrome in humans and animals. MPP+ declines the Trx1 expression (7). MPP+ inhibits complex I mitochondrial respiration and induces apoptosis through a cysteine-dependent aspartate-directed protease 12 (caspase-12)-dependent pathway in vitro and in vivo. The overexpression of Trx1 completely suppresses MPTP neurotoxicity in vivo, suggesting its therapeutic potential in the treatment of PD (7, 9).
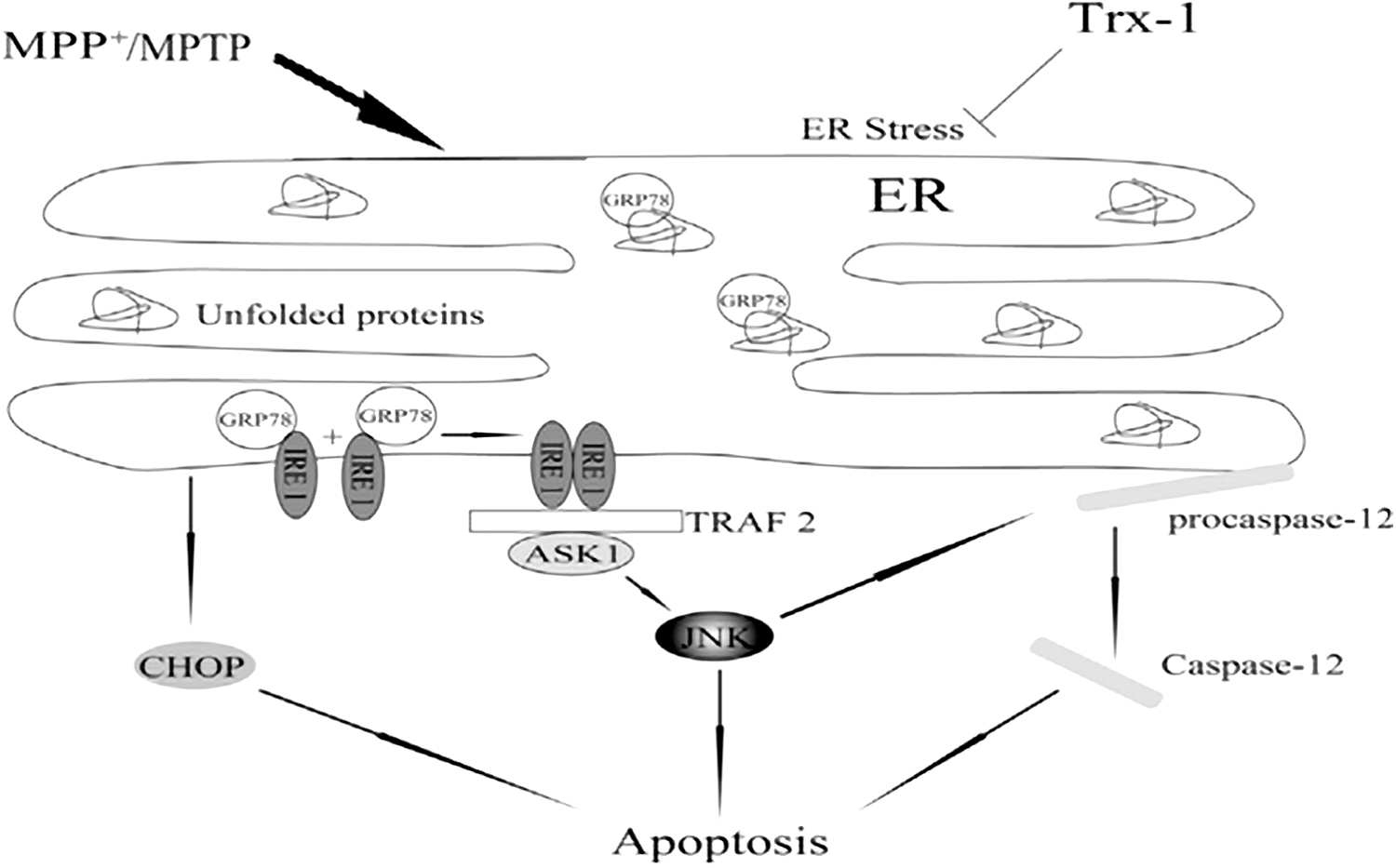
ER stress has been linked to neuronal death in PD in a number of studies (9, 82). Glucose-regulated protein (GRP78), an ER-resident chaperone, is a marker for ER stress and is involved in the regulation of ER dynamic homeostasis (28). GRP78 interacts with and suppresses ER transmembrane proteins such as inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) under nonstressed conditions. GRP78 is dissociated from these transmembrane proteins in response to ER stress, causing them to activate and triggering the unfolded protein response (UPR) (75, 138). IRE1 is thought to trigger apoptosis (134) and is closely linked to cell death (74) when inositol-requiring enzyme 1α (IRE1α) is activated, and then recruited the adaptor molecule, tumor necrosis factor receptor-associated factor 2 (TRAF2) (128). IRE1 binds to TRAF2 and activates c-Jun N-terminal kinase (JNK) (101, 128). MPP+ increases IRE1, TRAF2, and p-JNK, implying that MPP+ results in ER stress. The activation of cysteine aspartic proteases, cysteine aspartates, or caspase-12 has been reported to be involved in ER stress-induced cell death (47). In PC12 cells, the expression of procaspase-12 was likewise considerably reduced after MPP+ treatment. Furthermore, JNK inhibited MPP+-induced procaspase 12 cleavage (141). The ER stress in PD was resisted by Trx1 overexpression (141). Trx1 plays a neuroprotective role in PD by suppressing ER stress through regulating the activation of GRP78, IRE1α, TRAF2, c-Jun N-JNK, caspase-12, and CCAAT-enhancer-binding protein homologous protein (CHOP) (Fig. 6) (141). So Trx1 may be a potential target for regulating ER stress in PD.
Chemical compounds MPP+, paraquat, and rotenone inhibit mitochondrial complex I, which contribute to dopaminergic degeneration, and induce PD cells in animal models. These compounds have been extensively used to investigate neurotoxic mechanisms (14, 126). Paraquat oxidizes Trx1, while rotenone oxidizes Trx2 in SK-DAT cells as a basic mechanism for cell injury (111). Although MPP+ decreased the expressions of Trx1 and Trx2 in PC12 cells, the ratio of oxidized versus reduced Trx1 and Trx2 was relatively increased (30). Trx1 improved motor skills, restored the decreased tyrosine hydroxylase (TH) in the SNpc in mice, and protected dopaminergic neurons by suppressing ER stress in PD (9, 20, 141). In hippocampus-dependent learning acquisition, the dopaminergic system plays a key function (21). D1/D5 receptor agonists, for instance, promote long-term potentiation and boost spatial memory (55). Trx1 overexpression attenuated memory deficits induced by MPTP in mice. The expressions of TH, dopamine D1 receptor (D1R), and N-methyl
The oxidative DJ-1 protein has been found in intermittent PD patients (32). DJ-1 has the capacity to function as an antioxidant as well as a transcription factor to shield mouse culture cells and nigra substance from oxidative stress by activating expression of Trx1 via nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor (64). Antioxidant expression and detoxifying enzymes such as Trx and TrxR are associated with the Nrf2 transcription factor (34). α-synuclein is commonly found to be more exposed to oxidation by methionines and tyrosines (27). Soluble oligomers may be continually formed through synuclein methionine oxidation, whereas oxidized synuclein and hetero-oligomers may have a toxic impact on the cell setting (129). DJ-1 and Trx1 as the common endogenous antioxidants provide a new strategy against neurodegeneration resulted from oxidized synuclein (26).
Some Other Neurodegenerative Diseases
HD is a neurodegenerative disease, and it can be traced by the depletion of up to 95% of striatum by GABAergic neurons (113). Oxidative stress in the HD cell death cascade is either a causative event or a secondary variable (17). Decreases in Trx1 and TrxR1 have been identified in the plasma and erythrocyte blood samples of HD patients, as well as the peripheral oxidative stress reaction to this neurodegeneration has been shown (114). Schizophrenia is implicated in many neurological deficits that are expected to play a significant role in cognitive deficits during aging due to oxidative stress (143). Several studies have found a higher level of Trx1 in the plasma and serum of patients with schizophrenia in the first episode, as well as a high level of Trx1 in patients with long-term schizophrenia (104, 143, 144). ALS postmortem spinal cords and family amyotrophic lateral sclerosis (FALS) erythrocytes are with constant variants of SOD-1 mutated protein and elevated expression of the Trx1 gene and protein (86, 115).
Conclusion
Trx1 is a multifunctional protein. It is a cytoprotective protein that prevents cells from injury and stress. Trx1 not only serves as a significant antioxidant protection and regulating redox cell homeostasis, but also impacts energy metabolism, controls immune responses, and modulates cell growth and survival. At this stage of development, these factors play a role and also lead to medical disorders such as cancer, metabolic syndrome, aging, and multiple neurodegenerative illnesses. The mechanism and functions of Trx1 in the brain such as co-cytokines and chemokines are essential for the study of the pathogenesis of different neurodegenerative diseases. Elevated morbidity and mortality are linked with acute and chronic neurodegenerative conditions, including stroke and TBI. For the treatment of these diseases, few appropriate therapeutic methods are available. Progressive disorders characterized by the destruction of neurons secondary to oxidative stress and neuroinflammation are involved in neurodegenerative diseases. The Trx1 strategy will hopefully mitigate the adverse effects on neurodegenerative diseases today, since neurodegenerative diseases have no cure and carry a high cost to the health care system and patient families. Further studies of the Trx1 can open up therapeutic routes or prolong progressive neurodegenerative disease harm at least.
Footnotes
Acknowledgments
We thank Xianwen Zhang and Zhizhou Shi for their comments on the article.
Authors' Contributions
M.U.N.A. wrote and prepared the article. F.Y. corrected the references. F.M. helped to write one part of this article. L.B. and J.L. helped to revise the article. J.B. guided and revised the article. All authors read and approved the final article.
Author Disclosure Statement
All the authors declare no competing financial interests.
Funding Information
The Forum Review Article was supported by the National Natural Science Foundation of China (Nos. U2002220, 81660222), the Yunling Scholar (No. 1097821401), and the key laboratory for oxidative stress damage and defense in the University of Yunnan Province (2018).