
Abstract
Significance:
The development and progression of renal diseases, including acute kidney injury (AKI) and chronic kidney disease (CKD), are the result of heterogeneous pathophysiology that reflects a range of environmental factors and, in a lesser extent, genetic mutations. The pathophysiology specific to most kidney diseases is not currently identified; therefore, these diseases are diagnosed based on non-pathological factors. For that reason, pathophysiology-based companion diagnostics for selection of pathophysiology-targeted treatments have not been available, which impedes personalized medicine in kidney disease.
Recent Advances:
Pathophysiology-targeted therapeutic agents are now being developed for the treatment of redox dysregulation. Redox modulation therapeutics, including bardoxolone methyl, suppresses the onset and progression of AKI and CKD. On the other hand, pathophysiology-targeted diagnostics for renal redox dysregulation are also being developed. Urinary thioredoxin (TXN) is a biomarker that can be used to diagnose tubular redox dysregulation. AKI causes oxidation and urinary excretion of TXN, which depletes TXN from the tubules, resulting in tubular redox dysregulation. Urinary TXN is selectively elevated at the onset of AKI and correlates with the progression of CKD in diabetic nephropathy.
Critical Issues:
Diagnostic methods should provide information about molecular mechanisms that aid in the selection of appropriate therapies to improve the prognosis of kidney disease.
Future Directions:
A specific diagnostic method enabling detection of redox dysregulation based on pathological molecular mechanisms is much needed and could provide the first step toward personalized medicine in kidney disease. Urinary TXN is a candidate for a companion diagnostic method to identify responders to redox-modulating therapeutics. Antioxid. Redox Signal. 36, 1051–1065.
Acute Kidney Injury and Chronic Kidney Disease Are Groups of Heterogeneous Diseases
Acute kidney injury (AKI) is not a single disease with a homogeneous etiology, but a heterogeneous group of ailments distinguished based on their rate of progression and severity (22). Chronic kidney disease (CKD) also has several different etiologies and pathogeneses (46). However, these diseases are currently diagnosed from the consequences of renal failure based on serum creatinine, urine volume (128), and proteinuria (21), which are independent of pathogenesis. Despite the many advances in molecular biological studies on the pathogenesis of AKI and CKD, very few effective and specific therapies are available, other than supportive treatment (40).
The pathogenesis of AKI is largely related to oxidative stress in the kidney (6 –8). The primary reason for the kidney's susceptibility to hypoxia is the high oxygen demand and anatomical limitation to oxygen delivery to the renal tubules (33). These factors include the large metabolic demand imposed by active reabsorption of sodium (98); limitations on oxygen delivery to cortical tissue caused by the density of peritubular capillaries (165); the poor capacity for angiogenesis in the adult kidney (10); diffusive oxygen shunting between arteries and veins in the cortex and descending and ascending vasa recta in the medulla (99); the physiological requirement for low medullary blood flow to facilitate urine concentration (36); and the topography of the vascular–tubular arrangements in the outer medulla, which limits oxygen delivery to the thick ascending limb of Henle's loop (17).
In CKD, there are some common pathways that progresses to end-stage renal failure (45). The canonical pathway entails proteinuria-inducing angiotensin II (116), which increases levels of transforming growth factor (TGF)-β1 and reactive oxygen species (ROS), leading to renal tubular epithelial apoptosis, epithelial–mesenchymal transition, and fibrosis (76). Recent studies have shown that AKI can be a transition to CKD (34). The mechanisms by which AKI transitions to CKD include hypoxia (143) and microvascular rarefaction (144), mitochondrial fragmentation (77), persistent chronic inflammation and tissue senescence (61), activation of the renin–angiotensin system (81), cell cycle arrest in the G2/M phase (166), altered phenotype and function of renal-resident cells (34), and Wnt signaling (75).
Current evidence suggests that hypoxia, oxidative stress, and subsequent redox dysregulation are common pathophysiological factors in AKI, CKD, and AKI-to-CKD transition.
Renal Injury from Oxidative Stress Related to ROS Toxicity
In a healthy kidney, oxygen supply and demand are precisely matched through regulation of medullary blood flow and tubular workload (98). Medullary blood flow is regulated by the balance between vasodilation induced by prostaglandin E2 (129), nitric oxide (NO) (29), and atrial natriuretic peptide (79) and vasoconstriction induced by endothelin (115) and angiotensin II (17). Tubular workload is regulated by tubuloglomerular feedback (98). However, if medullary blood flow slows down beyond the compensatory mechanism due to various AKI triggers such as septic shock (117), invasive surgery (14), organ dysfunction such as heart failure (31), obese (86), or contrast medium (53, 156), the supply–demand balance for oxygen is disrupted, and the resultant hypoxia causes tubular damage and the development of AKI (114).
The major sources of ROS related to ischemia–reperfusion and hypoxia are mitochondrial complex I and complex III, respectively (26, 42), whereas the ROS source related to high glucose is nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4, a member of the NADPH oxidase (NOX) mitochondrial NADPH oxidase family (130) (Fig. 1). Because hydrogen peroxide (H2O2) is a small, uncharged molecule, it easily passes through the lipid bilayers that constitute cell and organellar membranes, diffusing throughout cells and tissues in vivo (19). The hydroxyl radical (OH・) produced from H2O2 is a highly reactive oxidizer that readily reacts with various biomolecules, leading to oxidation of lipids (134), deoxyribonucleic acid (139), and proteins (37), which can be damaging to kidney cells (Fig. 1).
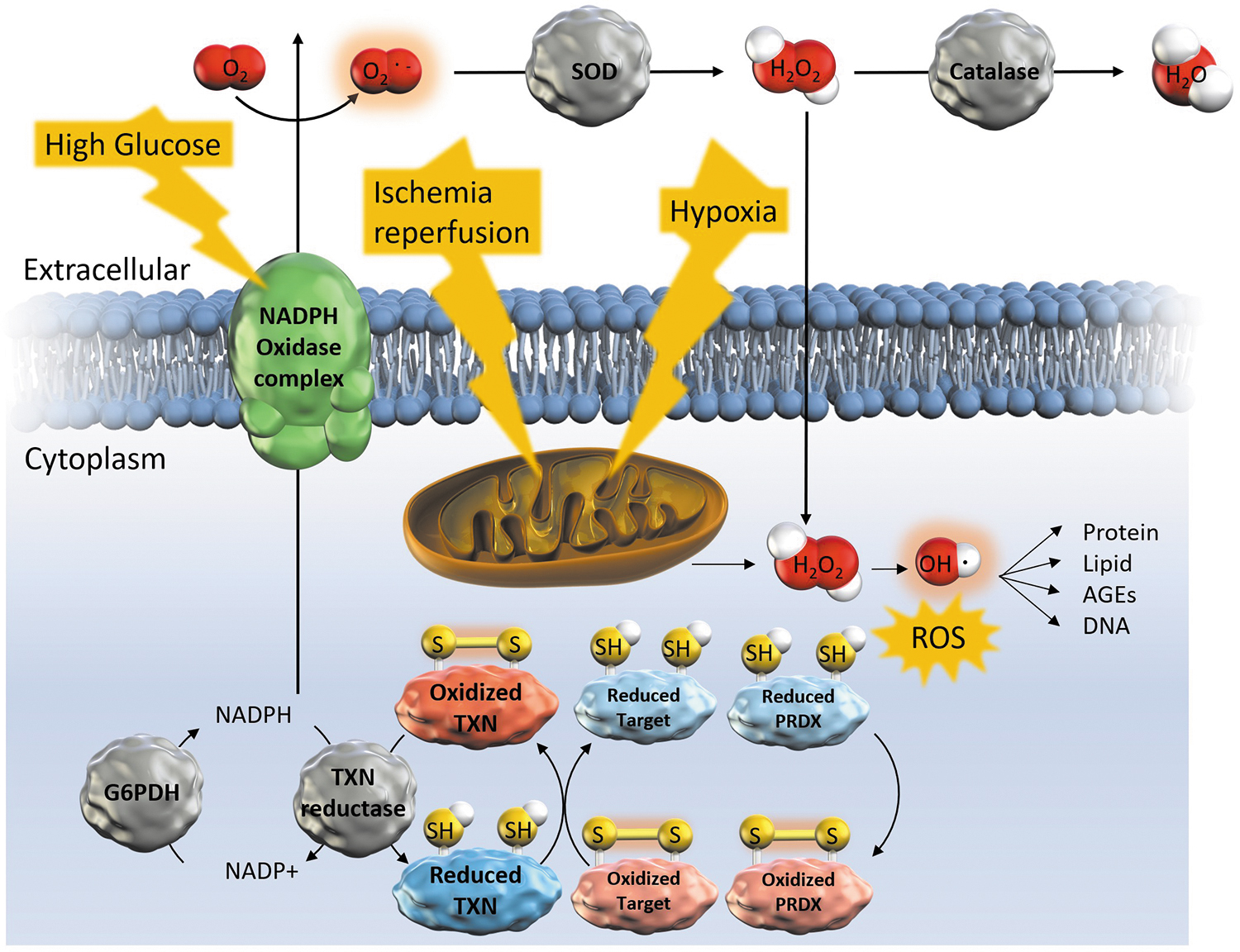
Excessive ROS generation is suppressed by Sirtuin (Sirt)3, the major nicotinamide adenine dinucleotide-dependent deacetylase in mitochondria, through activation of antioxidant enzymes (11, 13). Catalase, an H2O2 scavenger in the renal proximal tubules, inhibits interstitial fibrosis and tubular apoptosis caused by diabetes (18) and hypertension (133). Superoxide dismutase also play a role in protecting renal function (141, 167).
In addition, peroxiredoxin (PRDX) is a thioredoxin (TXN)-dependent scavenger of H2O2. As such, PRDX-6 transgenic mice suppress oxidative stress via p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) activation, inflammatory cell infiltration, renal damage, and mortality after lipopolysaccharide challenge (68). Infusion of exogenous PRDX-6 also suppresses renal ischemia–reperfusion injury, promotes the recovery of renal function, and increases the survival rate (41). Conversely, PRDX-5 knockout mice exhibit enhanced tubular damage and apoptosis, and reduced renal function (104), and PRDX-3 deficiency promotes fibrosis and inflammation with mitochondrial oxidative stress in obstructed and diabetic kidneys (48). Moreover, in patients with AKI, high tubular PRDX-3 expression is associated with a high probability of recovery of renal function (159).
These findings suggest that the TXN-dependent ROS scavenger PRDXs are specific therapeutics targeting ROS-mediated acute and CKDs.
Renal Injury Due to Oxidative Stress Targeting Cysteine Residues and Redox-Modulating Therapeutics
Cellular redox regulation is a specific mechanism that modulates signal transduction by targeting cysteine residues in proteins. Cysteine residues are the target of redox-modulating therapeutics (16). Cysteine residues are modified by ROS such as H2O2 to form disulfide bonds (105) (Fig. 1). They are also targets of hydrogen sulfide (H2S), which is recently discovered to be synthesized in the body and acts as a signaling molecule (84, 164). H2S production is decreased in AKI (161), CKD (8), obesity (157), and type 2 diabetes (157). On the other hand, H2S relieved the mouse kidney injury by ischemia reperfusion (152), cisplatin (56), or obstructive kidney injury (57).
The other key molecules involved in cysteine redox modulation are 8-nitro cyclic guanosine monophosphate (cGMP) and transcription factor NF-E2-related factor 2 (Nrf2). The ROS superoxide targets NO and carbon dioxide, producing nitroso-peroxocarboxylate (124), which reacts with guanine triphosphate to produce 8-nitro cGMP (3, 4). This 8-nitro cGMP induces S-guanosylation of cysteine residues in Kelch-like ECH-associated protein 1 (Keap1), a cytoplasmic anchor and ubiquitin ligase that activates Nrf2 (125), leading to its translocation into the nucleus to activate target genes (3, 4).
The target genes of Nrf2 include TXN, TXN reductase (TXNR), PRDXs (146), glucose-6-phosphate dehydrogenase (146), glutathione transferase (50, 55), quinone oxidoreductase (NQO1) (69), heme oxygenase (HO-1) (5), drug transport genes (52), and anti-inflammatory genes (69). Inhibition of protein disulfide isomerase, a TXN chaperone protein, reduces nuclear translocation of Nrf2, leading to mitochondrial dysfunction and renal cell apoptosis (109). Conversely, TXN, TXNR1, and the glutathione pathway are major components mediating stabilization of Nrf2 via redox regulation of cysteine residues in Keap1 (128). Thus, Nrf2 is regulated by the interaction of TXN-based enzymes with oxidants (44), and cysteine residues in Keap1 are potential therapeutic targets (121).
Nrf2 inducers targeting the cysteine residues in Keap1 include bardoxolone methyl (CDDO), sulforaphane, dimethyl fumarate (Tecfidera®), and 3-(dimethylamino)-4-((3-isothiocyanatopropyl)(methyl)amino)cyclobut-3-ene-1,2-dione as well as N-acetylcysteine and cysteamine combined with a pro-glutathione molecule (27, 35, 80, 107, 169). CDDO ameliorates oxidative stress-induced tubular injury (92) and improves glomerular filtration rate (GFR) in patients with type 2 diabetic nephropathy (108) or autosomal dominant polycystic kidney disease (38). CDDO protects against tubular damage caused by proteinuria through inhibition of mitochondrial ROS generation, and suppression of inflammation (91). Nrf-2 activation also inhibits AKI to CKD transition (73). These suggest that redox-modulating therapeutics target cysteine residues in redox-sensitive proteins.
Renal Expression of TXN and TXN-Related Proteins
TXN gene is transcriptionally activated by constitutive binding of the Nrf2/small musculoaponeurotic fibrosarcoma (Maf) complex to the antioxidant response element in its promoter region (63). Its product, TXN protein, is a 12 kDa cysteine-modifying enzyme that acts as an intracellular redox control protein that maintains a reducing environment within cells (Fig. 2). Oxidative stress leads to cellular damage resulting from the loss of intracellular NADPH (151), H+ donor of TXN. In catalyzing the reduction of various redox-sensitive proteins within cells, TXN is itself oxidized. The oxidized TXN is, in turn, reduced by TXNRs, making it available for reuse as a reducing agent (137).

The reduction of TXN by TXNR plays an important role in the pathophysiology of AKI, since the inhibitor of TXNR, ifosfamide caused AKI dose dependently (170). TXN scavenges oxygen radicals via TXN-dependent PRDXs. TXN is also required for intrinsic production of H2S (82). The first study examining the role of TXN in kidney disease was published in 1996 by Minakawa et al. (83), who reported that TXN expression is upregulated during rejection of transplanted kidneys. The next year, it was reported that intraperitoneal administration of ferric nitrilotriacetate, an oxidative stress inducer, elicited a 2.5-fold increase in TXN expression within the proximal tubules of mice, and that tubules strongly expressing TXN were less damaged by the oxidative stress (145).
Within kidneys, TXN and TXNR1 are present in the cytoplasm of all cell types, whereas TXNR2 is detected mainly in mitochondria. The TXN-dependent radical scavengers PRDXs -3 and -5 are also located within mitochondria, whereas PRDX-6 is found in lysosomes (96). The organs exhibiting the highest expression of TXNR messenger ribonucleic acid (mRNA) are the liver and kidney in proximal tubules of medullary rays (119). TXN mRNA and protein are localized specifically in the proximal and distal tubules. TXN-interacting protein (TXNIP) is most abundant in the glomerulus, distal tubules, and collecting ducts, and levels are significantly elevated in kidneys exhibiting diabetic nephropathy. Transgenic mice overexpressing TXN show resistance to AKI caused by ischemia/reperfusion injury (58).
Extracellular TXN Excretion by Oxidative Stress, Including AKI
TXN is secreted from several cell types through a leaderless pathway in response to various stimuli (32,118). For example, antigen-presenting dendritic cells release TXN, which is then able to enter T cells and provide the reducing environment necessary for their activation (32). Release of TXN is oxidative stress-dependent and is induced via its redox active center (67). Levels of TXN extracellular circulating plasma in the blood are known to be elevated in oxidative stress-related diseases, such as myocardial infarction (101).
In the kidney, extracellular TXN release increases in the urine with oxidative stress in AKI (59) (Fig. 2). Urinary TXN increases as early as urinary neutrophil gelatinase-associated lipocalin (NGAL) from 1 to 2 h after the initiation of cardiopulmonary bypass (59). Urinary TXN is unaffected by extrarenal oxidative stress-related diseases, such as hypoxemia or myocardial infarction (168). Moreover, urinary TXN is increased about 100-fold in response to a decrease in TXN in the renal parenchyma in AKI. In patients with AKI, the urinary TXN fraction in an oxidized state is higher than in healthy subjects. This suggests that urinary TXN is a potentially useful redox dysregulation-specific biomarker of AKI that could contribute to the establishment of oxidative stress specific AKI treatment strategies (59) (Fig. 3).

Urinary TXN and Pathology-Specific Diagnosis
The poor prognosis of AKI may be attributable to its delayed diagnosis, its complex etiology, and the heterogeneity of its pathogenesis (90). Attempts are being made to achieve earlier diagnoses by using novel biomarkers. These include NGAL (85, 89), kidney injury molecule-1 (KIM-1) (49), L-type fatty acid binding protein (110), and interleukin (IL)-18 (103), among others. To further improve prognosis, AKI biomarkers should provide valuable insight into the molecular mechanisms underlying this complex and heterogeneous disease and information for selecting appropriate clinical interventions (6). However, there is no diagnostic test that measures the redox state in vivo (122).
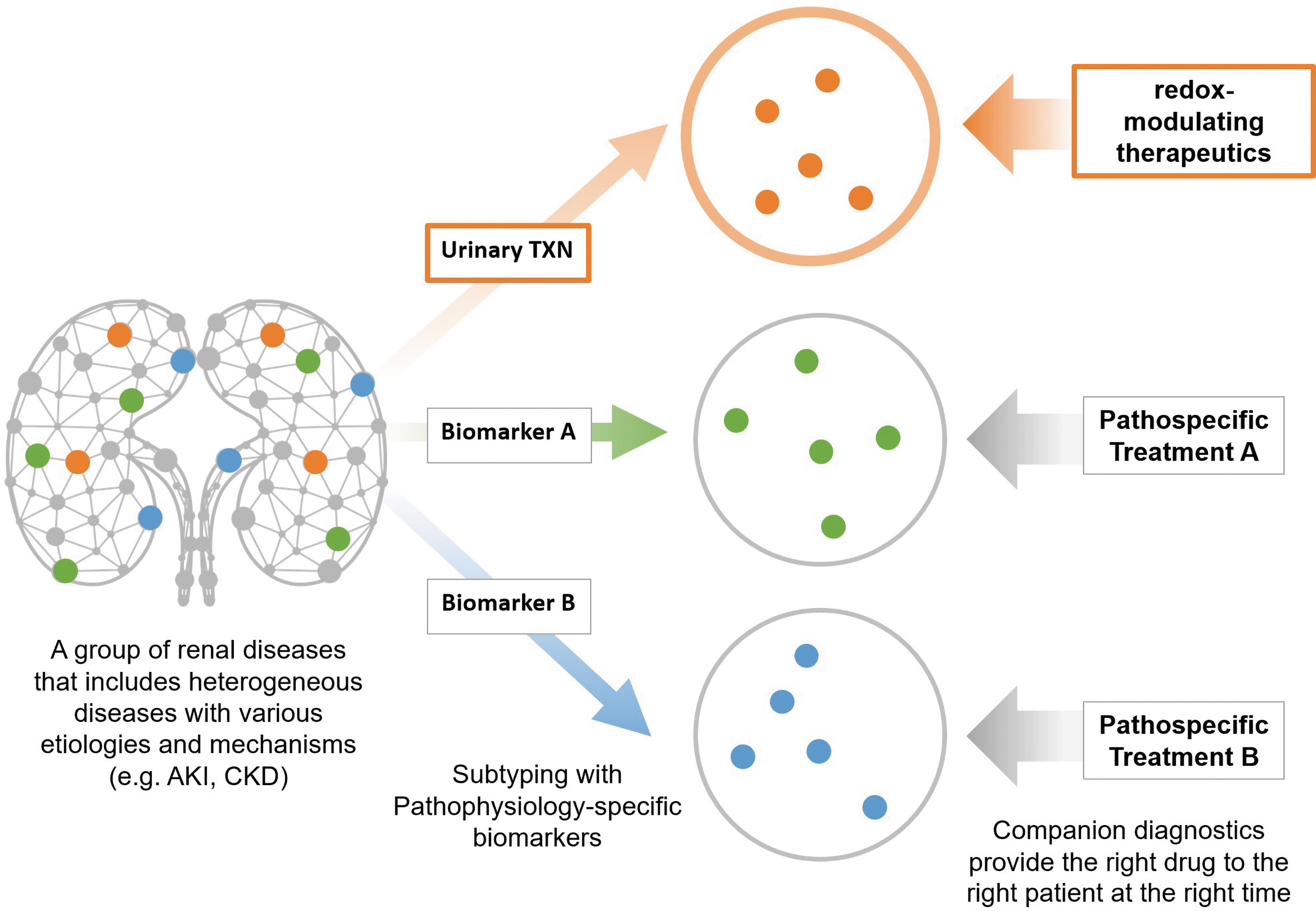
Urinary TXN is a potential biomarker that is able to capture the oxidative stress-induced redox dysregulation in AKI and CKD. TXN is secreted from tubular epithelial cells into the urine in response to oxidative stress, which depletes intratubular TXN. Urinary TXN is superior to conventional AKI biomarkers because of its direct relationship to redox dysregulation by oxidative stress. Urinary TXN makes it possible to distinguish patients who have developed redox disorders from among a heterogeneous population and to then treat them with redox-modulating therapeutics, which is expected to improve the prognosis of renal diseases (Fig. 3).
Apoptosis Signal Regulating Kinase1 as a Therapeutic Target in Kidney Disease
Apoptosis signal-regulated kinase 1 (ASK1) is a member of the mitogen-activated protein (MAP) kinase kinase family, which induces apoptosis by activating the JNK/p38 MAP kinase pathway. In the steady state, ASK1 is directly inhibited by TXN; however, oxidative stress induces S-nitrosylation of TXN and activation of ASK1 (138). ASK1+/+ mice have more severe renal dysfunction in AKI via JNK p38K and monocyte chemoattractant protein-1 (MCP-1) (147). ASK1 is also involved in the activation of Toll-like receptor 4 (TLR4), which is responsible for innate immune signaling in renal ischemia–reperfusion injury (12, 87) and in renal fibrosis in the mouse unilateral ureteral obstruction (UUO) model (74). In humans, activation of ASK1 is observed when renal allograft tubular cells experience rejection (7).
Based on those findings, ASK1 inhibition is thought to be a potentially useful therapeutic target for treatment of fibrosis in both AKI and CKD (148). Selonsertib (GS-4997) and GS-444217, orally administered ASK1 inhibitors, are effective for the treatment of diabetic kidney disease (25, 71) and liver fibrosis (100, 171).
TXN and TXNIP in Kidney Diseases
CKD is defined as proteinuria or abnormal renal function lasting more than 3 months. CKD can be caused by a variety of disorders, including diabetes, hypertension, chronic glomerulonephritis, and AKI. The onset and progression of CKD are mediated via multiple mechanisms, including the TXN redox system. Systemic overexpression of TXN in mice prevents the onset of diabetic nephropathy (43). Urinary excretion of TXN correlates positively with the rate of annual estimated GFR (eGFR) decline in patients with diabetic nephropathy (150). H2S converts oxidized TXN to reduced TXN and promotes dissociation of TXN from TXNIP; it inhibits cell injury (78).
TXNIP, also known as vitamin D3 upregulated protein 1 (VDUP1) or TXN binding protein-2 (TBP-2), was first identified in 1999 by Nishiyama et al. (95) as a negative regulator of TXN (54). Nrf2 represses TXNIP expression by binding to the antioxidant response element (−1286 to −1276) of the TXNIP promoter, which prevents the induction of TXNIP by high glucose (44). TXNIP plays a key role in the induction of NOX4 via an ROS signaling pathway under conditions of hyperglycemic stress (131). High glucose increases TXNIP transactivation via a TGF-β1-independent pathway (111), but it is mediated by the transcription factors Krüppel-like factor 6 (KLF6) and peroxisome proliferator-activated receptor γ (112) as well as epigenetic mechanisms (30) and several microRNAs (153).
High glucose increases TXNIP expression and decreases TXN expression, and TXNIP knockdown inhibits the progression of diabetic nephropathy (2, 132). TXNIP knockout suppresses hyperglycemia-induced tubular transformation (155). The allergy medication tranilast suppresses TXNIP and oxidative stress in an animal model of diabetic nephropathy (142). Elevated levels of TXNIP in urinary sediment from patients with type 1 diabetes are associated with decreased eGFR and diabetic kidney disease (88). TXNIP also regulates collagen expression in mesangial cells in patients with CKD (131). Advanced glycation end products also increase TXNIP (149).
TXNIP Targets Inflammasome in CKD
The specific mechanism by which TXNIP contributes to the pathophysiology of diabetic nephropathy and CKD was clarified in a recent study (172). TXNIP binds to the nod-like receptor protein 3 (NLRP3) inflammasome complex (172), resulting in increased proteinuria and albuminuria as well as podocytosis and glomerulopathy (1). These effects can be ameliorated via the mammalian target of rapamycin inhibition achieved through TXNIP suppression (132, 136, 137).
Inflammasomes are the center of an innate immune pathway in which sterile inflammation occurs in a pathogen-free environment. TXNIP is involved in the NLRP3 inflammasome. Pattern recognition receptors such as NLRP3, absent in melanoma 2 (AIM2) and NLR family CARD domain containing protein 4 (NLRC4), undergo structural changes on recognition of specific stimuli (agonists) and associate with proteins such as apoptosis-associated speck like protein (ASC) and caspase-1 to form a complex called an inflammasome. In other words, the TXN-TXNIP system serves as a conduit between oxidative stress and innate immunity. Abais et al. used molecular-weight exclusion chromatography and confocal microscopy to observe cultured podocytes with and without verapamil-induced TXNIP inhibition. Their results showed that stimulation with high homocysteine mobilizes TXNIP into the NLRP3 inflammasome complex, which is then involved in increasing caspase-1 activity and IL-1β production (1). TXNIP also plays a role in the induction of tubular autophagy (47).
Activation of TXNIP and NLRP3 is associated with renal ischemia/reperfusion-mediated induction of AKI via production of cleaved caspase-1, IL-1 β, and IL-18 (163). Inhibition of TXNIP suppresses production of type IV collagen and interstitial fibrosis in rats with streptozotocin-induced diabetic nephropathy (140). Moreover, TXNIP knockout in UUO model mice suppresses the renal fibrosis; macrophage invasion; apoptosis; α-smooth muscle actin (α-SMA), TGF-β1, connective tissue growth factor (CTGF) and MCP-1 expression; NLRP3 inflammasome formation; and Smad3, p38 MAPK, ERK1/2, nuclear factor-kappa B (NF-κB), 8-hydroxy-2'-deoxyguanosine (8-OHdG), hemoxigenase-1, and Nox4 activation seen in wild-type mice (160). These findings suggest that the preventive effect on diabetic nephropathy of targeting TXNIP may be mediated via an NLRP3 inflammasome-autophagy pathway.
AKI and Multiple Organ Failure
Systemic infections, including severe acute respiratory syndrome coronavirus 2 (coronavirus disease 2019 [COVID-19]), are associated with AKI and cause multiorgan dysfunction (23, 106, 117). The flavonoid quercetin suppresses the NLRP3 inflammasome via regulators such as TXNIP, SIRT1, and Nrf2 and it is a potential treatment for severe inflammation in patients with COVID-19 (120). It has become clear that kidney failure leads to failure of multiple organs (126). The high mortality rates in AKI (24, 70) are thought to be the result of multiple distant organ failure, not the kidney dysfunction per se (126). AKI leads to insufficient clearance of soluble IL-6, which has been shown to increase vascular permeability and cell inflammation via dysregulation of salt and water channels in the lung (113), brain (72), heart (60), liver, bone marrow, and gastrointestinal tract.
Notably, TXN is reportedly effective against this AKI-multiorgan system linkage. For example, a long half-life human albumin-TXN fusion protein (HSA-TXN) reduces lung injury caused by bilateral renal ischemia/reperfusion injury, suppresses increases in plasma IL-6 levels, and reduces macrophage migration inhibitory factor (MIF) expression and IL-6-CXC chemokine ligand (CXCL)1/2-mediated neutrophil infiltration into the lung (93). In addition, HSA-TXN significantly increases the survival rate among mice subjected to renal ischemia/reperfusion injury (93). Inhibitors of ASK1 are potential drug targets to suppress multiorgan dysfunction (100). These findings suggest that redox-modulating therapeutics may be useful for treatment of the multiorgan-renal syndrome caused by AKI. The heterogeneity of the AKI patient population can be solved by diagnosing AKI from the viewpoint of redox dysregulation (Fig. 4).

TXN and Macrophage MIF in Kidney Diseases
HSA-TXN suppresses AKI induced by contrast medium (65), cisplatin (64), or rhabdomyolysis (94). HSA-TXN inhibits the production of 8-nitro-cGMP (39) and suppresses renal ischemia/reperfusion injury-induced expression of MIF (94, 95). Intracellularly, MIF directly binds to and interacts with TXN (135) and TXNIP (62), and has been implicated in various diseases ranging from inflammatory diseases such as sepsis, rhinitis, and rheumatoid arthritis, to heart failure, myocardial infarction, AKI, organ fibrosis, and numerous malignancies (51). The MIF transgenic mice exhibit progressive increases in matrix within the mesangium with collagen IV accumulation. Mice showing a high expression of MIF often died of renal failure at 8 weeks, but most survived, despite significant proteinuria and progressive renal failure. Podocytes from these transgenic mice frequently exhibited characteristic ultrastructural changes, including cell flattening, contracted foot processes, and chorionic transformation (123). The MIF is also reported to be a cystic growth factor in autosomal dominant polycystic kidney disease (20).
TXN and Personalized Medicine in Kidney Disease
Personalized medicine aims at providing the right drug to the right patient at the right time (28). Personalized medicine also has advantages for drug development (158). Oncology is at the forefront of personalized medicine (102) because the specific genetic mutation is, itself, the pathophysiology that is the target of the therapeutic agents and companion diagnostics (9). On the other hand, many difficulties remain before personalized medicine in kidney diseases (28). Our current understanding of the interactions between the genetic and environmental factors that determine the appropriate targeted therapies for treatment is incomplete (97) (Fig. 5). For example, the latest high-throughput sequencing technologies revealed hundreds of gene mutations and genetic variants associated with renal disease (162). However, important gaps remain in our understanding of the mechanisms of action of the genetic components of CKD (97). In addition, CKD is often not due to expected environmental causes such as diabetes or hypertension, and no other conditions have exhibited a cause–effect relationship with CKD (154).
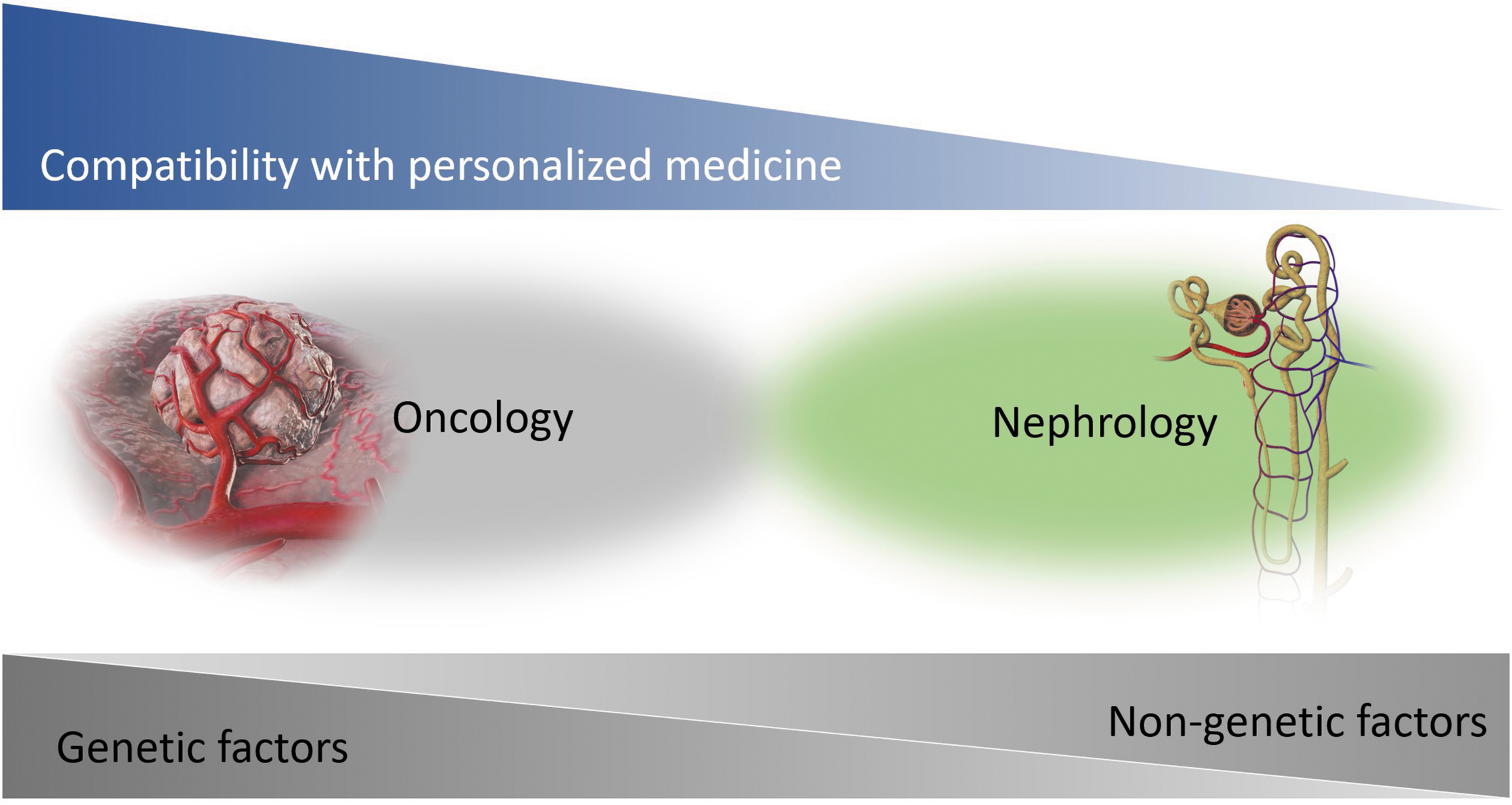
At present, therefore, the pharmacogenomics are not a suitable basis for personalized medicine in kidney diseases. As the second-best alternative, pathophysiological causes that are neither genetic nor environmental should be targeted for companion diagnostics (15). The evidence summarized in this article suggests that redox dysregulation resulting from oxidative stress, hypoxia, or ROS is a common etiology in AKI and CKD and would be an effective new target for diagnosis and treatment of these kidney diseases. Renal redox dysregulation increases urinary TXN (59), which could serve as a useful companion diagnostic biomarker to identify responders to redox-modulating therapeutics (Fig. 6). For right-time diagnoses, rapid measuring equipment has been developed for urinary TXN (168). Chemiluminescent enzyme immunoassays for TXN can be completed in <6 min, as compared with the more than 4 h required to complete conventional enzyme-linked immunosorbent assays (66). Urinary TXN could give physicians the ability to provide redox-modulating therapeutics to the right patients at the right time, enabling the first step toward personalized medicine in kidney disease.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This article is prepared with funding from the Japan Society for the Promotion of Science (JSPS KAKENHI grant nos. JP21K08252, JP20K09143, JP20H03696, JP20K08630, and JP18K08203), research grants from the Life Science Innovation Center, University of Fukui to K.K. and from the Headquarters for Innovation Society-Academia Cooperation, University of Fukui to K.K. Dr. William F. Goldman, MST Editing Company, provided writing and editorial assistance. Mr. Takashi TSUJINO, Science Graphics. Co., Ltd., provided assistance in the preparation of high-resolution images based on authors' detailed directions.