
Abstract
Significance:
The epithelial/mesenchymal transition (EMT) is commonly associated with tumor metastasis. Oxidative and nitrosative stress is maintained in cancer cells and is involved in the EMT. Cancer cells are endowed with high levels of enzymatic and nonenzymatic antioxidants, which counteract the effects of oxidative and nitrosative stress. Thiol-based antioxidant systems such as the thioredoxin/thioredoxin reductase (Trx/TrxR) and glutathione/glutaredoxin (GSH/Grx) are continually active in cancer cells, while the thioredoxin-interacting protein (Txnip), the negative regulator of the Trx/TrxR system, is downregulated.
Recent Advances:
Trx/TrxR and GSH/Grx systems play a major role in maintaining EMT signaling and cancer cell progression.
Critical Issues:
Enhanced stress conditions stimulated in cancer cells inhibit EMT signaling. The elevated expression levels of the Trx/TrxR and GSH/Grx systems in these cells provide the antioxidant protection necessary to guarantee the occurrence of the EMT.
Future Directions:
Elevation of the intracellular reactive oxygen species and nitric oxide concentrations in cancer cells has been viewed as a promising strategy for elimination of these cells. The development of inhibitors of GSH synthesis and of the Trx/TrxR system together with genetic-based strategies to enhance Txnip levels may provide the necessary means to achieve this goal. Antioxid. Redox Signal. 36, 1037–1050.
Introduction
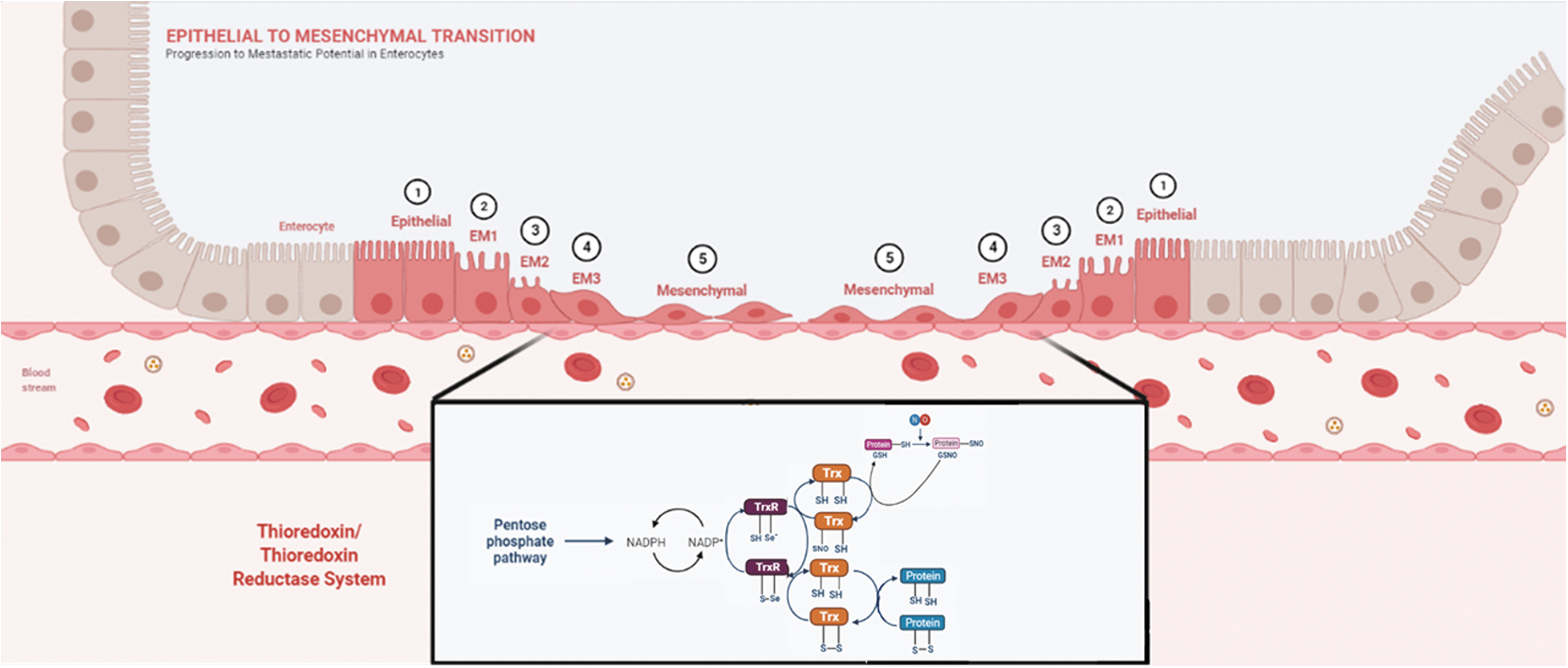
The epithelial/mesenchymal transition (EMT) and its reverse, named the mesenchymal/epithelial transition (MET), are critical events that occur during embryogenesis, and in cancer cells commonly associated with metastasis (43b, 73). Cancer cells of epithelial origin that undergo the EMT become less differentiated fibroblastic-type mesenchymal cells. The process involves the disruption of cell/cell adhesion and cellular polarity, elevated expression of matrix-degrading enzymes, remodeling of the cytoskeleton, cell/matrix adhesion changes, and resistance to apoptosis (37, 91). The EMT has become an interesting target for anticancer therapy (105).
The occurrence of the EMT and the formation of cancer stem cells (CSC) are associated events (68). Overexpression of the EMT-related transcription factors, Snail, Slug, ZEB1, Twist1, and Twist 2, is accompanied with stemness in cancer cells (reviewed in Ref. 93). Upon induction of the EMT, cell proliferation is inhibited in most cases, becoming an impediment for the establishment of metastatic lesions. The appearance of proliferating CSC among cancer cells undergoing the EMT may provide the necessary means for formation of established metastatic lesions (111, 57).
Cancer cells' mesenchymal-like features are associated with low cellular differentiation and a poor prognosis. The role of the EMT has been related to metastatic dissemination, which may be counteracted by senescence through key shared signaling points that entwine both processes. The EMT is incomplete in cancer cells, leading to multiple transitional states (29).
At the molecular level, the EMT is associated with the transcriptional repression of the cell adhesion molecule E-cadherin and elevation of the expression levels of N-cadherin and vimentin (21, 42, 53). Transcription factors, such as nuclear factor-kappa B (NF-kΒ), SOX4, Snail, Slug, and Twist1, also play an essential role in EMT activation (41, 79). The constitutive activation of the canonical signaling pathways driven by the EGFR, Src kinase, Ras/extracellular regulated kinases1/2 mitogen-activated protein kinases (ERK1/2 MAPK), and the phosphatidylinositol-3 kinase (PI3K)/Akt in cancer cells, upregulates the transcription factors associated with the EMT (21, 96).
EMT inducers include hypoxia, cytokines, and growth factors secreted by the tumor microenvironment, innate and adaptive immune responses, and treatment with antitumor drugs (91). The EMT and inflammation are associated through the production of proinflammatory factors in cancer cells. In addition, inflammation is a potent inducer of the EMT in cancer cells, consolidating a self-sustaining system (103).
Transforming growth factor-ß (TGFB) is a proinflammatory factor and a major EMT inducer that is frequently overexpressed in cancer cells. TGFB promotes tumor invasion and metastasis and controls the production of reactive oxygen species (ROS) (7, 55, 112a). In pancreatic cancer cells, TGFB induces NADPH oxidase (NOX) 4 gene expression along with the elevation in intracellular ROS levels (90). TGFB can also induce NOX2 gene expression, and its activation is dependent on the subunit NCF4 (p40phox) in HeLa human cervical cancer cells (52).
Another important oxidant that is generated at inflammatory sites is the signaling free radical nitric oxide (NO). NO is produced by the three isoforms of the enzyme NO synthase in normal and tumor cells (31, 75). The inducible isoform (NOS2) is a major source of NO in various types of cancer and its elevated expression is indicative of a poor prognosis (63). The generation of NO and ROS plays an essential role in maintaining oxidative/nitrosative stress in cancer cells, acting directly or indirectly during the EMT (19, 31). ROS and NO participate in metastasis and in the induction of the EMT (75). To counteract the effects of oxidative and nitrosative stress, cancer cells are endowed with high levels of enzymatic and nonenzymatic antioxidants.
Oxidative/nitrosative stress may be described as the disruption of the equilibrium between the production and elimination of ROS/NO in an organism. At the onset of this balanced disruption, specific thiol-based proteins, and the tripeptide glutathione (GSH) may alter the cellular redox state, modifying the conformation and activity of target proteins, reestablishing homeostasis (47, 28).
Signaling processes in cancer cells must be protected against intracellular oxidative and nitrosative stress by thiol-dependent and thiol-independent antioxidant systems. In mammals, there are two primary thiol-dependent antioxidant systems, thioredoxin/thioredoxin reductase/thioredoxin/thioredoxin-interacting protein (Trx/TrxR/Trx/Txnip) (29) and GSH/glutaredoxin (GSH/Grx) (28, 36, 39, 84). This review presents evidence concerning the supportive role played by both systems to maintain EMT signaling and cancer cell progression.
Trx/TrxR and the epithelial/mesenchymal transition in cancer cells
Trx is a 12-kDa multifunctional protein that regulates redox homeostasis in cells. Two isoforms of Trx are present in mammalian cells: cytosolic Trx1 and mitochondrial Trx2 with two conserved Cys residues (Cys32 and Cys35 for Trx1, and Cys31 and Cys34 for Trx2) at their redox-active site.
The effects of Trx2 on the regulation of the EMT have been scarcely explored. However, the study on NMuMG mouse mammary epithelial cells undergoing EMT stimulated by TGFB deserves attention. There was an increase in the cytoplasmatic and mitochondrial levels of ROS in these cells stimulated by TGFB concomitant with increased levels of fibronectin and high-mobility group AT-hook 2 protein, an EMT marker and a mediator of the EMT and metastatic progression, respectively. Overexpression of Trx2 impairs the expression of both markers (43), suggesting the existence of adequate expression levels of thiol-based antioxidants to maintain redox homeostasis and the occurrence of the EMT.
Trx1 stimulates cell proliferation, cell cycle progression, and angiogenesis (4, 5, 30, 78). Trx1 plays a major role in cellular redox balance and signaling in normal cells and cancer cells (66). Trx1 is highly expressed in many human primary cancers including in pancreatic cancer and hepatocarcinoma (HCC) (77, 78).
Treatment of HCC with sorafenib, a multikinase inhibitor, targets RAF kinases and downregulates specific EMT-related pathways such as integrin signaling. Trx1 downregulation in sorafenib-treated SNU475 HCC cells weakens cell resistance. Sorafenib induces reduction of cysteine residues in critical signaling proteins of the oncogenic pathways in HCC cell. Trx1 downregulation may act in synergy with the thiol reductive effect of sorafenib on STAT3, turning advanced HCC cells into sorafenib-sensitive cells (65). These findings support the notion that a combination of sorafenib with Trx inhibitors could prove effective as a therapeutic approach in certain cancers.
TXNDC12, a Trx-like protein, is upregulated in highly metastatic HCC cells, and in portal vein tumor thrombi and lung metastasis tissues of HCC patients. Induction of expression of TXNDC12 stimulates the metastatic behavior of HCC cells through activation of β-catenin and upregulation of ZEB1, which leads to the EMT. These findings corroborate the potential of TXNDC12 as a prognostic factor and
Fractionated ionizing radiation (FIR) promotes the EMT in human colorectal cancer (CRC) cell lines HCT116, HT29, SW480, and SW620. Overexpression of the signaling protein suppressor of cytokine signaling 1 in these cells upregulates the expression of Trx1 and blocks the FIR-induced EMT (51a).
In CRC and other cancer cells, the calcium-binding protein S100P plays an important role in tumor growth, invasion, and metastasis (23). Trx1 acts in concert with the protein S100P in promoting CRC cell invasion and metastasis. Overexpression of Trx1 and S100P in SW480 cells, a CRC cell line obtained from the primary tumor, which originated from the SW620, a cell line derived from a lymph node metastasis, promotes the expression of the protein, S100A4, and the phosphorylation of Akt. These events are associated with the occurrence of the EMT. The expression of S100A4 is higher in human CRC tissues compared with the normal adjacent tissues. Elevated expression of S100A4 in human CRC tissues is correlated with lymph node metastasis and low survival. Silencing S100A4 or inhibition of Akt phosphorylation abolishes the S100P- or Trx-1-mediated CRC cell EMT, migration, and invasion (64).
Trx is part of a thiol-based antioxidant complex system that is involved in critical physiological functions. The Trx system plays an important role in cell growth, cell proliferation, DNA synthesis, and apoptosis. It is made up of the pentose phosphate pathway, NADPH, Trxs, and the TrxRs, and is highly conserved.
The enzymes TrxRs belong to the pyridine nucleotide-disulfide oxidoreductase family. They are selenium-containing flavoenzymes and within their catalytic site have a conserved sequence—Cys-Val-Asn-Val-Gly-Cys—an NADPH binding site, and a Cys-selenocysteine sequence at the C-terminal domain that interacts with the catalytic site to perform their redox activity. They undergo reversible oxidation/reduction, existing as homodimers in normal and cancer cells. Three TrxRs isoforms have been characterized in mammalian cells: cytosolic TrxR1 with a molecular weight of 54 kDa, a mitochondrial TrxR2 with a molecular weight of 56 kDa, and a specific Trx GSH reductase with a molecular weight of ∼66 kDa, which is expressed in human testis (66, 74). TrxRs are highly active enzymes that consume NADPH and maintain Trxs in their reduced state (74).
The Trx system provides reducing equivalents for many proteins, performing essential functions in the defense against oxidative stress (50, 66). Regarding the NO-derived oxidative modifications in proteins, S-nitrosylation is by far the best understood. S-nitrosylation is a posttranslational modification controlled by the opposing actions of nitrosylases and denitrosylases (102). The action of Trx1 as a denitrosylase is found in normal and tumor cells (5, 9, 10) (Fig. 1).
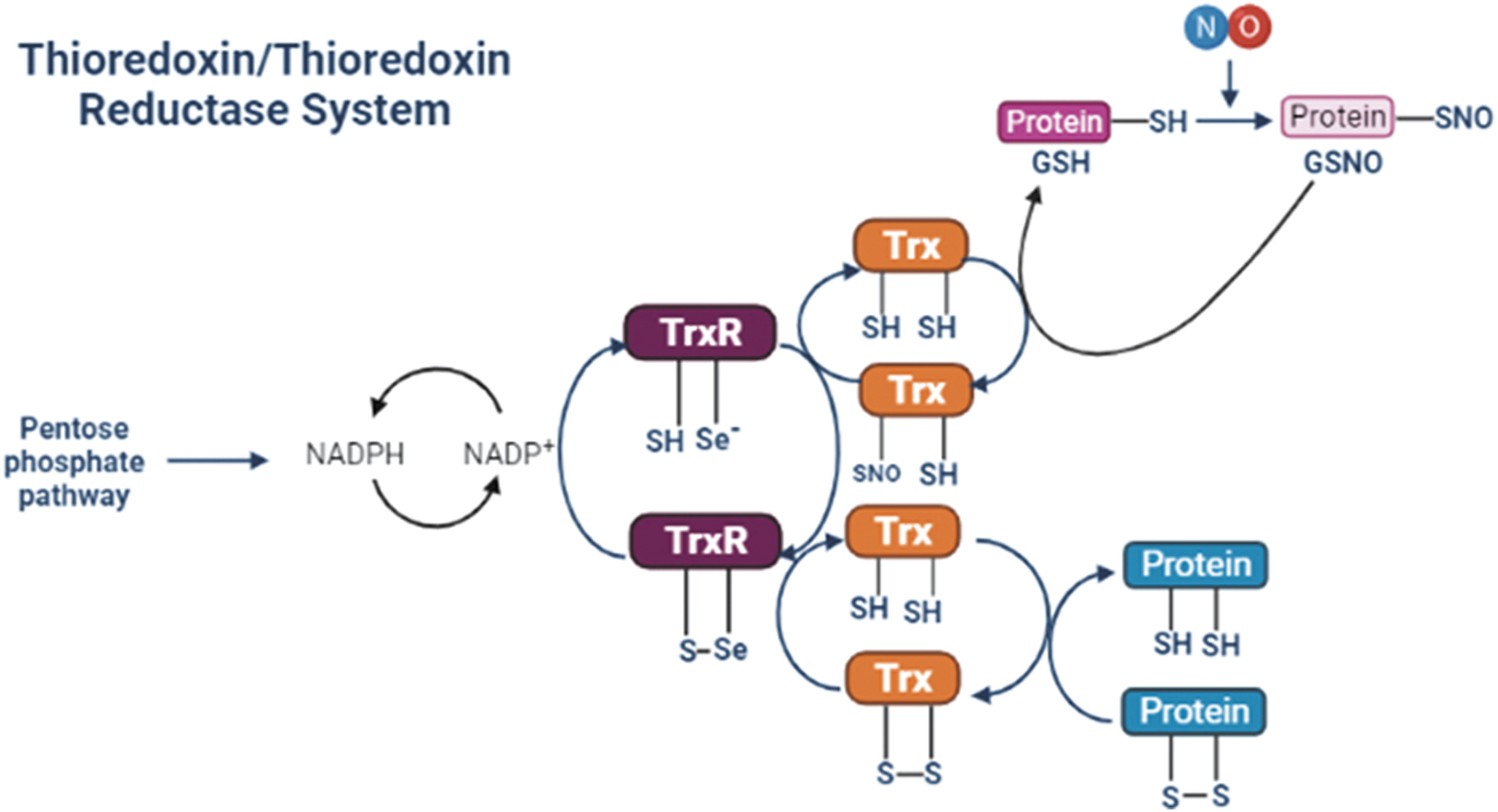
Considering the role of Trxs/TrxRs in important cellular functions and that redox signaling is involved in almost all “Hallmarks of Cancer” (35), it is anticipated that Trx/TrxR activity is associated with cancer cell development. The capacity of cancer cells to survive is related to their function in upregulating antioxidant defenses, which is correlated with progression in different cancer cell types (8, 22, 74, 117a). High expression levels of Trx/TrxR may promote tumor growth, invasion, and metastasis (95, 117a).
Overexpression of TrxR is a prosurvival adaptive change counteracting permanent oxidative stress in cancer cells, favoring tumor development (22). Inhibition of TrxR decreases proliferation, migration, and invasion of malignant tumor cells (72). The inhibition of TrxR1 has been suggested as a potential chemotherapeutic strategy for several types of cancer (27, 107, 117, 117a).
Several specific TrxR inhibitors, including auranofin, have been compared in human tumor xenografts and syngeneic mouse tumors regarding adaptation to oxidative stress. Tri-1, a specific inhibitor of TrxR1, exhibits high efficiency in impairing growth and viability of human tumor xenografts and syngeneic mouse tumors. Tri-1 is better tolerated than auranofin and has minor mitochondrial toxicity. Selective inhibition of TrxR1 by small molecules might become a potential anticancer therapy strategy (100).
The expression of Trx and TrxR correlates with the survival rate of patients diagnosed with salivary adenoid cystic carcinoma (SACC). An inverse correlation has been found in patients having lower survival rates that express higher levels of both proteins (43c).
Inhibition of TrxR1 by 1, 2- [bis (1, 2-benzisoselenazolone-3 (2H) -ketone)] ethane (BBSKE/ethaselen) reduces the rates of invasion and migration in SACC cells. BBSKE is a potent inhibitor of the EMT mediated by the TGFB-driven signaling pathway (43c). In human breast cancer cell lines, BBSKE has a moderate inhibitory effect on the EMT (22). Two distinct TrxR inhibitors have been shown to sensitize glioma cells to chemotherapy, decreased cell proliferation, migration, invasion, and increased cell death (48). Transformed MCF-10A (MCF-10AT) cells undergoing chronic treatment with H2O2, exhibit malignancy upon tumorigenicity evaluation, reversed by BBSKE. BBSKE partially reverses some malignant phenotypes, including the EMT of MCF-10AT cells (120).
Another class of compounds, the methylene-cyclohexanone derivatives, have a potent inhibitory effect against TrxR1. These compounds inhibit proliferation and metastasis of human breast cancer cells and abrogate the EMT induced by TGFB (72). Another compound having TrxR1 inhibitory activity, named tricyclohexylphosphanegold (I) η-mercaptobenzoate, suppresses the EMT and cell invasion in breast and ovarian cancer cells by modulating p53-related genes (3).
Silencing TrxR1 is another strategy that illustrates the importance of the Trx1/TrxR1 antioxidant system in the maintenance of the EMT and tumor progression. Decreased migration, invasion, and lower expression levels of HER2, CD44, MMP-9, VEGFR2, and PD-L1 are found in MCF-7 breast cancer cells with TrxR1-knockdowned and LoVo CRC metastatic cells (6, 91, 117).
In a similar manner to TrxR inhibition, the knockdown of methionine sulfoxide reductase suppresses proliferation and the EMT, by increasing E-cadherin, while decreasing N-cadherin, Slug, TGFB, Snail, vimentin, fibronectin, and β-catenin expression in human osteosarcoma epithelial cells (61).
The evidence presented in this section supports an important role for the thiol-based antioxidant system, Trx1/TrxR1, in the maintenance of an adequate intracellular redox status in cancer cells that allows for the occurrence of the EMT and cancer cell progression. This hypothesis is described in Figure 2.
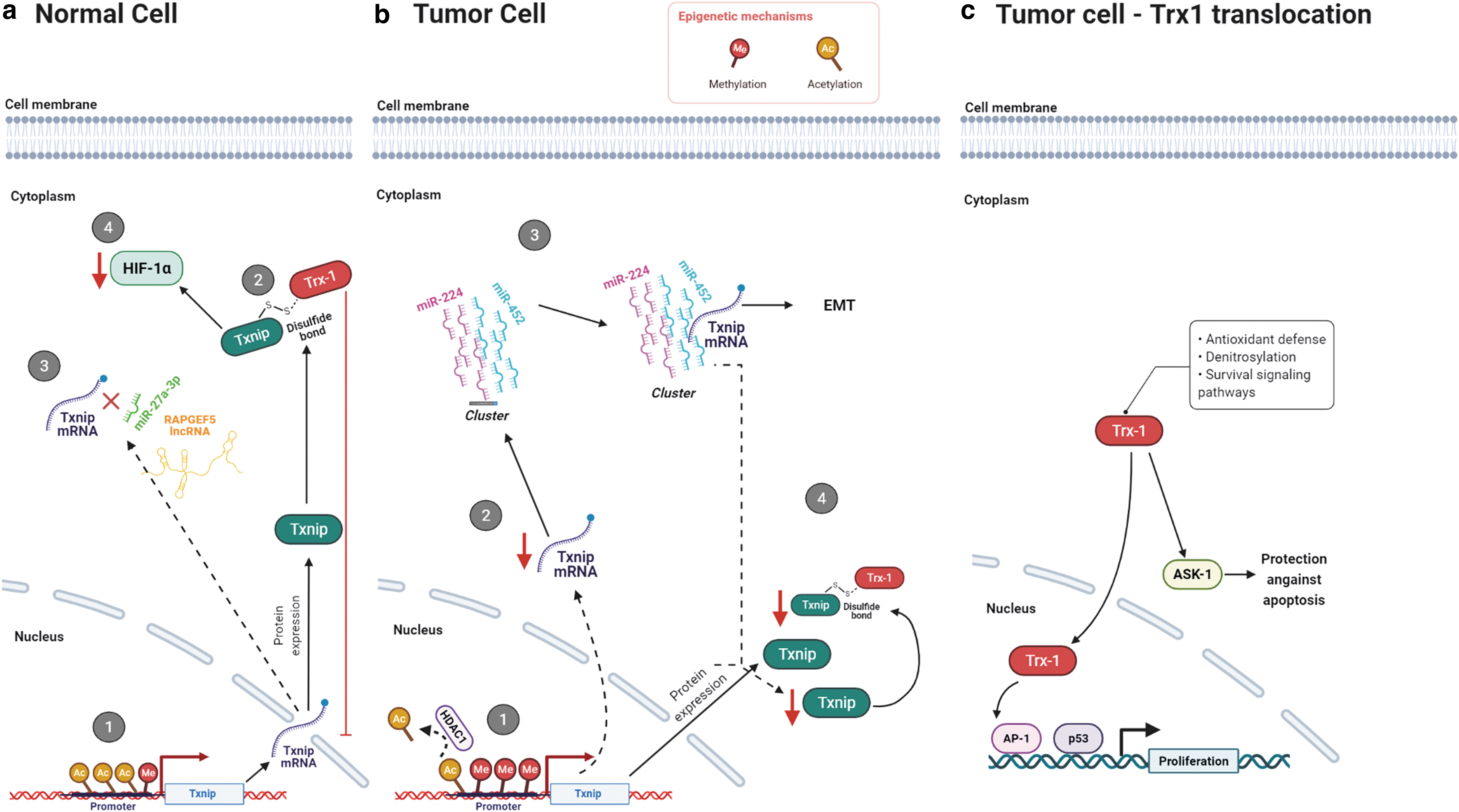
Txnip and the epithelial/mesenchymal transition in cancer cells
Although Trx1 is an essential part of the thiol-based antioxidant system Trx1/TrxR1, Trx1 is negatively regulated by its interaction with the protein Txnip. Txnip was proposed as the physiological inhibitor for Trx1. It was initially described as a vitamin D-upregulated protein 1 (VDUP-1) in 1994 (15). The same protein has been identified in a yeast two-hybrid assay as bait for Trx1 in its reduced form; hence, it was referred to as the thioredoxin binding protein-2 (81), which subsequently has been characterized as having a negative regulatory action on Trx1 activity in vitro (49). Active-site mutants of Trx1 have been used as bait in a two-hybrid screening system and positive colonies have been isolated. These colonies present the open reading frame of a binding partner to the reduced form of Trx1. The characterized protein is homologous to VDUP-1 (112).
Since the Trx1/TrxR1 system is involved in cellular proliferation and tumor biology (12), studies involving Txnip were a logical next step to be taken. Samples from patients with gastrointestinal cancer have decreased expression of Txnip. These observations suggest the participation of Txnip in the pathogenesis of gastrointestinal cancers (69).
Txnip has been characterized as a negative regulator of Trx1-mediated pro-survival signaling pathways in cancer cells (121). Downregulation of Txnip stimulates cancer progression, and its expression is reduced or even suppressed in various human cancer cells (9, 76, 82, 94).
Txnip is downregulated in renal cell carcinoma by ubiquitin-like, containing PHD and RING finger domains-1. This protein can recruit HDAC1 to the TXNIP promoter and deacetylate the histone H3K9, suppressing its expression (44).
In HCC induced by chronic hepatitis B virus infection, Txnip suppression
In a breast cancer model, induced by N-methyl-N-nitrosourea treatment, a significant decrease in TXNIP expression can occur within 6 weeks. The expression is compared within different organs and tissues and is more pronounced in mammary tissue. Tumor growth could be partially reversed by treatment with 1,25-dihydroxyvitamin D3 (113).
In a mouse model, TXNIP deletion
Methylation in the CpG islands of TXNIP's promoter region found in renal carcinogenesis reduces its expression (25). This epigenetic event has been observed in human T cell leukemia virus type I (HTLV-I)-positive T cells, the causative agent of adult T cell leukemia. Its reversal occurs after sequential treatment with 5-aza-2′-deoxycytidine and a histone deacetylase inhibitor (1).
An inverse correlation has been observed between the expression levels of Txnip and the intracellular redox status in melanomas transfected with antisense cDNA for TXNIP (99). These cells have increased melanin production, reduction in the expressions of the Fas ligand and CD44, decreased ROS levels, and increased Trx1 activity. Conversely, various tumor cell lines treated with the histone deacetylase inhibitor suberoylanilide hydroxamic acid have increased Txnip and decreased Trx1 expression (13, 51).
Trx1 nuclear migration is associated with oxidative/nitrosative stress in various cancer cell lines. ROS-inducing agents such as ionizing radiation, UV light, phorbol esters, and cisplatin (2, 37, 38, 106), and the NO donor S-nitroso-N-acetyl-penicillamine (SNAP), promote Trx1 nuclear migration (4). Once in the nucleus, Trx1 functions as a positive regulator of the transcription factors NF-kB, AP-1, p53, the glucocorticoid receptor, and the polyomavirus enhancer-binding protein 2 (2, 37, 38, 67, 106). Indirect evidence indicates that the positive actions of nuclear Trx1 on transcriptional activity are associated with the EMT (14). Txnip deficiency in A549 human lung carcinoma cells and in MDA-MB-231 human triple-negative breast cancer cells is associated with the upregulation of the expression of Snail or Slug in TGFB-driven EMT (70).
To explain the regulatory role of Trx1/Txnip on transcriptional events associated with tumor progression, a mechanism describing the cellular compartmentalized actions of Trx1 and Txnip under nitrosative and oxidative stress has been proposed by our group (83). In HeLa human cervical cancer cells exposed to elevated concentrations of SNAP, Trx-1 accumulates in the nuclear compartment due to S-nitrosylation of p21Ras and activation of downstream ERK1/2 MAP kinases. The nuclear translocation of Trx1 is triggered by downregulation of Txnip. The oxidizing environment stimulates nuclear migration of the ERK1/2 MAP kinases whose transcriptional activity down-regulates Txnip. In the presence of the mitogen-activated protein kinase kinase (MEK) inhibitors (PD98059 or UO126), or in cells transfected with the 15 kDa protein enriched in astrocytes, a cytoplasmic anchor of ERK1/2 MAP kinases, Trx1 nuclear migration and Txnip downregulation are no longer observed in cells exposed to oxidants. Overexpression of Txnip inhibits Trx1 nuclear migration under nitrosative/oxidative stress, whereas gene silencing of Txnip facilitates Trx1 nuclear migration even in the absence of nitrosative/oxidative stress. Changes in Trx1 compartmentalization under nitrosative/oxidative stress are dependent on the expression levels of Txnip, which are regulated by cellular compartmentalization and activation of the ERK1/2 MAP kinases (4, 83).
Txnip interacts with the transcriptional corepressors, Fanconi anemia zinc-fingers and histone deacetylase 1 (34), thereby negatively regulating hepatocarcinogenesis by suppressing TNF-α-induced NF-kB activation (56). Txnip mediates the nuclear export of the von Hippel–Lindau protein/HIF1α to destabilize HIF1α and to promote its degradation in the cytoplasm under normoxia in HeLa and HEK293t cells (98).
The ectopic overexpression of Txnip in HTLV-I-positive T cells results in the suppression of tumor growth. Although these findings confirm Txnip's tumor suppressor function, they do not correlate with the expression levels of Txnip and Trx1 in HTLV-I-positive T cells, nor do they exert reciprocal regulation against Trx1 in the antioxidant system of some cancer cells (80).
Txnip due to its actions as a tumor suppressor has been associated with the MET. The TXNIP gene in pheochromocytomas has been identified as one of the six suppressor metastasis genes (85). These genes have been selected due to their participation in cell growth regulation and apoptosis. Overexpression of TXNIP in a murine thyroid tumor model results in decreased tumor growth and metastasis (76). In melanoma characterized by loss of genetic material on chromosome 6 (30a), a nonmetastatic melanoma model has been generated by subtractive hybridization. An increase in the expression of TXNIP (gene locus on chromosome 1) and a consequent upregulation of the metastasis suppressor gene KISS1, also located on chromosome 1, inhibit metastasis. Such a shift in melanomas depends on the transcription factor E2F1; among the genes activated by it, the miR-224/miR-452 cluster is one of the most prominent, with the TXNIP gene as one of its targets. The decrease in the miR-224/miR-452 cluster expression induces the EMT and cancer cell invasion (54, 122). MicroRNAs are also involved in the EMT in breast cancer and pancreatic cancer cells, so that increasing the expression of miR-373 and miR-125b, respectively, decreases the expression of Txnip and enables the activation of the signaling pathway that culminates in HIF1α transcriptional activity (14, 109, 110). The decrease in RAPGEF5 CircRNA in renal cell carcinoma, which binds to miR-27a-3p and no longer binds to Txnip mRNA, results in reducing the expression of Txnip and a consequent increase in proliferation and migration in vitro and in vivo (16). The decrease in the expression of another circular RNA CircDCUN1D4 is involved in the metastatic process in pulmonary adenocarcinoma. The circular RNA serves as a framework for the interaction between the HuR protein and Txnip mRNA, increasing the stability of this nucleic acid (62).
Summarizing the data discussed in this section, cellular compartmentalization of Txnip at the protein level and the epigenetic control of TXNIP gene expression are important ways to negatively regulate the Trx1/TrxR1 antioxidant system (Fig. 3). Therapeutic strategies involving the enhancement of the expression of Txnip in cancer cells may prove beneficial.
GSH/Grx and the epithelial/mesenchymal transition in cancer cells
The thiol-based antioxidant system GSH/Grx comprises GSH, GSH reductase, NADPH, and Grx. Grx is a 12 kDa redox-active protein that utilizes the reducing power from GSH and has been characterized as an electron donor for the ribonucleotide reductase in a Trx-deficient E. coli strain (39). Four isoforms, Grx1, Grx2, Grx3, and Grx5, have been described. Grxs catalyze GSH-dependent redox regulation through glutathionylation, the conjugation of GSH to a substrate, and its reverse reaction, deglutathionylation (84).
The synthesis of GSH driven by the modifier subunit of the enzyme glutamate cysteine ligase is required for cancer initiation (36). However, the role of the thiol-based antioxidant system GSH/Grx on cancer development has not been fully explored (84).
Cancer cells must undergo major changes to detach from the tumor mass at the primary site and colonize distant organs. Changes include a decrease in cellular junctions and adhesion, and an increase in cell mobility via remodeling of the cytoskeleton and degradation of the extracellular matrix (ECM). Redox regulation via glutathionylation and oxidative/nitrosative stress potentially play a key role in these changes.
The initiation of the EMT diminishes cell junctions and promotes the deconstruction of epithelia-formed permeability barriers. Loss of tight junctions and cell/cell communication requires regulation of protein expression with the decrease of proteins such as occludin, claudin, connexin, and E-cadherin (43a). Among the transcription factors involved in this signaling pathway, NF-kB and HIF1α are redox regulated. Activation of NF-kB by oxidation promotes survival, proliferation, and metastasis by upregulating mesenchymal-associated genes and downregulating epithelial-associated genes (60).
Cell adhesion is mostly controlled by the organ-specific availability of adhesion molecules (integrins, selectins, and cadherins). Integrins are a broad family of proteins, which are involved in colonizing the new environment because they mediate cell adhesion in a series of steps. First, cells contact the substratum and integrins are newly synthesized to ligate to the ECM proteins. Second, the cells spread and finally organize the actin filaments into stress fibers for mechanical resistance.
Redox regulation of integrin signaling may occur during cell adhesion (18). Grx regulates the mobilization of neutrophils in vivo and in vitro via deglutathionylation of integrin α4 (115). This implicates ROS as second messengers in the organization of the cytoskeleton in response to integrin engagement.
Two sites on integrin α7 have been identified that are modulated by H2O2. This results in a conformational change that activates integrin α7β1 and its binding to laminin (11). The role of glutathionylation in the process and how it can be enzymatically modulated by Grx must be explored.
An important aspect of the EMT is strongly associated with cytoskeletal remodeling. The EMT regulates cell elongation and alterations of cellular protrusions (60). Treatment with polymerized actin modulators promotes hypoxia-mediated EMT and the consequent invasion and metastasis of HCC cells in a xenograft mouse model (88). S-glutathionylation of cytoskeletal proteins inhibits their polymerization and alters chemotaxis and adhesion (17, 24, 92, 108).
The EMT is characterized by increased formation of actin stress fibers and actin rearrangement, which contributes to cell directional motility. The redox state of the fast-reacting Cys374, next to the C-terminal region, modulates actin polymerization (20, 26). Among the different nitrosative and oxidative modifications, S-glutathionylation of Cys374 decreases the capacity of actin to polymerize and inhibits filament elongation (20). This process of depolymerization of actin filaments is a necessary step for the cytoskeletal rearrangement during the EMT. Actin glutathionylation is a reversible modification and Grx efficiently catalyzes deglutathionylation of actin. The removal of the GSH group leads to about a sixfold increase in the rate of actin polymerization (108).
Microtubules together with actin are the other major components of the cytoskeleton. High tubulin expression is significantly associated with an advanced depth of tumor infiltration, however, like actin, oxidation and S-nitrosylation decrease tubulin polymerization and microtubule assembly (40, 58, 59). GSH modulates tubulin aggregation indirectly leading to the formation of amorphous structures. Glutathionylated tubulin has been detected in vivo (33) (Fig. 4).

In a xenograft model using different melanoma cells isolated from patients, the GSH level was lower in circulating melanoma cells and metastatic nodules (growing in different organs) compared with subcutaneous (primary) tumors. This may indicate that low levels of reduced GSH due to oxidative/nitrosative stress are necessary for the cytoskeletal remodeling during the EMT.
In cancer cells undergoing the EMT, high intracellular NO concentrations rapidly react with the cysteine thiol group of GSH to form S-nitrosoglutathione and S-nitrosylated proteins, consuming GSH in the process (75). GSH is also consumed during the EMT by Grx to mediate protein S-glutathionylation. S-glutathionylation protects critical cysteine residues from oxidizing in structural and signaling proteins (86). Synthesis and consumption of GSH by the thiol-based antioxidant systems must be finely regulated according to the different stages of cancer development.
In this section, the involvement of the GSH/Grx system in cytoskeletal remodeling during the EMT has been discussed. Although some recent advances have been made, further investigations are necessary to understand the role of glutathionylation/deglutathionylation in regulating cancer progression, growth, and dissemination.
Conclusion
The central mechanism by which the thiol-based antioxidant systems participate in the EMT in cancer is hypothetically explained by the maintenance of signaling processes dependent on redox-active elements. The use of Trx1/TrxR1 inhibitors, inhibitors of GSH synthesis, and genetic-based strategies to enhance intracellular Txnip levels may provide the tools for upregulating the intracellular levels of ROS and NO in cancer cells. Manipulation of the intracellular ROS and NO levels in tumor cells has become a promising strategy for eliminating cancer cells (32, 89). Future studies and discoveries in this field may pave the way for novel individualized anticancer therapies.
Footnotes
Acknowledgment
We gratefully acknowledge Ms. Ana Caroline de Sousa Teodoro for producing the figures found in this review article.
Authors' Contributions
F.T.O. reviewed the literature on Trx/Txnip and EMT and drafted this part of the article. A.Y.S.S. and R.J.A. reviewed the literature on Trx/TrxR and EMT and drafted this part of the article. L.C. and F.T.O. reviewed the literature on GSH/glutaredoxin and drafted this part of the article. H.P.M. conceptualized the content of the article and wrote the article. H.P.M. and A.I.S. discussed and edited the article. All authors reviewed and approved the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Financial support to the studies published by our research group and reviewed here was provided by: Fundação de Amparo à Pesquisa do Estado de São Paulo/Brazil (FAPESP) Proc. 2007/59617-6; 2009/52730-7; 2018/15038-7; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Conselho Nacional de Desenvolvimento Científico e Tecnológico/Brazil (CNPq) Proc. 481154/2013-2; 305649/2015-9; 303832/2019-3.