
Abstract
Significance:
Hydrogen sulfide (H2S) is an endogenous signaling molecule, regulating numerous physiological functions from vasorelaxation to neuromodulation. Iron is a well-known bioactive metal ion, being the central component of hemoglobin for oxygen transportation and participating in biomolecule degradation, redox balance, and enzymatic actions. The interplay between H2S and iron metabolisms and functions impacts significantly on the fate and wellness of different types of cells.
Recent Advances:
Iron level in vivo affects the production of H2S via nonenzymatic reactions. On the contrary, H2S quenches excessive iron inside the cells and regulates the redox status of iron.
Critical Issues:
Abnormal metabolisms of both iron and H2S are associated with various conditions and diseases such as iron overload, anemia, oxidative stress, and cardiovascular and neurodegenerative diseases. The molecular mechanisms for the interactions between H2S and iron are unsettled yet. Here we review signaling links of the production, metabolism, and their respective and integrative functions of H2S and iron in normalcy and diseases.
Future Directions:
Physiological and pathophysiological importance of H2S and iron as well as their therapeutic applications should be evaluated jointly, not separately. Future investigation should expand from iron-rich cells and tissues to the others, in which H2S and iron interaction has not received due attention. Antioxid. Redox Signal. 36, 275–293.
Introduction
Iron and hydrogen sulfide (H2S) are important chemical species that lie on the opposite ends of the redox spectrum. Where excess of iron creates oxidative stress, H2S offers antioxidant protection. The metabolisms of H2S and iron in mammalian cells are also intertwined. Through a nonenzymatic process, iron participates in H2S production. On the contrary, H2S regulates iron uptake, transport, and accumulation. The interaction between H2S and iron affects cellular homeostasis and constitutes important signaling cross talk. Although with great efforts, our understanding of the intertwined metabolisms of H2S and iron in mammalian cells and their respective and integrated roles in redox signaling has been limited.
This review article aims at identifying the relevant knowledge gaps in iron and H2S biology. Multiple mechanisms are presented to interpret the reported metabolisms of H2S and iron, respectively, and their impacts to each other under different physiological and pathophysiological conditions. We further reviewed differential impacts of iron and H2S on cellular redox balance, considering one being a prooxidant and the other being an antioxidant. Finally, the functional consequences of the interaction between H2S and iron are discussed, focusing on the cardiovascular system, the liver, and the brain.
H2S Metabolism
H2S is produced in eukaryotes, enzymatically from three main enzymes: cystathionine γ-lyase (CSE; EC 4.2.1.22), cystathionine β-synthase (CBS; EC 4.4.1.1), and 3-mercaptopyruvate sulfurtransferase (MST; EC 2.8.1.2). Where CSE and CBS can use both

Nonenzymatic sources of H2S in eukaryotes include cysteine reaction with iron and vitamin B6 (200) or reduction of elemental sulfur in which protein-bound sulfur or sulfur containing compounds releases H2S (74, 98, 142). Vitamin B6 initiates a nucleophilic attack on cysteine, forming a cysteine-aldimine. Furthermore, free or heme-bound iron reacts with the cysteine-aldimine to form a cysteine-quinonoid. Ferric ions react with cysteine-quinonoid to eliminate the thiol group as H2S and the resulting desulfurated aldimine undergo hydrolysis to yield vitamin B6, ammonia, and pyruvate (Fig. 2A). A classic example of nonenzymatic H2S production is the reduction of elemental sulfur using NADPH (98). Searcy and Lee showed that RBC lysates can react with reduced glutathione (GSH), NADH, and NADPH to form H2S nonenzymatically (142). They further showed that H2S may be formed through inorganic sulfur using reducing equivalents from glucose oxidation in the phosphogluconate pathway in RBCs [Equation (1) in Fig. 2B]. The phosphogluconate pathway represents 5%–10% of glucose metabolism in RBCs but becomes more active in oxidative stress (142). Searcy and Lee predicted that 2 glucose molecules reacting with 6 elemental sulfur and 3 water molecules yield 3 lactate, 6 hydrogen sulfide (H2S), and 3 carbon dioxide molecules [Equation (1) in Fig. 2B]. However, their empirical data showed a ratio of 6 H2S to 2 carbon dioxide. They theorized this with the release of pyruvate instead of lactate but did not give a balanced reaction. A balanced equation would be achieved with 4 molecules of sedoheptulose [Equation (2) in Fig. 2B], a metabolite in the phosphogluconate pathway that coincidentally is also increased in oxidative stress (29, 192). This phenomenon also hints an antioxidative role of H2S. Similarly, thiosulfates can undergo reduction by interacting with GSH to release H2S (Fig. 2C). Leskova et al. showed this phenomenon in human umbilical vein endothelial cells (86).
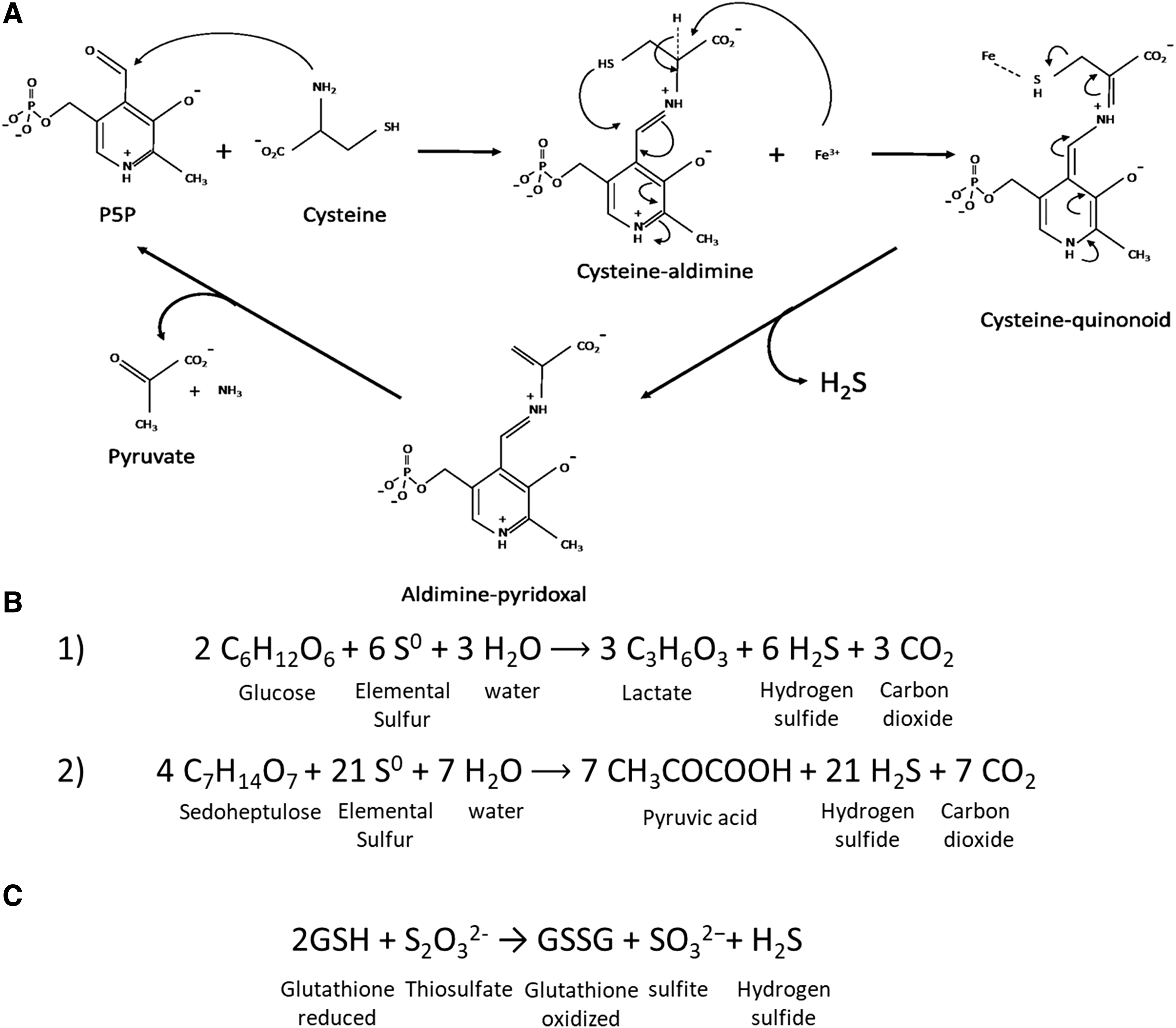
Where enzymatic production of H2S is part of a complex series of reactions in cysteine metabolism and the expression of H2S-producing enzymes can be induced or inhibited, nonenzymatic production of H2S is less regulated via nonspecific reactions. There would be no difference in the biological functions of H2S whether it is produced enzymatically or nonenzymatically as it is the same molecule. The functional differences, if any, may be accounted for by the differences in the concentrations of H2S produced and the coproducts from different pathways. Nonenzymatically produced H2S may contribute to basal levels of H2S in the blood stream (200) where H2S-producing enzymes are less expressed. Moreover, iron-dependent nonenzymatic production of H2S may offer a protection mechanism against iron-induced oxidative stress. This mechanism is discussed in detail later in this article.
H2S can be exhaled or passes through urine or feces as sulfate or thiosulfate (56, 177). H2S can also be oxidized in mitochondria to thiosulfate and further to sulfite or sulfate catalyzed by sulfate detoxifying enzymes, such as rhodanese (20, 126). Methylation of H2S to a nontoxic compound, dimethylsulfide, by thiol S-methyltransferase is another possible fate of H2S (181). Lastly, H2S can be scavenged by different proteins containing metallic groups or disulfide molecules, such as methemoglobin, which is a hemoglobin derivative with oxidized ferric irons (Fe3+) (165). The neutral H2S molecule rapidly binds with Fe3+ in methemoglobin in vitro and in vivo with an association constant of 3.5 × 103 M −1·s−1. This association is pH independent, but its dissociation is pH dependent. The dissociation of H2S increases with an increased acidity and the dissociation curve shifts to the right with an increase in pH (26). This is similar to Bohr's effect for oxygen, which governs loading and unloading of oxygen from hemoglobin depending on partial pressure of CO2 and local pH. RBCs may unload oxygen and H2S to tissues with increased metabolic activity to enhance local oxygen and increase vasodilation (59). Figure 3 summarizes transport of H2S via methemoglobin. Other molecules, which may bind H2S, include oxidized glutathione, catalase, and horseradish peroxidase (21, 27).

H2S moves intracellularly by free diffusion. It can also diffuse out of the cell for paracrine signaling. Bearden et al. found the presence of extracellular H2S, produced by CSE and CBS in the blood as part of plasma proteome (9). Moreover, H2S can bind to oxidized, ferric hemoglobin (methemoglobin) and be transported in the circulation as a potential endocrine signaling molecule (59).
Iron Metabolism
Human body contains 2.5–4.0 g of iron, predominantly present in hemoglobin and in lesser quantities in myoglobin, and in various enzymes and transport proteins (16). Iron is involved in a wide array of physiological functions, such as erythropoiesis, oxygen transport, and degradation of lipids, proteins, and nucleic acids. It is also used in myocardial and muscle metabolism, function of thyroid gland, central nervous system, and immune system (57). Iron exists in multiple states and forms in vivo, including Fe3+ (ferric), Fe2+ (ferrous), bound iron in the heme group, or in iron/sulfur (Fe-S) clusters such as 2Fe2S or 4Fe4S (83, 172). Each form is tightly regulated by complex mechanisms. The most abundant form of iron is the one bound to heme, followed by transferrin and ferritin (1). Free ionic forms of iron are kept at a very low concentration to avoid toxicity by free radicals (32). Transferrin levels in plasma are sufficient to bind more than 99% of iron, which leaves the rare possibility of free iron. Any free iron is usually loosely complexed with molecules such as citrate or serum albumin. However, in diseases such as atransferrinemia, hemochromatosis, and thalassemia, plasma free iron levels overwhelm the transferrin binding capacity (131).
Free iron enters the cell as Fe2+ through ion channels or transporters such as divalent metal transporter 1 (DMT1) and ZRT/IRT-like protein 14 (ZIP14). Whereas DMT1 chiefly prefers and transports iron in the cell, it can also transport copper, zinc, and manganese. DMT1-deficient mice showed decreased iron concentrations in blood, liver, spleen, and heart, but not other metal ions (144). ZIP14 is a zinc transporter, but it transports iron as well (71). Moreover, iron may enter through L-type Ca2+ channels as well (45, 96, 119). Ionic Fe3+ gets reduced to Fe2+ by ferrireductases such as six-transmembrane epithelial antigen of prostate 2 (STEAP2), stromal cell derived receptor 2 (SDR-2), and duodenal cytochrome B (Dcytb) outside the cell (15). Dietary heme enters the enterocyte by heme carrier protein 1 (HCP1) transporter protein (145). Transferrin-bound iron binds to a transferrin receptor 1 (TfR1) and is internalized as an endosome. Furthermore, the iron is released from transferrin as Fe3+ and reduced to Fe2+ by the neighboring STEAP2 ferrireductase in the endosome before being transported into the cytoplasm through DMT1 (6). Free iron as Fe2+ constitutes the labile iron pool (LIP), which may increase through breakdown of heme by heme oxygenase (HO) (188). Furthermore, Fe2+ can bind to apoferritin to become ferritin and be stored as Fe3+ (6). Free iron inside the cell may be incorporated as a cofactor for iron-containing proteins involved in processes such as reactive oxygen species (ROS) generation and degradation of biomolecules (57, 81). Free iron can be exported out of the cell via ferroportin (Fpn) transporter along with ferroxidase enzymes, hephaestin, and ceruloplasmin. Ceruloplasmin converts Fe2+ to Fe3+ outside the cell (15). Most of the iron requirements for cellular metabolism and function are fulfilled by recycling existing iron. An example of this process is heme recycling by macrophages, which engulf senescent RBCs and break them down into free iron or heme, which is moved into the cytoplasm via natural resistance-associated macrophage protein (NRAMP1) or heme responsive gene 1 (HRG1) transporter (76, 201). The heme can be broken down in the cytoplasm by HO or exported out of the cell via feline leukemia virus subgroup C receptor-related protein 1a (FLVCR1a) (201).
H2S can interact with a wide variety of ion channels (177). For example, H2S is a known KATP channel opener through which it mediates vasorelaxation (61). One possible interaction between H2S and iron is that H2S can inhibit L-type Ca2+ channels by S-sulfhydration as observed in mouse pancreatic beta cells (159). Iron can enter the cell through L-type Ca2+ channels as observed in cardiomyocytes (118). Inhibition by H2S may decrease the amount of iron entering in the cell.
Total iron concentration inside the body is largely controlled through the protein hepcidin. It is secreted by the liver into circulation when plasma iron concentration is high (Fig. 4). Hepcidin blocks Fpn by binding to it, causing its ubiquitination followed by internalization and degradation (134). This results in the blockade of iron movement into circulation from enterocytes, macrophages, or hepatocytes (43, 113). If there is an abnormal increase in hepcidin in circulation, such as in the case of inflammation, a higher level of hepcidin may lead to iron deficiency (113). Conversely, abnormally low hepcidin, as in the case of certain hereditary hemochromatosis (HH), leads to iron overload (91, 114). Human body loses 1–2 mg of iron daily by skin and enteric desquamation or through blood loss (171).

Iron status in vivo can be determined by different parameters, such as hemoglobin concentration, serum iron, serum ferritin, tissue ferritin, liver iron concentration, and nontransferrin-bound iron (NTBI) levels in the blood or tissues. Each parameter changes with respect to different physiological conditions and can be used as an indicator for disease. However, careful considerations need to be made before making a conclusion based on iron values as they tend to be different between different age groups, gender, and disease models (18, 38, 44, 125, 162). Table 1 summarizes total iron concentration (free and bound irons) in different biological samples.
The Concentrations of Total Iron and Selective Iron Proteins in Different Organs
SI units were converted.
One milligram of ferritin is equal to 8 mg of stored iron (111).
Whereas hepcidin controls overall iron in the body, intracellular regulation of iron level depends on iron response elements (IRE) and iron response element binding proteins (IRE-BP). IREs are short sequences on untranslated regions (UTR) of messenger RNA (mRNA), which can bind to IRE-BP (83). For example, mRNA of ferritin contains an IRE near 5′ UTR and mRNA of TfR1 has multiple IREs near 3′ UTR. If the cellular iron content is low, IRE-BP becomes active and binds to IRE of ferritin mRNA to destabilize it, resulting in decreased storage and more free form of iron. Conversely, IRE-BP binding to IRE of TfR1 mRNA stabilizes it, resulting in increased expression of TfR1 and increased intake of iron. With high cellular iron concentrations, IRE-BP becomes inactive and results in the exact opposite effect on ferritin and TfR1 (83, 108).
Iron and H2S in Redox Signaling
Oxidative stress is the accumulation of prooxidants, which overwhelms the body's natural antioxidant systems (50). The word “oxidative stress” has a frightening connotation, but events of oxidative stress also occur at a homeostatic level, which is essential for redox signaling, known as oxidative eustress (149). Redox signaling encompasses cellular processes involving reactions where electron transfer occurs through free radicals, redox-active metals, or related species. Overproduction of ROS and reactive nitrogen species in oxidative stress leads to widespread damage of biomolecules such as proteins, lipids, and nucleic acids (104). The reactive species of note include the superoxide (O2 •), hydroxyl (•OH), alkoxyl (RO•), peroxyl (ROO•), and nitroxyl radical (NO•), along with hydrogen peroxide (H2O2), organic hydroperoxides (ROOH), and hypochlorous acid (HOCl) (152). The most notable source of ROS includes mitochondria (mitoROS), where the electron transport chain may generate O2 •, •OH, and H2O2 (39). Complex I releases O2 • when molecular O2 reacts with reduced FMN (flavin mononucleotide) when there is a high NADH/NAD+ ratio in the matrix. Complex III produces O2 • in the presence of respiratory inhibitors such as antimycin; and complex II produces O2 • when complex I and II are inhibited (39). Other sources of mitoROS include enzymes such as the α-ketoglutarate dehydrogenase complex and pyruvate dehydrogenase complex (97, 155). Cytoplasmic production of ROS (cytoROS) includes ROS generation through the NADPH oxidase (NOX) family and nitric oxide synthases (eNOS, iNOS, and nNOS) (39).
Superoxide and H2O2 generated either in cytoplasm or mitochondria are freely membrane permeable and can react with iron to form the highly reactive and toxic hydroxyl radical through Fenton reaction (110). Under normal metabolic conditions, superoxide leads to the more favorable oxidation of Fe2+ to Fe3+ along with ROS generation. If superoxide levels are high, however, the less favorable reduction of Fe3+ to Fe2+ along with ROS generation takes place (25, 104). The source of the free Fe2+ or Fe3+ is the LIP in cytoplasm (cLIP) and mitochondria (mLIP) (17). Iron metabolism and formation of LIP are discussed in the previous section and summarized in Figure 4. There are several conditions that can lead to iron overload. HH is an autosomal recessive disorder that leads to increased absorption of dietary iron. There are four types of HH: type 1 due to a fault in HFE gene required for iron sensing; type 2 due to a fault in hemojuvelin, a regulator of hepcidin or hepcidin itself; type 3 due to a fault in TfR2; and type 4 due to a fault in Fpn (127). Downregulation of hepcidin is common in HH, which leads to iron overload (182). The increased iron is accumulated in parenchyma of organs such as the liver and heart and is responsible for widespread organ damage due to oxidative stress (182). Similar to HH, iron overload is also observed in diseases where repeated blood transfusion is required, such as thalassemia (49), sickle cell anemia (49), chronic kidney disease (133), liver disease (107), and certain cancer types such as colon-rectal (164) and liver cancer (54). Iron overload-catalyzed ROS can induce generalized damage to biomolecules such as carbohydrates, lipids, proteins, and membranous structures. A secondary effect of iron catalyzed-ROS is the induction of stress and inflammation signaling pathways such as Janus kinase/signal transducer and activator of transcription (JAK/STAT), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and p38/mitogen-activated protein kinase (p38/MAPK) (19, 26, 33, 151).
Oxidative stress can be countered through enzymatic and nonenzymatic antioxidant mechanisms (14, 132). The enzymatic antioxidants include the enzymes superoxide dismutase (SOD), catalase, glutathione peroxidase, and HO, and redox proteins such as glutaredoxins, thioredoxins, and peroxiredoxins. The nonenzymatic antioxidants include low-molecular-weight compounds such as GSH, NADPH, and vitamin C and E along with trace metals such as selenium (14, 132). Any O2 • that is converted to H2O2 by SOD can be reduced to water and oxygen by catalase or water only by glutathione peroxidase utilizing GSH molecules (183). Collectively, SOD, catalase, and glutathione peroxidase constitute first-line antioxidant defense, whereas other enzymes and molecules such as glutathione, ascorbic acid, and vitamins are considered second line of defense (55).
Another species critical to redox signaling are reactive sulfur species (RSS). Although sulfur is part of antioxidant mechanisms, as explained in subsequent paragraphs, its role in “reductive stress” has been recently brought forward (116). Sulfur has lower electronegativity than oxygen, which is why it can exist in more oxidation states. RSS can exist as hydrogen persulfide or polysulfide (H2S2 or H2Sn); protein sulfide or polysulfide [PSH, PS(S)nH, PS(S)nP]; small-weight polysulfides [RS(S)nH or RS(S)nR]; sulfenic acids or nitrosothiols (RSOH or RSNO) (75). RSS generation involves a loss of electron from 2 H2S molecules to form 2 HS•, which can recombine to form persulfides or polysulfides (116). Whether the formation of persulfide can be categorized as a redox process remains to be determined. Recently, Akaike et al. have shown the strong potency of cysteinyl-tRNA (transfer RNA) synthetases in forming cysteine polysulfides in mitochondria and cytoplasm during redox signaling (3).
The key difference in ROS and RSS is their ability to be preserved. When peroxide (ROS) attacks a sulfhydryl group of cysteine in a protein, the peroxide is reduced to oxygen and water. When persulfide attacks the sulfhydryl group, however, the persulfide can be reversed by thiol exchange between the protein-RSS and a low-molecular-weight thiol group as carrier. Consequently, the original persulfide is preserved (116). Coincidentally, oxidation of H2S produces a stronger antioxidant, that is, persulfide, which, if unchecked, may lead to reductive stress (116).
H2S itself has a significant antioxidant capacity but is not as recognized. At high concentrations, H2S can create reducing environments and cytotoxicity, but at low concentrations or from endogenous sources, it offers protective and beneficial effects (34, 176). H2S can inhibit ROS via diverse mechanisms (92, 123, 156, 187, 195). It is known to directly scavenge O2 •, H2O2, and other ROS (46, 184, 195). It can upregulate SOD and glutathione peroxidase, reducing oxidative stress in conditions such as diabetic nephropathy, and intestine or lung damage (30, 64, 90, 130). It also upregulates GSH as a potential mechanism to reduce oxidative stress (64). Pathways associated with antioxidant activity of H2S include activation of nuclear factor erythroid 2-related factor 2 (NRF2) pathway involved in cardioprotection (22, 158), and inhibition of p38/MAPK and NF-κB pathways, lowering ROS, oxidative stress, and inflammation (28, 37, 80, 89, 173). These effects of H2S are largely observed with exogenous H2S-based therapies. CSE and CBS knockout (KO) mice show reduced GSH levels and are more prone to oxidative stress (137, 198, 204, 208), proving a relationship between oxidative stress and endogenous H2S. Regarding the relationship between iron, H2S, and oxidative stress, it has been recently shown by Zhou et al. that CBS is required for iron homeostasis in mice (208). They noticed that CBS-deficient mice had increased iron content in the liver, serum, heart, and spleen along with other markers of oxidative stress. Homozygous CBS-deficient mice could not survive past 4 weeks (208). Hepcidin was upregulated in the liver and circulation due to a decrease in transcriptional factor, NRF2. Increased hepcidin caused a loss of Fpn and increased iron retention, severely damaging the liver. This phenotype was reversed by reintroduction of CBS through the adenoviral vector (208). Figure 5 summarizes interaction between H2S and Iron on cellular functions at a molecular level.

The Effects of Iron and H2S on Erythrocytes
The human body produces ∼200 billion RBCs per day with each containing 300 million hemoglobin molecules with single iron moiety. This feat requires 20 mg of iron daily, which is mostly sourced through macrophage recycling of senescent RBCs, body iron stores, and dietary iron (161). Transport of oxygen by RBCs is the most prominent role of iron inside hemoglobin in the body. Low oxygen delivery is sensed by peritubular cells in the renal cortex and outer medulla in kidney, which release erythropoietin (EPO) in response (143). The EPO drives erythropoiesis by proliferation and differentiation of erythrocytes, which requires a large reserve of iron to produce hemoglobin (161). Transferrin-bound iron is kept high during each stage of nucleated erythroblast differentiation (141). Once the hemoglobin production ceases, iron uptake is reduced, and TfRs are released as final steps of erythroid differentiation (2).
Iron metabolism and erythropoiesis are linked, and defect in erythropoiesis often leads to iron overload (112, 136). Hepcidin plays an important role in regulating iron and its expression can be controlled by erythropoiesis (93). Hypoxia-induced factor (HIF) stimulates EPO and suppresses hepcidin, resulting in an increase in iron concentration. Liu et al. also found that hepcidin suppression indirectly requires EPO-induced erythropoiesis (93). Erythroferrone (ERFE), released from erythroblasts, can suppress hepcidin in hepatocytes, strengthening the link between erythropoiesis and iron metabolism (69).
H2S and iron may interact on erythropoiesis in different ways. It has been recently demonstrated that chicken RBCs, which contain an intact nucleus and mitochondria, produce H2S, mainly from MST enzyme (63). Human RBCs, which lack the nucleus and mitochondria, may produce limited H2S by sulfur reduction nonenzymatically (12, 142, 170). Jennings demonstrated that deprotonated H2S in the form of HS− can be transported into the RBCs in exchange for a chloride ion through AE1 anion exchange protein as part of the Jacobs/Stewart cycle. The HS− can be converted to H2S in cells, which can take part in cellular functions or diffuse out (58).
H2S is a regulator of erythropoiesis. CBS-deficient mice had systemic iron overload due to decreased erythropoiesis and iron utilization (204). The absence of CBS caused a lower expression of delta-aminolevulinate synthase 2, ferrochelatase, FLVCR, HIF-2α, and EPO, resulting in low erythropoiesis and anemia (204). Exogenous H2S donor, GYY 4137, can stimulate erythropoiesis through HIF (85). Mouse lacking CSE had reduced EPO and hemoglobin in hypoxia, and patients with anemia associated with chronic kidney disease had low H2S and EPO (84).
Regulation of Cardiovascular Functions by Iron and H2S
Iron deficiency or overload has been associated with increased cardiovascular risk with different mechanisms (82). Iron deficiency can be caused by either underutilization of circulating iron or failure to absorb dietary iron (202). As a result of iron deficiency-induced anemia, heart workload increases along with elevated ischemia and myocardial cell death (82). Elevated renal ischemia can activate the renin/angiotensin system, causing increased salt and water retention and vasoconstriction, further putting stress on the already stressed out heart (51, 82). Anemic mouse models, IRP1/2 KO or TfR1 KO, showed decreased cardiac function and mitochondrial respiration, which were reversed with aggressive iron therapy (202). These findings have a connotation that iron deficiency due to underutilization or improper regulation may result in cardiovascular disturbance.
Iron overload can also produce detrimental effects on the cardiovascular system primarily through ROS generation and enhanced oxidative stress (82). After having tested adult and neonatal cardiomyocytes as well as H9c2 cells (rat myoblasts), Sweeney et al. demonstrated that iron overload increased ROS via mitochondria and xanthine oxidase. Moreover, iron overload decreased the autophagic reflux as seen by an increase in microtubule-associated protein 1A/1B-light chain 3-II (LC3-II) and p62 protein expression, which are markers for autophagy (160). This caused a decrease in insulin sensitivity in cardiomyocytes, which was confirmed in autophagy-deficient H9C2 cells prepared by overexpressing dominant negative form of autophagy-related 5 protein (Atg5) (157).
Júnior et al. measured effects of chronic iron overload on mesenteric resistance arteries isolated from rats. Chronic iron overload increased the wall to lumen ratio and thickness of the blood vessel, along with decreased lumen and external diameters. Decreased distensibility and increased stiffness and pulse wave velocity were also observed. With wire myography recording, Júnior et al. found that iron overload, induced by repeated injections of iron-dextran, increased the vasoconstrictor response in isolated mesenteric resistance arteries, but did not affect relaxation responses by acetylcholine and sodium nitroprusside. They concluded that iron overload-induced free radicals decreased nitric oxide bioavailability. This conclusion was supported by the observation that L-NG-Nitro arginine methyl ester (L-NAME), an inhibitor of nitric oxide synthase, had no effect on vasoconstriction of mesenteric resistance arteries with iron overload, whereas L-NAME with SOD resulted in typical shifting of the phenylephrine contraction curve toward left. In vitro assays also showed a decrease in NO amount in iron overload tissues (65). Interestingly, the same group showed similar results with aortic tissues (103). Kuang et al. reported on the acute effects of iron overload on rat aortic tissues (77). They found that ferric ammonium citrate can transiently contract isolated aorta with an increased contraction amplitude at higher calcium concentrations. They also note that incubating tissues with ferric ammonium citrate impair vasoconstriction by phenylephrine, and this decrease is abolished by adding catalase or glutathione before ferric ammonium citrate. This suggests that iron overload-caused contraction dysfunction is due to accumulation of ROS (77). Sangartit et al. report an increase in blood pressure and impaired vascular function in iron-overload mice (139). Nitric oxide and glutathione levels in the blood were decreased by iron overload but reversed by tetrahydrocurcumin and deferiprone, compounds with antioxidant and anti-inflammatory properties (139). Iron overload increased calcification in human aortic smooth muscle cells and increased interleukin (IL)-24, thereby accelerating arteriosclerosis (70). Similarly, iron overload accelerated atherosclerosis and induced endothelial dysfunction in apolipoprotein E-deficient mice due to oxidative stress (102). Hypertension as a complication or comorbid factor is also found in patients with sickle cell anemia (4), thalassemia (41), kidney disease (168), and liver disease (36). Recent research has shown that hypertension in sickle cell anemia and thalassemia patients is due to the toxic effects of chronic iron overload rather than anemia (4, 41). An increased cardiac output, a decreased heart rate variability, and vascular damage due to oxidative stress were observed in both diseases (4, 41, 49, 185). These insights warrant further research into the mechanisms of iron overload and vascular damage.
ROS may induce vascular damage and activation of p38/MAPK pathway. The consequential endothelial dysfunction, elastin remodeling, and vascular stiffness can result in hypertension (105, 166, 179). Inhibitors of p38/MAPK are known to ameliorate vascular damage and reduce endothelial dysfunction and hypertension (179). Iron overload has been shown to stimulate ROS production and p38/MAPK activation in MC3TC-E1 preosteoblast cells (26). Another mediator of inflammation, NF-κB pathway, also increases vascular damage and is believed to be pivotal in the pathogenesis of hypertension and can be activated by iron overload and ROS (19, 33).
One of the most prominent effects of H2S is vasorelaxation (198). The discovery that H2S caused vasorelaxation invoked significant interest into H2S biology worldwide. One of the earliest articles by our laboratory found that H2S at physiologically relevant concentrations opens KATP channels, which leads to vasorelaxation (206). We subsequently demonstrated that mouse lacking CSE-gene had hypertensive phenotype because CSE is the major enzyme for H2S generation in vasculature (198, 206). Vascular response to H2S is also endothelium dependent, not inhibited by 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ) or NS2028, two potent inhibitors of guanylate cyclase pathway (205). H2S can mediate the phosphorylation of endothelial nitric oxide synthetase through p38/MAPK and Akt-dependent pathway, increasing bioavailable NO for vasorelaxation (5). The interaction of H2S and NO on vasorelaxation also involves the redox regulation of soluble guanylyl cyclase (sGC). Zhou et al. suggested that H2S reduced ferric (Fe3+) heme center of sGC to its ferrous (Fe2+) form in cultured rat aortic smooth muscle cells. By doing so alone, H2S would not activate sGC. In the presence of H2S, however, sGC can be conditioned for activation by NO since the latter binds to ferrous (Fe2+) heme of sGC to increase its catalytic activity. Consequently, cGMP level was increased and vasorelaxation ensued (209).
H2S also suppresses the development of atherosclerosis (100) by inhibiting adhesion molecule expression and vessel intimal proliferation. We also observed that decreased endogenous H2S leads to atherosclerosis by vascular remodeling (99). Estrogen and H2S both inhibit atherosclerosis in CSE-wild-type (WT) mice fed with atherosclerotic diet, but estrogen cannot inhibit atherosclerosis in CSE-KO mice fed with the same diet. Markers of atherosclerosis, such as intracellular adhesion molecule 1 (ICAM1) and NF-κB, also increased more in CSE-KO than CSE-WT on atherosclerotic diet. Finally, the observation that estrogen increases H2S concentration in livers of CSE-WT but not CSE-KO leads to the conclusion that estrogen interacts with H2S to inhibit atherosclerosis (87). We recently reported that insulin-like growth factor 1 receptor (IGF-1R) interacts with estrogen receptor-α (ER-α) to cause atherosclerosis, but this interaction is inhibited by H2S, as the latter can inhibit IGF-1R by S-sulfhydration (147). Zheng et al. demonstrated that H2S donor, GYY4137, decreased atherosclerotic plaques in aorta by decreasing ICAM1, vascular cell adhesion molecule 1 (VCAM1), and inflammatory markers such as tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein 1 (MCP1), IL-6, and IL-1β in diabetes-accelerated atherosclerotic mice (207). It was concluded that H2S inhibits NLR family pyrin domain containing 3 (NLRP3) inflammasome, which ultimately led to a decrease in atherosclerosis (207).
The role of H2S as an inhibitor of smooth muscle proliferation and viability is well documented by our research team (100, 147, 148, 194, 197, 199) and others. The absence of CSE expression causes vascular smooth muscle cells (VSMCs) to overproliferate (197). Exogenous addition of 100 μM H2S significantly increased VSMC apoptosis and phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 in CSE-deficient mice. ERK regulates apoptosis, growth, and cellular response to stress. H2S also decreased cyclin D1 and increased p21Cip/WAF-1 expression, which arrests cell cycle in G1 phase (197). These findings have great implications in the pathogenesis of atherosclerosis. We recently demonstrated that estrogen increased endogenous H2S production to regulate smooth muscle cell proliferation (87). Iron is also associated with atherosclerosis, but this role of iron is often debated (191). Mueller et al. reported that the addition of ferrous ions decreased proliferation of smooth muscle cells (110). Conversely, Porreca et al. report that iron chelation and deficiency also inhibit proliferation of VSMCs (129). Although iron accumulation is observed in atherosclerotic plaques, epidemiological data are conflicting regarding the association between iron overload and atherosclerosis in patients (169). H2S can offer protection against iron overload, and H2S may be released as a consequence of iron overload. This may very well be the reason why it has been difficult to prove the link between iron and atherosclerosis, whereas a strong link between H2S and antiatherosclerosis already exists. For example, it was found that iron overload can stimulate ERK1/2 in SH-SY5Y neuroblastoma cells (8). Whether the activation of ERK results from iron overload or from the consequential increase in H2S is unsettled. Epidemiological data also suggest that females are at lesser risk of cardiovascular diseases and iron overload (31). The discovery that estrogen can induce H2S production may offer a valid explanation on why females are at lesser risk of iron overload and cardiovascular diseases.
H2S also regulates vascular endothelial proliferation and function (178). H2S stimulates angiogenesis and endothelial repair (121). H2S is also an endothelium-derived hyperpolarizing factor (EDHF), along with KATP channel opener, which establishes the role of H2S in regulation of vascular tone (178). Jian et al. reported that iron deficiency is associated with increased angiogenesis due to upregulation of vascular endothelial growth factor (VEGF) and stabilization of hypoxia-inducible factor-1α (60). Iron overload, on the contrary, is associated with increased oxidative stress and MAPK activation (60). Interestingly, H2S is proangiogenic under normoxic conditions through phosphorylation of PI3K/AKT, ERK, and p38/MAPK pathways, but it is antiangiogenic under hypoxic conditions by inhibiting translation of HIF-1α (177, 186). Liu et al. reported that H2S promoted angiogenesis by stabilizing HIF-1α in hypoxia by using cobalt to induce a chemical hypoxia (94). The conflicting conclusions may be due to different models of hypoxia, along with differences in chronic and acute hypoxia. Regardless, there seems to be a link between iron deficiency-induced angiogenesis and H2S-induced angiogenesis. Figure 6 summarizes potential avenues for interaction between H2S and iron in the cardiovascular system.

Molecular Mechanisms of Interaction Between H2S and Iron
As summarized in the previous sections, iron causes oxidative stress and increases markers of inflammation, whereas H2S can inhibit p38/MAPK, NF-κB, and ROS (38a, 156, 158, 173). These are all possible mechanisms where H2S can potentially decrease the deleterious effects of iron overload and ameliorate hypertension. Cysteine can undergo catalysis under the influence of ferric ions to release H2S as explained before (200). This nonenzymatic H2S generation is believed to be a protective mechanism to decrease excess iron as H2S can react with iron to form acid labile iron sulfide, quenching and inhibiting the excess iron from participating in oxidative stress (200). H2S has been shown to have a cardioprotective role through activation of NRF2 (22), but this role is yet to be established in vasculature. The increased NRF2 upregulates Fpn1, which will lead to efflux of iron and ultimately decrease iron overload (40, 203). CBS KO mice showed increased iron content in the heart, decreased Trf1, Fpn1, and IRP1 by a decrease in hepcidin (208). The effect of H2S on TfR1 and Fpn1 has only been demonstrated in macrophages and spleen tissue. Should the similar effect occur in the cardiovascular system, it may mitigate detrimental effects of anemia (203).
To reiterate, iron deficiency or overload causes cardiovascular diseases and H2S can potentially ameliorate the detrimental effects of abnormal iron metabolism. H2S can accomplish this through direct regulation of iron and transport and indirectly through suppressing downstream targets of iron dysfunction such as the renin/angiotensin system, ROS generation, p38/MAPK, and Nf-κB regulation. Furthermore, H2S can offer enhanced protection through Nrf2 signaling to lower iron-induced oxidative stress.
Iron and H2S Interaction in the Liver
The liver is the command center for iron metabolism, chiefly through expression of hepcidin. High levels of hepcidin can inhibit Fpn, decreasing iron release into the circulation through enterocytes and iron recycling by macrophages (13). The liver produces a significant amount of H2S (21, 66) and excretes H2S-producing enzymes, such as CSE and CBS, into the blood stream (9, 177). Iron and H2S can interact through different pathways, including the BMP/SMAD pathway, which governs apoptosis, cellular differentiation, embryogenesis, organogenesis, and osteogenesis (115). Another important pathway is the JAK/STAT pathway, which can affect genes responsible for apoptosis, cell differentiation, proliferation, and migration (135). Hepcidin expression is controlled by BMP/SMAD signaling in the liver sinusoidal endothelial cells (150). Activin receptor like kinase 2 and 3 (ALK2 and ALK3) can activate BMP6 and BMP2, respectively, in response to increased tissue iron or basal expression of hepcidin. The BMP-SMAD pathway also interacts with JAK/STAT3 pathway, which regulates hepcidin produced due to the inflammatory response (150).
Hepcidin is also regulated by erythropoietic drive, which stimulates the kidney to produce EPO in response to hypoxia. This increased EPO results in erythroblast differentiation, leading them to secrete ERFE to suppress hepcidin (175). Although direct regulation of ERFE by H2S is yet to be established, H2S may increase it indirectly by increasing EPO. Similarly, inflammatory cytokine IL-6 can upregulate hepcidin by the JAK-STAT3 pathway interacting with SMAD1/5/8 pathway and Activin B (174). It has been shown that H2S reduced hepcidin expression through JAK-STAT pathway by suppressing pSTAT3 leading to decreased IL-6 (203). A similar report revealed that H2S promoted SIRT1 expression, which stabilizes SIRT1-STAT3, reducing STAT3 acetylation. The latter event lowered hepcidin level (190).
It is evident that hepcidin expression is affected by a JAK/STAT and BMP/SMAD axis. The fact is that H2S can also affect these pathways, and ultimately, hepcidin expression points toward a key role of H2S in regulating physiological iron levels.
Iron and H2S Interaction in the Brain
As discussed earlier, iron is important for energy metabolism because it is bound to complexes in electron transport chain such as flavin adenine dinucleotide. Iron/sulfur clusters and heme also serve as cofactors for different enzymes. In the brain, iron serves as a cofactor in synthesis of neurotransmitters, such as tryptophan hydroxylase and tyrosine hydroxylase (109). Absorption of iron through the blood/brain barrier (BBB) is analogous to dietary iron absorption in the duodenum. Once crossing the BBB, it is oxidized to Fe3+, bound to transferrin, and released into the brain circulation. Transferrin-bound iron is thought to be either broken down in endosomes and released into the cytosol by DMT1 and exported by Fpn or to be transported into the endosomes from luminal to abluminal surface where endosomes release iron between brain endothelial cells and astrocytes. Furthermore, Fe2+ can bind to ATP or citrate to be transported as NTBI in oligodendrocytes and astrocytes, which do not express TfR1 (11). Iron concentration in the brain is tightly regulated. Abnormal iron concentrations are associated with neurodegenerative diseases, such as Parkinson's disease (PD), Alzheimer's disease (AD), Huntington disease, and neurodegeneration (62).
The physiological role of H2S in the nervous system has evolved to include neuroprotection, neuromodulation, and nociception (95). H2S also induces long-term potentiation and increases the sensitivity of NMDA receptor-mediated memory response. Administration of NaHS elicited an anti-inflammatory response to counter LPS-induced inflammation in microglia by inhibiting p38/MAPK (53). H2S increases [Ca2]i for neuronal health and function (95). Moreover, H2S can inhibit NOX and decrease oxidative stress in the brain (138). Studies have also shown the importance of H2S in peripheral nerve regeneration and degeneration, significant for Schwann cell proliferation and dedifferentiation (120). The role of H2S in various neurodegenerative diseases has been reviewed by Paul and Snyder (122).
With PD, the substantia nigra region of brain has considerable dopaminergic neuronal cell death (153). Symptoms of this disease include postural instability, tremors, and rigidity. The neuropathological characteristics include Lewy bodies, which are intracytoplasmic inclusions, widely distributed in paralimbic and neocortical regions of substantia nigra. Neuromelanin level, associated with iron storage, decreases with age, leading to high iron accumulation and oxidative stress. DMT1 isoform was upregulated in PD with increased iron uptake and dopaminergic cell death. A decrease in ceruloplasmin in PD is also observed in substantia nigra, which leads to increased iron deposition (7). Iron chelation therapy has shown to be effective in decreasing neurotoxicity in PD (10, 68). H2S offers a protective role in neurotoxin-induced PD in rat models by reducing oxidative stress and inflammation by regulating different pathways, including PKC, p38, JNK, PI3K/Akt, and ERK-MAPK (163). Endogenous H2S was downregulated in substantia nigra by 6-hydroxydopamine (6-OHDA), whereas exogenous administration of H2S reversed 6-OHDA-induced neuronal death, suggesting a protective role of H2S in PD (23).
AD is a neurodegenerative disease with impairment of memory, reasoning, and perception as its prominent features. Pathological features include insoluble amyloid beta protein (ab) aggregates and neurofibrillary tangles (NFTs). NFTs contain hyperphosphorylated tau proteins, which can reduce Fe3+ to Fe2+, increasing NFT production (193). Amyloid precursor protein (APP) is a transmembrane protein 1 ubiquitously found in the brain. It is upregulated in the presence of iron and causes iron deposition in amyloid plaques. APP is like ceruloplasmin and aids in the efflux of iron and converts Fe2+ to Fe3+. High iron can upregulate paired amino acid cleaving enzyme, resulting in an increase in amyloidogenic peptides (117).
H2S levels in brain are significantly decreased along with the expression of CBS in patients with AD (35). Exogenous H2S administration slowed down AD progression in mouse and rat models (47, 48). In these animal models, H2S lowered ab plaques in the hippocampus and proteins associated with AD progression. Also, H2S was found to modulate inflammation and apoptosis of hippocampus cells and with mice of different ages with AD (167). H2S may protect against AD by directly reducing HOCl-induced cytotoxicity, protein and lipid peroxidation, along with homocysteine-induced oxidative stress (180). There is a positive association with homocysteine and iron level and deposits in arterial plaques, suggesting iron/homocysteine interaction (50). H2S has been reported to inhibit homocysteine-induced AD-like pathology (67). This raises an interesting question whether the iron/homocysteine/H2S axis exists in regulating brain functions under physiological and pathophysiological conditions.
Conclusion
The interactions between iron and H2S impact each other's metabolism and functions. Whereas excess iron produces ROS, H2S can quench excess iron and ROS altogether. Conversely, whereas low iron causes anemia, H2S can downregulate hepcidin to release more iron. The interaction is also seen at the molecular level where iron and H2S can act on same cellular pathways but with opposite effects. For example, iron catalyzes ROS-activated p38/MAPK and NF-κB and H2S can counter this effect. Given that excess iron can increase H2S production nonenzymatically, the effect of iron on H2S signaling pathways and targets is much less known. The interactions between iron and H2S have been observed in numerous systems, including the brain, blood, liver, and cardiovascular system. Atherosclerosis is a case in point. Iron can increase H2S and the latter would protect from atherosclerosis. On the contrary, iron overload accelerated the development of atherosclerosis. The net outcome of iron overload and induced H2S production on atherosclerosis development is difficult to speculate yet. The similar dilemma also challenges the contributions of iron overload and H2S level, respectively, and/or integrated, to the development of hypertension. While having acknowledged these challenges, there is no doubt that a better understanding of the interactions between iron and H2S would shed light on the pathogenesis of and treatment strategies for many diseases. In the future, physiological and pathophysiological importance of H2S and iron as well as their therapeutic applications should be evaluated jointly, not separately. Future investigation should also expand from iron-rich cells and tissues to the others in which H2S and iron interaction has not received due attention.
Footnotes
Authors' Contributions
All the authors have equally contributed to the writing, editing, and revising of the article and figures.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This study has been supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada to R.W.