
Abstract
Aims:
The skeletal muscle maintains glucose disposal via insulin signaling and glucose transport. The progression of diabetes and insulin resistance is critically influenced by endoplasmic reticulum (ER) stress.
Results:
Innovation and Conclusion:
The study observations indicate that
Introduction
Insulin resistance in the skeletal muscles is a crucial factor contributing to glucose dysmetabolism in type 2 diabetes mellitus. Transportation of glucose in the skeletal muscles is driven by the activation of phosphatidylinositol-3 kinase (PI3K)/protein kinase B (Akt), which essentially participates in the translocation of glucose transporter 4 (GLUT4) from the intracellular pool to the plasma membrane. The impairment of GLUT4 translocation or reduced GLUT4 activity in the muscles results in systemic insulin resistance (52, 56), and overproduction of reactive oxygen species (ROS) is a key triggering factor (26). The endoplasmic reticulum (ER), mitochondria, and NADPH oxidase are responsible for excessive ROS production, and superoxide production in adipocytes and myotubes is a typical characteristic observed in insulin resistance mouse models (21, 57).
Innovation
The study observations provide strong evidence to support a preclinical rationale and molecular basis to explore potential therapeutic applications of oral
An increase in the activity of NADPH oxidase enhances the production of superoxides, leading to an increase in oxidative stress with hyperglycemia. NADPH oxidase-associated ROS are assumed to be the cause of the reduced insulin responsiveness (14, 44), ER dysfunction, and ROS production in the ER (4, 57). The ER is an important cellular organelle that facilitates protein folding. The chaperone-based electron transfer system is directly involved in this process (48, 53).
Unlike the adaptive ER stress response, redox imbalance or ROS accumulation is linked to prolonged or pathological ER stress, which influences post-translational modification of proteins and subsequent modifications of protein function (11, 20, 51). Protein dysfunction is influenced by sulfonation, which involves the oxidation of specific cysteine residues mediated by endogenous or exogenous ROS (23). ER-derived H2O2 is produced from NADPH-dependent oxidoreductase (Nox)4 and leads to ER stress (54). IRE1α sulfonation is activated by the Nox4-ER-ROS axis and is linked to cellular dysfunction (25).
The IRE1α post-translational modification-linked signaling axis, known as regulated IRE1-dependent decay (RIDD), has diverse substrates, including proteins associated with glucose metabolism.
Another study showed that
Moreover, animal models efficiently demonstrate impaired glucose tolerance when subjected to a glucose challenge test, and these models are well accepted for investigating type II diabetes. However, compared with mice models showing high blood glucose levels (>500 mg/dL), blood sugar levels of diabetic patients are consistently higher than the target range (200–350 mg/dL in adults and 200–240 mg/dL in children) (42).
Typically, patients manage their blood sugar level to stay within the optimal range and make all possible efforts to adjust the levels with drugs. Physiological differences may also be present in animals. However, db/db and high-fat diet (HFD)-fed mice models show characteristics of overt insulin resistance, progressive development of hyperglycemia, and β cell dysfunction with age. These models have been considered clinically more relevant than other animal models. In addition, muscle cells can consume up to 70% of glucose, indicating the suitability of the cells for use as in vitro diabetes models. Thus, we applied db/db and HFD-fed mice and human skeletal myoblasts (HSkM) for in vitro investigations. These models are relatively relevant for exploring the molecular mechanism underlying the anti-diabetic effect of
Results
d -Allulose controls glucose metabolism in db/db mice
Diabetic db/db mice were administered various doses of
Regarding post-prandial and fasting blood glucose levels, mice that were administered
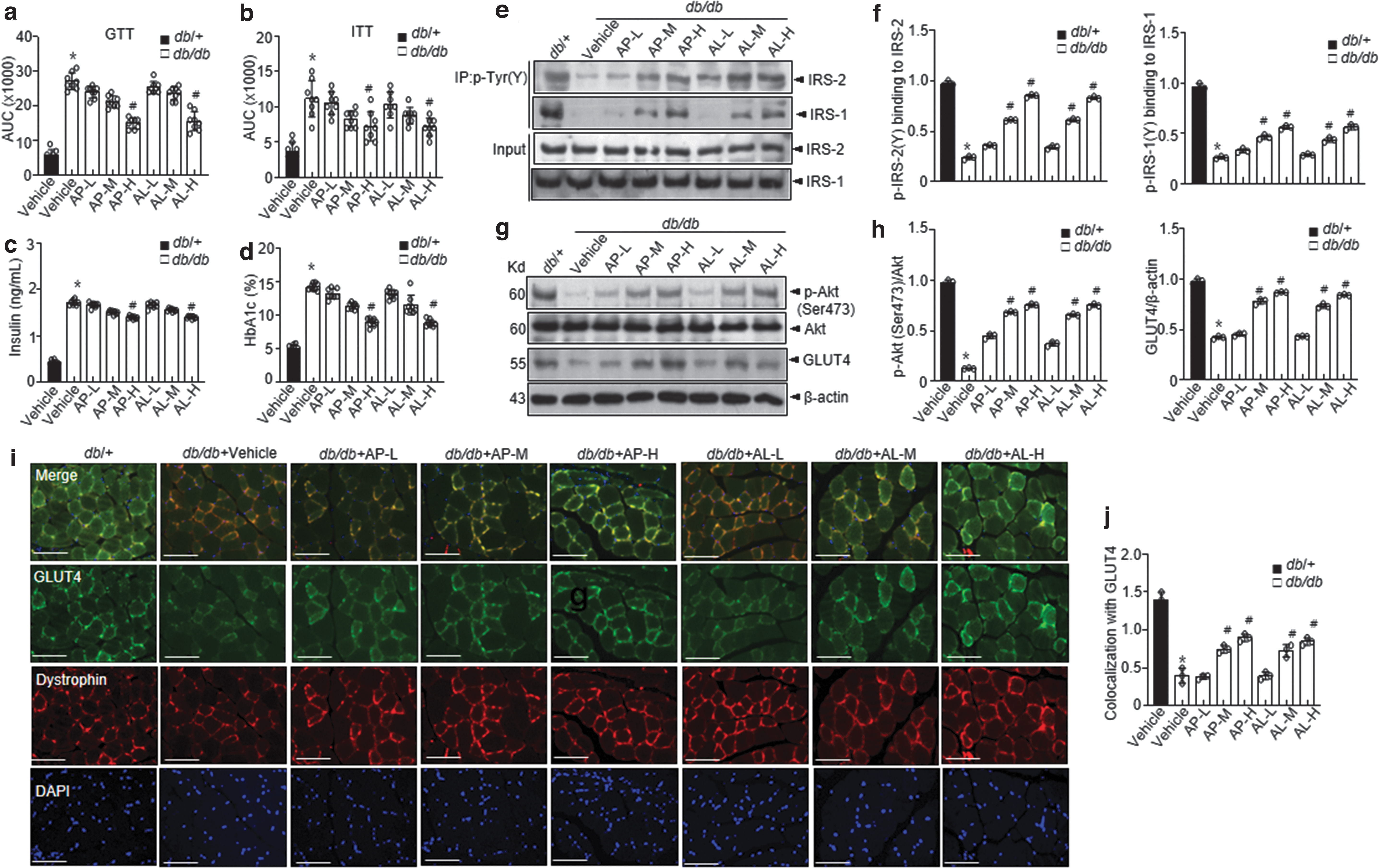
Insulin levels and hemoglobin A1c (HbA1c) scores were markedly decreased in the
Akt phosphorylation and its downstream GLUT4 expression were consistently increased in the
d -Allulose regulates skeletal muscle oxidative stress and ER redox imbalance under hyperglycemic conditions
Accumulating evidence has demonstrated a significant association of the overproduction of ROS (primarily superoxide anions) with diabetes and associated complications (8, 43). We first examined ROS levels in the skeletal muscles of
Glucose metabolism-related ROS production is highly affected by the conversion of NADPH to NADP+, which is controlled by NADPH oxidase (55). Therefore, the NADP+/NADPH ratio and NADPH oxidase activity were examined in
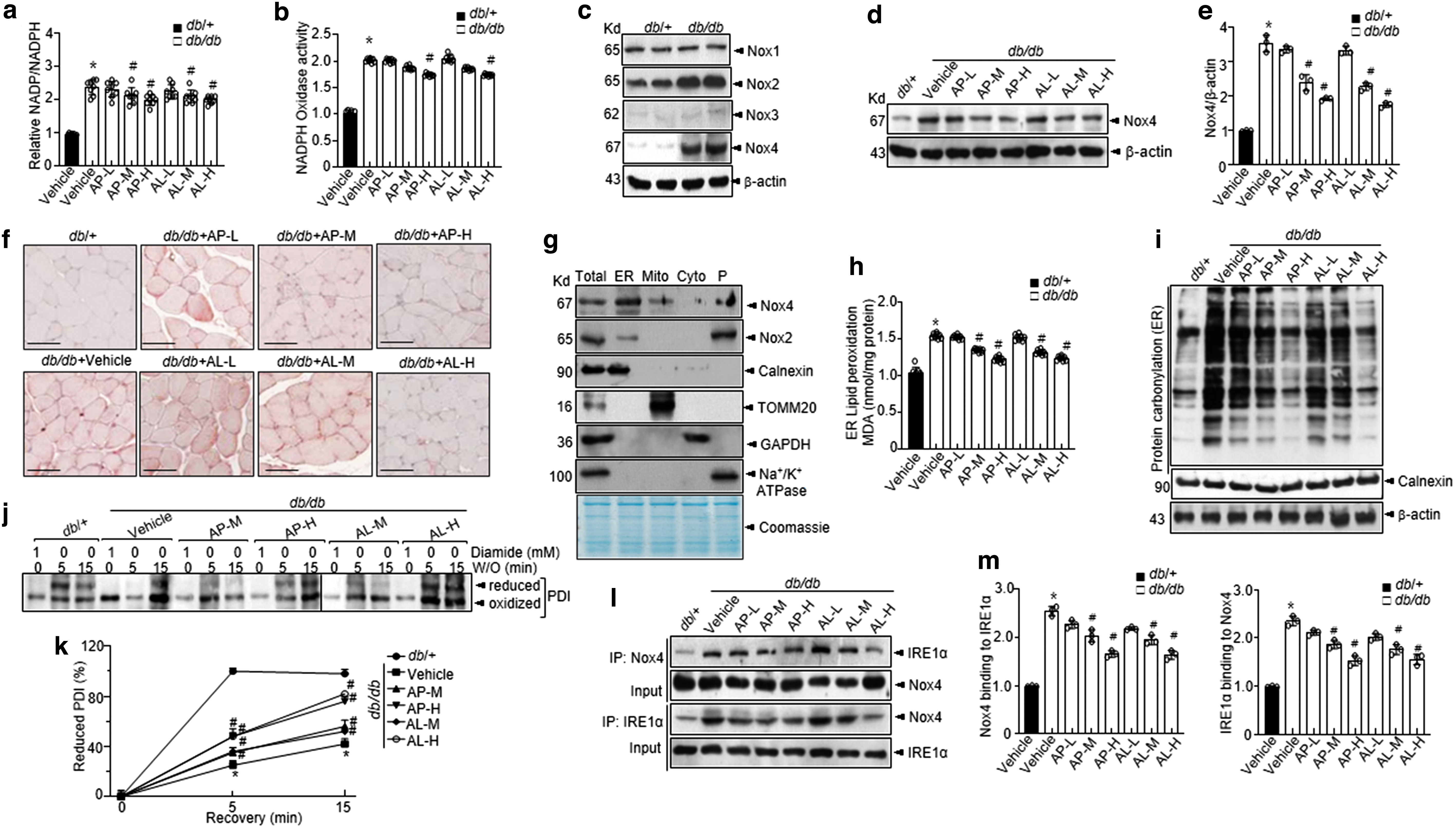
The levels of these proteins were significantly increased in the vehicle-treated db/db mice than those in db/+ mice, whereas supplementation with
The intra-ER redox state was examined by confirming the redox status of protein disulfide isomerase (PDI). The oxidized form of PDI continued to exist under diabetic conditions, whereas it was converted to a reduced form with ease under conditions involving washout in the control group (Fig. 2j, k). Notably, the conversion of PDI was recovered with
As shown in Figure 2l and m, Nox4 is bound to IRE1α, and this interaction was inhibited on treatment with
d -Allulose regulates the IRE1α sulfonation-RIDD-Sirt1 decay axis in skeletal muscles under hyperglycemic conditions
The ER stress response is a potential therapeutic target for chronic metabolic disorders, including diabetes (16). First, we determined the levels of ER stress markers (Fig. 3a, b). The levels of ER stress markers (p-IRE1α, GRP78, and CHOP) were markedly elevated in the vehicle-treated db/db mice than those in the vehicle-treated db/+ mice. We observed that phosphorylation of IRE1α, subsequent Xbp1 splicing, GRP78, and CHOP were inhibited in
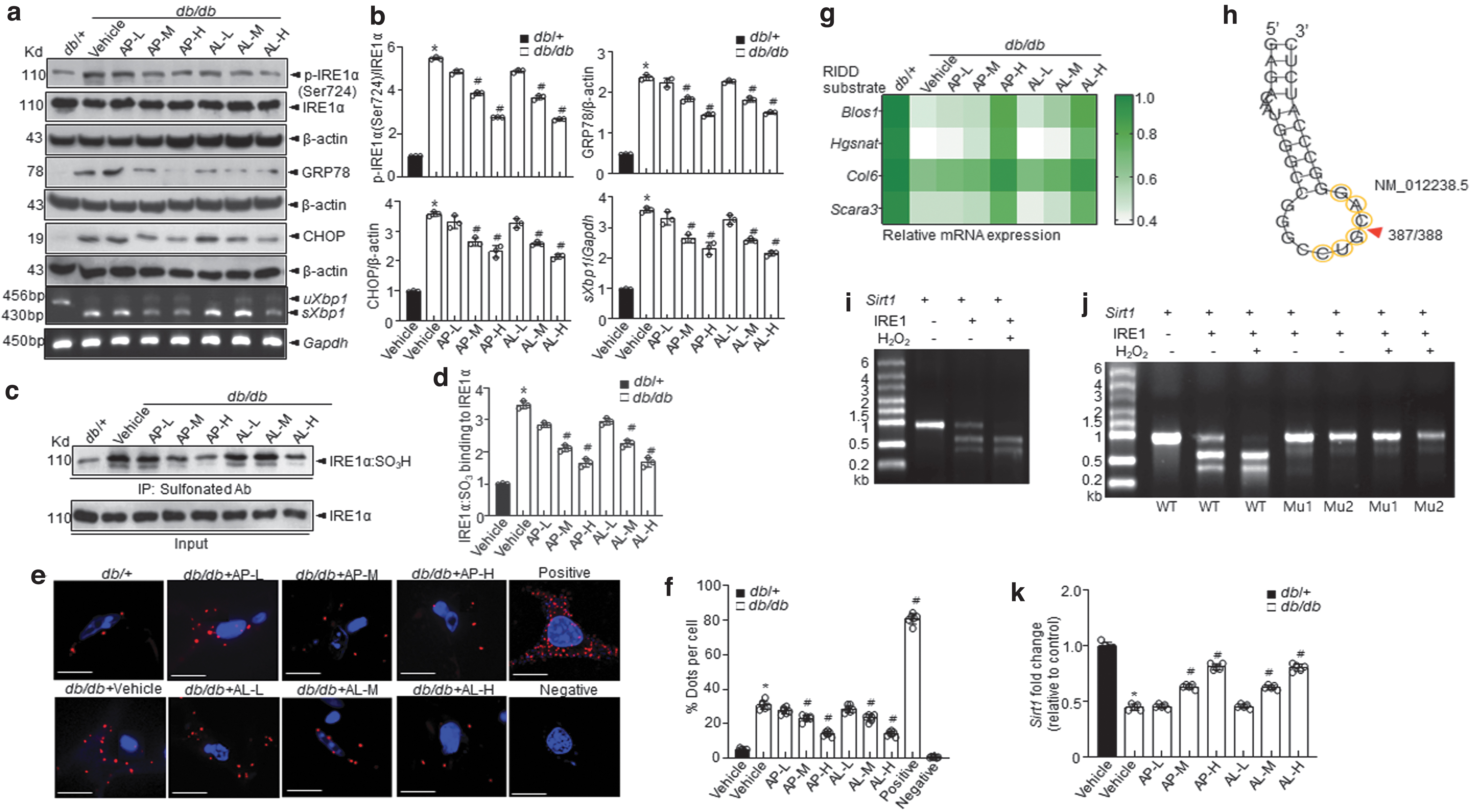
Among the ER stress response arms, IRE1α post-translational modifications, including phosphorylation, s-nitrosylation, sulfenylation, and sulfonation, play a crucial role in metabolic disorders (5). Especially, IRE1α sulfonation attributed to ROS production was reported to originate from an imbalance in the ratio of NADP+/NADPH and the associated activation of NADPH oxidase linked to glucose dysmetabolism (55). In addition, immunoprecipitation (IP) was performed with an anti-sulfonate antibody to further detect the oxidized form. IRE1α sulfonation (IRE1α:SO3) was also increased under diabetic conditions, whereas
Next, to assess the status of IRE1α sulfonation, we conducted a proximity ligation assay (PLA) using IRE1α antibody and cysteine-sulfonate with skeletal muscle samples. The interaction between IRE1α and cysteine-sulfonate observed in vehicle-treated db/db mice was attenuated in
Sirtuins are known to exert a variety of effects on gluconeogenesis, glycolysis, insulin secretion, and sensitivity and could demonstrate therapeutic potential for a variety of metabolic diseases (38). The sirtuin gene family comprising SIRT 1–7 genes was screened as a target for RIDD using RNA structure, a web server for RNA secondary structure prediction. Among the Sirt family members, Sirt1 had a specific RIDD target sequence, “CUGCAG” (Fig. 3h and Supplementary Fig. S4).
To assess whether IRE1α sulfonation affects the decay of Sirt1, we performed an in vitro cleavage assay and found that the cleavage of Sirt1 mRNA by IRE1α peptide increased in a time-dependent manner (Fig. 3i, j and Supplementary Fig. S4), leading to impairment of IRE1α-mediated decay of Sirt1. Sirt1 mRNA expression was decreased under diabetic conditions and was recovered on treatment with
Treatment of diabetic mice with
Next, the expression of the downstream target genes of PGC-1α, such as nuclear respiratory factor (NRF)-1, NRF-2, and mitochondrial transcription factor A (TFAM) genes, was examined (17, 47). The protein expression of the mitochondrial function-enhancing proteins NRF-1, NRF-2, and TFAM was significantly reduced in the skeletal muscles of vehicle-treated db/db mice than those observed in the skeletal muscles of db/+ mice; the expression was recovered on treatment with
Further,
d -Allulose controls Nox4-IRE1α-RIDD-Sirt1 decay, ameliorating the diabetic state under conditions involving administration of HFD
We next examined another metabolic stress mouse model. The HFD-fed mice treated with
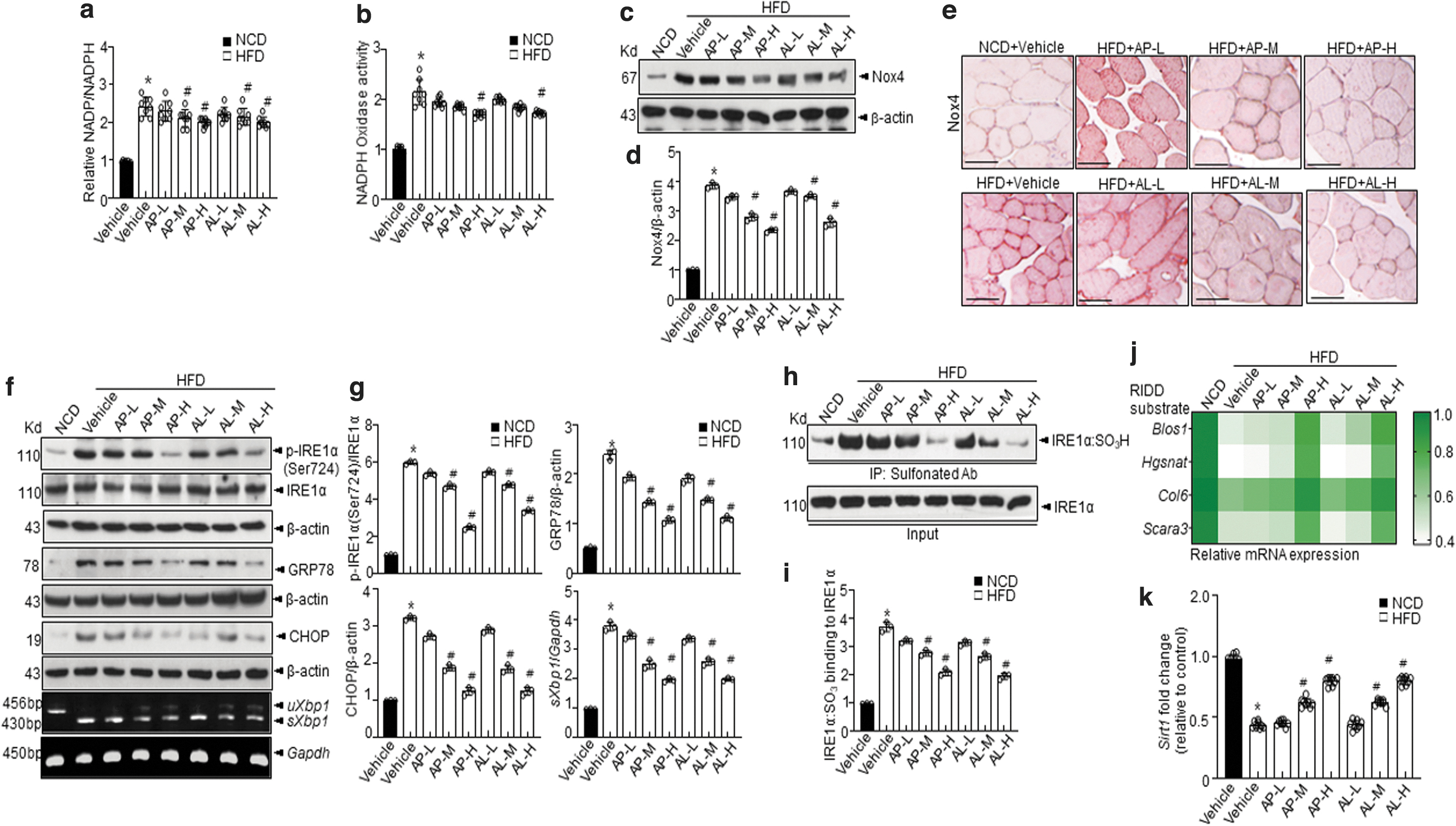
This finding indicates that
IRE1α-RIDD target genes, including Sirt1, were regulated by
Moreover, increased Akt phosphorylation was observed in
d -Allulose controls Nox4-IRE1α-RIDD-Sirt1 decay, enhancing glucose intake in HSkM cultured under high glucose conditions
Cells grown with a medium containing high levels of glucose mimic the diabetic condition and are linked to glucose dysmetabolism-based oxidative stress (34, 45). In this study, HSkM cultured with physiological glucose (5.5 mM) were incubated with a 30 mM glucose medium to mimic the diabetic glucose concentration. Glucose enhanced cellular ROS production and Nox4 expression in a time-dependent manner (Supplementary Fig. S11a–c). IRE1α sulfonation and the resultant RIDD were significantly increased under high glucose-culture conditions (Supplementary Fig. S11d–h).
The expression of the novel RIDD target gene, Sirt1, was also decreased under high glucose-culture conditions (Supplementary Fig. S11i, j).
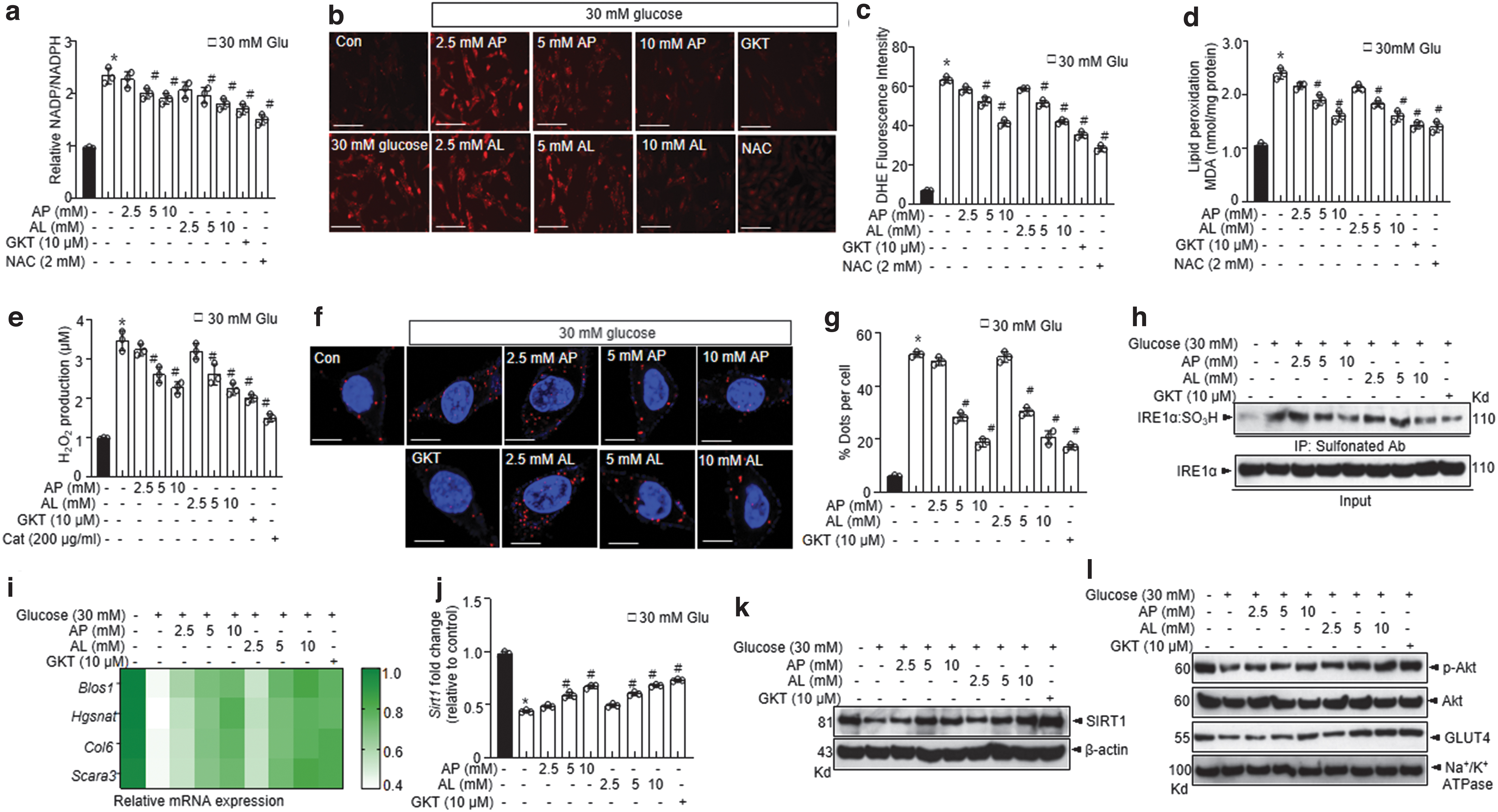
IRE1α sulfonation and the resultant expression of RIDD target genes including Blos1, Hgsnat, Col6, Scara3, and that of the Sirt1 gene were significantly inhibited by
In in vitro experiments, inhibitors of Nox4 (GKT137831 and GLX351322) showed effects similar to those of

The decay of Sirt1 RNA leading to decreased protein expression was significantly recovered under conditions involving Nox4-knockdown (Fig. 6i, j). Akt phosphorylation and the expression of its downstream target GLUT4 in the plasma membrane were consistently increased under conditions involving treatment with Nox4 siRNA (Fig. 6k, l). Nox4 silencing showed similar effects as

Discussion
This study demonstrated that diabetes-induced ROS production mediated by Nox4 and upregulation of IRE1α sulfonation, a master ER stress response signal, and associated RIDD-Sirt1 degradation could be controlled by
In this animal and cell-based study, Nox4 and associated ER stress were studied. This study demonstrated upregulation of Nox4 expression under diabetic conditions (Figs. 2 and 4). Under conditions involving glucose dysmetabolism and overnutrition, ER stress is associated with hyper-activated intra-ER electron uncoupling and electron leak with delayed ER oxidative folding process (19). Among the membrane-bound enzymes, Nox family proteins and ER-localized Nox4 (33) play a major role in the ER stress-linked ROS accumulation (46).
Phagocyte-like NAD(P)H oxidases are also a source of ROS in diabetes (1, 18). High glucose levels mediate ROS production via Nox4 in the rat kidney glomerular mesangial cells (18). In db/db and HFD-fed mice, the expression of Nox2 and Nox4 was high (Fig. 2d, e and Supplementary Figs. S3e and S9e). Nox2 expressed by plasma membrane is considered a typical ROS signal but not an ER-specific redox transducer. Nox4, specifically expressed by ER, clearly explains the ER stress-based GLUT4 disturbance, and it is highly associated with ER-localized NADPH oxidase-ROS signaling.
This phenomenon is linked to IRE1α sulfonation-RIDD-Sirt1 decay. Under physiologically stress conditions involving low ROS levels, the expression of GLUT4 is enhanced, indicating hormesis between ROS and GLUT4. Under hyperglycemic conditions, Nox4-induced ROS production and linked IRE1α sulfonation were reversed by
Sirt1, a major metabolic enzyme, was shown to be one of the IRE1α sulfonation-linked RIDD targets. Among the Sirt family, Sirt1 harbors a target sequence for RIDD (Fig. 3h–j and Supplementary Fig. S4). The effect of
Moreover, Sirt1 gene expression is decreased in patients with glucose intolerance (31). Hence, there is a direct relationship between Sirt1 transcription and measurements of insulin sensitivity (13). In this study, we found that Nox4 is controlled by
Thus, we assessed Sirt1 levels in the skeletal muscles of db/db mice and HFD-fed mice. In response to
SIRT1 in the skeletal muscles is essential for the PGC-1α-mediated induction of the expression of fatty acid oxidation-associated genes in response to fasting and exercise (9, 15). This specific observation suggests that during energy-demanding processes such as fasting and physical activity, PGC-1α is activated by SIRT1-mediated deacetylation.
The study dosage range seems to match the reported human equivalent dose range, “5 and 10 g” in diabetic or prediabetic patients (22, 41). With respect to the two formulations, liquid and powder, no differences concerning the anti-diabetic effects were observed, indicating that
In summary, the decay of Sirt1 as an RIDD target gene is suggested to be one of the mechanisms involved in the attenuation of diabetic conditions along with a well-established SIRT1-PGC-1α-linked GLUT4-associated glucose controlling mechanism. In addition,
Materials and Methods
d -Allulose products
Cell culture and transfection
HSkM (A12555; Thermo Fisher Scientific) were grown in a growth medium containing 10% (v/v) fetal bovine serum in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% penicillin/streptomycin under a standard humidified atmosphere. On reaching 85%–95% confluence, cells were stimulated with DMEM containing 2% horse serum for 2 days. HSkM were treated with 2.5, 5, or 10 mM AP or AL, 10 μM GKT137831, 2 mM N-acetylcysteine (NAC; Sigma-Aldrich, St. Louis, MO), and 10 μM GLX
Animal and experimental design
Diabetic db/db mice (C57BLK-Leprdb/Leprdb) and non-diabetic db/m mice (C57BLK-Leprdb/+) were purchased from the Jackson Laboratory (Bar Harbor, ME), and they were housed in the laboratory animal care facilities of Jeonbuk National University Hospital under standard SPF conditions. After acclimatization, 7-week-old male mice were randomly divided into eight groups (n = 8/group): normal control (db/m; water), negative control (db/db; water), three test groups (db/db; AP-low, -medium, and -high), and three test groups (db/db; AL-low, -medium, and -high).
Similarly, for developing another metabolic stress animal model, 7-week-old male mice were fed with HFD or a normal diet. All mice were randomly divided into eight groups (n = 8/group): normal control (normal diet; water), negative control (HFD; water), three test groups (HFD; AP-low, -medium, and -high), and three test groups (HFD; AL-low, -medium, and -high).
Blood samples were collected from the truncal vein, tibialis anterior muscle sections, and liver. The tibialis anterior muscle and the liver sections were used for Western blotting, real-time polymerase chain reaction (RT-PCR), and histological analysis. All dissected tissues were immediately frozen in liquid nitrogen and stored at −80°C until further analysis.
Glucose and ITT
For OGTT, mice fasted overnight were administered glucose at 1 g/kg via oral gavage. For ITT, mice fasted for 6 h were intraperitoneally injected with insulin (0.75 IU/kg BW: Actrapid Penfill Insulin human; Novo Nordisk, Bagsværd, Denmark). After administration of glucose/insulin, blood samples (∼30 μL) were derived from the tail vein at 0, 15, 30, 45, 60, 90, and 120 min, and blood glucose levels were determined by using a fresh capillary tube (Accu-Check glucometer; Roche Diagnostics). For checking glucose tolerance and sensitivity, we measured the total area under the curve (AUC) to show glucose tolerance test (GTT) and insulin tolerance test (ITT).
Biochemical analysis
The collected serum was used to measure the biochemical parameters. Insulin and HbA1c levels were measured with commercially available assay kits (Insulin: K4271; HbA1c: E4657; Biovision, San Francisco, CA). All protocols were performed according to the manufacturer's guidelines.
Histological analysis and immunohistochemistry
The tibialis anterior muscle was excised and frozen in liquid nitrogen-cooled isopentane (Nacalai Tesque). Transverse sections (5 μm) were cut by using cryostats (CM3050S; Leica Microsystems, Wetzlar, Germany) and collected onto MAS-coated glass slides (Matsunami). Hematoxylin and eosin (H&E) staining was carried out by using these sections. Immunohistochemistry (IHC) was performed as previously described (32). Briefly, hydrogen peroxide-fixed islets were embedded and cut into sections with a thickness of 4 μm.
To prevent nonspecific binding, sections were incubated with a blocking reagent (UltraTech HRP; Beckman Coulter, Brea, CA). The sections were subsequently incubated with antibodies against Nox4, Troponin T type I (Novus Biologicals, Littleton, CO), and MY-32 (Abcam). HRP-conjugated biotin-streptavidin complexes were used to assess antibody reactivity, and staining color was developed by substrate diaminobenzidine tetrahydrochloride.
Immunofluorescence staining
The immunofluorescence staining protocol was optimized for the analysis of GLUT4 (6). Briefly, the tibialis anterior muscle sections were fixed and washed with phosphate-buffered saline (PBS). The sections were exposed to a primary antibody against GLUT4 (MA5-17176; dilution: 1:200; Invitrogen) in combination with a primary antibody against dystrophin (PAS-32388; dilution: 1:400; Invitrogen) for 2 h at room temperature. After incubation with a primary antibody, the sections were washed thrice for 5 min in PBS and were then incubated in 1:200 dilution of secondary antibodies for 30 min at room temperature.
The sections were washed thrice for 5 min in PBS, and glass coverslips were mounted with 20 μL aqueous-mount solution (Scytek Laboratories). Images were captured by using an inverted confocal microscope (Leica DMIRE2; Leica Microsystems) with a 63× oil immersion objective lens. AlexaFluor 488-conjugated goat anti-rabbit IgG secondary antibody (F0382; Sigma) was used to detect the GLUT4 primary antibody, whereas AlexaFluor 594-conjugated goat anti-mouse IgG2b (T5393; Sigma) was used to detect the dystrophin primary antibody. Images were captured by using the Zeiss LSM 880 inverted confocal microscope (Carl Zeiss, Inc.). All the images were captured with the same laser intensities. For quantitating, colocalization of GLUT4 and dystrophin was calculated from each image by using ImageJ.
Immunoblotting and IP
Immunoblotting was performed as previously described, with minor modifications (32). Briefly, 20–40 μg of protein samples (tibialis anterior muscle, liver, and cell lysates) was separated on a polyacrylamide gel and transferred onto PVDF membranes. Later, blocked membranes were incubated with primary antibodies against p-IRE1α (No. ab124945; Abcam), IRE1α (No. 3294; Cell Signaling), p-eIF2α (No. 3597; Cell Signaling), eIF2α (No. 9722; Cell Signaling), ATF4 (No. 11815; Cell Signaling), CHOP (No. 2895; Cell Signaling), GRP78 (No. 13539; Santa Cruz), β-actin (No. sc-47778; Santa Cruz), Nox4 (No. PA5-72816; Invitrogen), Nox1 (No. PA5-38031; Invitrogen), Nox2 (No. ab129068; Abcam), and Nox3 (No. ab81864; Abcam). The blots were then washed, and corresponding species-specific secondary antibodies were added.
The signals were detected with enhanced chemiluminescence reagents. For IP, 100 μg of protein samples was incubated with corresponding antibodies overnight at 4°C. To pull the antibody/antigen complex, samples were incubated with protein A/G Sepharose beads. Then, the immunoprecipitates were washed with PBS-T and analyzed by immunoblotting with corresponding antibodies.
DHE staining
DHE was used to evaluate intracellular ROS production in the tibialis anterior muscle and HSkM cells. Fixed tibialis anterior muscles and cells were exposed to DHE (ThermoFisher Scientific, MA) at a concentration of 10 and 20 μM, respectively, for 30 min/37°C, followed by washing with PBS. The images were captured by using confocal microscopy. Relative fluorescence intensity was analyzed with ImageJ (National Institutes of Health, Bethesda, MD).
ROS measurement
Quantitative measurement of H2O2 levels was performed by using the Amplex® Red hydrogen peroxide/peroxidase assay kit (A22188; Molecular Probes, Thermo Fisher Scientific, CA) according to the manufacturer's instructions. Briefly, 50 μL of the lysate was incubated with 50 μL of a solution containing 0.05 M NaH2PO4 (pH 7.4), 0.2 U/mL of HRP, and 25.7 mg/mL of the Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) reagent for 2 h at 37°C.
After incubation, the absorbance of the reaction mixture was measured at 560 nm. Absorbance readings were compared with those obtained to develop a standard curve for H2O2 (0–40 μM). Results are expressed as H2O2 concentrations in μM.
Oxyblot assay
The levels of carbonylated protein in the tibialis anterior muscle were measured via OxyBlot Protein Detection Kit (Millipore, Billerica, MA) according to guidelines provided by the manufacturer. Briefly, the skeletal muscle lysate samples were incubated in a 1:1 ratio in 2,4-dinitrophenylhydrazine (DNPH), followed by the addition of 2-mercaptoethanol for protein denaturing. Later, a neutralization reagent was added to stop the reaction, and samples were loaded for immunoblotting as described in previous sections.
IRE1α sulfonation
Sulfonation was performed as previously described (30). The total tibialis anterior muscle and cell lysates (∼500 μg) were prepared by using lysis buffer (Cell Signaling) comprising protease and phosphatase inhibitor cocktail (Sigma). To detect sulfonation of IRE1α, IP was performed by using anti-cysteine-sulfonate (ab176487; Abcam) derived from lysates, followed by application of the anti-IRE1α antibody (3294; Cell Signaling). Protein A/G Sepharose beads (Sigma) were added and incubated for an additional 1 h.
Immunoprecipitates were washed five times with PBS-T buffer or PBS before being resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and were immunoblotted with the indicated antibodies.
Proximity ligation assay
Duolink in situ reagents (Sigma-Aldrich) were used to perform the PLA. The protocol was performed according to the manufacturer's instructions. Briefly, the tibialis anterior muscle sections (5 μm) and the cells were fixed with 4% paraformaldehyde (pH 7.4) in PBS for 10 min at room temperature. Permeabilization was performed by using 0.1% Triton X-100 in PBS for 10 min at room temperature. Blocking was realized with 0.1% Triton X-100 in PBS with 5% bovine serum albumin (BSA) for 1 h at room temperature. Incubations with primary antibodies (cysteine sulfonate and IRE1α, 1:100 in 0.1% Triton X-100 in PBS with 5% BSA) were performed overnight at 4°C in a humidity chamber.
Then, the cells were rinsed with Wash Buffer A. Next, PLUS and MINUS secondary PLA probes comprising both rabbit and mouse immunoglobulins diluted in 0.1% Triton X-100 in PBS with 5% BSA were added for 1 h at 37°C. The incubation was followed by washing for 5 min with two changes of Wash Buffer A. Subsequently, the slides were incubated with the Duolink ligation mix for 30 min at 37°C and thereafter washed with two changes of Wash Buffer A for 5 min each.
The Duolink amplification mix was then applied to the slides for 100 min at 37°C. Subsequently, the slides were washed twice for 10 min each time with Wash Buffer B. The nuclei were stained with DAPI and mounted on microscope slides, sealed with an anti-fading solution, and examined under a Widefield microscope Zeiss Axiovert 200M. HSkM were treated with H2O2 for 6 h and used as a positive control. Negative control was developed without incubation with primary antibodies. Quantification of the PLA red fluorescent dots was performed by using ImageJ.
NADPH-dependent oxidoreductase activity assay
NADPH oxidase activity was evaluated by analyzing superoxide production via lucigenin-enhanced chemiluminescence (28). Tibialis anterior muscle tissues or cells were washed with ice-cold PBS and homogenized in cold lysis buffer (20 mM sodium phosphate buffer [pH 7.0], 1 mM EDTA, 1 mM PMSF, 0.5 mM leupeptin, and 0.5 mM pepstatin) by sonication on ice. The homogenates (5 μL) were added to 0.5 mL of assay buffer (50 mM sodium phosphate buffer [pH 7.0], 1 mM EDTA, 150 mM sucrose, and 50 μM lucigenin).
After the addition of 0.1 mM NADPH, luminescence was measured as relative light units (RLUs) for 20 s at 1 min intervals in a Skanlt RE 6.1 (Thermo Fisher Scientific, Waltham, MA). The results were expressed as RLUs/mg protein and compared between groups.
Quantitative RT-PCR
The tibialis anterior muscle tissue was homogenized with the POLYTRON® homogenizer Kinematica Polytron™ PT2100) in 700 μL of QIAzol® Lysis Reagent. Total RNA was purified from the homogenate by using the miRNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Reverse transcription was performed by using 1 μg of RNA following the instructions of the iScript™ complementary DNA (cDNA) synthesis kit (Bio-Rad).
Experimental transcript levels were analyzed by using the iQTM SYBR Green Supermix (Bio-Rad) on StepOnePlus (Applied Biosystems). All quantitative RT-PCR data were normalized to β-actin expression analyzed in separate reactions.
In vitro transcription and cleavage assays
The synthetic Sirt1 gene sequence was inserted into pMA-RQ (GeneArt). To produce 5′ mRNA fragments, pMA-RQ Sirt1 plasmid DNA was linearized by BglII (Promega). In vitro transcription of the 5′ region of Sirt1 was performed with a T7 transcription kit (AM1333; Thermo Fisher Scientific). In vitro cleavage assay of Sirt1 mRNA was performed following a protocol previously described (27). The RNA products obtained by in vitro transcription were incubated with or without recombinant IRE1 (E31-11G; SignalChem) at 37°C. The RNA fragments were separated on a 1.5% denaturing agarose gel.
Statistical analysis
All immunoblots for in vitro studies were developed in triplicate. Data analysis was performed by using GraphPad Prism 5.02 (San Diego, CA). One-way analysis of variance performed by using Tukey post hoc comparison was used to compare groups. Basic data were presented as mean ± standard error of the mean. Statistical significance was set at p < 0.05. An electronic laboratory notebook was not used.
Footnotes
Authors' Contributions
H.-Y.L. and G.-H.L. planned the experiments, conducted the studies, analyzed the data, and were primarily responsible for writing the article. T.H.H., S.-A.P., J.W.L., and J.H.L. conducted the studies and analyzed the data. S.S., G.E.K., and J.S.H. reviewed and edited the article. J.K. and H.-J.C. is the guarantor of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Author Disclosure Statement
The authors have read the journal's policy and declare conflict of interests. There are no patents, products in development, or marketing products to declare.
Funding Information
This work was supported by Samyang Corp.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Supplementary Figure S14
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.