
Abstract
Aims:
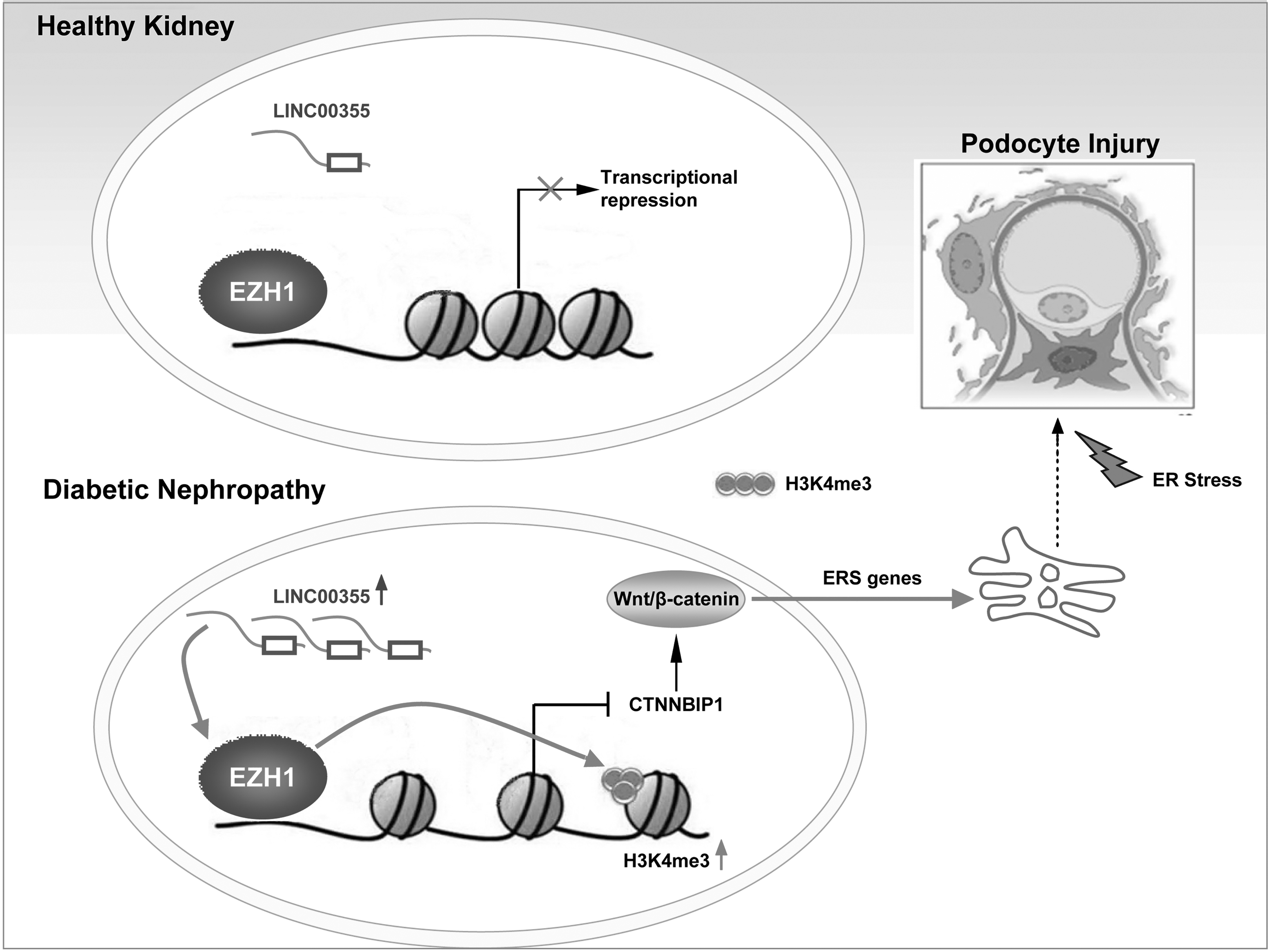
Endoplasmic reticulum stress (ER stress) plays an important role in podocyte injury in diabetic nephropathy. Wnt/β-catenin signaling modulates ER stress, yet the epigenetic regulation of β-catenin in ER stress and podocyte injury remains largely unknown. Herein, we tested the hypothesis that LINC00355 recruits EZH1 to the promoter region of CTNNBIP1 and trimethylates H3K4 to regulate ER-stress induced podocyte injury in DN.
Results:
LINC00355 is upregulated in podocytes and correlates with renal function decline in DN patients. LINC00355 localizes in the nucleus and exerts biological functions by directly binding EZH1, which epigenetically targets CTNNBIP1 through repressive trimethylation of H3K4 and activates Wnt/β-catenin signaling and ER stress. Further, we provide mechanistic evidences that LINC00355 recruits EZH1 to the promoter region of CTNNBIP1 and regulates ER-stress induced podocyte injury in DN.
Innovation and Conclusion:
Our data reveal a major role of LINC00355/EZH1/CTNNBIP1 network in triggering podocyte injury, providing new evidences for understanding the role of ER stress in DN. Antioxid. Redox Signal. 39, 225–240.
Introduction
The incidence of diabetes is on the rise globally, with diabetic nephropathy (DN) being the second most common cause of end-stage renal disease (Cheng et al., 2021). It is estimated that by 2040 the number of diabetic patients in the world will reach 642 million (Perkovic et al., 2016). DN is one of the most common microvascular complications of diabetes mellitus and the main cause of death and disability in diabetic patients. The proportion of DN-related dialysis population is increasing year by year (Liu, 2013), having become a global public health issue.
Long non-coding ribonucleic acids (lncRNAs) are a class of non-coding RNAs with a transcriptional length of more than 200 nucleotides. They regulate gene expression through epigenetic, transcriptional, and post-transcriptional mechanisms. Previous studies have detected and analyzed the expression profile of lncRNAs in diabetic animal models, showing that taurine up-regulated1 (TUG1) can regulate oxidative stress in podocytes through epigenetic mechanism and participate in the development and progression of the disease (Long et al., 2016).
Innovation
The epigenetic regulation of β-catenin in ER stress and podocyte injury is unclear. Herein, we demonstrate that LINC00355 directly binds EZH1 through epigenetic regulation of β-catenin in the podocytes of diabetic nephropathy.
Our previous study has shown the role and function of LINC01619 as a competing endogenous RNA in regulating miR-27a/FOXO1-mediated endoplasmic reticulum (ER) stress and podocyte injury in DN (Bai et al., 2018). Further, an epigenetic role of Wilm tumor gene 1 (WT1) in attenuating podocyte injury via the WT1/EZH2/β-catenin axis has also been elucidated (Wan et al., 2017).
The enhancer of zeste homolog 2 (EZH2), the histone methyltransferase enzyme, belongs to the family of the catalytic subunit of polycomb repressive complex 2 (PRC2). EZH2 has been found to catalyze the histone H3 lysine 27 trimethylation (H3K27me3) mark and plays a critical role in the methylation process (Cao et al., 2002). As a variant of the core catalytic subunit, EZH1 is a paralogue of EZH2 and likely arises from an EZH2 gene duplication event.
They share 63% overall identity and 94% identity of their SET domain. EZH1-containing PRC2 includes all the other three core components, EED, SUZ12, and RbAp46/48 (Grau et al., 2021). Studies have shown that systemic EZH2 inhibition accelerates kidney injuries in diabetes (Siddiqi et al., 2016) and alterations in H3K27me3 levels have been shown to affect podocyte development in glomerular diseases (Majumder et al., 2018). Podocytes are post-mitotic cells and their dedifferentiation in glomerular diseases, such as DN, brings research to evaluate the role of epigenetic modulation in these quiescent cells.
In the present study, to more accurately and objectively clarify the biological role of lncRNAs in DN, we detected their expression profiles in renal tissues from DN patients with the lncRNA chip and analyzed related biologically functions using bioinformatics analysis. Of note, we found that LINC00355 was upregulated in DN compared with the control. To this end, we designed the present study to investigate the role of LINC00355 in podocyte injury in the pathogenesis of DN (Fig. 1).
Results
LINC00355 is upregulated in podocytes and correlates with renal function decline in DN patients
To demonstrate the biological function of LINC00355, the expression level of LINC00355 in renal biopsy samples from DN patients was analyzed and its correlation with patients' clinical characteristics was examined.
Metabolic and anthropometric data of the patients whose renal biopsies were evaluated were presented in Tables 1 and 2. The sequencing data were submitted to the NCBI GEO database (Gene Expression Omnibus database,
Metabolic and Anthropometric Data of Diabetic Nephropathy Patients
p < 0.01; *** p < 0.001; versus TBMN.
BUN, blood urea nitrogen; DBP, diastolic blood pressure; DN, diabetic nephropathy; SBP, systolic blood pressure; Scr, serum creatinine; TBMN, thin basement membrane nephropathy.
Clinical Data of Patients from Diabetic Nephropathy and Thin Basement Membrane Nephropathy
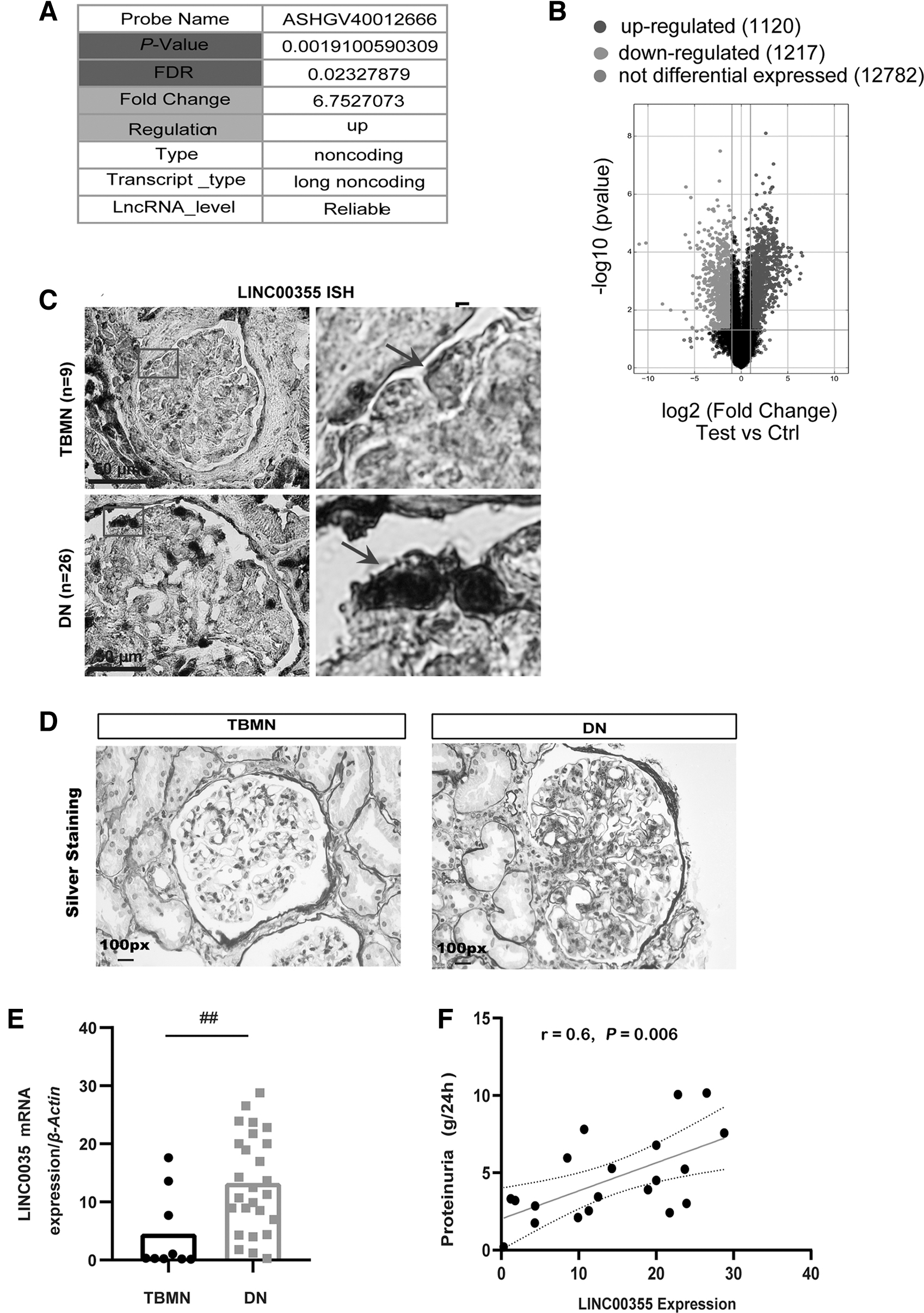
Further, it was shown in the UCSC database that LINC00355 is located on chromosome 13, containing 6 exons and a total length of 1878 bp. The volcano plot for differential expressed lncRNAs revealed 1120 upregulated genes, 1217 downregulated genes, and 12,782 not differential expressed genes (Fig. 2B).
Second, the expression of LINC00355 in 26 cases of DN and 9 cases of thin basement membrane nephropathy (TBMN) kidney tissues was detected by in situ hybridization (ISH) analysis. The results have confirmed upregulated expression of LINC00355 in DN and the positive signal was mainly located in the podocyte nucleus (Fig. 2C). The function of lncRNAs is related to its intracellular localization, for example, cytoplasmic expression of lncRNAs plays a role in regulating messenger ribonucleic acid (mRNA) stability and translation, whereas nuclear expression of lncRNAs is involved in epigenetic regulation (Holoch and Moazed, 2015).
Further, we analyzed the mRNA expression level of LINC00355 in renal biopsy samples from 26 cases of DN and 9 cases of TBMN as controls (Fig. 2D). As shown by quantitative real-time reverse transcription–polymerase chain reaction (qRT-PCR) analysis, LINC00355 expression was upregulated in DN compared with TBMN (p < 0.001) (Fig. 2E).
Third, we further analyzed the correlation between the expression level of LINC00355 and DN patients' clinical parameters. The results have shown that LINC00355 expression level was positively correlated with the level of 24-h urinary protein (r = 0.60, p = 0.005, Fig. 2F), and it was not correlated with serum creatinine (Scr) (Supplementary Fig. S1).
Taken together, these data suggest that the upregulation of LINC00355 in DN is correlated with the level of patients' proteinuria, suggesting the involvement of podocytes in the disease progression. LINC00355 may serve as a molecular marker in podocyte injury and could play an important role in the development of DN.
LINC00355 upregulation is involved in ER stress signaling pathway in DN
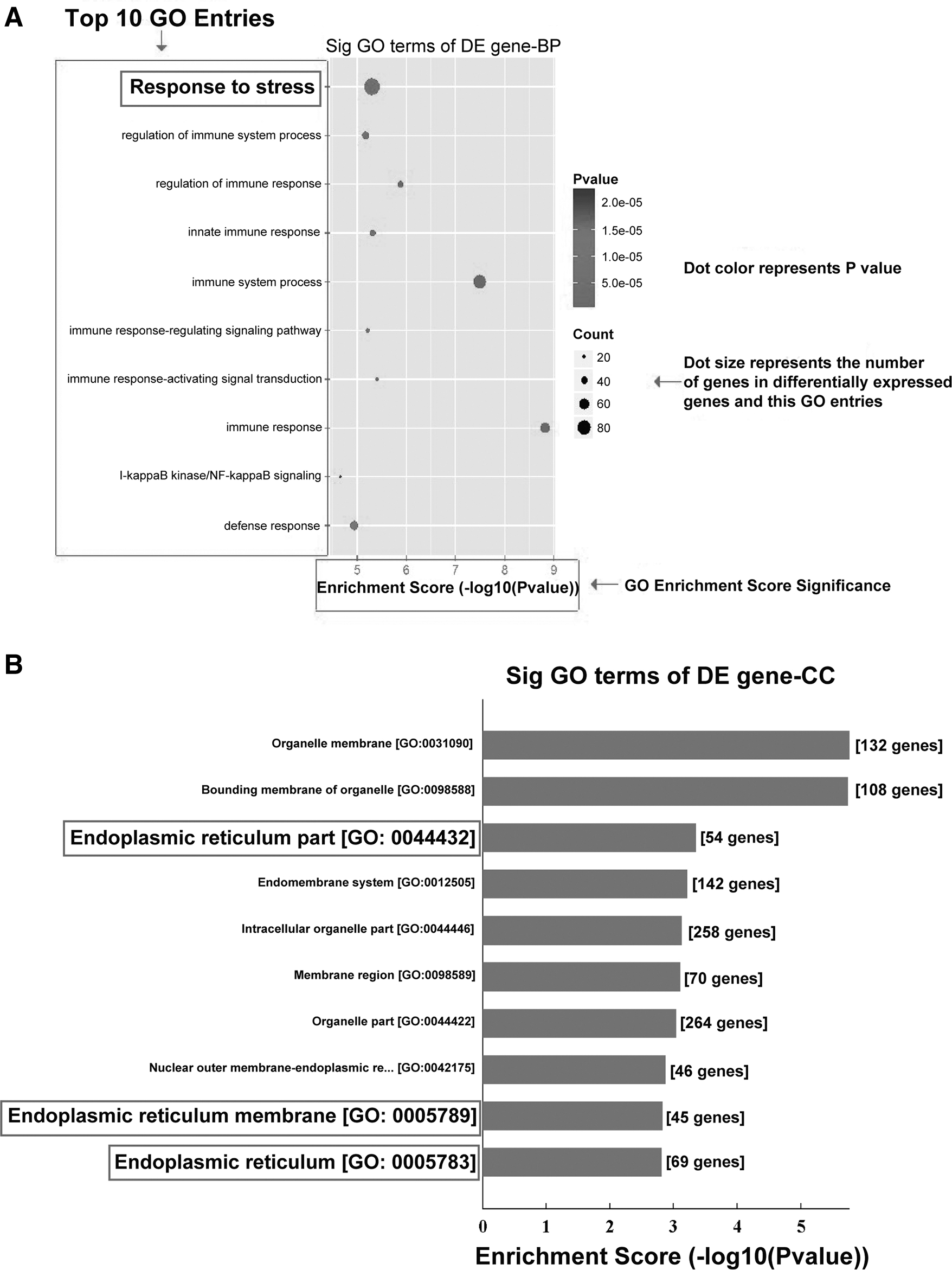
To elucidate the role of LINC00355 and its significance in DN, we first used bioinformatics technology and analyzed the gene ontology and signaling pathways related to the upregulation of lncRNA expression. The results have shown that stress response (Fig. 3A) and ER-related signaling pathways (Fig. 3B) were located in the top 10 enriched items, indicating that LINC00355 upregulation is involved in the ER stress signaling pathway in DN.
Second, to elucidate the relationship between LINC00355 and ER stress, we conducted in vitro experiments to investigate the function of LINC00355. We found that high glucose stimulated the expression of LINC00355 in cultured podocytes, suggesting that LINC00355 plays a role in high glucose condition (Fig. 4A). Third, LINC00355 expression was knocked down using small interfering RNA (siRNA) and the downstream effects were examined.
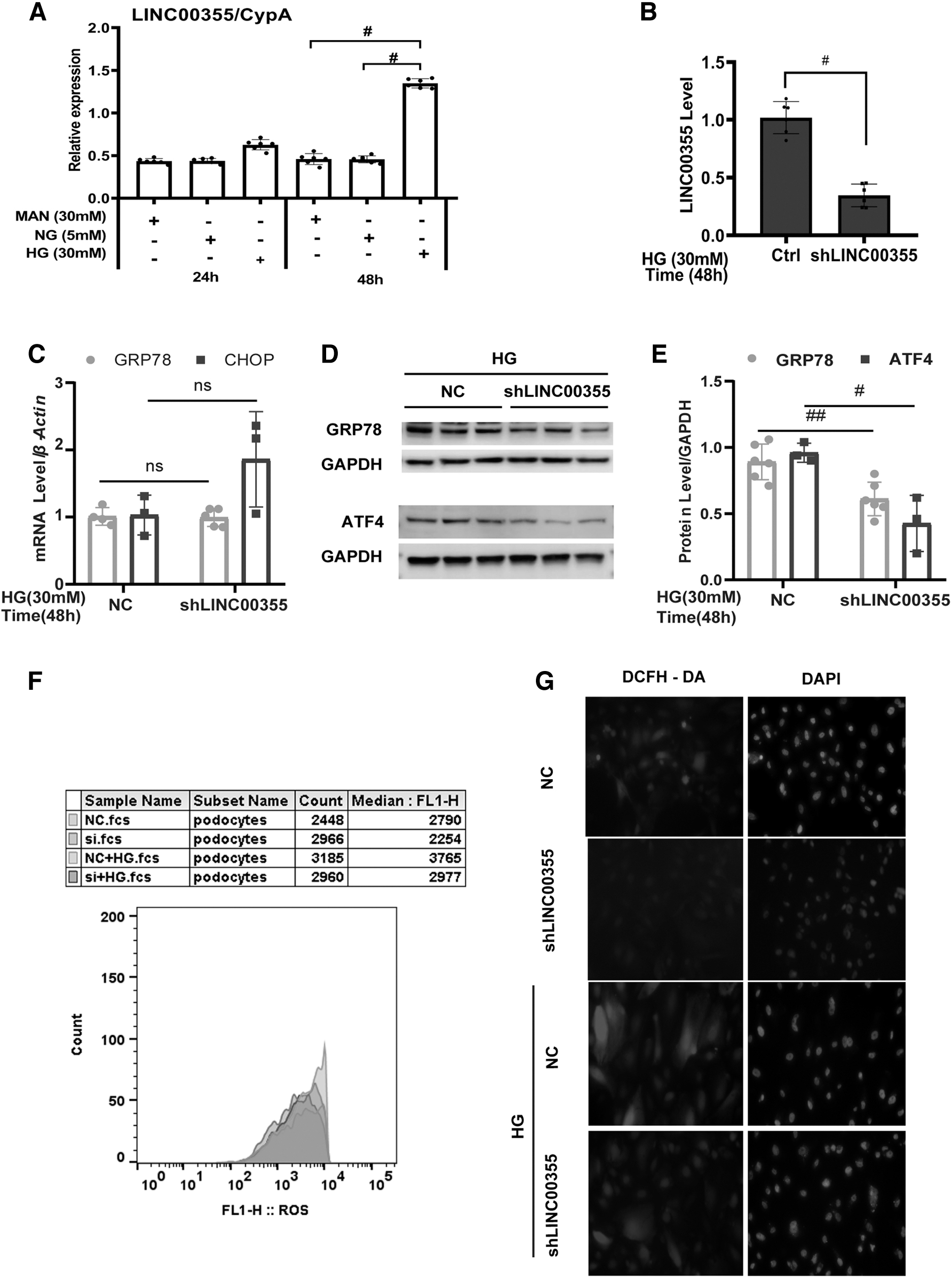
We used three siRNA sequences to knock down the expression of LINC00355 and found that siRNA #3 significantly downregulated the level of LINC00355 to 85% as examined by real-time qRT-PCR analysis (Fig. 4B). Of note, LINC00355 silencing did not have any effects on the mRNA level of glucose-regulated protein 78 (GRP78) and CCAAT/enhancer binding protein homologous protein (CHOP) (Fig. 4C). However, knocking down LINC00355 significantly reduced the protein level of GRP78 and AMP-dependent transcription factor 4 (ATF4) as detected by Western blot analysis (Fig. 4D, E). Further, LINC00355 silencing decreased the production of reactive oxidative stress as detected by 2′-7′dichlorofluorescin diacetate (DCFH-DA) assay (Fig. 4G) and the immunofluorescent staining (Fig. 4F). In addition, LINC00355 overexpression in cultured podocytes transfected with LINC3355 overexpression plasmid attenuated the production of ROS as detected by immunofluorescent staining (Supplementary Figs. S6 and S7).
These results altogether reveal that LINC00355 upregulation is related to the activation of the ER stress pathway in the diabetic condition. LINC00355 may mediate the ER stress signaling through epigenetic modulation.
LINC00355 induces podocyte injury in DN
Next, we set out to investigate the biological function of LINC00355 in podocytes both in vitro and in vivo. In high glucose cultured podocytes, LINC00355 silencing significantly reduced the number of apoptotic cells as detected by flow cytometry (Fig. 5A, B). LINC00355 knockdown increased the expression level of apoptosis molecules B cell lymphoma-2 (Bcl-2) by Western blot and semi-quantification analysis (Fig. 5C, D). In db/db diabetic mice in vivo, LINC00355 overexpression with HBAAV2/9-h-LINC00355 increased the number of podocytes in the glomeruli as shown by TUNEL staining (Fig. 5E). Significantly, LINC00355 overexpression worsened the renal function of db/db mice as demonstrated by an increase in Scr (Fig. 5F) and urinary albumin excretion rates (Fig. 5G and Table 3).
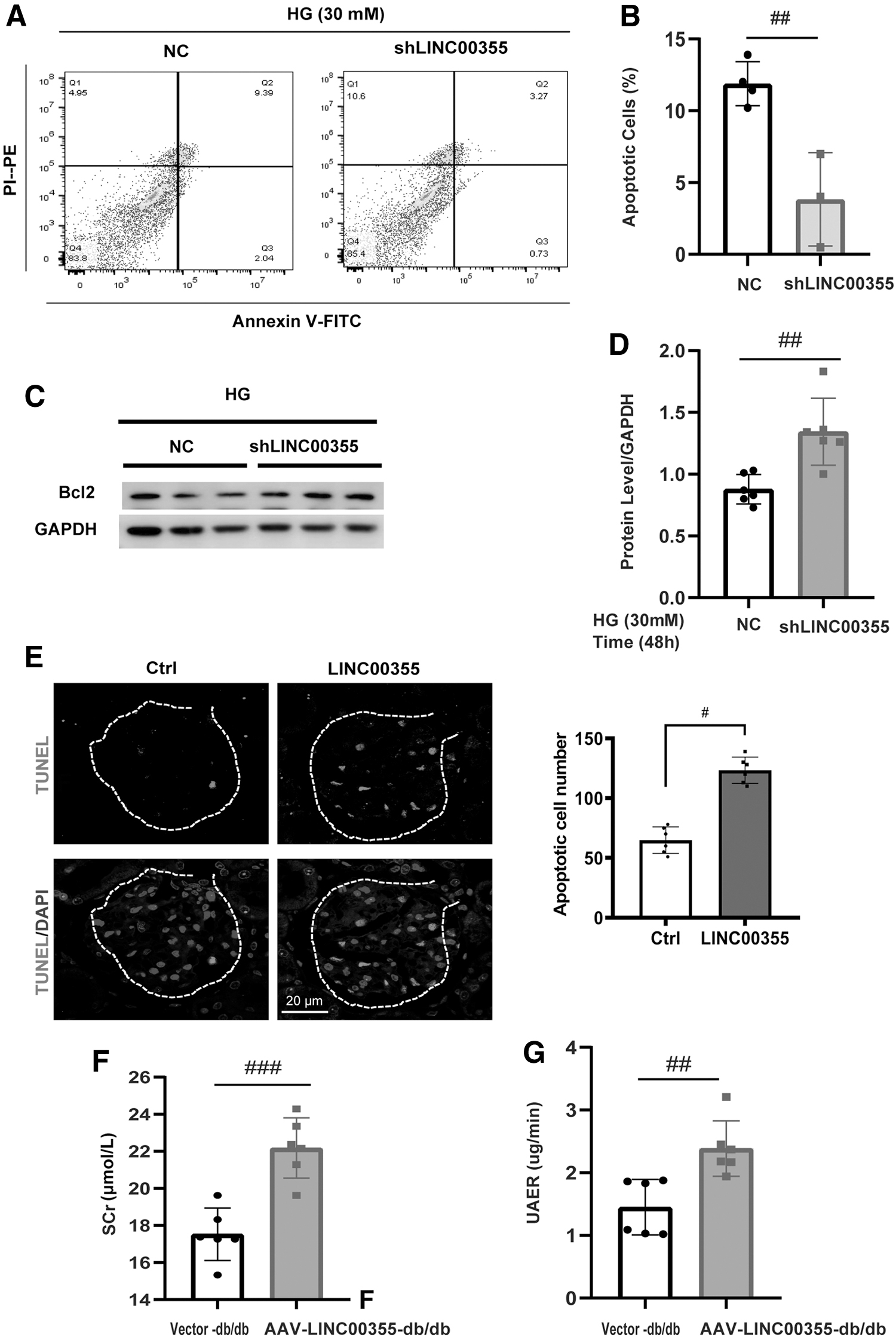
Biological Parameters for db/db Diabetic Mice Treated with HBAAV2/9-h-LINC00355 or Vector Control
p < 0.01; *** p < 0.001; **** p < 0.0001 versus vector control group.
UACR = urine albumin to creatinine ratio.
Collectively, these results suggest that LINC00355 exerts a deleterious effect on podocytes and promotes the progression of DN.
LINC00355 inhibits CTNNBIP1 and activates oxidative stress via β-catenin signaling pathway
To investigate the mechanisms of how LINC00355 regulated podocyte oxidative stress, we first conducted the coding-non-coding gene co-expression bioinformatics analysis to predict potential target genes in the regulation of LINC00355. Noteworthy, we found that CTNNBIP1, the inhibitor of β-catenin, was the target gene of LINC00355 (Fig. 6A). Second, in high glucose cultured podocytes in vitro, knocking down LINC00355 did not have effects on the mRNA level of CTNNBIP1 and CTNNB1 by RT-PCR analysis (Fig. 6B).
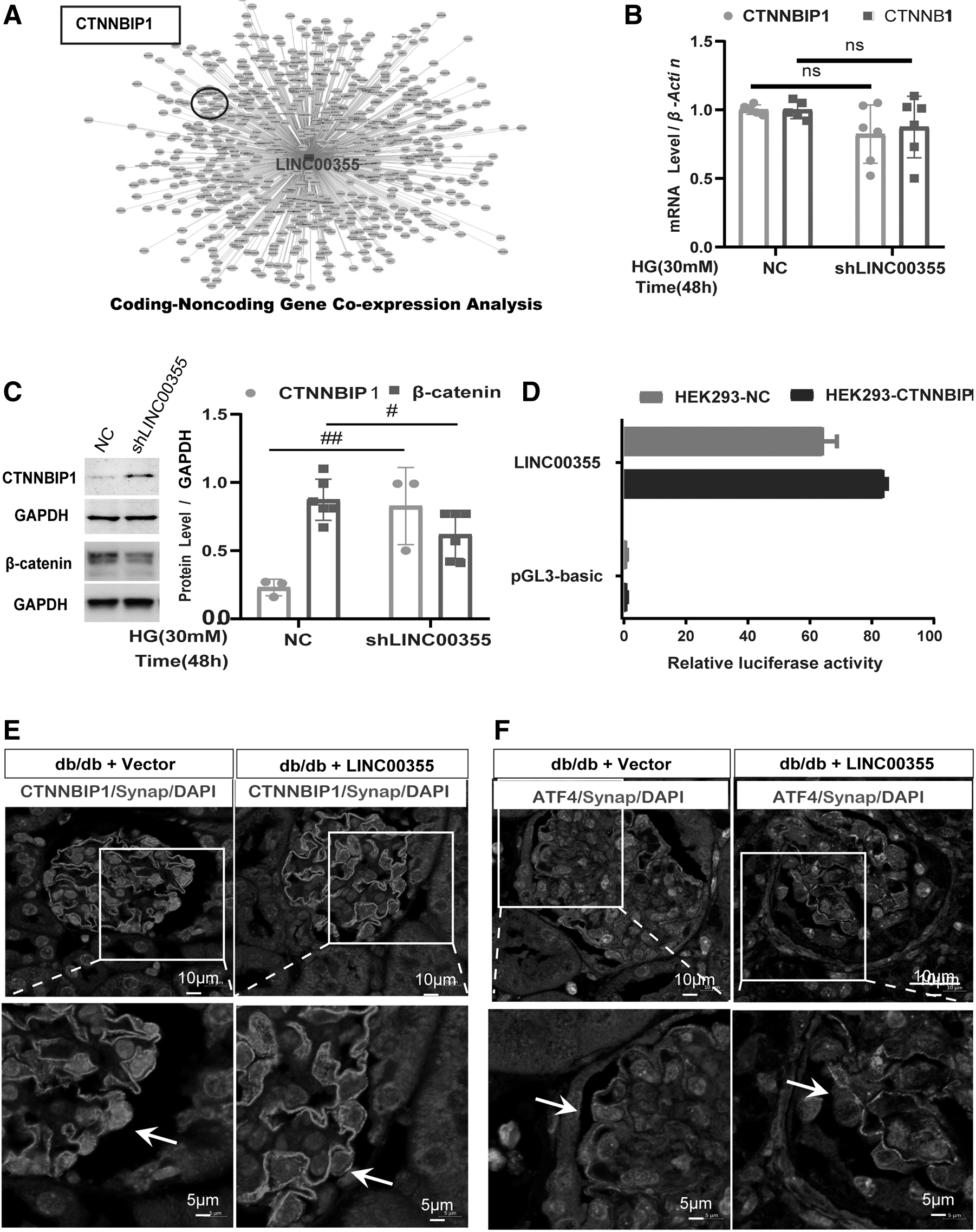
However, silencing LINC00355 significantly increased the protein level of CTNNBIP1 and decreased the level of β-catenin, as detected by Western blot analysis (Fig. 6C). In addition, by the dual-luciferase reporter gene assay, we found that silencing LINC00355 effectively increased the luciferase activity of CTNNBIP1 (Fig. 6D). These results suggest that LINC00355 may regulate the target gene expression through epigenetic mechanisms.
Further, in db/db diabetic mice, by immunostaining analysis, we found that overexpression of LINC00355 with HBAAV2/9-h-LINC00355 decreased the level of CTNNBIP1 (Fig. 6E) and increased the level of ATF4 (Fig. 6F) in podocytes as detected by immunofluorescent microscopy. In addition, by immunohistochemical staining, we also found that β-catenin was increasingly expressed in podocytes (Supplementary Fig. S5).
These data together demonstrate that LINC00355, by targeting CTNNBIP1, is involved in podocyte injury in high glucose condition. LINC00355 plays an inhibitory effect on the expression of CTNNBIP1 and may activate oxidative stress via the β-catenin signaling pathway.
LINC00355 recruits EZH1 to the promoter region of CTNNBIP1 to inhibit CTNNBIP1 expression
To gain molecular insights into the mechanisms of LINC00355 in regulating the expression and function of CTNNBIP1, we analyzed the correlation between the expression of LINC00355 and CTNNBIP1. First, by immunohistochemistry staining, we found that the expression level of EZH1 was increased in the podocyte nucleus of diabetic mice (Fig. 7A) and renal biopsy samples from DN patients (and Supplementary Fig. S2). Third, we found that LINC00355 targeted EZH1 as detected by lncRNA-mRNA pull-down assay (Fig. 7B).
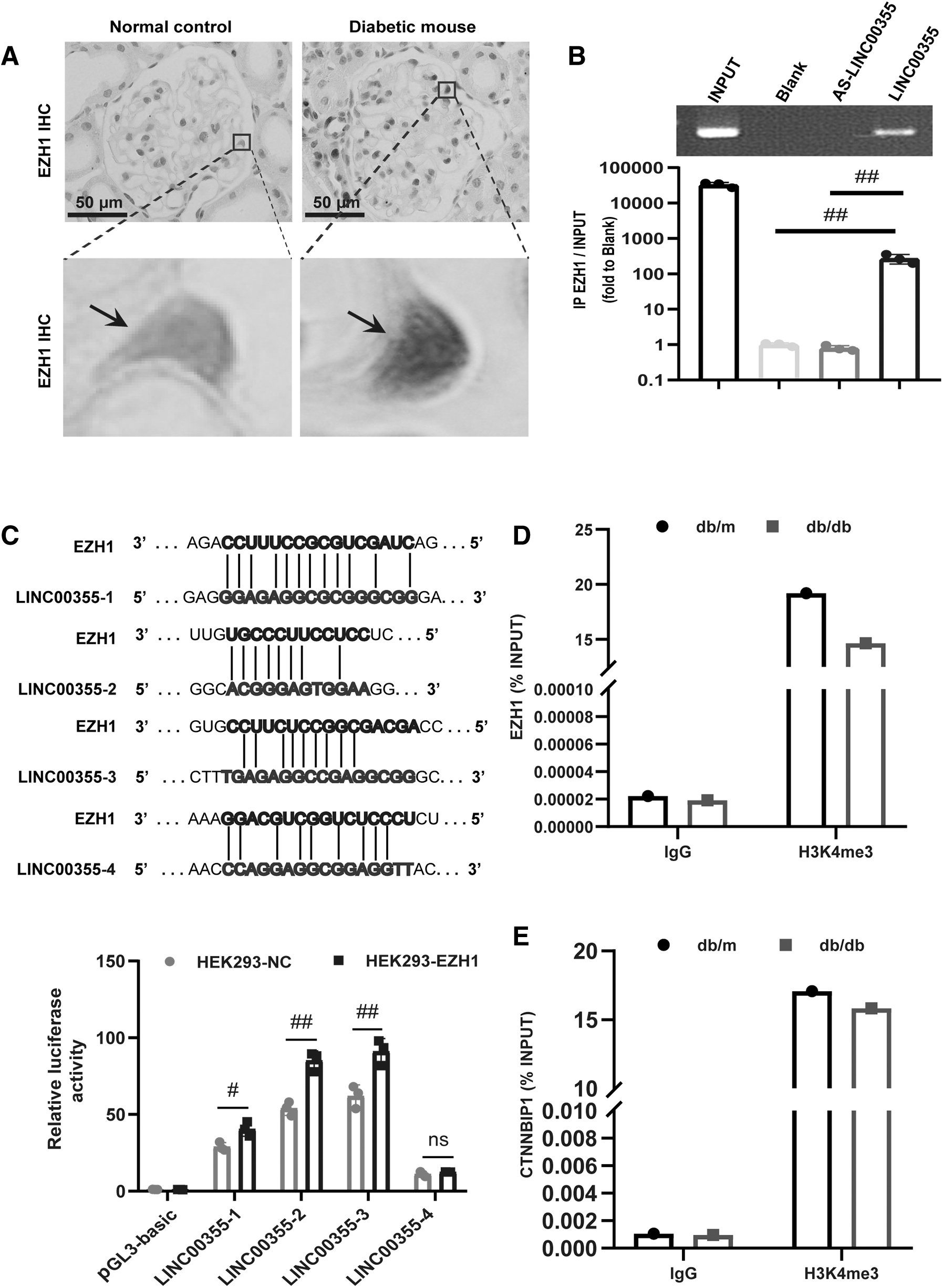
Next, to verify the bioinformatics results, we constructed the luciferase reporter plasmid pGL3-EZH1 to examine the effect of LINC00355 silencing on EZH1. Co-transfection of pGL3-EZH1 and reporter gene plasmid was conducted in human embryonic kidney 293 (HEK 293) cells, and the fluorescence signal was increased. These results suggest that LINC00355 can directly bind to EZH1 (Fig. 7C).
To further elucidate the molecular mechanisms of LINC00355 in regulating EZH1, we finally used the chromatin immunoprecipitation (ChIP)-qPCR assay and confirmed the presence of H3K4 methylation in the EZH1 promoter region in renal tissues of db/db diabetic mice (Fig. 7D, E).
Taken together, these data illustrate that LINC00355, by recruiting EZH1 to the promoter region of CTNNBIP1, activates the β-catenin signaling pathway in DN. The epigenetic regulatory mechanism plays a pivotal role in ER stress and podocyte injury in the development of DN.
Discussion
Podocyte injury is the key initiating factor of DN. High glucose microenvironment, excess ROS, and oxidative stress are critical mechanisms of podocyte injury (Bai et al., 2018; Li et al., 2019). The phenomenon of “metabolic memory” induced by diabetic microenvironment indicates that epigenetic modification is involved in the pathogenesis of DN (Kato and Natarajan, 2019), an important mechanism causing podocyte injury (Cao et al., 2021).
The effect of “metabolic memory” is a bridge between diabetes and its complications, posing a new challenge to the blood glucose management of type 2 diabetic patients. The mechanism of “metabolic memory” effect has been studied extensively since the theory was pointed out two decades ago. High glucose-induced oxidative stress is the central link of many factors related to “metabolic memory.”
Oxidative stress may ultimately lead to vascular endothelial dysfunction through multiple pathways, including polyol pathway, formation of glycosylation end products, activation of protein kinase C pathway, hexosamine pathway, and gene expression (Miller and Orchard, 2020). Undoubtedly, elucidating the molecular mechanism of podocyte injury under the condition of “metabolic memory” is one of the most important questions in the research and treatment of DN.
Podocyte injury and oxidative stress contribute to the development and progression of DN (Kopp et al., 2020), mechanisms of which, however, still remain incompletely understood. Podocyte is the research focus for the development of new preventive and therapeutic strategies in treating chronic kidney disease, including DN. A series of scientific integrative approaches have vastly advanced our understanding of podocyte biology, function, and metabolisms in the field of glomerular diseases, especially in DN (Hernandez-Diaz et al., 2019).
Lines of evidence have shown that LINC00355 triggers malignant progression in several malignant cancers. In gastric cancer, LINC00355 induces proliferation and invasion through promoting ubiquitination of P53 (Zhao et al., 2020). In hepatocellular carcinoma, LINC00355 triggers proliferative rate and inhibits apoptosis by activating the Wnt/β-catenin signaling (Luo et al., 2021).
In bladder cancer, LINC00355 promotes the EMT and metastasis through the miR-424-5p/HMGA2 axis (Li et al., 2021). Though multiple studies have illustrated the role of LINC00355 in cancer, so far, there is still no report demonstrating its role in ER or oxidative stress in podocytes or other cell types. In the present study, based on our in vitro and in vivo findings, we illustrate that LINC00355 promotes the ER stress and podocyte injury via epigenetic regulatory mechanisms in DN.
Our conclusions are supported by several key observations. First, LINC00355 was found to be upregulated in podocytes and positively correlated with proteinuria in DN patients. Further, using bioinformatics and functional studies, we demonstrate that LINC00355 upregulation is involved in the ER stress signaling pathway and silencing LINC00355 alleviates podocyte injury in both high glucose cultured podocytes and diabetic mice.
Second, we found that LINC00355 inhibits CTNNBIP1 and activates oxidative stress via the β-catenin signaling pathway. Previous studies have confirmed the role of non-coding RNAs in oxidative stress and the development of DN, including TUG1, MALAT1, and LOC105374325 (Hu et al., 2018; Long et al., 2016; Majumder et al., 2019). In the present study, we propose a unique mechanism that LINC00355 epigenetically inhibits CTNNBIP1 and plays a critical role in activating ER stress in DN.
Third, we provide mechanistic evidence that LINC00355 recruits EZH1 to the promoter region of CTNNBIP1 and inhibits its expression, thus activating the β-catenin signaling pathway and oxidative stress. A series of recent studies have shown that non-coding RNAs regulate podocyte pathophysiology through various mechanisms (Long et al., 2016). In the context of the diabetic microenvironment, our findings provide the novel molecular insights into the epigenetic modulation of LINC00355 in triggering oxidative stress and podocyte injury in diabetic condition.
Mechanistically, epigenetic processes are associated with podocyte dedifferentiation and the development of glomerular diseases. Findings have delineated that alterations in the H3K27me3 mark in podocytes affect Notch pathway activation and susceptibility to glomerular diseases (Allison, 2018). Histone protein modifications determine podocyte fate during the normal development and disease. In DN, deletion of the histone methylating enzyme EZH2 decreased H3K27me3 levels and sensitized podocytes to injuries causing deteriorated renal function (Majumder et al., 2018), suggesting the role of the epigenetic reprogramming in controlling the disease outcome.
Our present data also revealed that LINC00355 had a direct binding site with EZH1 and H3K4me3 methylation was in the promoter region of EZH1. Noteworthy, ChIP-qPCR assay showed that compared with control db/m mice, in kidney tissues of db/db diabetic mice, there was a reduced level of H3K4me3 methylation in the promoter region of EZH1 and CTNNBIP1, indicating the critical role for EZH1 in mediating LINC00355-induced podocyte injury.
In summary, the present study demonstrates new insights into the epigenetic role of non-coding RNA LINC00355, by recruiting EZH1 to CTNNBIP1 promoter region and activating the β-catenin signaling pathway, promotes ER stress-induced podocyte injury in DN, offering potential opportunities for innovative therapeutic treatments for the disease.
Materials and Methods
Electronic laboratory notebook was not used.
Patients and specimens
Data collection followed the STROBE statement for observational studies. Type 2 diabetic patients undergoing renal biopsy at the Department of Nephrology in the First Affiliated Hospital of Guangdong Medical University in Dongguan from 2016 to 2021 were chosen for the study. In total, 26 patients were included according to the criteria as follows: (i) DN patients with no history of taking renal toxic or herbal medicine; (ii) DN patients without microscopic hematuria and fast drop in renal function; and (iii) DN patients with no complications of other renal diseases.
Archival renal biopsy samples from the type 2 diabetic patients mentioned earlier were included for analysis. Twenty-six snap frozen fresh renal tissues from these samples were used for transcripts analysis. Written informed consent was obtained from each patient, and tissue specimens were processed according to the protocols of the Ethics Committee from the first affiliated hospital of Guangdong Medical University.
In all specimens, the morphological diagnosis of DN was made by two individual renal pathologists. Each diagnosis was confirmed by the past medical history of diabetes and electron microscopic examination. Nine cases of renal biopsy samples from TBMN were used as controls.
Microarrays, histology, and ISH analysis
Snap-frozen fresh renal tissues from three DN patients and three TBMN were selected and processed for microarray analysis according to the manufacturer's instructions previously described. In brief, we used the ArrayStar non-coding RNA microarray chip (No. H1611087A; Shanghai Kangchen Biotechnology) to detect the expression profiles of lncRNAs. For histology analysis, paraffin-embedded sections were cut for histological examination and ISH analysis according to the protocol previously described (Bai et al., 2018).
For ISH staining, after deparaffinization with xylene and a graded series of ethanol, 5-μm thick tissue sections were digested and hybridized with a digoxin-labeled probe at 55°C overnight. Probe localization was visualized with nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate and imaged. Cells with dark blue nuclear staining were regarded as positive. The sections were evaluated semiquantitatively by systematically selecting 30 podocytes, and staining intensity was evaluated with ImageJ analysis software (ImageJ 1.44; National Institute of Health, Bethesda, MD).
Cell culture and studies
A conditionally immortalized human podocyte cell line (AB8/13; provided by Moin Saleem) was cultured as previously described (Saleem et al., 2002). Podocytes were maintained in normal glucose (5 mM) for 1 week, grown to 75%–85% confluence, and made quiescent by incubation overnight in a serum-free medium. Cells were then exposed to mannitol (30 mM) or high glucose condition (30 mM) for indicated time periods according to each individual experiment. Experiments were performed using passages 10–18.
HEK 293 cells were maintained in fetal bovine serum (Cat. No. SH30087.01; Hyclone), Dulbecco's modified Eagle's medium high sugar and penicillin (Cat. No. SH3022.01B; Hyclone) for the Dual-Luciferase reporter gene assay.
Oligonucleotides and transfection
Three LINC00355-specific siRNAs (si-LINC00355-001, si-LINC00355-002, and si-LINC00355-003) were used to knock down LINC00355, and a non-silencing siRNA (si-NC) oligonucleotide was used as a negative control (Ribobio Co., Ltd., Guangzhou, China). To evaluate the effect of LINC00355 overexpression, the full-length human LINC00355 sequence with 1878-bp DNA fragment was amplified and subcloned into a pcDNA3.1 vector (Invitrogen, Carlsbad, CA) to generate the vector pcDNA-LINC00355 (Hanheng Biotechnology Co., Ltd., Shanghai, China). The empty pcDNA3.1 vector (pcDNA-NC) was used as a control.
For transfections, 1 × 106 cells (per well) were plated into a six-well plate, and the plasmids were transfected into the cells using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. The transfected cells were harvested after 48–72 h, and the transfection efficiency was validated by qRT-PCR. Sequences for LINC00355-specific siRNAs are listed in Table 4.
Sequences Used for Knockdown or Overexpression of LINC00355
si, short interfering.
Dual-luciferase reporter assay
For analysis of LINC00355-EZH1 interactions, the predicted 3′-UTR sequence of EZH1 interacting with LINC00355 was synthesized and inserted into the pGL3-basic control vector (Promega, Madison, WI). Wild-type EZH1 cDNA (containing LINC00355 binding site) was cloned into pGL3-basic vector (Promega) and resulting vectors were designated, respectively, as pGL3-wt EZH1 and pGL3-LINC00355.
HEK 293 cells were transfected with 0.5 μg/mL plasmids or negative controls, followed by co-transfection with wild type or EZH1 plasmid using 4 μL Lipofectamine 2000 in Opti-MEM (Invitrogen). Thirty-six hours after transfection, cells were lysed with 100 μL of passive lysis buffer and the luciferase assay was carried out on cell extracts and measured with the GloMax bioluminescence detector (Promega) according to the protocol previously described. An internal control was used by co-transfecting with pGL3-basic expressing Renilla luciferase. Data were normalized by the ratio of firefly and renilla luciferase activities measured at 36 h post-transfection. All experiments were repeated in triplicate.
LncRNA-mRNA pull-down assay
Pre-cold phosphate-buffered saline (PBS, pH = 7.4) was used to wash cells twice followed by lysis buffer and centrifugation at 4°C, 15,000 g for 15 min, as described in detail elsewhere (Trendel et al., 2019). LINC00355 and AS-LINC00355 were transcribed from the vector pcDNA3.1, using T7 RNA Polymerase (D7069; Beyotime Biotechnology Co., Ltd., Shanghai, China), and then transcribed into RNA with the Pierce's 3′ end desthiobiotinylation kit (20163; Thermo Scientific Pierce).
RNeasy Mini Kit (Qiagen, Valencia, CA) was used for RNA purification. RNA-pull down assay was performed using Streptavidin agarose-bead kit (20164; Thermo Scientific Pierce). The RNA mixture was obtained and purified by RNeasy Mini Kit (Qiagen). cDNA was reversed using an RNA reverse transcription kit (#4387406; ABI). The expression of RNA in the pull-down product was detected by qRT-PCR and PCR analysis. The original image of RT-PCR Agarose Gel were shown in Supplementary Fig. S4.
Intracellular ROS measurement
The fluorescent probe DCFH-DA (S0033; Beyotime Biotechnology Co., Ltd.) analysis was used to measure the level of intracellular ROS. After incubation with DCFH-DA (10 μM) at 37°C for 15 min, the cells were mixed upside down to allow full contact with the probe. The cells were then washed three times with serum-free cell culture medium to adequately remove DCFH-DA that did not enter the cells. Intracellular ROS levels were determined by measuring 10,000 events per sample by Beckman CytoFLEX (Beckman Couliter Life Science).
After excitation with a 488-nm laser, flow cytometry data were analyzed with the FlowJo software (v10.6.2) and results were evaluated to indicate the level of ROS. Immunofluorescent images in each individual group were taken using Zeiss LSM 800 Confocal Laser Scanning Microcope (Zeiss Co., Ltd., Germany).
Apoptosis assay using flow cytometry
Podocytes in different groups and corresponding controls were harvested after 48 h and used for apoptosis analysis. Single-cell suspensions were fixed and stained with the Annexin V-FITC/PI apoptosis kit (KGA108; Jetway Biotech, China), and apoptosis was evaluated by examining the percentage of apoptotic cells. Data acquisition and analysis were performed using Beckman CytoFLEX (Beckman Couliter Life Science). The results were analyzed with the FlowJo software (v10.6.2) and viable, dead, early apoptotic, or late apoptotic cells were identified.
Animal experiments
Eight-week-old C57BL/KsJ-db/db mice were purchased from the Model Animal Research Center of Guangdong Provincial Animal Experiment Center (Guangzhou, China) and were individually housed in the specific pathogen-free grade animal facility of Guangdong Provincial Animal Experiment Center with a 12-h light/dark cycle, free access to food, and water and standard conditions according to the policy of the Committee for Animal Usage. The study protocols conform with the Guide for the Care and Use of Laboratory Animals, eighth edition (2011) and was approved by the Animal Ethics Committee at Guangdong Provincial People's Hospital, Guangzhou, China.
To investigate the effect of LINC00355 on disease progression, HBAAV2/9-h-LINC00355 or the vector control (1 × 1012, 150 μL/each; Hanheng Biotechnology Co., Ltd.) was injected through the tail vein of the mice. The db/db mice were randomly assigned to two groups, with six to seven mice each: db/db mice treated with HBAAV2/9-h-LINC00355 or vector control constructs. Blood samples and spot urine at 9:00–10:00 AM were collected and prepared for analyzing glucose, creatinine, and blood urea nitrogen.
At 8 weeks after the treatment, mice were anesthetized, and left kidneys were fixed by retrograde aortic perfusion using PBS for 3 min. Cortex from the left kidneys were harvested and embedded in paraffin for histological analysis. The non-perfused right kidney was snap-frozen and stored at −80°C for further analysis. Mice were euthanized, and all efforts were made to minimize suffering.
TUNEL assay
Kidney tissue samples from diabetic mice were fixed with 4% paraformaldehyde and detected using TUNEL assay with an Apoptag Peroxidase In Situ Apoptosis Detection kit (Chemicon International, Temecula, CA) as previously described. The nuclei were counterstained with hematoxylin and coverslipped with Aqueous mounting medium (JX0286; Jingxin Biotech, Guangzhou, China). Images were taken with a Leica DM6B Microscope Imaging System (Leica, Germany). Slides were evaluated, the number of TUNEL-positive cells in 20 fields was counted, and the sum was recorded as the total number of apoptotic cells in the sample. PBS instead of primary antibodies served as a negative control.
Measurement of biochemical parameters
Blood samples from the DN patients and mice were collected for analysis of biological parameters. Serum samples were obtained by standard venipuncture and centrifugation using EDTA-coated Vacutainer tubes (Becton-Dickinson, Franklin Lakes, NJ). Serum and urine samples were collected and used for determining the level of Scr and 24-h urinary protein with a Beckman Coulter AU480 Chemistry Analyzer (Beckman, CA). Urine albumin was determined with an enzyme-linked immunosorbent assay kit specific for mouse albumin (E99-134; Bethyl Laboratories, Montgomery, TX). All experiments were repeated in triplicate. Biological parameters for diabetic mice treated with HBAAV2/9-h-LINC00355 or vector control constructs are listed in Table 3.
qRT-PCR analysis
Total RNA from podocytes, kidney tissue samples from diabetic mice and normal controls was isolated using TRIzol Reagent (Invitrogen) and measured by a NanoDrop ND-1000 Spectrophotometer (Agilent, CA) following the manufacturer's protocol. cDNA was synthesized using the PrimeScript RT reagent kit (Takara, Dalian, China). Quantitative real-time PCR was performed in triplicate with Power PCR SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA) using the ABI PRISM 7500 FAST Real-TIME PCR System (Applied Biosystems) with results normalized to β-actin expression. PCR cycling conditions were as follows: 95°C for 30 s; 40 cycles of 95°C for 5 s and 60°C for 30 s; and dissociation at 95°Cfor 15 s, 60°C for 60 s, and 95°C for 15 s. Results were analyzed using the 2−ΔΔCt method. Primer sequences used in RT-PCR are shown in Table 5.
Primer Sets Used in the Quantitative Real-Time Reverse Transcription–Polymerase Chain Reaction
Western blot analysis
Cell lysates from cultured podocytes or renal cortex were used for Western blot analysis. In brief, mouse renal cortical tissue, placed in 200 μL Radio-Immunoprecipitation Assay lysis buffer containing 1 × phenylmethylsulfonyl fluoride, was ground with a tissue grinding rod and lysed with an ultrasonic crusher (Zhixiang, Guangzhou, China). Cultured podocytes were incubated on ice for 10 min after being washed three times with ice-cold PBS and then lysed with 100 μL lysis buffer.
After centrifugation at 4°C for 15 min at 12,000 g, soluble supernatant was carefully transferred to a fresh eppendorff tube and the protein concentration was determined using BCA Protein Assay Kit. Lysates from each experimental group were separated in parallel on two nitrocellulose membranes, blocked with 5% non-fat milk in 0.1% tris buffered saline with Tween-20, and probed at 4°C overnight using the following primary antibodies: CTNNBIP1 (1:500, bs4095R) from Bioss; β-catenin (1:2000, 51067-2-AP), GRP78 (1:1000, 11587-1-AP), and ATF4 (1:1000, 10835-1-AP) from Proteintech; bcl-2 (1:1000, D17C4) from cell signaling; and GAPDH (AC002) from ABclonal.
After rinsing, the secondary antibody (horseradish peroxidase-labeled IgG anti-rabbit/mouse antibody; Jackson ImmunoResearch) was used at 1:5000 dilution for 1 h at room temperature. The supersignal-enhanced chemoluminescent substrate (New Cell and Molecular Biotech) was applied to the probed membrane and exposed for 1–10 min before the protein bands were visualized on radiograph films (ImageQuant LAS). The original images of western blot are seen in Supplementary Fig. S3.
Immunofluorescence and immunohistochemical analysis
Podocytes and mice tissue samples were labeled with primary antibodies, respectively. For immunofluorescence staining, Alexa Fluor 555-conjugated goat anti-mouse IgG and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:500; Cell Signaling) were used for secondary antibodies, nuclei were counterstained with 4′,6-diamidino2-phenylindole and coverslipped with aqueous mounting medium (F6057; Sigma Aldrich).
For immunohistochemistry, EnVision™ Detection Systems peroxidase/diaminobenzidine Rabbit/Mouse kit (K4065; Dako, Carpinteria, CA) was used. Primary antibodies used in the immunostaining are CTNNBIP1 (1:100, bs4095R), EZH1 (1:100, 1bs-6528R) from Bioss, and ATF4 (1:100, 10835-1-AP) from Proteintech. Nuclei were counterstained with hematoxylin and coverslipped with Permount mounting medium (JX0286; Jingxin Biotech).
Samples were analyzed by systematically selecting 20 fields without bias and evaluated semiquantitatively for the staining intensity with ImageJ analysis software. Leica DM6B Microscope Imaging System (Leica, Germany) was used to take images with appropriate filters. PBS instead of primary antibodies served as a negative control.
Chromatin immunoprecipitation-qPCR
ChIP was performed as previously described (Gorkin et al., 2020). In brief, tissue samples were cross-linked with 1% formalin and lysed in SDS buffer and the sonication method was used to fragment the DNA. In total, the chromatin was immunoprecipitated using 1 μg rabbit anti-Histone H3 (tri methyl K27) (ab6002; Abcam), 1 μg rabbit anti-Histone H3 (tri methyl K4) antibody (ab8580; Abcam), or 1 μg non-specific immunoglobulin G (Santa Cruz).
Immunoprecipitated DNA and the input were subjected to reverse cross-linking and purification. Eluted DNA fragments were detected by quantitative real-time PCR analysis using ViiA™ 7 Real-time PCR System (Applied Biosystems) by KangChen Bio-tech (Shanghai, China). One-third of the reaction product was electrophoresed on a 5% polyacrylamide gel, followed by quantification with a PhosphoImager (Molecular Dynamics) and Image Quant software. Primers used in the ChIP-qPCR assay are listed in Table 6.
Primer Sets Used in the Chromatin Immunoprecipitation-Quantitative Polymerase Chain Reaction
Statistical analysis
Data are presented as mean ± standard deviation values. Normally distributed data were justified using parametric tests, including Independent-Samples t-test and one-way ANOVA. Non-normally distributed data were then followed with Student-Newman-Keuls post hoc test for statistical significance between groups. Pearson correlation analysis was used to test the correlations between the expression of CTNNBIP1 and LINC00355. All statistical tests were performed using SPSS 12.0 (SPSS, Inc., Chicago, IL). The significance level was set as p ≤ 0.05.
Footnotes
Authors' Contributions
X.B. and H.C. contributed to the experimental design. T.Z. performed the confocal microscopy. T.Z., Y.Z., H.X., J.L., Z.F., and R.H. performed in vivo animal studies and in vitro experiments on podocytes. T.Z. analyzed human renal biopsy samples. X.B. interpreted the data, wrote the manuscript, and approved the final version of the manuscript for publication.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (No. 81873616, No. 82170730), GDPH Supporting Fund for NSFC Program (No. 81873616, No. 82170730), and High-level Hospital Construction Project (No. KJ012021013) to Xiaoyan Bai, and “Group-type” Special Support Project for Education Talents in Universities (4SG21227G).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.