
Abstract
Significance:
Acute responses to hypoxia are essential for the survival of mammals. The carotid body (CB), the main arterial chemoreceptor, contains glomus cells with oxygen (O2)-sensitive K+ channels, which are inhibited during hypoxia to trigger adaptive cardiorespiratory reflexes.
Recent Advances:
In this review, recent advances in molecular mechanisms of acute O2 sensing in CB glomus cells are discussed, with a special focus on the signaling role of mitochondria through regulating cellular redox status. These advances have been achieved thanks to the use of genetically engineered redox-sensitive green fluorescent protein (roGFP) probes, which allowed us to monitor rapid changes in ROS production in real time in different subcellular compartments during hypoxia. This methodology was used in combination with conditional knockout mice models, pharmacological approaches, and transcriptomic studies. We have proposed a mitochondria-to-membrane signaling model of acute O2 sensing in which H2O2 released in the mitochondrial intermembrane space serves as a signaling molecule to inhibit K+ channels on the plasma membrane.
Critical Issues:
Changes in mitochondrial reactive oxygen species (ROS) production during acute hypoxia are highly compartmentalized in the submitochondrial regions. The use of redox-sensitive probes targeted to specific compartments is essential to fully understand the role of mitochondrial ROS in acute O2 sensing.
Future Directions:
Further studies are needed to specify the ROS and to characterize the target(s) of ROS in chemoreceptor cells during acute hypoxia. These data may also contribute to a more complete understanding of the implication of ROS in acute responses to hypoxia in O2-sensing cells in other organs. Antioxid. Redox Signal. 37, 274–289.
Adaptive Acute Responses to Hypoxia
Oxygen (O2
Sustained hypoxia (hours or days) triggers a generalized transcriptional response, which stimulates glycolysis to increase energy production anaerobically, increases red blood cell number, and activates angiogenesis. The regulated expression of O2-sensitive genes depends on prolyl hydroxylases (PHDs), which are oxygenases that use O2 as a substrate to hydroxylate hypoxia inducible transcription factors (HIFs) and thereby inhibit their activity in normoxic conditions. In hypoxia, HIF stabilization induces the expression of a broad cohort of genes. The PHD-HIF pathway regulates more than 2500 transcripts, many of them critically involved in pathophysiological processes such as stem cell fate and differentiation, tissue regeneration, cancer, and inflammation, among others (20, 55, 72, 111, 139).
However, the survival of mammals under hypoxia depends on acute cardiorespiratory reflexes, such as hyperventilation and increase in cardiac output, which rapidly (in a few seconds) increase O2 uptake by the lungs and its distribution to the tissues. These responses play a fundamental role in adaptation to high altitude (54, 141) and are also needed for physiological compensation in patients with dysfunctions of gas exchange in the lung (119).
The detection of hypoxia and the initiation of adaptive acute reflexes depend on a series of specialized tissues or organs, such as the carotid body (CB), adrenal medulla (AM), pulmonary/systemic arteries, and neuroepithelial bodies in the lung airways, which together form a homeostatic acute O2-sensing system (71, 86, 137). Most of the current knowledge on hypoxia-induced chemoreception was obtained from studies initially performed in the CB, the main arterial chemoreceptor and prototypical acute O2-sensing organ (50, 91). The CB is essential for O2 regulation of breathing, as its surgical removal results in individuals who are unaware of hypoxemia and thus exhibit a complete absence of hypoxic ventilatory response (HVR) (122).
In this review, recent advances in molecular mechanisms of acute O2 sensing by CB glomus cells are discussed, with a special focus on the signaling role of mitochondria through regulating the cellular redox status.
CB Chemoreceptor Activation by Hypoxia and Mitochondrial Inhibitors
Electrical properties of CB cells and mechanism of chemotransduction
The CB is a paired organ located at the carotid artery bifurcation that is organized in clusters of cells, denominated as glomeruli. Each glomerulus contains a few O2-sensitive glomus cells (also called type I cells), which establish chemosensory synapses with afferent fibers transmitting sensory information via the glossopharyngeal nerve to the brain stem respiratory and autonomic centers (Fig. 1A). Glomus cells are surrounded by type II cells and immature progenitors, which provide structural support and contribute to the hypertrophy of the organ during acclimatization to hypoxia (98). Type II cells also contribute to paracrine signaling during chemotransduction, potentiating activation of sensory fibers by glomus cells (87, 127). The CB is rich in blood vessels and is considered one of the most irrigated organs in the body (24). The CB functions as a multimodal sensory organ, which, in addition to changes in O2 tension, can detect changes in CO2 tension, pH, glucose, and lactate in the blood (91, 124). In addition, the CB can be activated by circulating hormones and cytokines (50, 51).
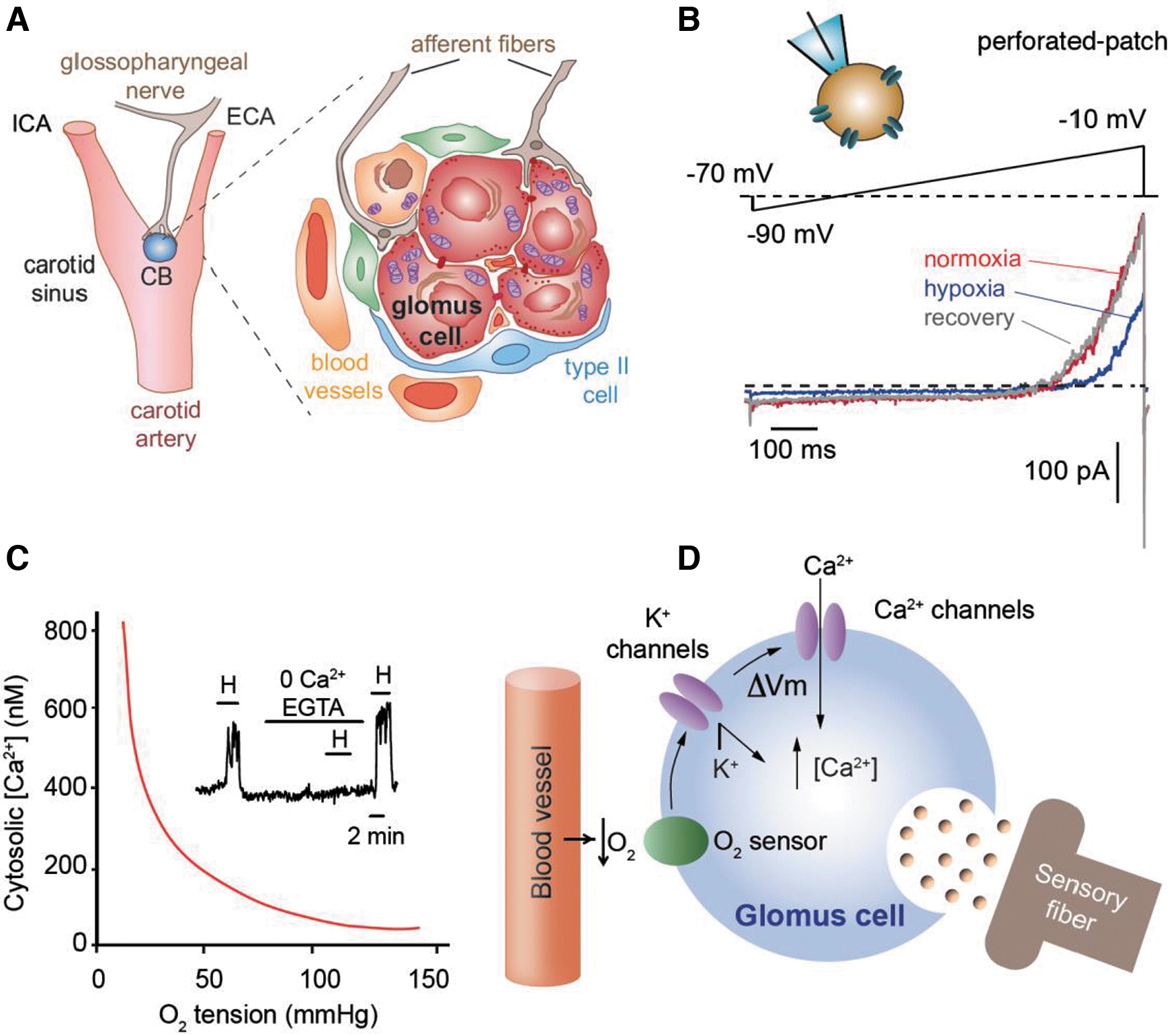
The basic cellular mechanisms underlying the O2 sensing properties of CB glomus cells were discovered several decades ago. It was shown that glomus cells, which derive from the neural crest (98), are excitable and can generate action potentials repetitively (32, 70). These cells contain O2-sensitive background and voltage-gated K+ channels, which are both inhibited by decreases in O2 tension (Fig. 1B) (12, 41, 70). The inhibition of these K+ channels leads to a depolarization of the plasma membrane, which in turn activates voltage-dependent Ca2+ channels causing Ca2+ influx. The increased cytosolic Ca2+ triggers neurotransmitter release by exocytosis, to stimulate afferent sensory fibers transferring information to the brain stem respiratory and autonomic centers (14, 68, 79, 130).
The rise in cytosolic Ca2+ required for glomus cell secretion under hypoxia depends on extracellular Ca2+, since it can be abolished with either Ca2+ free recording solution or addition of Ca2+ chelator EGTA to the recording solution (Fig. 1C). In glomus cells, the changes in intracellular Ca2+ induced by O2 tension follow a hyperbolic curve (Fig. 1C) similar to the relationship existing between ventilation and the level of O2 tension in arterial blood (38). Therefore, the electrophysiological and O2-sensing properties of single glomus cells, summarized in Figure 1D, are the major determinants of O2 regulation of breathing in experimental mammals and in humans (79, 93).
Occlusion of responsiveness to hypoxia by rotenone
Although there is a consensus regarding the basic process of sensory function in chemoreceptor cells during hypoxia, the molecular mechanisms underlying this process have remained not fully understood. Since the late 1990s, several hypotheses have been proposed to address the nature of the molecules that detect hypoxia and the signaling pathway leading to inhibition of K+ channels on the plasma membrane. However, most hypotheses were discarded eventually due to lack of solid support from experimental data.
A detailed discussion about these hypotheses is beyond the scope of the current review and has been addressed recently [for reviews see Iturriaga et al. (50), Lopez-Barneo et al. (69), Rakoczy and Wyatt (105), and Yoo and Kim (144)]. Mitochondria have classically been considered to be implicated in acute O2 sensing (78, 140). These organelles are the largest consumer of cellular O2 for energy production by oxidative phosphorylation, which makes them a natural candidate to detect changes in O2 tension and to initiate adaptive responses to hypoxia.
Supporting this hypothesis are the high O2 consumption (24) and the sensitivity of CB glomus cells to inhibitors of mitochondrial electron transport chain (ETC) (82, 92, 143). Similar to hypoxia, inhibition of ETC complexes also stimulates secretory activity and Ca2+ influx from extracellular space, and inhibits K+ channels in glomus cells (Fig. 2A, B) (92).
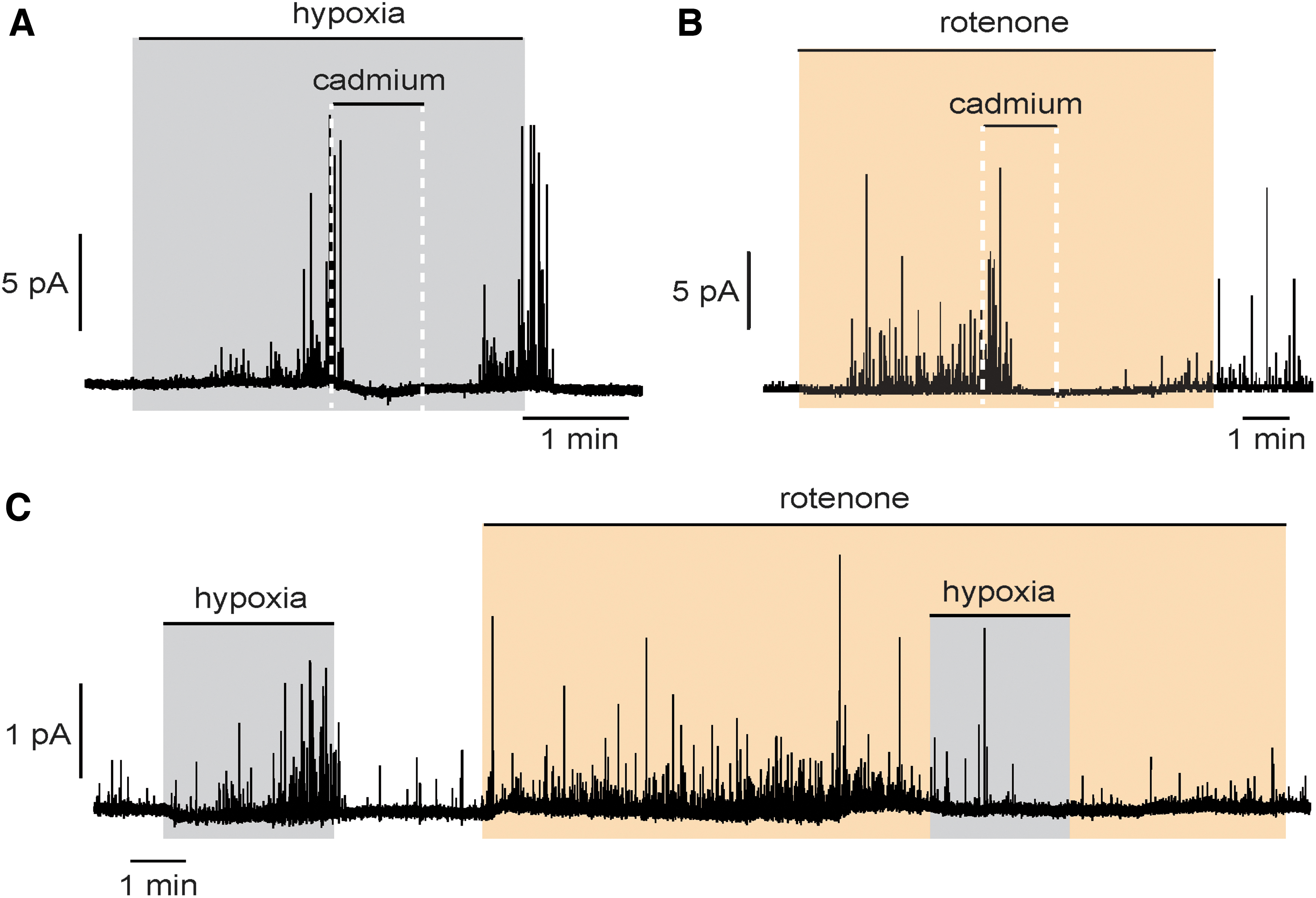
Although all ETC inhibitors mimic and impair further hypoxic responses (92, 143), rotenone, a mitochondrial complex I (MCI) inhibitor, is particularly efficient in blocking hypoxic responses in rat CB glomus cells. Treatment with rotenone induces catecholamine secretion from glomus cells, which, similar to the response elicited by hypoxia, is inhibited by cadmium, a blocker of voltage-dependent Ca2+ channels on the plasma membrane, indicating the dependence of extracellular Ca2+ (Fig. 2A, B). Incubation with rotenone abolishes catecholamine secretion from glomus cells during hypoxia (Fig. 2C). This inhibitory effect of rotenone is specific to hypoxia, since rotenone has no effect on hypoglycemia-induced catecholamine secretion in glomus cells (44).
In addition to CB glomus cells, rotenone also blocks the sensitivity to hypoxia in rat (121) and ovine (56) AM chromaffin cells. These results suggest that the inhibitory effect of rotenone on acute O2 sensing is not limited to the CB, but it is a rather general phenomenon in chemoreceptor cells. Another MCI inhibitor 1-methyl-4-phenylpyridinium (MPP+) also blocks hypoxic responses in glomus cells, whereas MCI proximal inhibitors, such as diphenyliodonium, present less effect than rotenone and MPP+ (92). Both rotenone and MPP+ bind to a distal site (Q site) of MCI, which is highly conserved from bacteria to mammals (5, 37, 147, 148), thereby preventing the binding of ubiquinone and inhibiting the MCI NADH/quinone oxidoreductase activity. These data suggest that mitochondria in chemoreceptor cells are highly sensitive to hypoxia and that a signaling pathway from mitochondria to plasma membrane exits during acute O2 sensing, in which MCI, especially the rotenone and ubiquinone-binding site, may have a special role. As shown below, this idea has been demonstrated in mice with complete genetic disruption of MCI activity (4, 36).
O2-sensitive mitochondrial signaling
The implication of mitochondria in acute O2 sensing implies that one or several classes of mitochondria-derived molecules must be generated during hypoxia to signal membrane ion channels. The signaling molecules proposed to be involved in hypoxia-induced chemotransduction are NADH, reactive oxygen species (ROS), and ATP. The potential implication of MCI in acute O2 sensing suggests that NADH, the MCI substrate, or NAD+/NADH ratio could signal changes in O2 tension from mitochondria to plasma membrane of chemoreceptor cells. Pyridine nucleotides have been reported to bind to voltage-gated K+ channel β subunits and modulate their function (57, 123). In addition, cytosolic NADH has also been suggested to modulate cationic channels in glomus cells (124).
Several groups have observed a reversible increase in NAD(P)H autofluorescence induced by hypoxia in glomus cells, which is proportional to decreases in O2 tension (4, 13, 31, 36). This hypoxic signal is absent in conditional knockout mice lacking NDUFS2, a conserved subunit participating in ubiquinone binding, which is essential for MCI structure and function (4, 36). Similar to hypoxia, MCI inhibitor rotenone induces an increase in NAD(P)H autofluorescence (4). Incubation of glomus cells with intracellular NADH prevents further modulation of voltage-gated K+ current by hypoxia (36). Taking together, these studies support the role of NADH as a signaling molecule in glomus cells during acute O2 sensing.
Bioenergetic deficiency has also been proposed to address mitochondrial function in acute O2 sensing, due to the contribution of mitochondria in energy production. A decrease in cytosolic ATP concentration or a decrease in ATP/ADP ratio in glomus cells during hypoxia has been considered a signal to regulate the function of K+ channels in the plasma membrane (131, 143). The possible role of ATP on acute O2 sensing in CB glomus cells as well as in pulmonary myocytes and chromaffin cells has been a matter of debate (15, 35, 48, 88, 136).
Data on single glomus cells point against a relevant role of ATP. Hypoxia-induced inhibition of voltage-gated K+ channels has been reported in glomus cells dialyzed with 3–5 mM MgATP (73, 102). Moreover, the effects of hypoxia and rotenone (or genetic MCI deficiency), both of which inhibit the ETC and presumably ATP synthesis, should be additive but they are not. In addition, rotenone blocks the effect of hypoxia but does not alter glomus cell activation by hypoglycemia (44). Glomus cells are activated by hypoglycemia and by lactate, which are conditions expected to have opposite effects on ATP production (36, 44, 124). Therefore, a signaling role of ATP under hypoxia is unlikely, although it cannot be discarded completely until more definitive experiments, including precise measurements of ATP changes in glomus cells, are performed.
A mitochondrial-based redox sensor of acute hypoxia was proposed back in the 1990s, especially in hypoxia-induced pulmonary artery vasoconstriction (1, 64, 117, 118, 133). ROS represent collectively a group of molecules derived from O2, which are formed by redox reactions or electronic excitations. Overproduction of ROS may lead to oxidative damage, such as chronic inflammation and other diseases (11, 22, 115). On the contrary, controlled ROS production in a low-concentration range plays a signaling role in a variety of physiological processes, which have been discussed extensively in the literature (11, 112, 115). Intracellular hydrogen peroxide (H2O2), with an overall concentration between 1 and 100 nM, is the major ROS in regulating intracellular redox status, and therefore, biological functions (115).
Mitochondria are the major source of ROS production, first identified almost 50 years ago (8, 17, 52). Electrons leaked from ETC reduce O2 to superoxide radical anion (O2 −), or to a lesser extent, directly to H2O2. O2 − can be converted to H2O2 by spontaneous dismutation or catalyzed by superoxide dismutases. Rotenone, which mimics hypoxic secretory effect, is also known to increase ROS production from MCI (129).
As discussed later in this review, recent studies support the signaling role of mitochondrial ROS during hypoxia in CB glomus cells, pulmonary artery myocytes, and neonatal AM chromaffin cells (1, 4, 36, 63, 121, 133 –135). In glomus cells, hypoxia induces a dose-dependent increase in ROS at the intermembrane space (IMS) and the cytosol (4, 36). In addition, intracellular dialysis of glomus cells with H2O2 increases input resistance (an electrical change compatible with the inhibition of background K+ channels) and blunts any further effect of hypoxia (36).
Mitochondrial ROS Signaling in CB Chemoreceptor Cells During Acute Hypoxia
Sites of mitochondrial ROS production
O2 − and H2O2 are the two primary ROS produced in mitochondria. Most O2 − generated in the matrix is rapidly converted to H2O2 by mitochondrial superoxide dismutase (SOD2). In the IMS, the conversion of O2 − to H2O2 is catalyzed by cytosolic superoxide dismutase (SOD1), which easily crosses the mitochondrial outer membrane. Mitochondrial H2O2 can diffuse to the cytosol, modulating cytosolic redox status and physiological processes, such as acute O2 sensing.
Until now, at least 11 specific sites in the ETC and associated dehydrogenases have been identified to produce and release O2 − (and H2O2) in either the mitochondrial matrix or the IMS [for detailed reviews see Brand (9 –11), Murphy (83), Waypa et al. (136), and Wong et al. (142)]. Quinone binding sites at MCI and mitochondrial complex III (MCIII) (IQ and IIIQ sites, respectively) contribute to the majority of ROS production from the mitochondrial ETC. O2 − produced from the IQ site is believed to be released to matrix (11).
However, as discussed later in this review, in O2-sensitive glomus cells, hypoxia-induced reversal electron transport (RET) of MCI may cause O2 − produced at the IQ site to be released to the IMS (4, 36). O2 − produced in the Q binding sites of MCIII can be delivered to both IMS and matrix (11, 136). O2 − produced at the FMN site in the peripheral arm of MCI (IF site), as well as in other NADH-related dehydrogenases, is released to mitochondrial matrix.
In addition, electrons leaked from the flavin site in mitochondrial complex II (MCII) (IIF site) and other FADH2-related dehydrogenases contribute to a small amount of O2 − delivered to mitochondrial matrix. Most H2O2 in the matrix is degraded by peroxiredoxin-3 (PRDX3) and glutathione peroxidases, such as glutathione peroxidase 1 (GPX1). Only a small percentage (∼10%) of H2O2 is either converted to other ROS or diffused to IMS and cytosol (11, 21). Inhibition of MCI by rotenone results in O2 − production from the IF site and other associated mitochondrial dehydrogenases (9, 10, 89, 125).
Real-time measurement of mitochondrial ROS with genetic probes
Monitoring changes in real-time ROS production during acute hypoxia has been a technical challenge, as most traditional techniques are not sensitive enough to record small transient variations in ROS levels in single chemoreceptor cells. The commonly used techniques either require ROS accumulation or detect ROS-induced modifications of lipids, proteins, and DNA (84). As a result, these techniques detect oxidative damages rather than rapid and subtle changes in ROS production during physiological processes, such as acute hypoxia.
To circumvent these limitations, redox-sensitive fluorescent probes (dichlorodihydrofluorescein, lucigenin, superoxide-selective hydroethidine and its mitochondria-targeted derivative MitoSOX, and Amplex Red, among others) have been developed (2, 3, 84, 136). However, these probes also have serious drawbacks due to their high susceptibility to artifactual side reactions, limitation in location, lack of specificity to specific oxidant, and modification of plasma membrane and mitochondrial membrane potentials.
In recent years, a new generation of redox-sensitive genetically engineered protein probes have been developed to detect specific oxidants with high sensitivity (84, 134, 136). These probes include redox-sensitive green fluorescent protein (roGFP) (18, 27, 30, 134), redox-sensitive yellow fluorescent protein HyPer (6), redox-sensitive fluorescence resonance energy transfer-probe (45), and peroxiredoxin-based probe roGFP2-Tsa2ΔCR (81). All these probes are specifically sensitive to changes in H2O2 levels. In addition to redox-sensitive fluorescent protein probes, other alternative probes have also been explored, such as small-molecule fluorescent probes with improved selectivity (28, 77, 116, 149). A dual-purpose mitochondrial O2 − probe (MitoNeoD) has been developed, which permits monitoring changes in O2 − production, especially in mitochondrial matrix, in vivo by mass spectrometry and in vitro by fluorescence (113).
Recently, we have set up a protocol to record in real time rapid changes in intracellular redox status of mouse glomus cells during acute hypoxia using roGFP probes in combination with microfluorimetric techniques (42). Redox-sensitive cysteine residues are incorporated in roGFP probes, which allow reciprocal changes in emission intensity when excited at two different wavelengths, and thus, a ratiometric quantification of redox status (Fig. 3A–D) (42). Genetically engineered roGFP probes can be designed to target specific subcellular compartments, such as cytosol, mitochondrial matrix, or intermembrane space (4, 36, 42, 134).
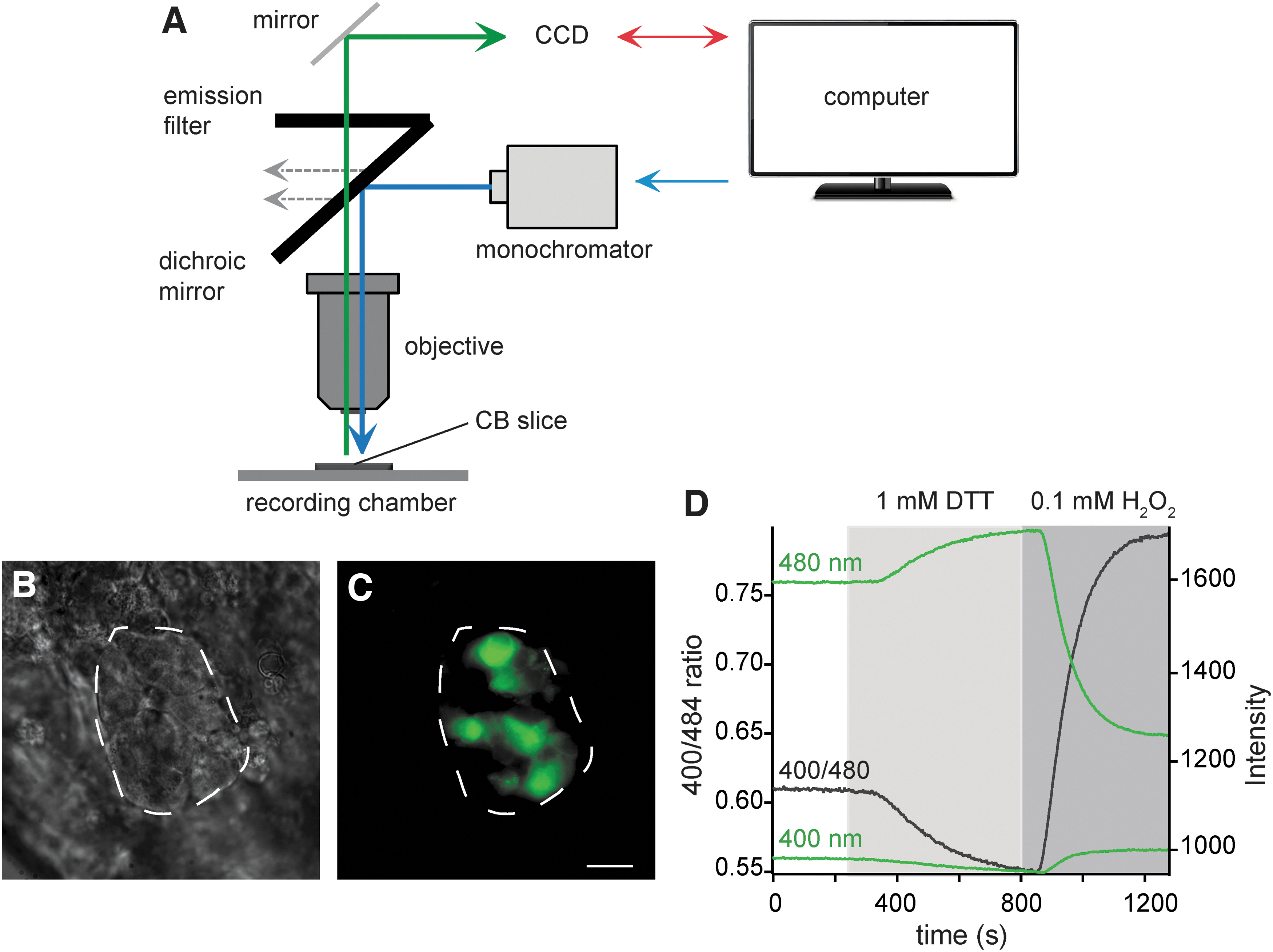
Compartmentalized mitochondrial ROS changes during acute hypoxia
Reversible changes in ROS production during exposure of CB glomus cells to hypoxia have been measured using roGFP probes designed to target specific subcellular compartments (42, 134). Using roGFP targeted to cytosol (36), a transient reversible increase in the ratio of fluorescent signals excited at 400 and 484 nm (400/484 ratio) is detected upon hypoxic treatment, indicating a low O2 partial pressure (PO2)-induced increase in the cytosolic H2O2 level in glomus cells (Fig. 4A).

A similar fluorescent signal can also be recorded using the roGFP probe targeted to mitochondrial IMS (Fig. 4B). Hypoxia-induced increases in 400/484 ratio in both the cytosol and mitochondrial IMS occur with a time course of seconds to minutes, which is similar to the dynamic range of cellular and systemic responses to hypoxia. The intensity of hypoxia-induced ROS signal in the IMS is proportional to the decrease in PO2 (Fig. 4B).
The relationship between IMS ROS production and PO2 follows a hyperbolic curve (Fig. 4C) that is similar to the increase in cytosolic Ca2+ (Fig. 1C) or secretory activity (79) induced by hypoxia in glomus cells, therefore suggesting the participation of the ROS signal in chemosensory transduction. However, in glomus cells transfected with a roGFP probe targeted to the mitochondrial matrix, hypoxia systematically induces a decrease in the 400/480 ratio, suggesting a decrease in H2O2 production. Treatment with exogenous H2O2 results in an increased 400/484 ratio in the matrix, indicating that the roGFP probe targeted to matrix is sensitive to H2O2 (Fig. 4D). Hypoxia-induced decrease in ROS production in mitochondrial matrix was also confirmed using MitoNeoD, an O2 − probe targeted to mitochondrial matrix (4, 113).
Previous pharmacological studies demonstrate that the MCI inhibitor rotenone blocks hypoxia-induced secretory activity in glomus cells (92). Rotenone binds to the ubiquinone binding site (Q site) at MCI and blocks both forward and reverse reactions of MCI. Rotenone is also known to increase ROS production from the IF site (the peripheral arm of MCI) and upstream dehydrogenases due to the backlog of electrons in MCI, with O2 − (and H2O2) being released to mitochondrial matrix (10, 11, 62, 83, 89, 125, 132).
As expected, in mouse glomus cells, rotenone stimulates a reversible increase in H2O2 in mitochondrial matrix (Fig. 4E) (4), which seems to diffuse to the IMS (Fig. 4F). Rotenone treatment strongly inhibits a further increase in H2O2 in IMS during hypoxia, but has no effect on hypoxia-induced decrease in H2O2 in mitochondrial matrix (Fig. 4E, F) (4). These data suggest that an increase in mitochondria IMS ROS is associated with sensing hypoxia by glomus cells. This signal is strongly inhibited by rotenone, indicating that it is mainly originated at the MCI IQ site. The hypoxic increase in IMS ROS contrasts with the decrease in matrix ROS during hypoxia, a phenomenon that is probably nonspecific and is due to the generalized lack of O2.
Loss of ROS signaling and responsiveness to hypoxia in genetically modified mice
The generation of conditional knockout mice deficient in MCI subunit NDUFS2 has allowed us to better understand the signaling role of ROS derived from MCI in acute O2 sensing. NDUFS2 forms the ubiquinone binding site of MCI together with NDUFS7 and ND1 (5, 106). In mice and glomus cells deficient in NDUFS2, hypoxic responses are selectively abolished, whereas responses to hypercapnia are maintained (4, 36). In addition, hypoxia-induced mitochondrial signals (NADH and ROS) are also lost in NDUFS2-deficient glomus cells (4, 36). Whereas a hypoxia-induced increase in H2O2 in the cytosol (36) and mitochondrial IMS is strongly reduced in amplitude in glomus cells lacking NDUFS2 (Fig. 5A), a hypoxia-mediated decrease in H2O2 in mitochondrial matrix is maintained (Fig. 5B).

These results are in agreement with those obtained from rotenone-treated wild-type glomus cells (Fig. 4E, F), and further suggest that the increase in H2O2 production in mitochondrial IMS is a signal associated with responsiveness to acute hypoxia.
CB glomus cells have intrinsic metabolic properties, which make their mitochondria highly sensitive to hypoxia. Studies of gene expression profiling demonstrate a constitutive high level of expression of Epas1 (the gene coding hypoxia inducible factor 2α [HIF2α]) and two atypical subunit isoforms of the catalytic core of mitochondrial complex IV (MCIV) (Cox4i2 and Cox8b) in glomus cells (43, 146). Although HIF2α is classically considered a transcription factor regulating gene expression during chronic hypoxia, these transcriptomic data along with studies using conditional knockout mice deficient in HIF2α (47, 80) demonstrate an essential role of this factor in acute O2 sensing by glomus cells. HVRs are diminished significantly in HIF2α-deficient mice. In addition, these mice show a loss of glomus cell and mitochondrial responsiveness to hypoxia (80).
In HIF2α-deficient glomus cells, the hypoxia-induced IMS ROS signal is inhibited, whereas the decrease in ROS production in mitochondrial matrix is not affected (Fig. 5C, D). In HIF2α-deficient mice, expression of atypical isoforms of MCIV subunits Cox4i2 and Cox8b is greatly suppressed. In addition, the hypoxic response is lost in conditional knockout mice deficient in COX4I2 (80). These results demonstrate that the enrichment of HIF2α results in an induction of gene expression of atypical isoforms of MCIV subunits, thus making glomus cells highly sensitive to hypoxia.
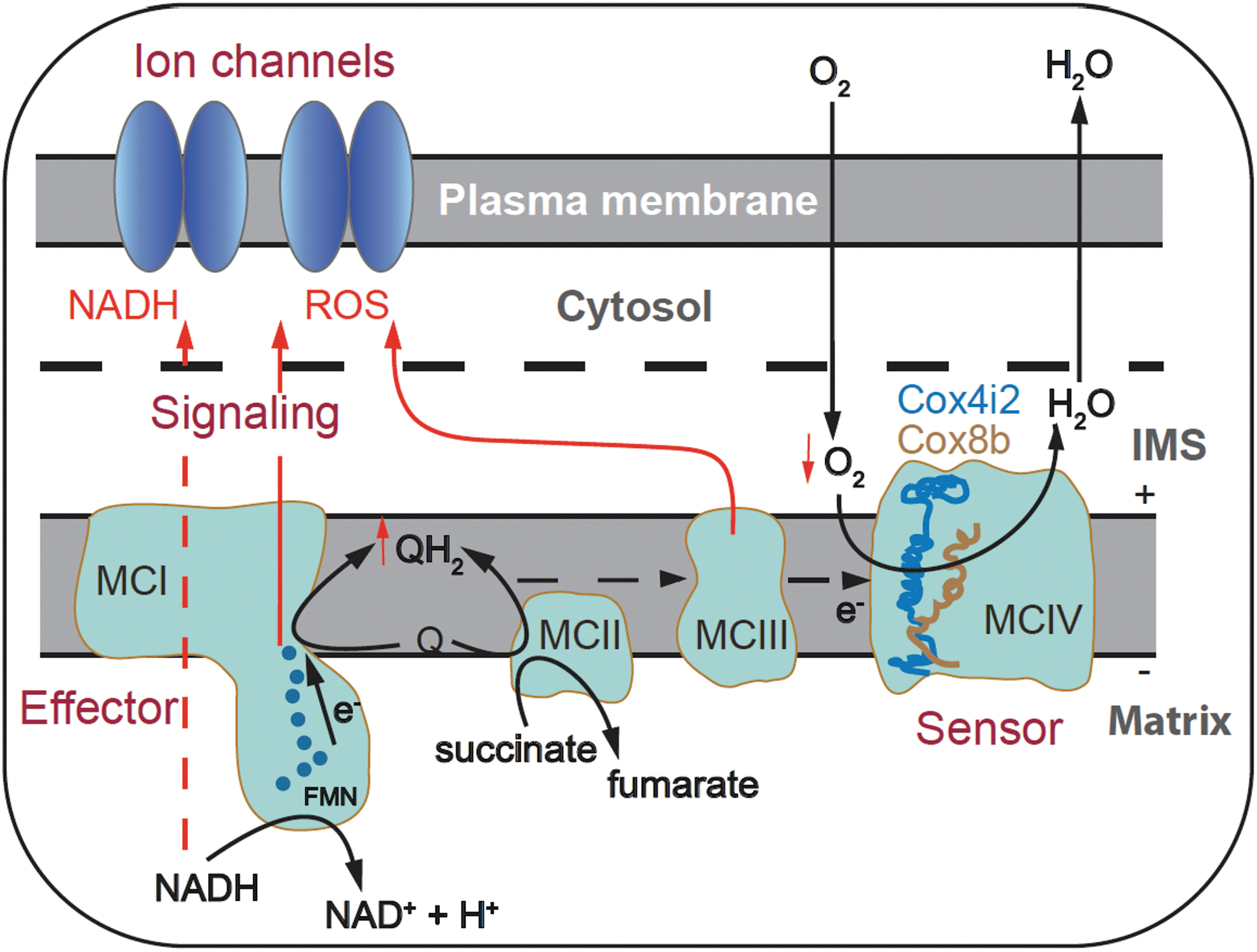
The data obtained from wild-type mice and mice with mitochondrial mutations have led us to propose a mitochondria-to-membrane signaling model of acute O2 sensing in CB glomus cells. In this model, MCIV functions as an O2 sensor with a low apparent affinity for O2 due to the atypical isoforms expressed in glomus cell cytochrome c oxidase (43, 80, 146). As mentioned above, genetic ablation of the gene coding COX4I2 strongly decreases sensitivity to hypoxia in glomus cells (80). Regarding this subunit isoform, it has recently been reported that genetically engineered HEK293 cells present a COX4I2-dependent MCIV affinity for O2 (P50 about 1 mmHg), lower than that with COX4I1 (P50 about 0.5 mmHg) (96).
Although this study supports our hypothesis of COX4I2-dependent low affinity for O2, the levels of O2 tension that modulate HEK293 cell mitochondria are too low compared with the physiological range of O2 that modulates glomus cells (Figs. 1C and 4C) (4). Therefore, it seems that other factors (e.g., expression of COX8B or pyruvate carboxylase-dependent Kreb's cycle anaplerosis) may influence the sensitivity of glomus cell mitochondria to O2. During hypoxia, the decrease in MCIV activity causes a backlog of electrons along the ETC and accumulation of ubiquinol (QH2) at the IQ site of MCI. This leads to not only a slowdown of MCI reaction in the forward direction, but most likely a switch to the reverse direction as well (Fig. 6A).
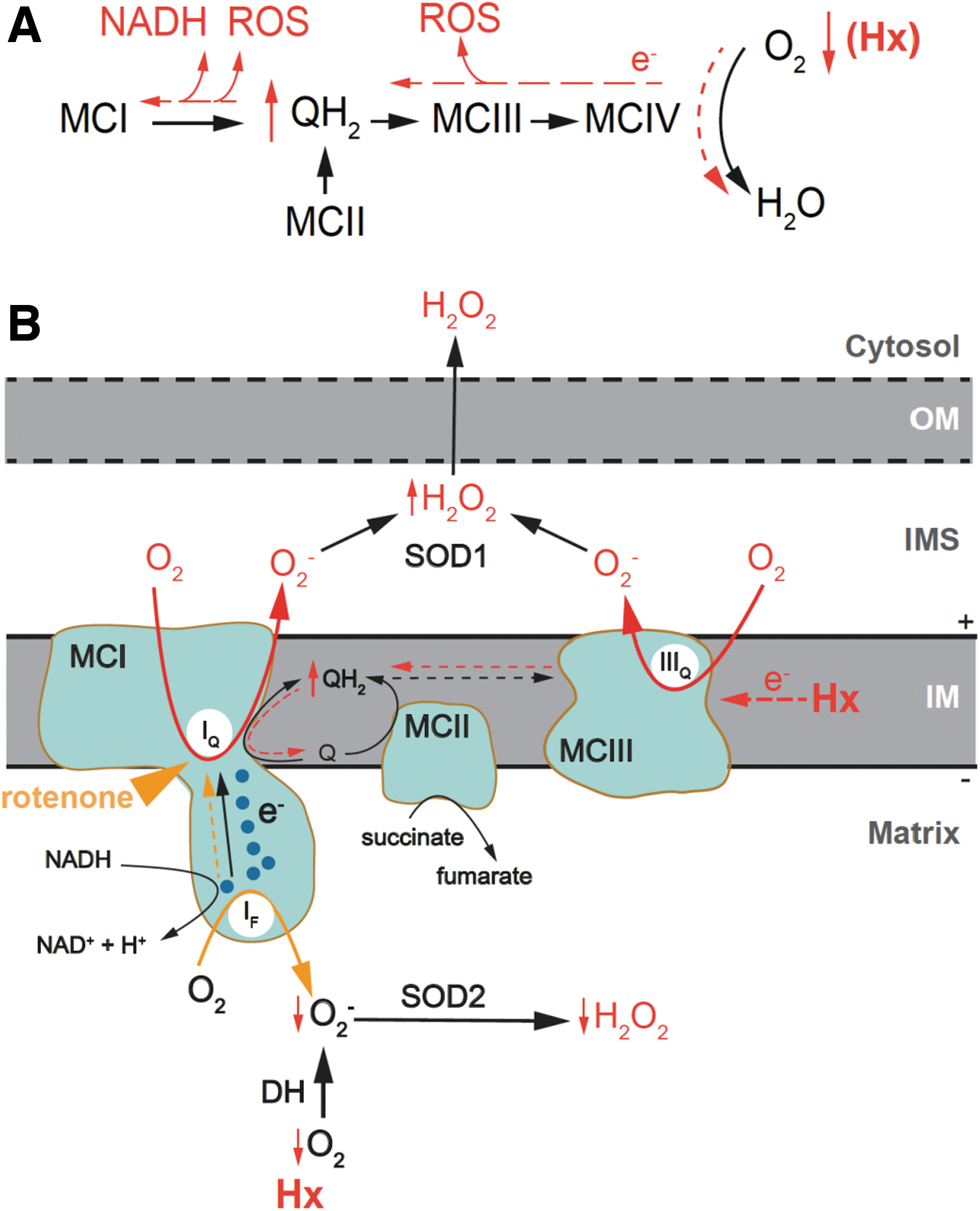
Reverse MCI function is an intriguing property of MCI and is associated with a succinate-dependent high level of ubiquinol, as it occurs in glomus cells (36, 43, 62, 66, 104, 132). The RET results in a burst of O2 − production at the IQ site and the reduction of NAD+ to NADH at the FMN site (19, 61, 83, 104). O2 − produced at the IQ site may be electrostatically repelled by the negative potential in the mitochondrial matrix and directly channeled to the IMS, where it is converted to H2O2 by SOD1. As a result, MCI functions as an effector, releasing mitochondrial signals (NADH and H2O2), which locally accumulate in the cytosol, near the plasma membrane, to inhibit neighboring K+ channels and trigger adaptive responses. The IMS hypoxic ROS signal may have also a component due to O2 − production at a highly reduced outer IIIQ site (Fig. 6A, B) (134).
In normoxic conditions, rotenone blocks electron transport at MCI and produces ROS at the IF site directed to the matrix (Fig. 4E). In the presence of rotenone, the IMS ROS signal induced by hypoxia is greatly suppressed due to blockade of MCI RET (Fig. 4F). However, hypoxia-induced decrease in ROS production in the matrix, possibly due to the inhibited activity of ETC and associated dehydrogenases, is unaffected by rotenone (Fig. 6B; Fig. 4E).
Modulation of membrane ion channels by ROS in CB glomus cells
The mitochondria-to-membrane signaling model predicts that mitochondria-originated NADH and H2O2 regulate K+ channels on the plasma membrane during acute hypoxia. Indeed, dialysis of glomus cells with NADH inhibits modulation of voltage-dependent K+ channels by hypoxia (36). On the contrary, modulation of ion conductance by intracellular redox status has been reported previously in a variety of ion channels on the plasma membrane, in which channel inhibition is frequently associated with cysteine oxidation (7, 16, 60, 97, 107, 108).
In CB glomus cells, intracellular dialysis with H2O2 (15–50 μM) or a thiol-oxidizing agent diamide (50–60 μM) inhibits background K+ channels, as demonstrated by an increase in input resistance, without affecting voltage-dependent K+ channels. These oxidants mimic the hypoxic effect and prevent further modulation of these channels by hypoxia (Fig. 7A–D) (36). N-acetyl-cysteine, a reducing agent, has no effect on the basal level of K+ current (Fig. 7D). This suggests that in normoxic conditions, glomus cells are in a relatively reduced state, as determined by the cytosolic roGFP probe (36). Taken together, these data support that during hypoxia, mitochondria-released H2O2 may inhibit background K+ channels on the plasma membrane of glomus cells to initiate cell depolarization.
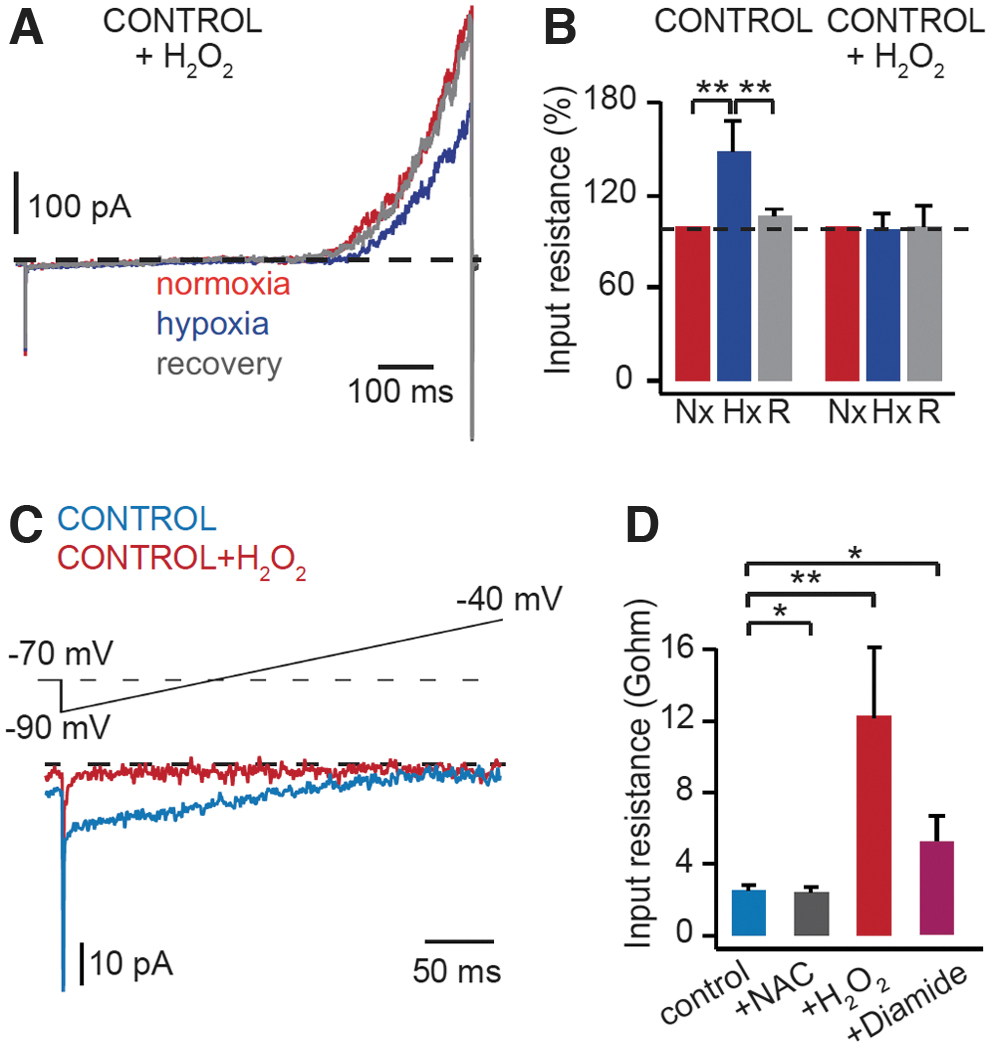
The molecular mechanisms underlying the redox regulation of K+ channels in glomus cells are not yet fully known. Background K+ channels TASK1 and TASK3 are highly expressed in CB glomus cells, which form mainly TASK3 homomers or TASK1/TASK3 heteromers (43, 58, 128, 146). TASK3 subunits contain cysteine residues, which are susceptible to cytosolic redox modulation by H2O2 (59). TREK2, K+ background channels closely related to TASK channels, are also inhibited by intracellular oxidants (99).
On the contrary, acute responses to hypoxia are intact in double knockout mice deficient in both TASK1 and TASK3 (90), indicating redundant functions among K+ background channels in CB glomus cells. In addition to acting directly on pore-forming subunits, ROS also interact with accessory β subunits of voltage-gated K+ channels (108). Moreover, redox regulation has been reported to modulate phosphatase and protein kinase activities (34, 114, 126). It is unclear whether this redox-dependent phosphorylation/dephosphorylation balance could indirectly regulate ion channel activities and, therefore, their responsiveness to changes in O2.
Role of ROS in Other Acute O2 Sensing Tissues
The mitochondria-to-membrane signaling model, summarized in Figure 8, which is established mainly based on results obtained from CB glomus cells, may also apply to other chemoreceptor cells. Chromaffin cells in AM are highly sensitive to hypoxia, especially in neonates, although its sensitivity decreases in adult animals. O2 sensitivity in chromaffin cells depends on MCI function, as rotenone mimics hypoxia-induced inhibition of K+ current and prevents further inhibition by hypoxia in rat chromaffin cells (121). Hypoxia-induced secretory activity is also abolished in chromaffin cells of adult mice deficient in MCI subunit NDUFS2 (36). ROS are also implicated in the chemotransduction pathway leading to the inhibition of K+ current in postnatal chromaffin cells.
However, a hypoxia-induced decrease in ROS production, rather than an increase in ROS production, is proposed, based on the measurement of luminol chemiluminescence and treatments with exogenous H2O2, antioxidants, and antioxidant enzymes (121). It is technically difficult to measure ROS production using roGFP probes in primary AM cultures due to the short survival time of chromaffin cells in these cultures. Further studies are necessary to clarify the changes of ROS production in AM cells during hypoxia.
Pulmonary resistance artery smooth muscle cells are sensitive to O2 tension, which produce Ca2+-dependent vasoconstriction during hypoxia to divert blood to better ventilated regions to maximize gas exchange. Although it is generally accepted that changes in mitochondrial ROS production are implicated in the hypoxic response of pulmonary arteries, it has been under debate since the 1990s whether ROS production is increased or decreased during hypoxia in these tissues (1, 10, 46, 75, 76, 83, 133 –136). Part of the dispute results from the difference in detection probes used and the location of measurement systems applied in each study.
Using roGFP probes, an increase in H2O2 production is detected in the IMS upon treatment of hypoxia for 10–30 minutes (134). This increase in H2O2 production appears to be dependent on MCIII, as it is absent in mice deficient in MCIII subunit RISP (135). On the contrary, using H2O2-specific probes (HyPer) targeted to the cytosol and mitochondria, a hypoxia-induced decrease in H2O2 production has been reported in pulmonary smooth muscle cells, which is associated with MCI based on results using siNdufs2 (33). Dysfunction of hypoxia-induced pulmonary vasoconstriction is also observed in mice with deficient MCII (95), further demonstrating the implication of mitochondrial ETC in acute O2 sensing in the lung.
In addition to mitochondrial origin, ROS produced from NADPH oxidase have also been proposed in pulmonary arterial vasoconstriction (39, 40, 65, 138), which may amplify the mitochondrial ROS signal.
CB ROS Production and Mechanisms of Disease
The CB has classically attracted clinical interest due to its critical role in adaptation of the organism to high altitude (acclimatization to hypoxia) or lung diseases characterized by gas exchange restrictions (54, 119, 141). More recently, the CB has regained medical attention due to its involvement in the pathogenesis of diseases prevalent in the human population. The CB chemoreflex has a strong autonomic component, and therefore, it has been suggested that the exaggerated sympathetic outflow characteristic of obstructive sleep apnea (OSA) or chronic heart failure (CHF) is due to CB overactivation secondary to chronic intermittent hypoxia (CIH) and hypoperfusion, respectively [see for reviews Iturriaga (49), Nanduri et al. (85), Schultz et al. (110), Zera et al. (145)].
Although the molecular bases for these maladaptive processes are still poorly understood, available data suggest that cumulative ROS production by CB cells may have a critical role. Several groups have shown that hypoxia/reoxygenation cycles, the main feature of OSA, produce oxidative and nitro-oxidative stress in the CB, which in turn enhances the CB chemosensory responses to hypoxia leading to cardiovascular and autonomic alterations. ROS may increase the expression of signaling pathways such as proinflammatory cytokines and ET-1, which may contribute to potentiating the CB chemosensory responses to hypoxia and other stimuli.
In addition, ROS could modify the O2-sensitive K+ channels, increasing the intracellular Ca2+ levels, which in turn evokes the release of excitatory transmitters (27, 94, 100, 101). CHF has also been shown to produce oxidative stress in the CB (29). In glomus cells from CHF rabbits, the outward voltage-gated K+ current has a smaller amplitude and its sensitivity to hypoxic inhibition is enhanced, resulting in the discharge of chemoreceptor afferents under normoxic state and an increase in discharge responsiveness to hypoxia (109). It has been shown that CB resection or deafferentation restores the sympathetic tone and improves associated cardiovascular alterations in an animal model of CIH and CHF (25, 26, 74).
However, the translation of these procedures to the clinical setting requires strict monitoring because bilateral CB removal may cause cardiovascular events, particularly during episodes of hypoxia and hypercapnia (49, 53, 103). In this scenario, antioxidants are potential therapeutic options to pharmacological modulation of CB overactivation. Systemic administration of low-molecular-mass antioxidant compounds has been shown to be ineffective to eliminate ROS in clinical trials. However, a more refined redox pharmacology could be developed to target specific signaling pathways in dysfunctional CB cells (23, 115). In this regard, it is noteworthy to stress a recent report indicating that mitochondrial antioxidants decrease CB afferent discharges in hypoxia and blunt the HVR in awake rats (120).
Concluding Remarks
The development of genetically engineered redox-sensitive protein probes has allowed us to monitor in real time rapid changes in ROS production in CB cells. This has helped us better understand the signaling role of ROS with low concentration in acute O2 sensing, a fundamental physiological process essential for the survival of mammals in hypoxic environments. In acute hypoxia, ROS produced in mitochondria diffuse to the cytosol and inhibit K+ channels in the plasma membrane of glomus cells, triggering depolarization and transmitter release. This mitochondria-to-membrane signaling mechanism (Fig. 8) may also apply to other O2-sensing chemoreceptor cells.
Several questions remain to be addressed in the chemotransduction pathway during acute hypoxia, such as the specific ROS implicated and their direct molecular target(s). During hypoxia, electron leakage in the mitochondrial ETC could result in increases in O2 − production, which may be converted rapidly to H2O2 by SOD2 in mitochondrial matrix or by SOD1 in IMS. It would be interesting to study whether manipulation of the mitochondrial H2O2 level, such as modification of SOD1 or SOD2 expression, and overexpression of peroxiredoxins (especially PRDX3), glutathione peroxidases (such as GPX1), or transgenic mitochondrial targeted catalase (mCAT) (22, 23), affects acute response to hypoxia in glomus cells. PRDX3, GPX1, and mCAT all remove H2O2.
It remains to be determined whether MCIII contributes to ROS signaling during acute hypoxia in glomus cells, as it happens in pulmonary artery smooth muscle cells. In vitro cell line models overexpressing recombinant K+ channels and thiol-redox proteomics may help us identify the direct target(s) of ROS during acute hypoxia.
Footnotes
Acknowledgments
The authors would like to thank all members of the laboratory who contributed to the experiments discussed in this review.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by grants from the Spanish Ministries of Science and Innovation and Health (SAF2016-74990-R and PID2019-106410RB-I00), from the Andalusian Government (FEDER Andalucía 2014–2020, 2018 Call, US-1255654), all cofunded by FEDER, and the European Research Council (ERC-ADGPRJ201502629).