
Abstract
Significance:
Uncoupling proteins (UCPs) are a family of proteins that allow proton leakage across the inner mitochondrial membrane. Although UCP1, also known as thermogenin, is well known and important for heat generation in brown adipose tissue, striated muscles express two distinct members of UCP, namely UCP2 and UCP3. Unlike UCP1, the main function of UCP2 and UCP3 does not appear to be heat production.
Recent Advances:
Interestingly, UCP2 is the main isoform expressed in cardiac tissues, whereas UCP3 is the dominant isoform in skeletal muscles. In the past years, researchers have started to investigate the regulation of UCP2 and UCP3 expression in striated muscles. Furthermore, concepts about the proposed functions of UCP2 and UCP3 in striated muscles are developed but are still a matter of debate.
Critical Issues:
Potential functions of UCP2 and UCP3 in striated muscles include a role in protection against mitochondria-dependent oxidative stress, as transporter for pyruvate, fatty acids, and protons into and out of the mitochondria, and in metabolic sensing. In this context, the different isoform expression of UCP2 and UCP3 in the skeletal and cardiac muscle may be related to different metabolic requirements of the two organs.
Future Directions:
The level of expression of UCP2 and UCP3 in striated muscles changes in different disease stages. This suggests that UCPs may become drug targets for therapy in the future. Antioxid. Redox Signal. 37, 324–335.
Introduction
In eukaryotic cells, mitochondria are cell organelles that generate the mass of adenosine triphosphate (ATP) by oxidative phosphorylation. However, mitochondria have additional functions in the cell and control thermogenesis, radical production and redox-dependent signaling, calcium homeostasis, and cell apoptosis. All these functions, as well as the regulation of substrate metabolism, require proper control mechanisms. Specifically, the exchange across the inner mitochondrial membrane of anions such as adenosine diphosphate, ATP, phosphate, and of intermediates of the citric acid cycle such as malate, citrate, glutamate and others, requires control mechanisms that are not well characterized.
Uncoupling Proteins: Nomenclature, Distribution, and Potential Function
Uncoupling proteins (UCPs) are a family of structural related proteins of the inner mitochondrial membrane that are involved in several steps of regulation of mitochondrial function. The names of these proteins originate from the UCP1 (UCP1, also known as thermogenin), which is almost exclusively expressed in brown adipose tissue or adjacent white adipose tissue that derives from the same cell linage as brown adipose tissue (19). The latter one can re-express UCP1, a process called “browning” of adipose tissue and the adipose tissue is then named “beige” or “brite” (3). In contrast, other types of white adipose tissue such as subcutaneous white adipose tissue or visceral white adipose tissue do not express UPC1 and they cannot be forces to re-express it. UCP1 acts as a proton carrier activated by free fatty acids (FFA) (8).
This creates a shunt through which protons are directed from the mitochondrial intermembrane space into the mitochondrial matrix (Fig. 1). This shunt uncouples the proton gradient for ATP synthesis leading to heat production. This mechanism is essential for thermogenesis of newborn mammalians and remains important in small rodents during adulthood. Proton transport activity of UCP1 is high (39, 81). Today, four additional members of the protein family have been identified, namely UCP2, -3, -4, and -5, based on the structural homology with UCP1. Among them, UCP2 and UCP3 are expressed in striated muscles (heart and skeletal muscle) (Fig. 1). All isoforms have a tissue-specific expression that is distinct from UCP1, although UCP3 is also expressed in brown adipose tissue (66).
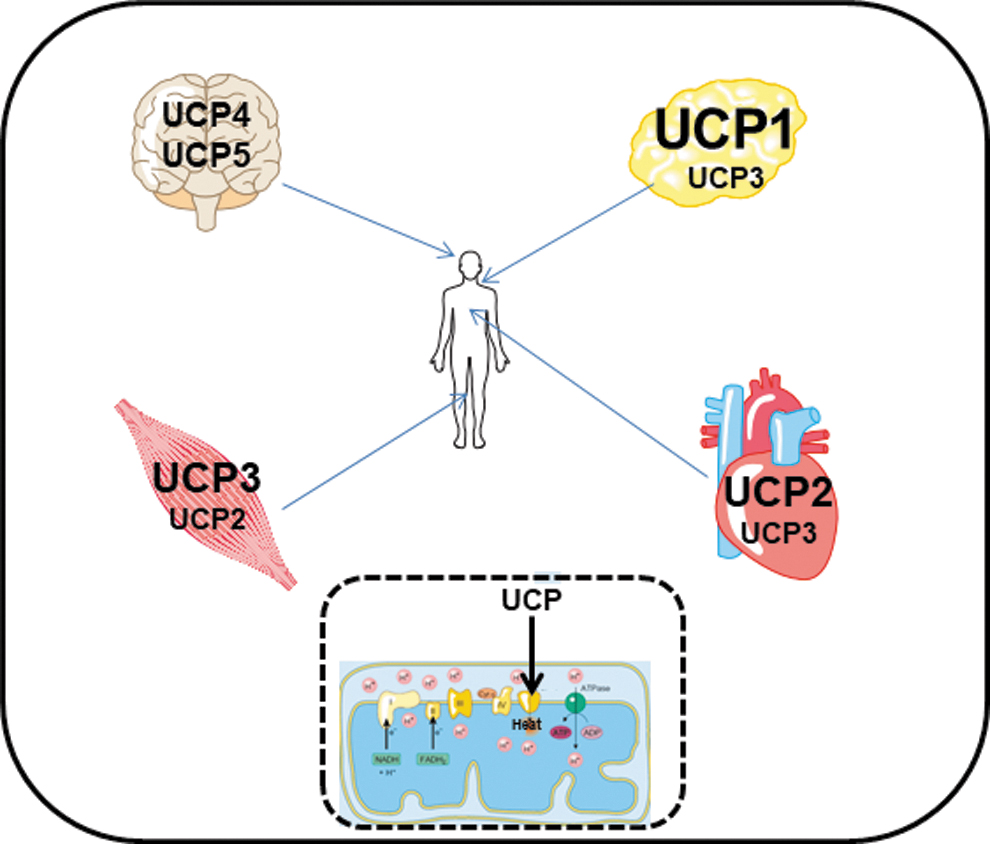
Moreover, these isoforms of UCPs have a much smaller transport capacity for protons than UCP1 (8, 55). A tissue-specific expression of these isoforms and their lower proton transport activity compared with UCP1 suggest that the main function of these members of the UCP family is not thermogenesis, as known for UCP1. Moreover, although UCP2 and UCP3 allow translocation of protons from the intermembrane space into the matrix, this requires an activation but does not occur under basal conditions (66).
Furthermore, the expression of UCP2 and UCP3 differs not only in tissue distribution but also in the level of expression. These UCPs are 100-fold less expressed than UCP1 in brown adipose tissue (63). These differences strongly suggest that novel UCPs play distinct roles in mitochondrial biology than UCP1. In this review, we will focus on two members of this protein family, UCP2 and UCP3, and their role in striated muscles.
Uncoupling Protein-2
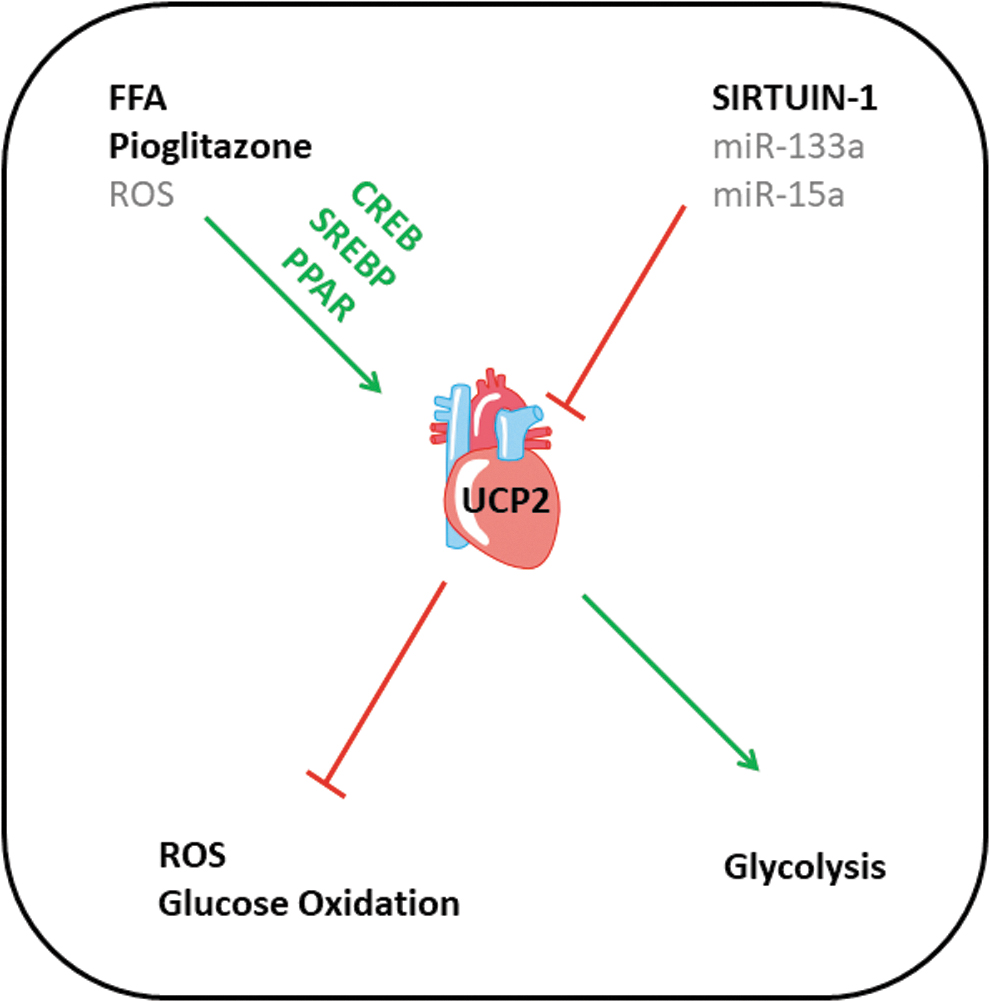
UCP2 (alternative name: solute carrier family 25 member 8; Slc25a8) displays a 59% sequence homology with UCP1. In contrast to UCP1, UCP2 is widely expressed in different tissues, including the heart and skeletal muscle. The gene coding the UCP2 protein is located on chromosome 11 in humans, chromosome 7 in mice, or chromosome 1 in rats, in close proximity to UCP3, indicating a potential gene duplication during mammalian evolution. UCP2 and UCP3 share 72% sequence homology. The UCP2 promoter contains three potential response elements linked to cyclic AMP response-element binding protein, sterol-responsive element-binding protein, and peroxisome proliferator-activator receptor (PPAR) (4, 29, 57, 58). However, little is known about the influence of these responsive elements for UCP2 expression in muscles.
Sirtuin-1 (Sirt-1) acts as a potential inhibitor of UCP2 expression (29). The expression of UCP2 and that of UCP3 seems to be sensitive to the muscle-specific miR-133a (29). In pancreatic cells, UCP2 expression is also sensitive to miR-15a (29). Proton transport activity of UCP2 is induced by FFA, but the exact mechanism by which FFA allow proton transport remains elusive (8). Several models are discussed how proton transport by UCP2 is coupled to FFA transport, as illustrated in Figure 2 (57). Cardiomyocytes can metabolize multiple substrates for energy production, that is, fatty acids and carbohydrates such as glucose. This requires control mechanisms by which the different substrates are preferentially utilized. FFA induce the activity and probably the expression of UCP2 (52, 57, 82, 84).
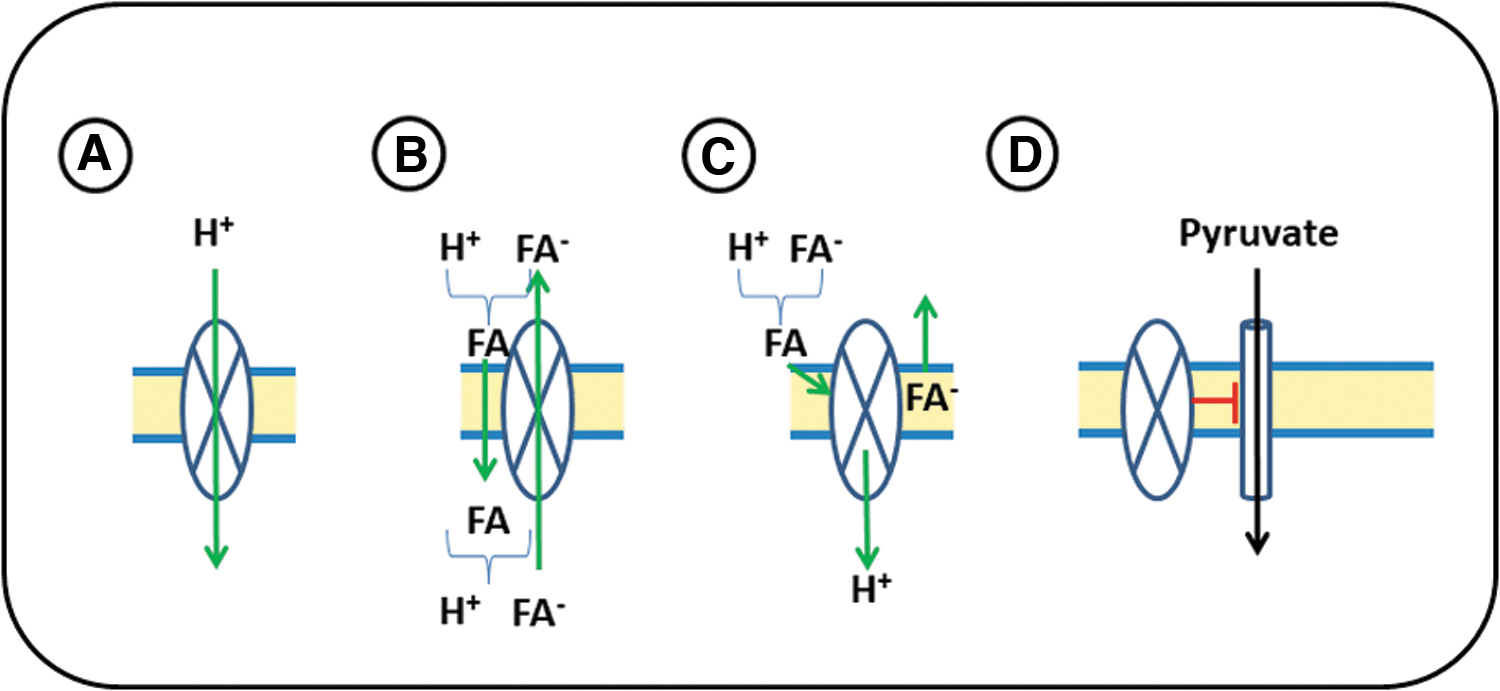
In principle, FFA-dependent activation of UCP2 should increase the amount of FFA that can be used for fatty acid oxidation in mitochondria. However, at the same time, higher expression of UCP2 would reduce the proton gradient and thus reduce fatty acid oxidation-dependent ATP formation. Furthermore, high transport rates of FFA into mitochondria enhance the risk of mitochondrial lipotoxicity. Therefore, it is unclear whether cardiomyocytes will benefit from an FFA-dependent activation of UCP2. Alternatively, UCP2 may inhibit the transport of pyruvate into the mitochondria, as illustrated in Figure 2D (13).
As a result of this, pyruvate can be metabolized in the cytoplasm to lactate and branch substrates of glycolysis (35). It is in line with these suggestions that FFA stimulate UCP2 expression. Increased amounts of UCP2 would then shift substrate utilization in mitochondria from glucose to fatty acid oxidation by inhibition of pyruvate uptake. At the same time, cells can use glucose for anaerobic metabolism. This is typically found in growing tissues even in the presence of high levels of oxygen (Warburg effect). The expression pattern of UCP2 in heart development is at least in part in agreement with such an assumption, as UCP2 expression is high in growing fetal hearts and low in fully differentiated, nonproliferating hearts (38).
UCP2 expression in the heart
In the past, there has been a discussion whether UCP2 is expressed on the mRNA and protein level in the heart. Some authors found no protein expression of UCP2, whereas others found constitutive expression of mRNA, protein, or both in the heart (38, 49, 57, 63, 71). Although it is not always clear why these differences occur, there is consensus that cardiac UCP2 expression is stronger during fetal development than during adulthood and increases at later time points or under pathophysiological conditions (38, 48, 49). The differential expression of UCP2 seems to be a requirement to adapt to altered metabolism (48, 49). The actual level of expression may also show some species-dependent differences (2, 49).
As the expression of UCP2 is differentially regulated during development, it is important to understand the mechanism of regulation. Figure 3 summarizes our current understanding about the regulation of UCP2 in the heart. As mentioned before, the UCP2 gene has a PPAR-responsive element in its promoter region (29). That PPARγ stimulation is capable to induce the expression of UCP2 in the heart was directly demonstrated by feeding mice with pioglitazone (18). The data show a significant increase of UCP2 expression and provide evidence that at least in vitro this is accompanied by UCP2-dependent radical scavenging (18). Exposure of adult rat ventricular cardiomyocytes to FFA induced the expression of UCP2 and subsequent apoptosis in vitro (52).
Also, the feeding of conjugated linoleic acid to adult mice induced the cardiac expression of UCP2 in vivo (94). Both experiments suggest that FFA induce UCP2 expression in the heart via PPAR-response elements. In neonatal cardiomyocytes, FFA regulate the expression of UCP2 in a PPARα but not PPARγ-dependent way only in the early phase of cardiac differentiation, but it has been suggested that this type of regulation is lost in adult hearts (82). Loss of fatty acid-dependent upregulation was not confirmed by another study showing induction of cardiac expression of UCP2 in adult lean Zucker rats (84). Collectively, data on induction and expression of cardiac UCP2 expression are heterogeneous in the literature, and this may either indicate large variability in different species and strains or experimental limitations.
The main inhibitor of UCP2 expression is Sirt-1 (29). This inhibitory effect was confirmed in cardiac tissue. Cold starvation of isolated rat hearts reduced the expression of Sirt-1 and subsequently increased the expression of UCP2 in these hearts. In vitro, this was accompanied by less cardiac performance (21). When Sirt-1 was activated by resveratrol, cold-induced changes in UCP2 expression remained absent. Similarly, Sirt-1 expression and its modulation by resveratrol reduced the expression of UCP2 in H9c2 cells (27). Collectively, these data confirmed that Sirt-1 is an endogenous repressor of UCP2 expression in the heart (Fig. 3).
Specific attention should be given to the observation that miR-133, a muscle-specific microRNA, that is abundantly expressed in cardiac tissue, can target UCP2 (87). Three separate studies show that overexpression of miR-133 can suppress the mRNA and protein expression of UCP2. In THP1 cells (derived from human lung adenocarcinoma), miR-133a-1 suppressed the expression of UCP2 that inhibits inflammasome activity (6). In C2C12 myoblasts, overexpression of miR-133a also suppressed the expression of UCP2, and this allows the cells to differentiate into myocytes (23). However, if this mechanism would be relevant for cardiac development, high expression of UCP2 in fetal hearts would block cardiac differentiation. Finally, overexpression of miR-133a reduced UCP2 expression in MCP-7 cells (human breast cancer) (92).
Moreover, miR-133a is differentially regulated in stress conditions. It is constitutively downregulated in postinfarcted hearts (12, 36, 91). Cardiac expression of miR-133a remains stable in hearts with cardioprotection, such as ischemic postconditioning (36). If miR-133 does really downregulate UCP2 expression in cardiac tissue as well, this would give an important information explaining improved postinfarct remodeling by cardioprotection. However, there are several findings that do not suggest that UCP2 can be targeted by miR-133 in cardiac tissue. At first, miR-133 has no consensus sequence linking miR-133 to UCP2 expression (reference: TargetScan). However, miR-133 may indirectly affect the expression of Mef2c and UCP2 by regulation of DNA methylation as suggested before (20).
Second, in postischemic hearts in which miR-133 is downregulated, there is no upregulation of UCP2 (74). Third, UCP2 is upregulated by ischemic postconditioning, but miR-133 is not reduced (74). miR-133 is a muscle-specific marker in conjunction with Mef2c and required for cardiac differentiation (87). Our own analysis in rat hearts shows a strict co-regulation of Mef2c and UCP2 (unpublished observation). This observation fits to the high expression of UCP2 in developing heart cells independent of miR-133 In summary, there is no evidence that miR-133 directly targets UCP2 in cardiac tissue. The observed correlations between miR-133 and UCP2 expression in other cells and tissues could not be confirmed in cardiac-specific tissues.
UCP2 and cardiac metabolism
In a transgenic mouse model in which the activity of acetyl CoA carboxylase-2 (ACC-2), the rate-limiting enzyme of fatty acid synthesis, is attenuated, cardiac expression of UCP2 is upregulated (1). Interestingly, ACC-2 is physiologically inactivated by epinephrine-dependent phosphorylation. Thus, inhibition of ACC-2 during severe exercise lowers the formation of malonyl-CoA, a co-regulator of the carnitine/palmitoyl-CoA shuttle system and thus fat storage. Consequently, ATP generation by fatty acid oxidation is induced. In short, inhibition of ACC-2 shifts fatty acid metabolism from fatty acid storage to fatty acid oxidation. Interestingly, UCP2 has been linked to inhibition of mitochondrial pyruvate uptake (13).
Therefore, the upregulation of UCP2 under these conditions will indirectly support fatty acid oxidation by inhibiting glucose oxidation through the inhibition of pyruvate influx into mitochondria. Via this mechanism, UCP2 may potentiate the effect of ACC-2 inhibition on fatty acid metabolism. These data suggest a key role for UCP2 in metabolite control. However, the upregulation of UCP2 has been shown in transgenic mice lacking ACC-2 activity and not in mice or rats under severe exercise. Therefore, the experiment cited above does not directly test whether severe exercise can induce a metabolic switch, but the data support the idea that UCP2 may function as a regulator of substrate consumption in the heart. In this line, it was observed that in newborn rats with intrauterine growth restriction, cardiac expression of UCP2 is regulated in a time-dependent way (50).
The authors suggest that downregulation of UCP2 in adult rats with prior intrauterine growth restriction goes along with the preference use of glucose, whereas high expression in the early postnatal period goes along with fatty acid metabolism. These rats also developed a hypertensive phenotype. Nevertheless, the conclusion was not supported by quantification of substrate utilization (50).
In patients undergoing coronary bypass surgery, FFA concentration correlated positively with the cardiac expression of UCP2, whereas the expression of the glucose transporter type 4 (Glut-4) was inversely downregulated (61). Again, high expression of UCP2 was associated with fatty acid metabolism and impaired glucose utilization. Similarly, our own studies conversely showed that silencing of UCP2 by siRNA in cardiomyocytes improved glucose metabolism and was associated with upregulated expression of Glut-4 (49).
In a rarely used animal model of postnatal overnutrition it was investigated, whether postnatal overnutrition leads to cardiac adaptations preceding obesity in adulthood (9). Interestingly, these authors describe increased oxidative stress in mitochondria (determined by trans-
The effect of high expression of UCP2 in cardiomyocytes was also tested in a more direct way by cellular overexpression of UCP2 (10). Normally, cardiomyocytes are cultured in glucose-containing media without fatty acids. If this was also done here, which is not specified in the article, overexpression of UCP2 should subsequently avoid pyruvate uptake of mitochondria. Therefore, the cells are expected to rely on the less effective anaerobic ATP production by glycolysis. Indeed, the authors observed a significant reduction of ATP content in these cells and acidosis as expected from anaerobic glycolysis. The data suggest that low expression of UCP2 favors glucose oxidation (10).
The same conclusions were drawn using neonatal and adult mice (48). In contrast, embryonic stem cells differentiating into cardiomyocytes have high expression of UCP2 and rely also on glycolysis (38). However, these cells may depend on branch products of the anaerobic glucose metabolism for growth and differentiation, but this was not further analyzed. Although the strict regulation of cardiac UCP2 expression at different stages of cardiac ontogenetic development and subsequent regulation in adulthood suggest an important role for UCP2 in cardiac development, it must be mentioned that UCP2−/− mice show normal development of heart structure and function, unless they are challenged by pressure overload, ischemia, or other models requiring cardiac adaptation.
In conclusion, high UCP2 expression is mostly associated with low oxidative glucose metabolism. UCP2 appears to be a switch that is required under stressed conditions and is involved in pathophysiological processes. In contrast, metabolic switches during development do not appear to be dependent on UCP2.
UCP2 and mitochondrial calcium uptake in cardiac cells
In addition to the possible role of UCP2 as a metabolic regulator in cardiac cells, UCP2 may also be involved in the regulation of mitochondrial calcium uptake. This suggestion is mainly based on studies with UCP2 knockout mice, but it remains unclear whether physiological changes in UCP2 expression are sufficient to modify this process. Nevertheless, in the absence of UCP2, Ca2+ uptake in mitochondria is reduced (51, 60). This has two consequences. At first, mitochondrial Ca2+ load is important for the activity of enzymes of the citrate cycle, thereby again coupling UCP2 expression to metabolism. Second, less Ca2+ uptake by mitochondria without compensation induces Ca2+ overload in the cytosol.
Under these conditions, an inhibitory effect on voltage-dependent Ca2+ channels (VDCC) was described, reducing the duration of action potential and subsequent alteration of ECG recordings as well (51).
Although this is an interesting observation in UCP2 knockout mice, a mechanism by which less Ca2+ uptake in mitochondria triggers VDCC open probability needs to be established. In contrast to these assumptions, studies with adenoviral overexpression of UCP2 in neonatal cardiomyocytes protected the cells against H2O2-induced apoptosis and increased the probability that Ca2+ sparks propagate into Ca2+ waves (77, 80). In both cases, reduced mitochondrial Ca2+ uptake by overexpression of UCP2 was observed. The question that arises from these studies is how overexpression of UCP2 in neonatal rat cardiomyocytes and lack of UCP2 expression in transgenic mice can reduce the capacity of mitochondria to reduce Ca2+ load in the cytosol.
Although in both cases the authors speculate how this contributes to pathophysiology, it must be critically claimed that both experimental settings do not directly reproduce the time- and differentiation-dependent regulation of UCP2 in vivo.
Uncoupling Protein-3
UCP3 (alternative name: solute carrier family 25 member 9; Slc25a9) shares strong sequence homology with UCP1 and UCP2. An important difference between UCP2 and UCP3 is the distinct pattern of expression. UCP3 is the dominant isoform of novel UCPs in the skeletal muscle but less commonly expressed in other tissues (86). UCP3 expression shows a strong association to fatty acid metabolism, suggesting a role in fatty acid oxidation or its regulation (66). However, an exact description how fatty acid oxidation and UCP3 expression and activity interact is still missing. Noteworthy, UCP3 knockout mice did not have a phenotype under basal and stressed conditions, indicating at least that the absence of UCP3 can be bypassed by other pathways.
UCP3 expression in the heart
Although UCP3 is constitutively expressed in the heart, too, less attention has been directed to the regulation of cardiac UCP3 expression and activity and its potential physiological role. This may be because UCP2 is more dominantly expressed in the heart, whereas UCP3 is the dominant isoform in the skeletal muscle (see UCP2 and UCP3 in the Skeletal Muscle). In normotensive rats, the cardiac expression of both isoforms decreased within 6 months after birth (28). Lipid infusion into rats directly stimulated the cardiac expression of UCP2 and UCP3 (84). In a streptozotocin (STZ)-dependent diabetes model, the expression of UCP3 but not that of UCP2 was increased. However, no functional deficit in cardiac function was observed. Exposure of rats to hypobaric hypoxic environment for 7 days significantly reduced the expression of UCP3 but not that of UCP2 (31).
In the same ventricles, the expression of PDK4, an inhibitor of glucose oxidation, was decreased, whereas the expression of mCPT1 (mitochondrial carnitine palmitoyltransferase I), the rate-limiting enzyme for fatty acid import into mitochondria, was also decreased. This type of regulation would favor glucose utilization. However, it remained unclear from that study, whether this regulation is dependent on UCP3 expression or not. Nevertheless, both examples clearly indicate that the mRNA expression of both UCPs is differentially regulated in dependence of the metabolic requirement. In stem cells undergoing differentiation into cardiomyocytes, the expression of UCP3 goes hand-in-hand with that of markers of fatty acid metabolism (38).
Both studies support the hypothesis that UCP3 expression is a marker of fatty acid metabolism. In principle, UCP3 shares this function with UCP2. However, as we have learned from the aforementioned studies, UCP3 expression can be modified independent of UCP2 expression. Similarly, transgenic expression of the mitochondrial membrane protein stanniocalcin-1 was associated with increased expression of UCP3 but not UCP2 (54). Induction of fibroblast growth factor (FGF) 21 release from cardiomyocytes increases the expression of UCP2 and UCP3 (65). However, Sirt-1, a direct inhibitor of UCP2 expression, induces the release of FGF21a finding directly contrasted by this study, which reported a Sirt-1-dependent upregulation of UCP2. The contrasting findings notify the need of additional studies. Figure 4 summarizes the current findings.
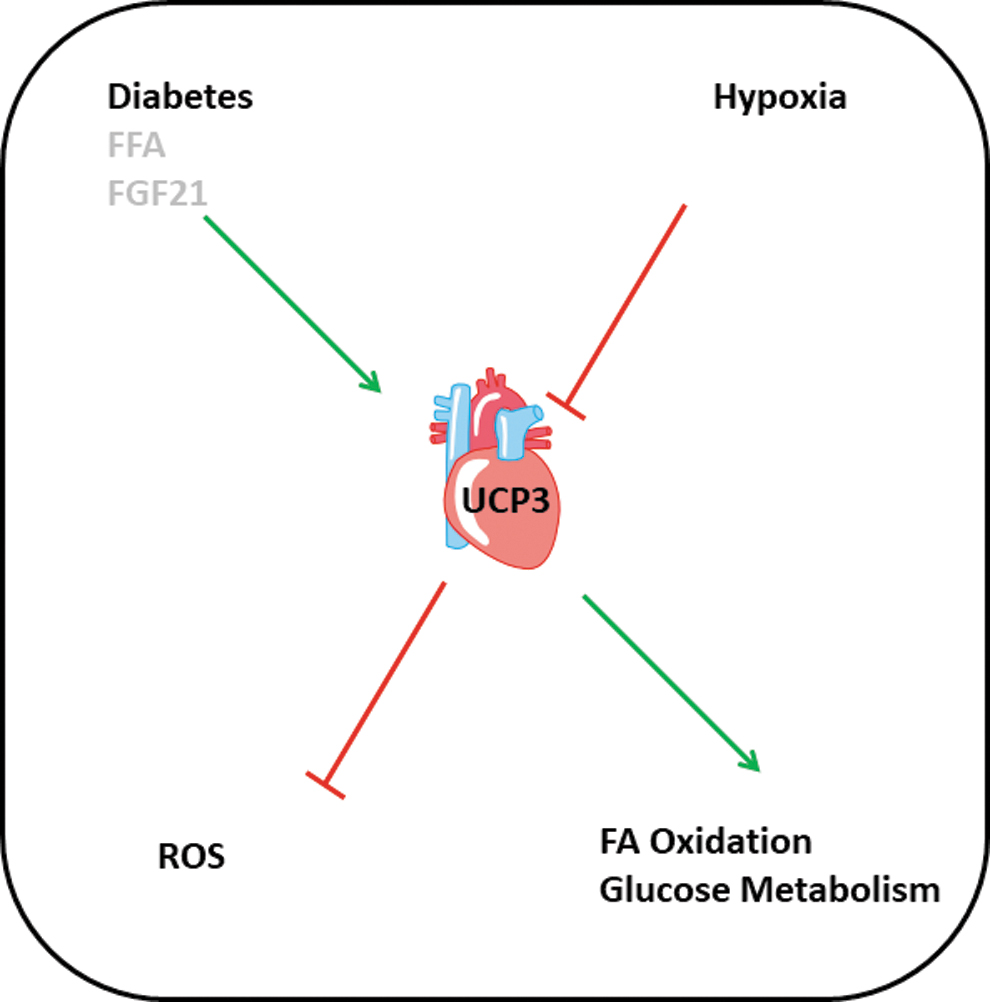
Expression of UCP2 and UCP3 in Heart Failure Models
Hypertension models
Maladaptive cardiac remodeling relies on an activation of the renin–angiotensin system in which angiotensin II induces several transcriptional alterations via activation of the angiotensin receptor type 1. Although stimulation of cardiomyocytes with angiotensin II is a standard cell culture model of cardiac hypertrophy, only few studies analyzed the relationship between angiotensin II and UCP expression. Studies with neonatal rat cardiomyocytes showed that angiotensin II induces the expression of UCP2 in these cells (88). Inhibition of the renin–angiotensin system reduces the cardiac expression of UCP2 vice versa (72). In line with these in vitro studies, the cardiac expression of UCP2 is induced in several models of pressure overload and myocardial hypertrophy (9, 10, 22, 33, 97).
The data suggest that UCP2 is either involved in metabolic adaptations (metabolic switch) or modifies other pathways such as mitochondrial ROS control and thereby modifies signaling. Our own studies showed a biphasic regulation of UCP2 in cardiac tissue in spontaneously hypertensive rats. In the early adaptation to hypertension, UCP2 expression was decreased and glucose uptake in cardiomyocytes improved, consistent with a metabolic switch toward glucose metabolism in hypertrophic myocardium. However, at later time points when hypertrophy loses its compensatory character, UCP2 was induced again and the related effect on glucose uptake was reduced. Importantly, blocking aldosterone in the late phase was sufficient to attenuate UCP2 induction and maintained function (49).
In a specific in vitro model using indoxyl sulfate as a typical protein bound uremic toxin and neonatal rat cardiomyocytes as cell system, it was found that toxification caused a downregulation of UCP2 via downregulating of adenosine monophosphate kinase (AMPK) activity and thereby induced hypertrophy (89). Here, the authors claimed that the toxin inactivates AMPK that then downregulates UCP2. However, Sirt-1 activates AMPK but decreases the expression of UCP2. Therefore, this mechanism is again puzzling as it contrasts with in vivo findings showing an increased expression of UCP2 in hypertrophic models and a different regulation by Sirt-1 and AMPK.
Ischemia/reperfusion models
Another important pathophysiological model is ischemia/reperfusion. Again, upregulation of UCP2 was observed in postischemic hearts (88). Whether this is functional relevant is still under debate and data are not conclusive. Whereas some authors show that downregulation of UCP2 via Sirt-1 increases ischemia tolerance of the heart, others report that ischemic conditioning, a method that protects the heart against ischemia/reperfusion injury, induces the expression of UCP2 (17, 27, 74). Genetic depletion of UCP3 increases reperfusion injury (62).
Diabetes models
In a diabetes model, the expression of UCP3 but not that of UCP2 increased (28). However, hyperglycemia in vitro reduces the expression of UCP2 (85).
Doxorubicin, smoking, and sepsis
Similarly, confusing results are reported on other models of heart failure. In doxorubicin-induced heart failure, the expression of UCP2 and UCP3 is decreased resulting in better mitochondrial coupling in vitro but more generation of ROS (16). In cold-exposed rodents, however, the expression of UCP2 increases but function drops in parallel (21). Smoking reduces the expression of UCP2 and authors speculate that this increases the ROS formation (64). The role of UCP2 has also been investigated in two models of sepsis. Whereas sepsis seemed to induce UCP2 expression, it was proposed that UCP2 can protect against sepsis-induced heart failure (70, 95). Finally, cardiac expression of UCP2 is decreased in alcohol-induced heart failure (93).
Collectively, differential regulation of UCP2 has been observed in several models of heart failure and that of UCP3 at least in a few studies. Most authors linked differential regulation of UCP2 to either metabolic control or ROS scavenging. While most studies showed an upregulation in hypertrophic myocardium at least at the time points when function declines, the regulation of UCP2 in other models of heart failure, including reperfusion injury, diabetes, and sepsis, do not give a clear picture so far (Fig. 5).
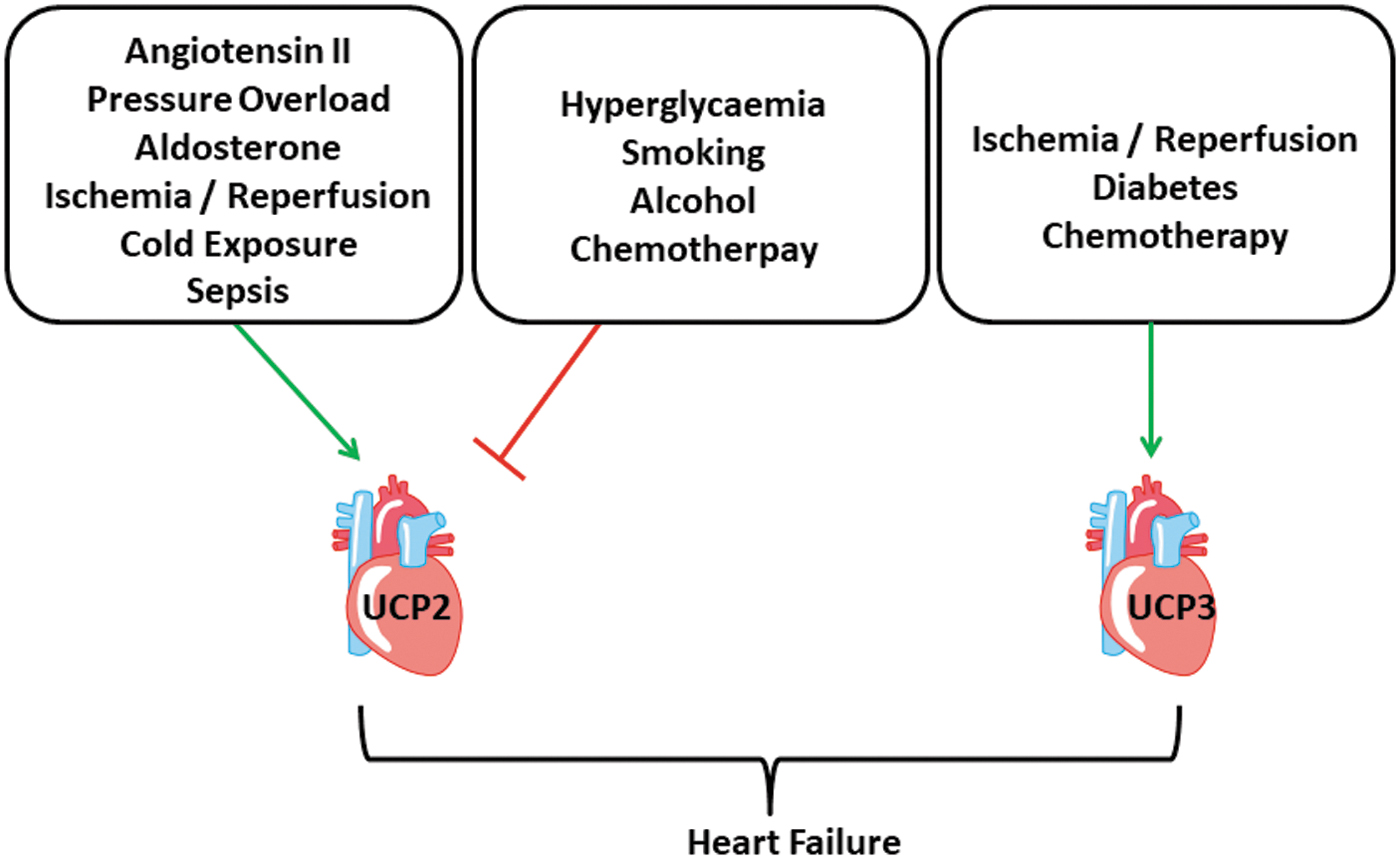
UCP2 and UCP3 in the Skeletal Muscle
The first and obvious difference between the heart and skeletal muscle is the different pattern of UCP expression, with UCP2 being the dominant isoform in the myocardium heart and UCP3 being the dominant isoform in the skeletal muscle. There are reports that show an increase of UCP3 expression in response to exercise in the acute phase (44, 96). Mechanistically, this seems to be linked to AMPK activation (78, 96). Direct activation of AMPK, that is, by administration of AICAR, can also induce the expression of UCP3 in skeletal muscles (68). However, in chronic exercise models such as voluntary running wheel exercise, induction of UCP3 is not affected, normal, or reduced (11, 26, 37, 67).
A recent study from our own group has further shown that the regulation of UCP2 and UCP3 in the skeletal muscle depends on the genetic background of the animals, as UCP3 mRNA was downregulated by voluntary running activity in the Musculus soleus in both strains (spontaneously hypertensive rats and normotensive Wistar) but in the Musculus gastrocnemius only in normotensive rats (86).
There were also differences in the basal expression of UCP2 between both strains in the skeletal muscle. Upregulation of UCPs during the acute phase of exercise may help to neutralize ROS that are generated by higher oxidative phosphorylation during exercise periods. Increase oxidative phosphorylation is required to scope the increased energy demand. However, as this lowers the amount of ATP generated by oxidative phosphorylation due to uncoupling of the electron transport chain, but also generates heat, this must be a transient phenomenon. This is a critical point. Based on preliminary observations in transgenic mice overexpressing UCP3 in the skeletal muscle, it has been suggested that exercise-induced expression of UCP3 may contribute to effective weight loss. However, if this is only a transient phenomenon, the efficiency will be limited.
Moreover, the expression of UCP3 in the skeletal muscle undergoes age-dependent downregulation (42, 45). However, under electrical stimulation, hyperactivation of AMPK and subsequently upregulation of UCP3 were found in skeletal muscles of animals with advance aged (78). Only few signal transduction pathways were proven to trigger UCP3 expression such as AMPK, PPARα but not PPARγ (14), thyroid hormones (69, 76), nonesterified fatty acids (NEFA) (43), or fish oil (5). The expression of UCP3 further depends on the activation by MyoD (myoblast determination protein D) and requires all-trans retinoic acid as a transcriptional activator of the gene (75). UCP3 expression in the skeletal muscle is activated by NEFA, but the level of regulation is not the plasma concentration of NEFA.
Instead of this, regulation occurs by lipoprotein lipase, an enzyme required for hydrolysis of plasma triacylglycerols in the skeletal muscle (46).
Similarly, it was shown that in the skeletal muscle thiazolidinediones can induce the expression of UCP3, independent of NEFA (15). Therefore, it remains unclear whether NEFA, as they accumulate in acute response to exercise, can directly affect the expression of UCP3. Collectively, it seems that there is some improvement in our current understanding how UCP3 expression is regulated in the skeletal muscle but not yet finally characterized. Some of the observed effects show a specific effect on UCP3 that are not seen for UCP2, that is, the role of NEFA (43), exercise (44), or the overexpression of Glut-4 in mice that is associated with induction of UCP3 expression in the M. gastrocnemius but not with induction of UCP2 expression (79).
Concerning the potential role of UCP3 in the skeletal muscle, different modes of action are discussed, such as improvement of fatty acid metabolism, ROS scavenging, and energy loss due to electrical uncoupling, but direct measurements of such effects are rare. In cancer patients, weight loss resulting in cancer cachexia is associated with an increased expression of UCP3 in the skeletal muscle (25). If this is causally linked to each other, upregulation of UCP3 in the skeletal muscle, by either exercise or pharmacological methods such as AMPK activation, will be attractive to normalize metabolism in diabetic and obese patients as well. Experimentally, this was investigated by transgenic overexpression of UCP3 in the skeletal muscle, leading to mice that were hypoglycemic, had less fat, and lower weight (24).
In a diabetes rat model (STZ-induced diabetes), it was found that the expression of UCP3 (and UCP2) is increased in the skeletal muscle and this induction could be normalized by insulin (40). However, in type 2 diabetic patients, the skeletal muscle expression of UCP3 was reduced (47). When mitochondria from diabetes patients were analyzed directly, state 3 respiration was impaired, but this was independent of the expression of UCP3 (59). In conclusion, although upregulation of UCP3 expression in skeletal muscles by exercise seems to be an attractive model to improve glucose metabolism and induce weight reduction, the data in clinical settings do not necessarily support the idea that this may occur via upregulation of UCP3 in the skeletal muscle.
UCP2 is less expressed in the skeletal muscle compared with UCP3 and compared with the cardiac muscle. The skeletal expression of UCP2 is increased in the elderly rodents but not in cancer patients (7, 25). Endurance training has been shown to reduce its expression (11). However, in other studies, exercise did not affect the expression of UCP2 in the skeletal muscle (37, 44). Hypoxanthine, a metabolic product of ATP, circulating during exercise, induced the expression of UCP2 (90). As also found in cardiac tissue, the expression of UCP2 and Glut-4 is counterregulated in some cases (30, 32, 41, 53, 73). However, ghrelin induces both genes in parallel (34).
As shown for UCP3 as well, thyroid hormones induce the expression of UCP2 in the skeletal muscle (56, 83). A specific function for UCP2 in the skeletal muscle has not yet been identified. An overview about the regulation of UCP2 and UCP3 is given in Figure 6.
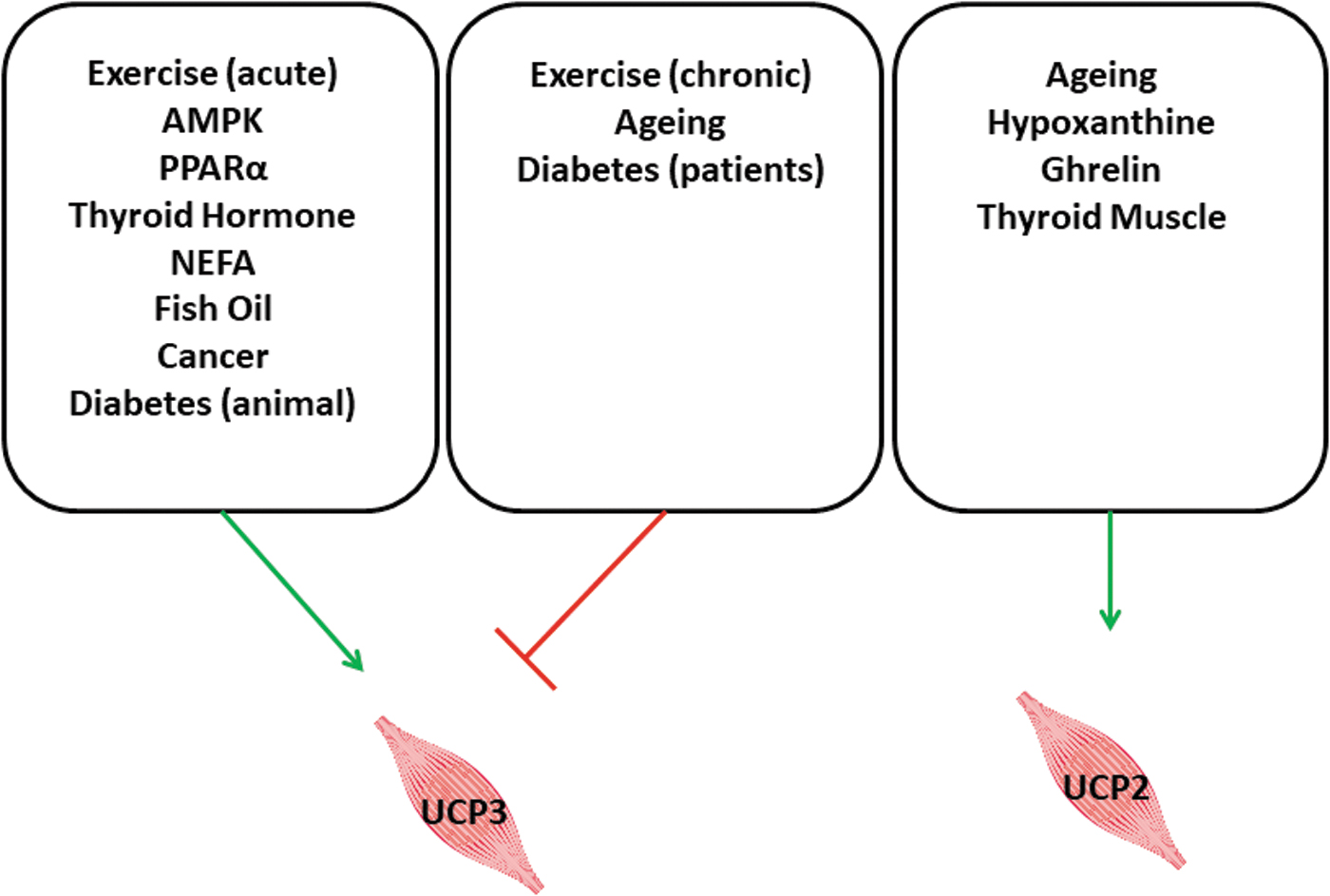
Conclusive Remarks
The observation that UCP2 and UCP3 are differentially expressed in different types of striated muscles and the additional notification that their tissue-specific expression is modulated under pathophysiological conditions has turned our interest to these two UCPs. In general, both UCPs seem to be linked to fine-tuning of metabolic requirements of the muscles, that is, the direction of either fatty acids or glucose as a dominant substrate for oxidative phosphorylation. However, a general description about the role of these two proteins in muscle physiology is largely limited by the fact that their precise function is still unclear. Several assumptions have been presented in the literature with quite different models but not leading to conclusive results.
Nevertheless, at least in the heart, a low expression of UCP2 in adulthood seems to favor glucose consumption, whereas higher expression favors the consumption of fatty acids. Another problem in our current understanding is species-dependent differences in the expression pattern of these two proteins in the cardiac and skeletal muscle. Nevertheless, the large requirement of energy for the skeletal muscle function due to the fact that it represents a large metabolic active tissue and the requirement of the heart to generate sufficient ATP for proper cardiac function indicate the large potential for UCP2 and UCP3 as potential drug target to improve of metabolic disorders.
Footnotes
Authors' Contributions
K.-D.S. wrote the article and was responsible for the overall direction and planning. H.S.K. and R.S. contributed to the writing of the article and reference selection.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors are supported by research grants from the Deutsche Forschungsgemeinschaft (DFG), CRC 1213, project B05.