
Abstract
Background:
Mitochondrial Na+ has been discovered as a new second messenger regulating inner mitochondrial membrane (IMM) fluidity and reactive oxygen species (ROS) production by complex III (CIII). However, the roles of mitochondrial Na+ in mitochondrial redox signaling go beyond what was initially expected.
Significance:
In this review, we systematize the current knowledge on mitochondrial Na+ homeostasis and its implications on different modes of ROS production by mitochondria. Na+ behaves as a positive modulator of forward mitochondrial ROS production either by complex III (CIII) or by decreasing antioxidant capacity of mitochondria and as a potential negative modulator of reverse electron transfer (RET) by complex I (CI). Such duality depends on the bioenergetic status, cation and redox contexts, and can either lead to potential adaptations or cell death.
Future Directions:
Direct Na+ interaction with phospholipids, proven in the IMM, allows us to hypothesize its potential role in the existence and function of lipid rafts in other biological membranes regarding redox homeostasis, as well as the potential role of other monovalent cations in membrane biology. Thus, we provide the reader an update on the emerging field of mitochondrial Na+ homeostasis and its relationship with mitochondrial redox signaling. Antioxid. Redox Signal. 37, 290–300.
Introduction
All metazoans require oxygen to produce energy through the oxidative phosphorylation system (OXPHOS). The OXPHOS system is located in the inner mitochondrial membrane (IMM), a lipid bilayer separating the outer mitochondrial membrane and the space between both—the intermembrane space—from the mitochondrial matrix and builds the crista. It comprises five complexes, the first four constitute the electron transport chain (mETC) and carry out the successive oxidoreduction of respiratory substrates, coupling the electron transfer to H+ pumping across the IMM.
The latter creates an electrochemical gradient, the proton motive force (μ), which comprises an electrical component, the mitochondrial membrane potential (Δψmt), negative inside, and a chemical component, the pH gradient (ΔpH), alkaline inside. The energy stored in μ is then used by the fifth complex to phosphorylate adenosine diphosphate (ADP) into adenosine triphosphate (ATP).
Complex I (CI) oxidizes NADH into NAD+, transferring two electrons to ubiquinone (CoQ) and pumping four H+ ions. In turn, complex II (CII) oxidizes succinate into fumarate and transfers two electrons to CoQ without pumping H+. The reduced form of CoQ, ubiquinol (CoQH2), transfers its two electrons to complex III (CIII), which performs a singular reaction to transfer the electrons to cytochrome c (cyt c), the so-called Q cycle. In this cycle, CIII, at its Qo site, takes one electron from CoQH2, converting it to semiquinone (Q•−) and transferring it to cyt c.
The second electron is directed to the Qi site of CIII where a new CoQH2 molecule will be formed. Noticeably, as Q•− is expected to be formed at the Qo and Qi sites, short circuits are expected to occur and single-gated and double-gated models preventing it have been proposed (21, 22, 65). Then, a second CoQH2 molecule performs the same series of reactions, donating its second electron to the Q•− previously formed at the Qi site and forming CoQH2. CIII pumps two H+ ions per CoQH2 molecule oxidized. Finally, cyt c transfers its electron to complex IV (CIV), which reduces oxygen to water and pumps four H+.
OXPHOS complexes can assemble into quaternary structures called supercomplexes. CI interacts with CIII and CIV, forming supercomplexes I + III2, I + IV, I + IV2, and the N-respirasome or I + III2+IV (1, 12, 61, 76) (Fig. 1A). CIII can also interact with CIV, forming the Q-respirasome or III2+IV (1, 12, 19, 27, 50, 76) (Fig. 1B), and CIV can form dimers and oligomers (12, 19). CV is also found as dimers and oligomers, which have been shown to promote bending of the IMM in cristae and to be involved in the mitochondrial transition pore (mPTP) (15, 24, 30, 35, 79) (Fig. 1C).

CoQ Channeling
The role of the association between CI and CIII has been debated for years. Initial studies suggested the existence of kinetic dependency between CI and CIII and proposed that their structural interaction could also confer a relevant functional association (8). Later, this hypothesis was supported by genetic data, proposing the existence of two functionally differentiated CoQ pools in the IMM, one of them being more dedicated to CI (CoQNAD) and the other dedicated to FAD-dependent enzymes in the IMM (CoQFAD) (50) (Fig. 2).
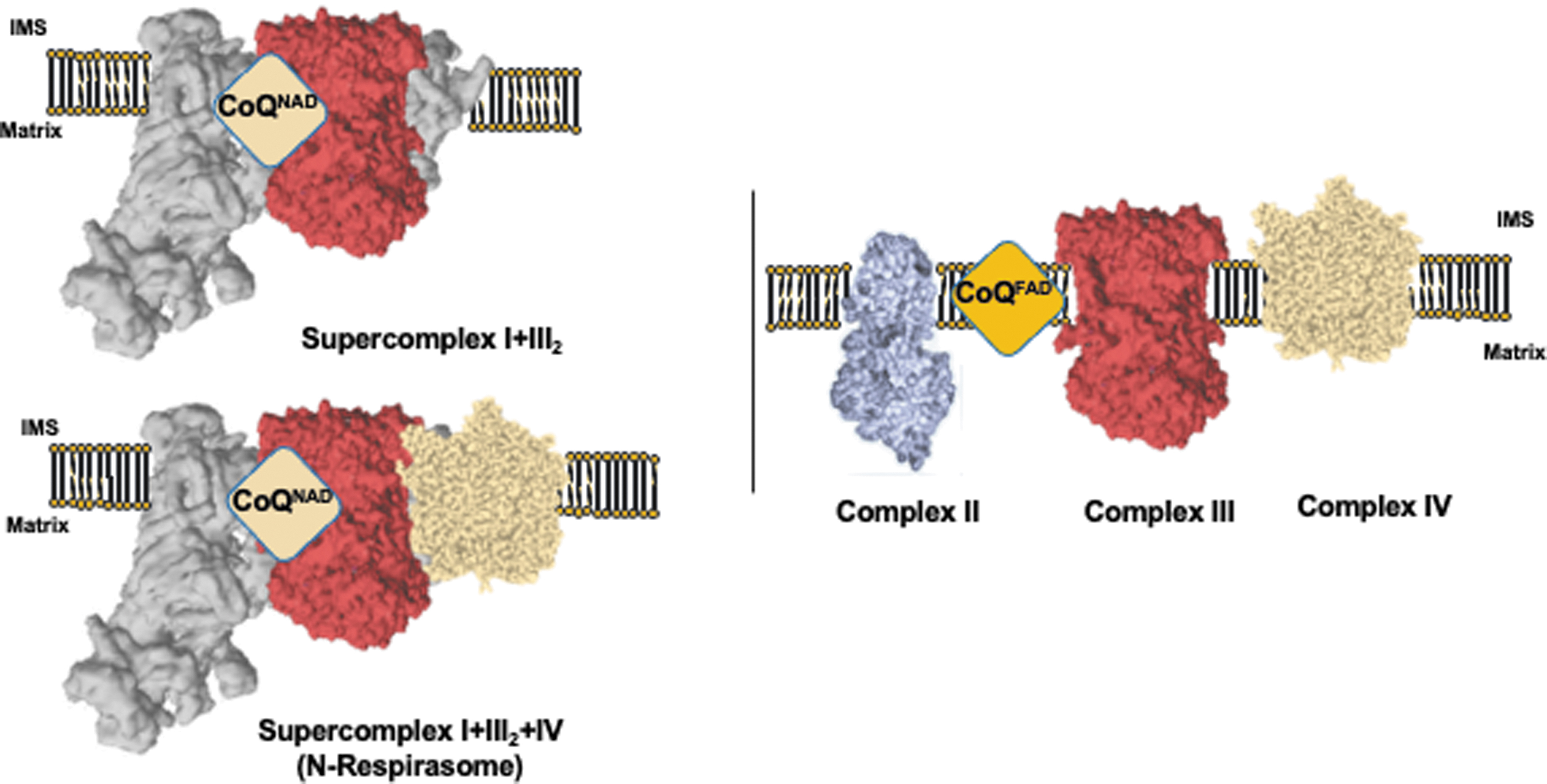
This hypothesis was challenged by the use of an alternative oxidase (AOX), which showed relevant NADH oxidation after CIV inhibition in submitochondrial particles (26). However, it was later demonstrated in vivo that AOX cannot accept CoQH2 from CI unless CIII is pharmacologically or genetically inhibited (12, 78). Thus, under normal circumstances, CoQ is functionally segmented into CoQNAD and CoQFAD pools, and supercomplexes containing CI perform the so-called CoQ functional channeling [see Ref. (37) for further discussion; Fig. 2].
The molecular mechanism for this segmentation is still poorly understood and several hypotheses are conceivable. It is possible that the proximity of CI and CIII CoQ reaction centers in the supercomplexes makes CoQ channeling possible. Although supercomplex structures show that CI CoQ cavities would permit diffusion of CoQH2 into the IMM, a gradient of lower CoQH2/CoQ toward the active CIII may be formed, facilitating diffusion of CoQH2 into CIII within the supercomplex. In parallel, a different phospholipid composition in the surroundings of the supercomplex may hinder CoQH2 diffusion outside of the supercomplex, thus enabling CoQ channeling within the same structure [see Ref. (37) for further discussion].
Another role ascribed to CI-containing supercomplexes and CoQ channeling is the limitation of the production of reactive oxygen species (ROS). Reconstitution of CI and CIII in liposomes yielded higher production of ROS than if CI and CIII were reconstituted as I + III2 (57). This has important implications as cellular control of supercomplexes can have an impact not only on ATP production and metabolic regulation but also on redox homeostasis.
Several examples have been observed supporting CoQ segmentation in physiology. Free CIs in astrocytes led to higher levels of ROS, whereas neurons, which have most of the CI superassembled, showed lower ROS levels. Interestingly, lowering CI association with CIII increased ROS production and dropped oxygen consumption rates (53). It would be very interesting to test whether increasing supercomplex levels in astrocytes may have an impact on their physiology and homeostasis. In addition, it has been shown that hypoxia promotes a specific interaction between Na+ and phospholipids in the matrix side of the IMM, which decreases its fluidity.
Very interestingly, Na+ could only decrease CoQH2 transfer between CII and CIII, but not between CI and CIII, again showing that CoQH2 transfer between CI and CIII is functionally differentiated from that of CII and CIII (36). Mitochondrial Na+ entry during hypoxia is allowed by the mitochondrial Na+/Ca2+ exchanger (NCLX) and potentiated by the mitochondrial Ca2+ uniporter (MCU; see section: Na+ Regulates Mitochondrial ROS Production in Several Modes), and the function is discussed below.
Mitochondrial Ca2+ and Na+ Homeostasis
mETC-driven Δψmt generates a deep electrical gradient, negative inside, which facilitates the entry of positively charged cations into the mitochondrial matrix. Ca2+ entry is enabled by the MCU, a protein located in the IMM, which is largely regulated by other regulatory subunits, such as the mitochondrial Ca2+ uptake protein (MICU) 1 and 2, and the essential MCU regulator (EMRE; 67, 75).
On the other hand, mitochondrial Ca2+ exit is regulated by NCLX (66), permitting 1 Ca2+ extrusion in exchange of (the generally accepted) 3 Na+ (4, 41, 45). In turn, Na+ entry into the matrix is mediated by the high activity of the canonical mitochondrial Na+/H+ exchanger (mNHE), a molecule yet to be identified in animal mitochondria, which catalyzes the exit of 1 Na+ and introduction of 1 H+ (60).
Given its coordination chemistry, Ca2+ has become a pleiotropic second messenger in the cell. Several properties, such as its valency, ionic radius, hydration energy, or polarizability, explain why this cation is readily accepted by very irregular surfaces such as those in proteins (13). These very properties of Ca2+ make it a very unsuitable cation for bioenergetics as it also has extremely high affinity for phosphate.
Thus, concentrations in most cellular compartments must be maintained low, usually in the nanomolar range. However, mitochondria can readily take up Ca2+ as cytosolic Ca2+ levels reach above 10 μM (23, 29, 72) and are able to assimilate hundreds of times the initial content under normal conditions, before their normal function becomes compromised (14, 44). This is in striking contrast with its propensity to react with phosphate as mitochondria require phosphate to make OXPHOS work.
Such contradiction can be easily explained by the fact that mitochondria accumulate Ca2+ in the form of calcium phosphate (Ca-P) precipitates. Indeed, on its way to the mitochondrial matrix, Ca2+ entry is facilitated by phosphate, which, in turn, enables precipitation (82). The stoichiometry of Ca:phosphate in the precipitates has been calculated as the ratio of 1.67 (73), which is exactly that of hydroxyapatite. This identity has been confirmed and complementary formulations, in addition, have been proposed such as whitlockite; these salts were found together with other organic compounds, sugars and adenine nucleotides (83, 84), explaining why ATP was required for the precipitation process (81).
Mitochondrial Ca-P precipitates can accumulate Ca2+ in amounts equivalent to 1 M (16) and can be dissolved upon matrix acidification (31). Thus, Ca-P precipitates can accumulate large amounts of Ca2+ in the mitochondria and can be regulated by physiological alterations in the matrix pH (36). The former provides the explanation for the paradox behind the apparent contradiction in the levels of Ca2+ and Na+ transport by NCLX, since whereas Ca2+ transport in mitochondria reaches levels around μM/s, Na+ transport rates range at mM/s (5, 41, 64). As recently reviewed, Ca2+ can be efficiently buffered within mitochondria and the measurement of its soluble fraction would not reflect actual changes in mitochondrial Ca2+ levels.
On the other hand, Na+ measurements may reflect real NCLX transport rates, which could be higher than expected (4). However, as the interest on mitochondrial Na+ is an emerging field, there are limited tools and probes to reliably measure mitochondrial Na+ in situ. Hopefully in the future, new probes with higher Na+ affinities will be generated and mitochondrial Na+ probes, such as CoroNa Red, will return on stock.
Given the limitations for its measurement in situ, Na+ concentration in the different cellular compartments has only been measured a few times. Whereas K+ concentrations in and out of mitochondria are very similar (i.e., ∼100 mM), Na+ concentrations vary (6, 62). Na+ concentration in the cytosol normally varies within the range of 8–10 mM in resting conditions, whereas mitochondrial Na+ content normally lies between 2 and 6 mM (43, 62). This difference is probably due to the highly active NHE in contrast to the low-activity K+/H+ exchanger (KHE) in the mitochondria (6, 25, 60). However, this will only be confirmed after the molecular identification of the NHE and development of new, fluorescent, mitochondria-targeted Na+ probes.
Na+ Regulates Mitochondrial ROS Production in Several Modes
In the last few years, Na+ has gained importance regarding mitochondrial (patho)physiology. Extracellular Na+ increases or alterations in plasma membrane Na+ currents and exchangers can modify mitochondrial activity. For instance, it has been recently found that moderate Na+ increment in the extracellular milieu lowers OXPHOS and alters cell activation and improves bactericidal function in M1-like macrophages and diminishes cell migration in M2-like macrophages (28).
In addition, chronic intracellular Na+ overload through partial inhibition of the plasma membrane Na+/K+ ATPase is linked to a metabolic shift, cardiac remodeling, and heart failure in mice. Inhibition of NCLX is enough to ameliorate metabolic changes (2). These studies highlight the importance of Na+ in modulating mitochondrial physiology and that the main Na+ entry pathway in mitochondria can regulate these processes.
In this line, store-operated Ca2+ entry (SOCE) has been shown to be accompanied by an increment in cytosolic Na+ levels, which in turn activate NCLX and mitochondrial redox signaling (64). Thus, the discovery of NCLX (66) has allowed the study of mitochondrial Na+ homeostasis, its molecular targets inside the mitochondria, and its roles in physiology and disease. Based on recent findings, Na+ can modify redox signaling in four ways, three of them being mutually exclusive.
First, extremely high levels of mitochondrial Ca2+ cause ROS production by mitochondria, which can be found after long-term deletion of NCLX, and extramitochondrial Na+ can clear such excess (Fig. 3). Ca2+ is a pleiotropic second messenger and its action in mitochondrial physiology mostly depends on its matrix concentration. Moderate increases in mitochondrial Ca2+ can enhance tricarboxylic acid (TCA) cycle dehydrogenases and boost OXPHOS (20), a process often necessary to match energy demand and expenditure (48, 51).
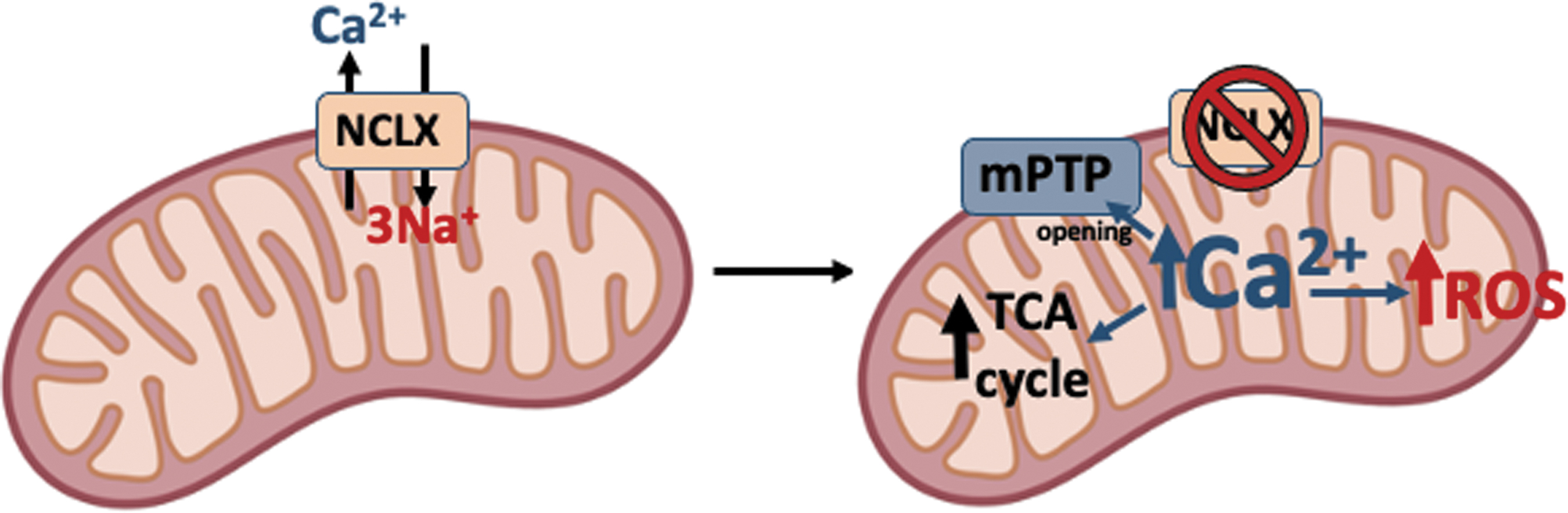
However, extreme Ca2+ overload can promote the opening of the mPTP, mitochondrial swelling, cyt c loss, OXPHOS uncoupling, and excessive ROS production, together with apoptotic cell death (6, 68). Thus, as NCLX is the major mitochondrial Ca2+ exit pathway (58), Na+ is able to regulate Ca2+ signaling and cell fate. Indeed, mouse hearts without NCLX showed remarkably high mitochondrial Ca2+ levels and ROS production and were much more susceptible to cell death upon ischemia–reperfusion than their wild-type counterparts (54).
In accordance, the same study revealed that mouse hearts overexpressing NCLX had lower mitochondrial Ca2+ levels and normal ROS production and were protected against ischemia–reperfusion injury. This concept was found in parallel by Sekler's group who discovered that the presence of NCLX in human cells promoted lowered mitochondrial Ca2+ and lowered ROS production. However, in the absence of NCLX, high ROS were responsible for Cys195 oxidation in Orai1, the plasma membrane Ca2+ entry channel during SOCE, which is the major Ca2+ influx route in nonexcitable cells (64).
In the same line, adrenergic stimulation of brown adipose tissue in NCLX-null mice induced profound mitochondrial Ca2+ overload, mitochondrial swelling, cyt c release, and apoptotic cell detachment, which are events associated with mitochondrial ROS production (3). In addition, mitochondrial Ca2+ overload contributes to Alzheimer's disease (AD) progression not only by activating mitochondrial dehydrogenases and augmenting cellular bioenergetics but also driving maladaptive alterations, leading to superoxide generation, mitochondrial dysfunction, and AD progression (42). All these studies point to the fact that mitochondrial Na+ entry through NCLX can ameliorate mitochondrial ROS production in situations of Ca2+ overload (Fig. 3).
Second, mitochondrial Na+ entry can induce ROS production through its interaction with phospholipids on the inner side of the IMM (Fig. 4). In the nineties, it was shown that hypoxia promotes mitochondrial ROS production and that it was a necessary event for hypoxic adaptation through the hypoxia inducible factor 1 (HIF-1) (17), a finding discussed for decades (11, 33, 34, 39, 56, 70).
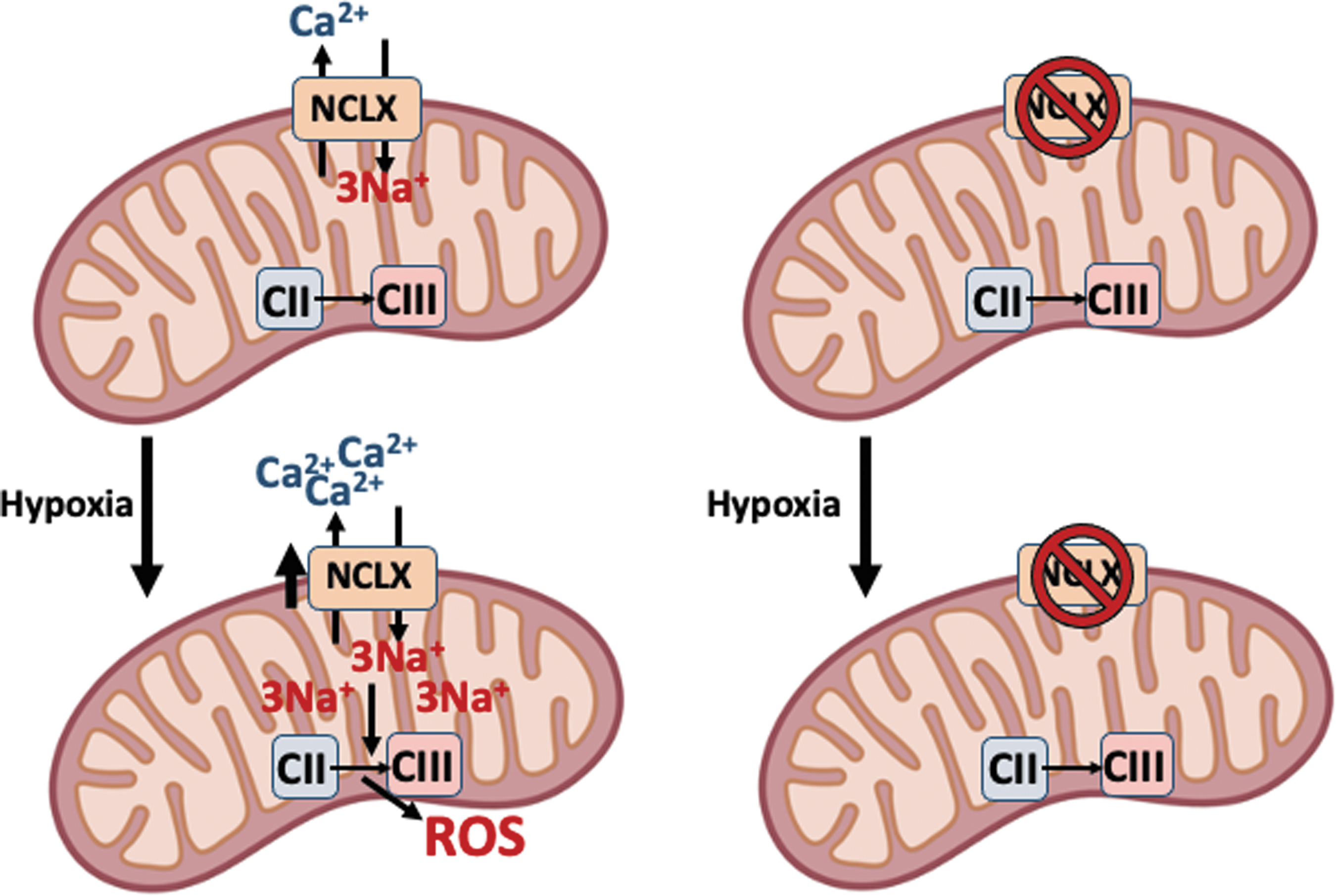
The mechanism behind the paradoxical ROS increase during hypoxia has not been solved until recently. CI undergoes active/deactive (A/D) transition by which the enzyme becomes inactivated and stops pumping H+. This slightly acidifies the mitochondrial matrix and partially dissolves the Ca-P precipitates, and the concomitant small increase in soluble Ca2+ activates NCLX and promotes Na+ entry. Na+ in the matrix interacts with the headgroups of phospholipids in the inner leaflet of the IMM, which highlights the importance of membrane asymmetry (see section: The Unexpected Interaction of Na+ with Mitochondrial Phospholipids, a Posible Role in other Membranes), forming 1 Na+:3 phospholipid clusters that reduce its fluidity and lower the CoQ transfer between CII and CIII, but not inside supercomplexes.
The decreased CoQ transfer uncouples the Q cycle in CIII and therefore the superoxide anion is produced at the level of the CIII Qo site (36, 38, 40). ROS production during hypoxia is necessary for metabolic adaptation through HIFs (34). In accordance with the relationship of NCLX with metabolic reprogramming, it has recently been found that intracellular Na+ elevation requires NCLX activity for cardiac metabolism rewiring through a switch from fatty acid to carbohydrate metabolism (2).
These findings provide evidence that mitochondrial Na+ can promote mitochondrial ROS production directly by interacting with phospholipids in the IMM and regulate cellular metabolism.
In line with this new role of Na+ in mitochondrial ROS production, it is known that elevated Na+ levels in failing cardiac myocytes can lower cellular antioxidant capacity. Increased cytosolic Na+ levels lower steady-state Ca2+ concentration in the mitochondria through the activity of NCLX. Decreased mitochondrial Ca2+ levels reduce Ca2+-dependent activation of the TCA cycle dehydrogenases and NADH generation.
This, in turn, impacts the regeneration of one of the most important antioxidant buffers in the cell, NADPH, which causes the levels of ROS to increase and produce oxidative stress (7, 47, 51, 52, 55). This pathway reflects the relationship between cation and redox homeostasis as changing levels of cytosolic Na+ and mitochondrial Ca2+ can impact redox balance also by diminishing the antioxidant capacity of the cell (Fig. 5).

Fourth, as mitochondrial Na+ reduces IMM fluidity, the CoQ transfer rate is diminished, and this may also apply in the transfer of CoQH2 from CII to CI. Reverse electron transfer (RET) is a mechanism whereby CI functions in its reverse reaction, accepting electrons from CoQH2 and generating superoxide anions. The high levels of CoQH2 necessary for RET are mainly produced by CII, although other FAD-containing enzymes in the IMM may also contribute (38).
Thus, increases in mitochondrial Na+ would reduce IMM fluidity and lower superoxide production by RET as COQH2 transfer from CII to CI would be hindered (Fig. 6). In this way, mitochondrial Na+ would be able to regulate the source of mitochondrial superoxide by the mETC as its increase would both promote CIII-produced ROS (i.e., hypoxic ROS) and lower CI-produced ROS (i.e., RET). On the contrary, lower mitochondrial Na+ levels would reduce CIII-derived ROS to a minimum and would exacerbate CI-derived RET.

According to this hypothesis, it is possible that NCLX knockout hearts displayed higher levels of ROS upon reperfusion due to the absence of Na+ interacting with IMM phospholipids, among other factors such as Ca2+ overload (see section: Na+ Regulates Mitochondrial ROS Production in Several Modes). On the contrary, NCLX-overexpressing hearts showed lower levels of ROS, which may be associated with an increased entrance of Na+ into the mitochondria during ischemia (54). Still, this tempting hypothesis needs to be specifically tested.
Altogether, the present discussion puts forward a disjunctive perspective on mitochondrial ROS production related to cation homeostasis. Mitochondrial ROS production occurring during Ca2+ overload may be considered the unspecific or toxic, and the ROS directly modulated by mitochondrial Na+ increase (i.e., hypoxic ROS and RET) may be considered signaling ROS. The former would be associated with the ability of Ca2+ to interact with many proteins and substrates, and any excess would flow into an imbalance in the targets' response and mitochondrial ROS production through metabolic instability, causing mPTP opening and cell death.
The other two would be viewed as the ROS involved in signaling (and adaptation) given the specificity of its origin and the molecular pathway leading to it. Nevertheless, under certain situations, adaptation through signaling ROS may provoke pathology-like phenotypes in specialized tissues and organs. For instance, this may happen in Parkinson's disease as the ROS-dependent glycolytic switch through HIF-1 in PINK1-deficient neurons leads to an unsatisfied energy demand, metabolic collapse, and cell death (71).
In addition, RET is not only used as a signaling mechanism for ROS production during macrophage metabolic reprogramming (59) but it is also the main mechanism leading to oxidative damage during ischemia–reperfusion (18). Under the risk of being simplistic, this view may help to elucidate the molecular mechanism underlying pathologies in which Na+, Ca2+, and ROS are involved.
The Unexpected Interaction of Na+ with Mitochondrial Phospholipids, a Possible Role in Other Membranes
The mechanism of mitochondrial redox signaling during hypoxia involved a remarkably unexpected interaction of Na+ with phospholipids in the inner leaflet of the IMM (36). Interestingly, this interaction allowed regulation of IMM fluidity due to two important aspects in mitochondrial and membrane biology: (i) the differential Na+ concentrations in and out of the mitochondria, which are between 2–6 mM in and 8–10 mM out (43, 62); and (ii) the asymmetry in the composition of phospholipids in biological membranes, including the IMM (49, 85).
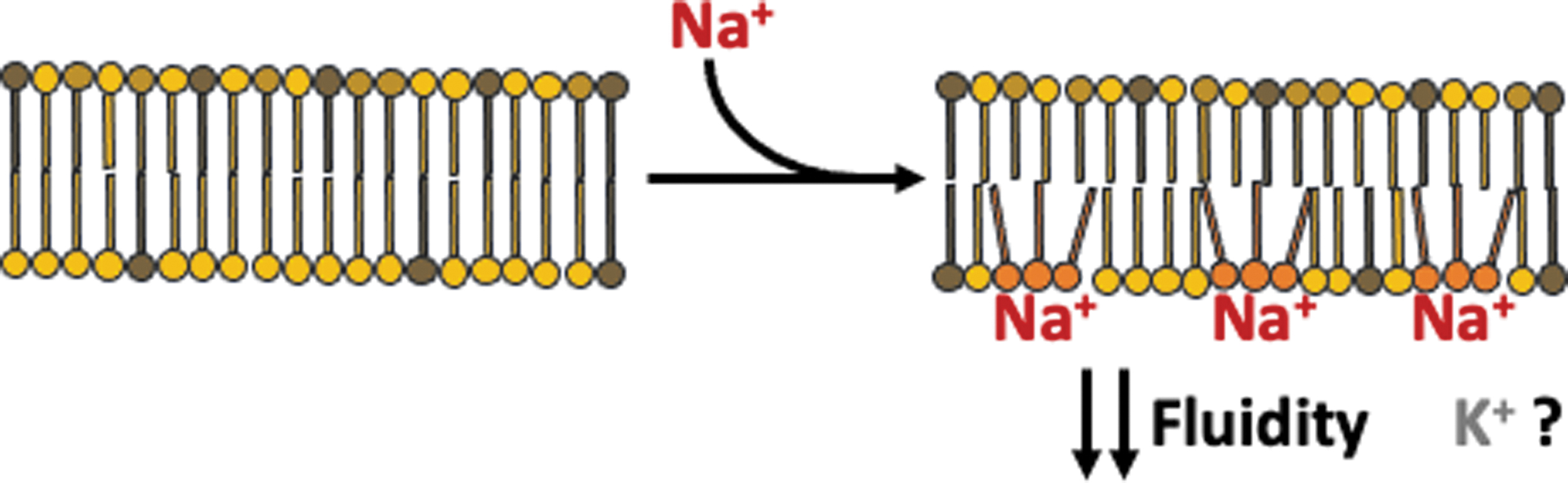
These features allow rather small changes in mitochondrial Na+ concentration to be sensed specifically by the inner leaflet of IMM. Electron spin resonance spectroscopy indicated that Na+ interacts specifically with the phospholipid headgroup, and Fourier transform infrared spectroscopy further showed that this interaction occurs specifically at the level of the carbonyl group. Given the atomic structure of carbonyls, in which the oxygen atom provides two lone electron pairs in sp2 orbitals, the interaction with Na+ will most probably involve them.
Such an electrostatic interaction promotes the formation of Na+:phospholipid clusters with a 1:3 stoichiometry, as measured by inductively coupled plasma mass spectroscopy. However, given that Na+ is typically six coordinated, although five and four coordination also exists, the Na+:phospholipid cluster must involve water molecules as well. Indeed, molecular dynamic simulations revealed that Na+ in the cluster was also coordinated with 1–2 water molecules (9), yielding a total coordination number of 4–5.
This larger new complex formed in the bilayer may display a peculiar geometry compared with the phospholipids before Na+ interaction, such a geometry would ultimately lie behind the observed reduced mobility of phospholipids in Na+:phospholipid clusters (Fig. 7).
Reduced mobility of phospholipids due to Na+ interaction produced a decrease in IMM fluidity and CoQ transfer between CII and CIII, but not between CI and CIII. This observation could be easily explained by the fact that CI is mostly found within supercomplexes in which CoQ channeling is enabled and thus slight alterations in membrane fluidity do not have such a profound effect as if both complexes were separated or had the same phospholipid composition as the rest of the membrane [see Ref. (37) for further discussion].
In this regard, it is important to note that CoQ is not only present in the IMM but also in most cellular membranes and that protein clusters in membranes, particularly of those involving CoQ and redox reactions, may form to enhance reaction efficiency in the presence of the “disturbing” agent, Na+. Indeed, NADH-cytochrome b5 reductase, a known constituent of lipid rafts, is a redox enzyme that uses CoQ as a cofactor (63, 74). NAD(P)H:quinone oxidoreductase-1, a wide range reductase in the plasma membrane, has also been found to be enriched in lipid rafts (77).
Thus, the presence of redox enzymes in protein clusters may serve to ensure efficient catalysis, particularly when mobile electron carriers are involved, in the presence of Na+, a cation normally found at 5–10 mM in the cytosol. This proposition makes possible that different concentrations of Na+ in various cellular compartments may impact cellular adaptations toward stress and, eventually, development of diseases.
However, we do not currently know the exact concentrations of Na+ in different cellular compartments, such as endoplasmic reticulum and intracellular vesicles, and thus we can only speculate on the role of Na+ in compartments other than the cytosol and mitochondria.
Another relevant aspect is the potential interaction of other monovalent cations with phospholipids and their characteristics, such as strength, stoichiometry, and geometry. It is well known that cytosolic and mitochondrial K+ concentrations are maintained remarkably higher than Na+. Isothermal titration calorimetry has revealed that Na+ binding to the phospholipid bilayer surface was much more than K+ (46) and molecular dynamic simulations of asymmetric membranes confirmed it (80).
It is possible that the weaker interaction of K+ with bilayer phospholipids may be due to the larger size of the K+ ion, compared with Na+ (32). Thus, regulation of Na+ and K+ levels may influence redox homeostasis by altering the probability of their interaction with phospholipids.
On the other hand, as Na+ and K+ levels are also modified by ROS production through regulation of cation transporters, as during hypoxia in hearts (10, 69), redox balance may counterpoise the excessive ROS production by modulating Na+ and K+ levels in the cell. In addition, another layer of complexity arises from the composition of the bilayer as the strength of cation:phospholipid interaction is largely determined by the phospholipid mixture (32, 36).
Concluding Remarks
Mitochondrial Na+ signaling has emerged as a novel field providing an unexpected layer of complexity to bioenergetics and redox homeostasis. Importantly, mitochondrial Na+ may be able to regulate diverse mechanisms of ROS production by different means, which will provide versatility to the researcher and new venues of investigation. The interaction of monovalent cations with phospholipids may lie behind the formation of protein clusters in membranes, as happens with CI-containing supercomplexes in mitochondria, to augment electron transfer efficiency and minimize ROS production.
Thus, tight regulation of Na+ in the cell and in mitochondria poses as a remarkably important feature for mitochondrial homeostasis, bioenergetics, and redox signaling.
Footnotes
Acknowledgments
The authors thank the whole GENOXPHOS group for suggestions and discussions. Figures were created using
Authors' Contributions
Conceptualization and writing was done by P.H.-A. and J.A.E. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by MINECO: SAF2015-65633-R, RTI2018-099357-B-I00, HFSP (RGP0016/2018), and CIBER (CB16/10/00282). The CNIC is supported by the Instituto de Salud Carlos III (ISCIII), the Ministerio de Ciencia, Innovación y Universidades (MCNU), and the Pro CNIC Foundation and is a Severo Ochoa Center of Excellence (SEV-2015-0505). This research has been financed by Spanish Government grants (ISCIII and AEI agencies, partially funded by the European Union FEDER/ERDF).