
Abstract
Significance:
How mechanical forces and biochemical cues are coupled remains a miracle for many biological processes. Integrins, well-known adhesion receptors, sense changes in mechanical forces and reduction-oxidation reactions (redox) in their environment to mediate their adhesive function. The coupling of mechanical and redox function is a new area of investigation. Disturbance of normal mechanical forces and the redox balance occurs in thromboinflammatory conditions; atherosclerotic plaques create changes to the mechanical forces in the circulation. Diabetes induces redox changes in the circulation by the production of reactive oxygen species and vascular inflammation.
Recent Advances:
Integrins sense changes in the blood flow shear stress at the level of focal adhesions and respond to flow and traction forces by increased signaling. Talin, the integrin-actin linker, is a traction force sensor and adaptor. Oxidation and reduction of integrin disulfide bonds regulate their adhesion. A conserved disulfide bond in integrin αlpha IIb beta 3 (αIIbβ3) is directly reduced by the thiol oxidoreductase endoplasmic reticulum protein 5 (ERp5) under shear stress.
Critical Issues:
The coordination of mechano-redox events between the extracellular and intracellular compartments is an active area of investigation. Another fundamental issue is to determine the spatiotemporal arrangement of key regulators of integrins' mechanical and redox interactions. How thromboinflammatory conditions lead to mechanoredox uncoupling is relatively unexplored.
Future Directions:
Integrated approaches, involving disulfide bond biochemistry, microfluidic assays, and dynamic force spectroscopy, will aid in showing that cell adhesion constitutes a crossroad of mechano- and redox biology, within the same molecule, the integrin. Antioxid. Redox Signal. 37, 1072–1093.
Introduction
Cellular interactions develop in the presence of mechanical forces and reducing or oxidative molecules (redox mediators) in their immediate environments. In the circulation, cells are exposed to mechanical forces, such as the “haemodynamic drag” from the blood flow and traction from attachment to extracellular matrices and surrounding cells, and to redox active molecules, such as reactive oxygen species (ROS) and thiol oxidoreductases. Transient changes in these components cause changes in receptor–ligand interactions. One of the most studied groups of mechano-sensitive and redox-sensitive receptors is the group of integrins.
Integrins are widespread cell surface glycoproteins, consisting of an α and β subunit, that control cell–cell and cell–matrix adhesion. The transition of integrins from their resting to activated state increases the affinity for their ligands. This is a mechanical event, as it requires a “swing-out” motion of the β relative to the α subunit (23, 24) (Fig. 1A). Activation of integrins is via two signaling pathways: inside-out and outside-in signaling (Fig. 1B).

For example, activation of platelets by adenosine diphosphate (ADP) (16), collagen (86), or thrombin initiates “inside-out signaling” pathways that converge on the binding of talin to the membrane proximal region of αlpha IIb beta 3 (αIIbβ3), unclasping of the α and β subunits and opening to its activated state (16). Talin couples αIIbβ3 with the actin cytoskeleton, which permits subsequent integrin clustering, the formation of focal adhesion points, and binding to fibrinogen (1, 58, 62). The binding of fibrinogen to αIIbβ3 stimulates “outside-in signaling,” which involves actin polymerization, platelet spreading, and clot retraction (107, 136).
Increased binding of integrins to their cognate ligands is found in conditions of perturbed mechano-redox balance such as vascular thrombosis and inflammation, termed thromboinflammation (Fig. 1C–I). Vascular thrombi cause mechanical disruption of the blood flow, whereas inflammation promotes the production of oxidative molecules in the vasculature.
Thrombus formation involves the accumulation of platelets and fibrin on the endothelial surface (105). Platelet integrin αIIbβ3 binds to fibrinogen, facilitating platelet/platelet and platelet/neutrophil bridging (Fig. 1C). Other platelet integrins involved in thrombus formation include integrins alpha 2 beta 1 (α2β1) (Fig. 1D) and alpha 6 beta 1 (α6β1), which bind to collagen and laminin respectively.
Endothelial integrins include the vitronectin receptors, alpha v beta 3 (αvβ3) and alpha v beta 5 (αvβ5), fibronectin receptors, alpha 4 beta 1 (α4β1), and alpha 5 beta 1 (α5β1) (Fig. 1E); the collagen receptors, alpha 1 beta 1 (α1β1), α2β1; the laminin receptors, alpha 3 beta 1 (α3β1), α6β1, and alpha 6 beta 4 (α6β4); and the osteopontin receptor, alpha 9 beta 1 (α9β1) (3, 4). The binding of the endothelial integrins to circulating or immobilized ligands creates the matrix for the capture of platelets during thrombosis.
During vascular inflammation, leukocytes (neutrophils and monocytes) are recruited to sites of tissue injury or infection to initiate wound repair or pathogen destruction. Leukocytes utilize selectin ligands and integrins to coordinate their recruitment. Leukocyte integrin alpha L beta 2 (αLβ2)-integrin (lymphocyte function-associated antigen 1 [LFA-1] or CD11a/CD18) is expressed by neutrophils, monocytes, and lymphocytes, whereas alpha M beta 2 (αMβ2)-integrin (macrophage-1 antigen [Mac-1] or complement receptor 3 [CR3] or CD11b/CD18) is found mainly on neutrophils (Fig. 1F, G) and monocytes (Fig. 1H) (33, 34, 104).
Other integrins include α4β1 (very late antigen [VLA]-4; CD49d/CD29) on monocytes; T lymphocytes, alpha 4 beta 7 (α4β7), αvβ3, and alpha X beta 2 (αXβ2) on macrophages (Fig. 1I); and alpha D beta 2 (αDβ2) on foam cells (lipid-laden macrophages found in atherosclerotic lesions) (113). During vascular inflammation, endothelial cells release chemokines, which activate integrins αMβ2 and αLβ2 to engage with the endothelial ligands: intercellular adhesion molecule 1 (ICAM-1), intercellular adhesion molecule 2 (ICAM-2), and the receptor for advanced glycation end products (RAGE) (103).
Integrins participate in the crosstalk of thrombosis and inflammation (44, 140). For example, leukocyte integrin αMβ2 binds to platelet receptor glycoprotein Ib alpha (GPIbα), promoting the formation of leukocyte-platelet aggregates. αMβ2 also binds to fibrinogen and fibrin, which are ligands of platelet integrin αIIbβ3. The αIIbβ3 integrin can directly interact with the choline transporter-like protein 2 (SLC44A2) on neutrophils, enhancing the release of neutrophil extracellular traps (30).
The simultaneous activation of thrombosis and inflammation is evident in disease pathologies such as atherosclerosis and immune-mediated thrombosis (i.e., the antiphospholipid syndrome) (108, 144) or more recently coronavirus disease (COVID19) (135, 154). These thromboinflammatory conditions can alter the mechanical and redox milieu.
In the following section, we will present our current understanding of mechanical and redox regulation of platelet, endothelial, and leukocyte integrins, separately, and the emerging evidence of mechano-redox coupling for integrin function.
Overview of Vascular Integrins
The primary function of vascular integrins is to mediate the adhesion of cells to cells and extracellular matrix proteins under conditions of blood flow. Integrins exist in different affinity states, resting, intermediate, and activated (16) with resting state having the least affinity for ligand and activated state having the highest affinity for ligand (Fig. 1A). The high affinity state is achieved following inside-out signaling after the cell is activated (i.e., by thrombin for platelets, by cytokines for leukocytes) (Fig. 1B).
Platelet integrins
The platelet-specific integrin αIIbβ3 integrin is the most abundant receptor on the platelet receptor (80,000 copies) (Fig. 1C). Patients with absent or defective αIIbβ3 on the surface of platelets (Glanzmann's thrombasthenia) develop life-threatening bleeding from defective binding to fibrinogen and fibrin under shear (45, 133).
Integrin α2β1 is a receptor for collagen on platelets and endothelial cells (Fig. 1D). On injury of the endothelial surface, basement membrane collagen fibers are exposed and attract platelet binding via their α2β1 integrin and glycoprotein VI (GPVI) receptor. Polymorphisms in the ITGA2, the gene encoding the α2 subunit of the integrin, or altered surface expression of α2β1 are linked to thrombotic or bleeding tendencies (81, 82).
Endothelial integrins
Endothelial cells line the vessel wall interior and are 10 times larger in length (20 μm) to platelets (2 μm), providing a surface for interaction with platelets and neutrophils (diameter 10 μm). Endothelial integrin α2β1 binds to collagen and laminin in the extracellular matrix, and αvβ3 binds to vitronectin. Integrin αvβ3 is highly expressed on activated endothelial cells but is not present in resting endothelial cells (4). Under hyperglycemic conditions, signaling of the αvβ3 integrin supports a pro-atherosclerotic phenotype (97).
Endothelial integrin α5β1 is the main receptor for the extracellular matrix protein fibronectin (Fig. 1E). Fibronectin increases under inflammatory conditions such as atherosclerosis. During atherosclerosis, α5β1 mediates increased fibronectin deposition in the fibrous caps of unstable atherosclerotic plaques (3). Oxidized low-density lipoprotein augments α5β1 integrin signaling in endothelial cells to a pro-atherosclerotic phenotype (170).
Leukocyte integrins
Leukocytes (neutrophils and monocytes) express αMβ2 and aLb2 integrins. αMβ2 is a multi-faceted receptor, with ligands including fibrinogen (34, 160), ICAM-1 (34), and GP1bα (120, 138, 159) (Fig. 1F). Patients with loss-of-function mutations in the ITGB2, encoding the β2 subunit, develop a debilitating condition known as leukocyte adhesion deficiency type I. Patients with this condition present with recurrent bacterial infections (153).
Integrins αMβ2 and αLb2 mediate tethering, rolling, and firm adhesion of neutrophils and monocytes on inflamed endothelium (33, 39, 140, 155) (Fig. 1G). These processes occur sequentially in a manner dependent on inside-out signaling. First, selectins on the surface of platelets (P-selectin), endothelial cells (P-selectin and E-selectin), or other adherent leukocytes (L-selectin) provide a point of initial weak attachment, or tethering, via the selectin counterreceptor P-selectin glycoprotein ligand-1 (PSGL-1).
The binding of selectins to PSGL-1 results in a signaling cascade that shifts αMβ2 to an intermediate affinity state that permits slow rolling of the cell without firm adhesion. Signaling from PSGL-1 and cytokines, chemokines, or other damage/pathogen-associated molecular patterns results in complete activation of αMβ2 (139, 165), allowing firm adhesion. Integrins αMβ2 and αLb2 play an important role in the development of atherosclerosis; studies with β2 knockout mice have shown that β2 integrins accelerate atherosclerosis in response to chronic dyslipidemia (6).
Integrins αXβ2 and α4β1 promote monocyte homing to endothelium and monocyte migration into the tissue, where they differentiate into macrophages (Fig. 1H). Hypercholesterolemia increases the expression of integrin αXβ2 on circulating monocytes in humans and mice. Signaling through αXβ2 in monocytes promotes α4β1 activation (43).
Integrins expressed on macrophages are important for retention of macrophages within the atherosclerotic tissue, including the collagen receptor α1β1, the laminin receptors α3β1 and α6β1, and the fibronectin-binding integrins α4β1, α5β1, and αvβ3, whereas αvβ3 also binds to osteopontin (Fig. 1I). Of interest, macrophages significantly upregulate fibronectin-binding integrin αDβ2 during macrophage foam cell formation (43).
To summarize, platelets, endothelial cells, and leukocytes express a multitude of integrins that bind to their respective ligands under shear conditions in the circulation. In many disease states, mechanical forces in the circulation can be dramatically different compared with the healthy state. For instance, atherosclerotic plaques alter the lumen diameter of blood vessels, creating a region of increased mechanical force at the apex of the plaque (98). Integrins have developed to sense and respond to these changes in mechanical force.
Mechanical Regulation of Vascular Integrins
Vascular mechanical forces
Mechanical forces are integral to protein–protein interaction in the vasculature. Spatiotemporal aspects of protein–protein interactions such as bond-lifetimes, binding site exposure, and signal transduction are regulated by mechanical force (23, 101, 177). Within the vasculature, blood flow is one of the main determinants of the mechanical force experienced by integrins.
Blood flowing within an enclosed system, such as blood vessels, develops a force parameter named shear. Shear is generally reported in the literature through two values: shear rate and shear stress (172). At the center of a vessel, blood flow velocity is fastest due to unobstructed flow. In contrast, the velocity of blood is slowest at the vessel wall due to friction forces (Fig. 2A). Shear rate is defined as the velocity gradient away from the center of blood flow and is expressed in terms of inverse seconds (s−1).

Shear rate is determined by the diameter of vessels. High shear rate develops when blood flow is fast and the vessel diameter is small (arteries); low shear rate develops when blood flow is slow and the vessel has a large diameter (veins). Shear stress is directly related to shear rate; shear stress is the product of the viscosity of the fluid and the shear rate and is typically expressed as dynes/cm2. In turn, blood viscosity is determined by plasma viscosity, hematocrit, and erythrocyte deformability (Fig. 2B) (22).
Intermolecular interactions between proteins under shear are usually described as one of three types of bonds; catch, ideal, or slip bonds (22, 23). Bonds, whereby affinity counterintuitively increases as force increases, are described as catch bonds. Examples of ligands that undergo catch bonds include selectins, collagen, and von Willebrand factor (vWF). Catch bonds occur below a certain shear rate threshold. When proteins are exposed to a larger force beyond this, the affinity for ligand decreases and is referred to as slip bonds. Interactions that are unaffected by force are called ideal bonds (Fig. 2C).
Mechanical regulation of platelet integrins
Shear plays a direct role in αIIbβ3 integrin function by modulating the binding of ligands and subsequent outside-in signaling. The αIIbβ3 integrin binds to fibrin(ogen) by a classical slip-bond; as shear rate increases, the interaction between αIIbβ3 and fibrin(ogen) is weakened (93, 131). Conversely, the binding of αIIbβ3 to vWF (131) follows catch-bond behavior; at low shear, the binding of vWF is weak. However, at higher levels of shear, the vWF binding site for the αIIbβ3 integrin becomes exposed, allowing firmer adhesion.
Catch bond behaviors have also been described for the binding of fibronectin and vWF to platelets (22, 23, 73). The importance of shear in regulating αIIbβ3 function is reflected in diabetes mellitus. Patients with diabetes have a prothrombotic tendency, which is partially due to platelet hyperreactivity (5). Diabetic platelets display increased αIIbβ3-dependent adhesion to fibrinogen compared with healthy platelets under the same levels of shear (74).
Indirect mechano-regulation of αIIbβ3 is achieved through vWF and collagen-dependent pathways. Capture of vWF by collagen, and subsequent unfolding of vWF under shear rates >1000 s−1, leads to exposure of the A1 binding site of vWF for platelet GPIbα (35, 73, 132). The binding of vWF to GPIbα induces a unique intermediate-affinity state of αIIbβ3 when compared with ADP-induced platelet activation (73, 90). GPVI, on the other hand, forms catch bonds with collagen under increasing shear rates. At low shear rates, GPVI mediates direct binding of platelets to collagen. As shear rate increases, the affinity of GPVI to collagen also increases, resulting in inside-out signaling, subsequent αIIbβ3 activation, and amplification of platelet aggregation (111, 130).
Intracellularly, talin binds to the cytoplasmic domain of integrin β-subunits to regulate integrin signaling. Talin is the linker of integrin to the cytoskeleton and is a force transducer. Talin rod domains unfold under stretching to maintain a physiological force range of only a few pNs in the integrin-talin force transmission pathway. Talin and integrin regulate the formation of focal adhesion complexes, which help cells sense their mechano-environment and transduce signals (19).
Mechanical regulation of endothelial integrins
The endothelial cell lining is exposed to shear from the blood's laminar flow. Laminar flow promotes endothelial cell alignment and the development of an anti-adhesive surface, whereas disturbed flows in vascular branches and curved regions are pro-atherogenic (137). Under shear stress, αvβ3 interacts dynamically with vitronectin, which generates downstream signaling events, such as Src homology and collagen homology (Shc) association and RhoA activation (137).
Mechanical regulation of leukocyte integrins
Similar to integrin αIIbβ3, αMβ2 is both directly and indirectly affected by shear. PSGL-1 undergoes a catch bond with selectins at shear rates between 50 and 150 s−1 (101), which are shear rates in veins and venules, respectively. The binding of PSGL-1 to selectins results in inside-out signaling, which, in turn, activates αMβ2 (56, 162). αMβ2 also develops a catch bond directly with its ligand ICAM-1, with maximum affinity at forces of 12 pN (127). Under venous shear rates, the binding of αLb2 and αMβ2 to ICAM-1 is markedly enhanced (22, 127). The binding of αMβ2 to complement fragment iC3b is also shear dependent (127). Together, these conditions provide an optimal mechanical milieu for the rolling and adhesion of circulating neutrophils to inflamed endothelial cells.
Redox Regulation of Integrins
Although there are multiple mediators of oxidation and reduction reactions in the circulation, this review will focus on the role of ROS and thiol oxidoreductases in the regulation of integrin functions.
Vascular ROS and regulation of integrin function
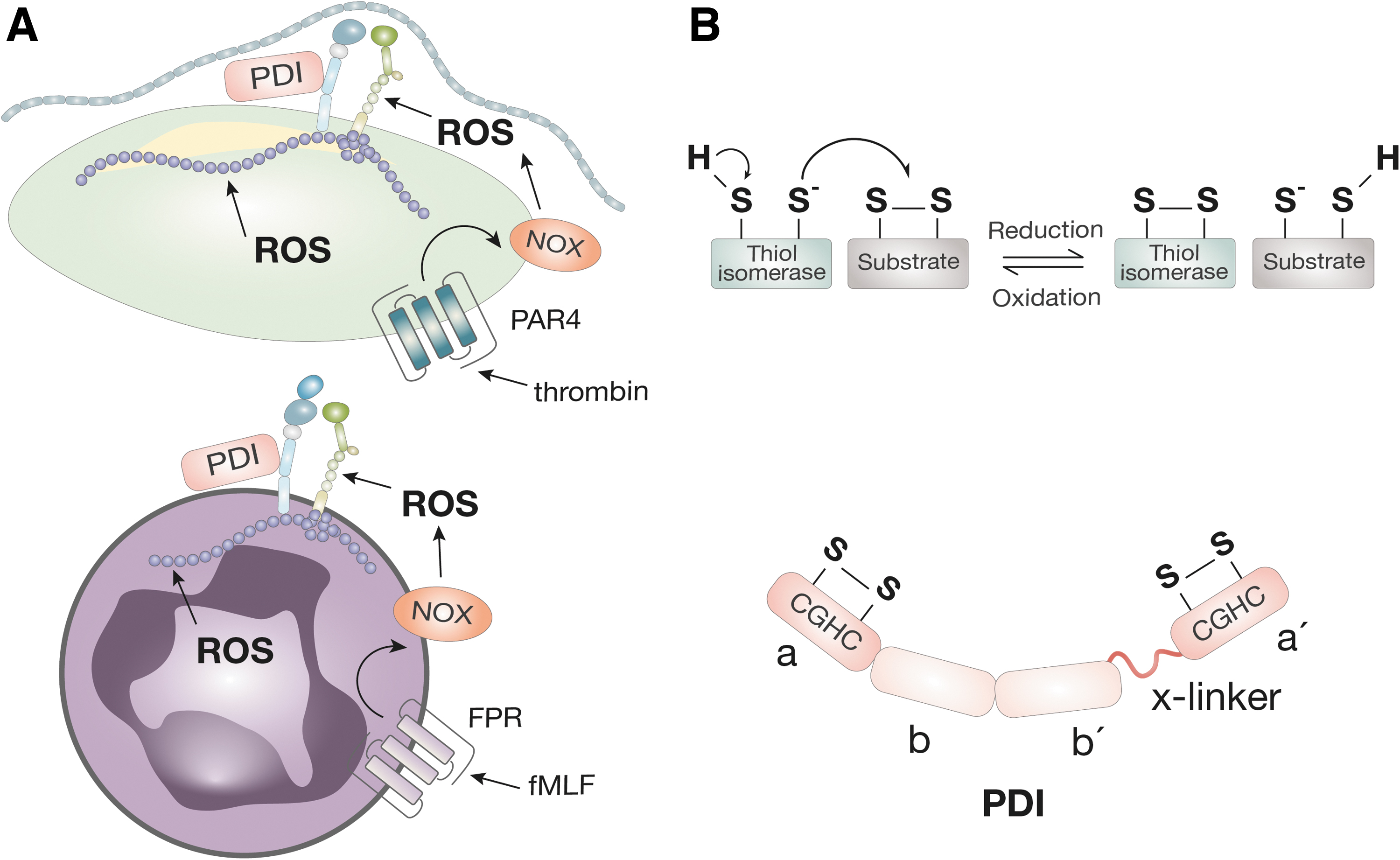
The ROS are key signaling molecules that play an important role in inflammation (50, 171). The ROS include superoxide anion (•O2 −), hydrogen peroxide (H2O2), hydroxyl radical (OH•), and hypochlorous acid (HOCl), which are constantly produced during normal metabolism and in response to external stimuli. H2O2 primarily oxidizes cysteine residues in proteins, such as protein tyrosine phosphatases (PEST), causing their inactivation. Inactivation of tyrosine phosphatases prolongs the signaling from tyrosine kinases. Thus, H2O2-mediated activation of tyrosine kinases (such as PEST and SH2 containing protein tyrosine phosphatase-2 [SHP2]) in integrins may enhance integrin signaling.
Neutrophils and monocytes produce a large amount of ROS controlled by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and myeloperoxidase (MPO) as part of the innate immune response. NOX is a multidomain complex including a Rho guanosine triphosphatase (GTPase), usually Ras-related C3 botulinum toxin substrate 1 (Rac1), and oxidase subunits, for example, p67phox. NOX catalyzes the transfer of an electron from NADPH to oxygen, producing NADP+ and superoxide free radical (•O2 −).
In response to invading pathogens, immune cells produce ROS to mediate pathogen destruction (114). In addition, pathogenic levels of ROS have also been known to drive inflammatory diseases such as atherosclerosis (87). The MPO-derived oxidants contribute to the formation of foam cells, endothelial dysfunction, and the expression of tissue factor that promote the development of vulnerable atherosclerotic plaque (146).
Cellular production of ROS can result from integrin-ligand engagement, which initiates cell signaling pathways. In platelets, the binding of αIIbβ3 to fibrin(ogen) under shear induces production of ROS by NOX2, amplifying in vivo thrombus formation (32, 163). In leukocytes, αMβ2 mediates the production of ROS on exposure to pathogens (8, 54, 171). Although integrin-dependent ROS production has been well characterized, there is some evidence that ROS reciprocally regulates integrin function.
Direct modification of β2 integrins by ROS has been shown to activate the β2 integrin; exposure of β2 integrin to H2O2 leads to the binding of mAb24, an antibody that binds to the activated conformation of β2 integrins (12) (Fig. 3A). The MPO-knockout neutrophils accumulate H2O2 and have increased αMβ2-dependent adhesion to endothelial cells (152). H2O2 generated by neutrophil NOX2 activates αMβ2, an effect reversible with the ROS scavenging enzyme catalase. Inhibition of NOX in platelets prevents the activation of αIIbβ3 (10). Moreover, the absence of NOX2 abrogates the formation of platelet–neutrophil aggregates and hepatic ischemia-reperfusion injury (77). These findings suggest a role for ROS in regulating integrin-dependent thromboinflammation.
In addition, ROS play an important role in the function of the actin cytoskeleton. In the absence of NOX in neutrophils, neutrophil chemotaxis is abrogated (63). The ROS produced by NOX directly modify the actin cytoskeleton (Fig. 3A) by mediating the glutathionylation of cysteines in monomeric actin, preventing polymerization, and inhibiting actin-dependent chemotaxis (129). This indirectly leads to the inhibition of β2 integrin focal adhesion formation and ICAM-1 binding (147, 148).
Redox role of thrombin
Thrombin binds to G protein receptors (protease-activated receptor 1 [PAR1] and protease-activated receptor 4 [PAR4]) on platelets to activate inside-out signaling of integrin αIIbβ3. It catalytically cleaves fibrinogen to form the fibrin clot. In addition, it exerts pro-inflammatory function, in part by increasing the activity of NOX (66).
Thrombin activates the guanine nucleotide exchange factor (GEF) p114RhoGEF, leading to the activation of Rac1 and promoting the membrane translocation of p67phox (112). Thrombin activation of NOX in vascular smooth muscle cells leads to the production of ROS and the expression of cell surface tissue factor (66). In platelets, thrombin activates the production of ROS via the thrombin receptor PAR4 (17).
Thrombin indirectly promotes ROS production by the activation of αIIbβ3. Recently, it was shown that the adhesion of αIIbβ3 to fibrinogen activated NOX activation (activation of the p47phox subunit) and ROS production, which was greatly enhanced by shear (163). Therefore, thrombin plays a pivotal role in the reciprocal redox regulation of thrombosis and inflammation via integrin activation by canonical G protein inside-out signaling and by activation of NOX.
Integrin engagement of ligand (outside-in signaling) is also important for redox regulation via the function of Rho-GTPases.
Redox regulation by Rho-GTPases
Integrin engagement of ligand initiates outside-in signaling, which activates Rho family GTPases. Rho-GTPases are mechano-sensors (100) that regulate the dynamic formation and disassembly of actin filament-based structures, including lamellipodia and filopodia (42). The activity of Rho GTPases is controlled by the reversible binding of guanosine-5′-triphosphate (GTP): When GTP-bound, Rho GTPases are active; when guanosine diphosphate (GDP)-bound, they are inactive. The GEFs catalyze the exchange of GDP with GTP to promote activation, whereas GTPase-activating proteins (GAPs) deactivate the Ras protein by stimulating hydrolysis of bound GTP to GDP.
There is bi-directional regulation of Rho GTPases by ROS. For example, the Rho GTPase Rac1 is part of the enzymatic complex of NOX in neutrophils, recruited to the cell membrane during neutrophil activation. Ligand binding to integrin results in the sequential activation of phosphatidylinositol 3-kinase (PI3K), which, in turn, generates lipid products that can activate GEFs. Activation of the GEF leads to increased RacNOX activity and production of ROS. Integrin engagement of ligand induces oxidative inactivation of focal adhesion kinase (FAK) phosphatase, promoting cell adhesion (25).
Conversely, ROS can directly modify the function of Rho GTPases; Rho GTPases contain redox-sensitive motifs, that is, Rac-1 contains the redox-sensitive cysteine Cys18, which can react with ROS to induce guanine nucleotide dissociation.
In summary, the production of ROS by NOX, thrombin, and RhoGTPases coordinates the redox control of integrin function. The ROS production can also be induced by thiol oxidoreductase protein disulfide isomerase (PDI) by the activation of NOX1 (41, 48, 53).
Vascular thiol oxidoreductases and regulation of integrin function
Thiol oxidoreductases are enzymes of the endoplasmic reticulum (ER) that catalyze the correct folding of native proteins. Thiol oxidoreductases contain cysteines in active site motifs Cys-X-X-Cys (where X is any amino acid) (CXXC) that allow disulfide bond reactions (Fig. 3B). Through reduction/oxidation, thiol oxidoreductases can reduce (cleave) or oxidize (form) disulfide bonds within other proteins, resulting in alterations in protein tertiary structure. Disulfide bonds are important structural features of integrins. However, disulfide bonds can also act as allosteric modulators of integrin function (118, 166, 167). It has been shown that the reduction of disulfide bonds in β1 integrins and in αIIbβ3 by dithiothreitol leads to activation of the integrin (26, 110). Integrins also display endogenous disulfide isomerase activity (156).
Although thiol oxidoreductases primarily localize within the ER, circulating and cell-surface expressed thiol oxidoreductases have been identified. Thiol oxidoreductases that are secreted extracellularly by platelets and endothelial cells are named vascular thiol oxidoreductases. Vascular thiol oxidoreductases include PDI, endoplasmic reticulum protein 57 (ERp57), endoplasmic reticulum protein 72 (ERp72), endoplasmic reticulum protein 5 (ERp5), and thioredoxin-related transmembrane protein 1 (TMX1).
Vascular thiol oxidoreductases interact with various plasma and cell surface proteins, that is, vitronectin (13) and tissue factor (84). However, integrins have, by far, been the most studied substrate of vascular thiol oxidoreductases, as summarized in Table 1.
Integrins Regulated by Thiol Oxidoreductases in Thrombosis/Inflammation
α2β1, alpha 2 beta 1; α5β1, alpha 5 beta 1; αIIbβ3, αlpha IIb beta 3; αMβ2, alpha M beta 2; αvβ3, alpha v beta 3; ApoA-IV, apolipoprotein A-IV; CD40L, CD40 ligand; ERp5, endoplasmic reticulum protein 5; ERp57, endoplasmic reticulum protein 57; ERp72, endoplasmic reticulum protein 72; GP1bα, glycoprotein Ib alpha; iC3b, complement fragment iC3b; ICAM-1, intercellular adhesion molecule 1; IGF-1, insulin-like growth factor-1; oxLDL, oxidized low-density lipoprotein; PDI, protein disulfide isomerase; RAGE, receptor for advanced glycation end products; TMX1, thioredoxin-related transmembrane protein 1; vWF, von Willebrand factor.
Regulation of platelet integrins by thiol oxidoreductases
PDI, ERp57, ERp5, and ERp72 have all been shown to bind and regulate αIIbβ3 integrin (28). Mice with PDI-null platelets have decreased platelet thrombus formation on vessel injury, without affecting fibrin formation or hemostasis (76). On the other hand, ERp57- and ERp72-null mouse platelets experienced a bleeding phenotype and decreased fibrin deposition on thrombi in vivo (174, 175).
On the contrary, TMX1 inhibits αIIbβ3 activation, platelet aggregation, and thrombus formation (173). Thus far, TMX1 has been the only thiol oxidoreductase displaying anti-thrombotic activity. ERp5 has the potential to display more than one functions in platelets, which may be determined by its redox status; the inhibition of ERp5 by an inhibitory anti-ERp5 antibody prevented thrombus formation and fibrin deposition in vivo (117). However, exogenously added, reduced ERp5 enabled detachment of αIIbβ3 from fibrinogen in vitro (116).
Platelet α2β1 is regulated by PDI; using PDI-inhibitory antibodies, platelet aggregation was inhibited in response to a collagen receptor peptide (82).
Regulation of endothelial integrins by thiol oxidoreductases
Immunofluorescent staining of PDI and αvβ3 integrin on endothelial cells reveals high colocalization of the two proteins (143). Small-molecule inhibitors of PDI inhibit vitronectin binding to αvβ3. An interaction between ERp57 and αvβ3 has also been identified; in muscle cells, increased ERp57 expression in injured muscle tissue promoted β3 integrin-dependent myoblast differentiation, supporting a role for ERp57 in myogenesis (158).
PDI can regulate β1 integrins; human embryonic kidney (HEK) cells and baby hamster kidney (BHK) cells, overexpressing redox active PDI, showed increased adhesion to fibronectin (125). In a rabbit iliac artery dilation model, inhibition of PDI was associated with a decrease in reduced β1 disulfide bonds and phosphorylation of FAK, which was associated with an increase in constrictive remodeling of the iliac artery (145).
Redox regulation of leukocyte integrin αMβ2
Hahm et al. showed that PDI increased free thiol content in αMβ2, supporting a direct reduction of disulfide bonds in αMβ2 (57). PDI promotes αMβ2-dependent neutrophil adhesion to inflamed endothelium and to fibrinogen (57). PDI reduces disulfide bonds Cys4-Cys17 and Cys209-Cys248 in the extracellular domain of platelet GPIba, which is associated with increased binding of vWF (88). The binding of αMβ2 to GP1bα potentiates thrombosis in mouse models of vessel injury (159) and thrombotic glomerulonephritis (67). The redox regulation of αMβ2 disulfides by PDI occurs under conditions of shear, introducing the concept of mechano-redox interaction in the control of integrin function.
Known Mechano-Redox Interactions
The effects of mechano-redox regulation on protein function are twofold: (i) by altering mechanical properties of proteins and (ii) by changing protein-ligand affinity.
For many proteins, tension generated from pulling forces is crucial to both maintain structural integrity of the protein and transduce mechanical forces along protein fibers. In both cases, disulfide bonds play a crucial role: Oxidized disulfide bonds improve protein tensile strength and maintain structure under force, whereas reduced disulfide bonds promote protein relaxation and decrease tensile strength (99). This mechanism of disulfide regulation has been shown to be important for the function of contractile proteins, such as titin, that require flexible disulfide bond redox states to transduce contractile forces during muscle contraction (38). Titin is the main functional protein that comprises the I-band of muscle sarcomeres and contributes to a large portion of muscle elasticity.
The demonstration of the mechano-redox control of disulfide bond reactions in titin was pioneered by Fernandez and group (161). The group employed single-molecule force-clamp spectroscopy to study the reductive activity of thioredoxin and PDI on titin, over a range of 25–600 pN. Force alone was unable to reduce titin's disulfide bonds; the rate of reduction decreased fourfold between 25 and 250 pN. (161). PDI oxidizes single disulfide bonds in the IgG domain of titin at 4.0 pN, generating a power of 6000 zW (39, 51).
Apart from PDI and thioredoxin, small thiol agents can enhance disulfide bond reduction under force. Using single-molecule dynamic force spectroscopy (DFS), it has been shown that under shear, poor nucleophilic organic thiols (such as
The DFS studies on artificial disulfide bonds in a protein revealed that changes occur in disulfide chemistry on protein unfolding induced by force, an effect that was recapitulated in molecular dynamics (78). Through in silico modeling of single-molecule disulfides, it has been proposed that increasing tensile force on a disulfide bond promotes its cleavage (7, 9). Other studies have also identified that force-dependent exposure of hidden disulfide bonds promotes disulfide-exchange reactions. Together, these findings reveal the importance of force in disulfide bond reduction.
Mechano-redox regulation of proteins is expected to play an important role in the hemodynamic and redox environment of our circulatory system. Highlighted next are the most prominent examples of proteins involved in mechanically regulated disulfide bond reactions in thrombosis and inflammation.
αIIbβ3
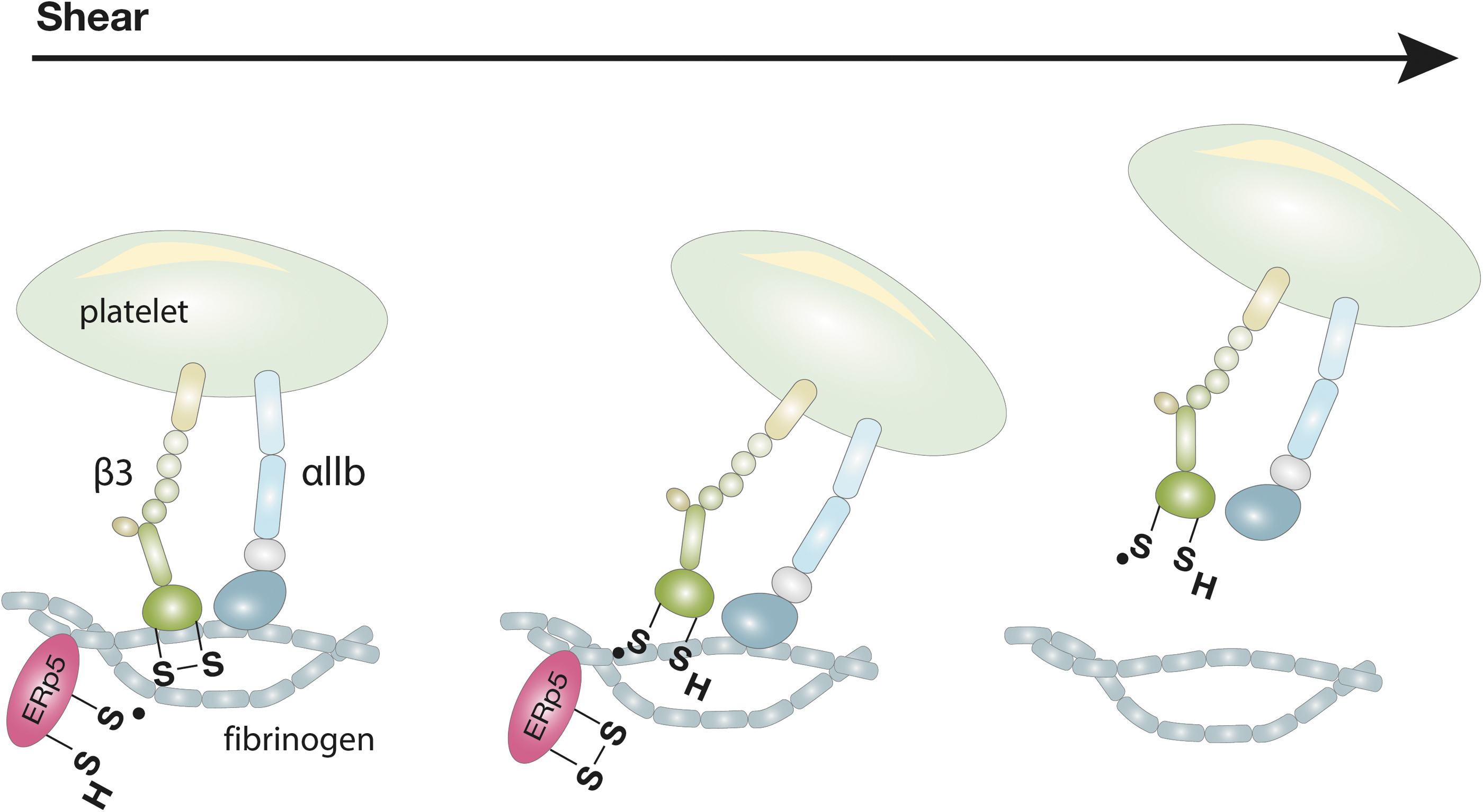
We have previously shown a direct mechano-redox regulation of αIIbβ3 integrin (116). ERp5 can reduce the disulfide bond Cys177-Cys184 within the β-propeller of the β3 subunit under shear. The Cys177-Cys184 disulfide bond is proximal to the fibrinogen binding pocket of activated αIIbβ3. The reduction of Cys177-Cys184 results in the release of fibrinogen. This results in increased detachment of platelets from fibrinogen under shear.
The application of force, by pulling fibrinogen away from αIIbβ3 with a biomembrane force probe (BFP), stretches the sulfur–sulfur bonds or changes the ligand-bound conformation of αIIbβ3 and increases its susceptibility to cleavage (Fig. 4) (118). Force-redox coupling enables platelet de-adhesion in the control of thrombus size.
von Willebrand factor
vWF is a large multimeric protein that circulates within the vasculature and is secreted by endothelial cells and platelets. vWF binds to platelets primarily through receptor GPIba and to αIIbβ3 (73, 131). The function of vWF is dependent on shear and redox regulation in a few ways: Increasing shear stress increases the release of ultra large (UL) vWF multimers from endothelial cells (46, 47). ULvWF, adhered to endothelial cells via P-selectin, experiences enhanced shear rates, compared with circulating vWF, causing vWF to unfold and expose the A1 domain for binding to platelet GPIba (59, 73). Shear also enables the enzymatic cleavage of vWF by the enzyme a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) (92) by unfolding vWF to reveal the scissile peptide bond (Tyr1605-Met1606) in the A2 domain.
In addition, ADAMTS13 has been shown to reduce disulfide bonds in vWF under shear (169). On the other hand, ROS inhibits ADAMT13 cleavage of vWF; neutrophil HOCl oxidized Tyr1605 and Met1606 of the cleavage site, preventing ADAMTS13-mediated cleavage (20). The formation of lateral disulfide bonds between Cys2431-Cys2453 of the C2 domain of vWF monomers supports multimerization (47), whereas PDI activity is required for dimerization of vWF in the ER (91). On the contrary, the reduction of disulfide bond Cys1669-Cys1670 within the A2 domain of vWF decreased shear-dependent adhesion of platelets to vWF (15).
Notably, a novel mechanism of vWF-force induced activation of αIIbβ3 has been described by Chen et al. Using BFP and microfluidics assays (see next in the Mass Spectrometry and Shear Systems to Quantify Redox Modifications in Proteins section), the authors showed that binding of the A1 domain of vWF to GPIba under shear induced an intermediate activation state of αIIbβ3. In contrast, the activation of αIIbβ3 in response to agonists, such as ADP, was shear-independent (24). This provides insight into the mechanical regulation of αIIbβ3 by vWF.
Fibrinogen
Fibrinogen is the primary ligand of αIIbβ3 that is proteolytically cleaved by thrombin to form fibrin. The binding of fibrinogen to αIIbβ3 occurs under shear force with forces of 80–100 pN that are sufficient to detach fibrinogen from αIIbβ3 (93). Fibrin unfolding and re-positioning of fibrin fibrils occurs under shear, leading to fibrin stiffening in response to stretching or compression (119). In addition to mechanical force, fibrinogen is subject to redox regulation. Oxidation of fibrin monomers by ROS increases the strength of activated factor XIII (fXIIIa)-mediated cross-linked fibers (126).
Exposure of fibrinogen and thrombin to a nitric oxide (NO) donor produced fibrin with a lower fiber density and thicker fibers (64). On the other hand, the reduction of disulfide bonds in fibrinogen prevents fibrin polymerization in the absence of calcium (122). The regulation of circulating fibrinogen by the coupling of mechanical force and disulfide bond reduction has recently been shown: Novel mass spectrometry analysis showed that the exposure of fibrinogen to shear of 10,000 s−1 for 5 min reduced five disulfide bonds in fibrinogen's E region and eight disulfide bonds in the D region.
Scanning electron microscopy of fibrinogen with reduced disulfide bonds showed decreased polymerization and lower density of fibrin fibers (14). This demonstrates how advanced tools can aid in characterizing the mechano-redox control of proteins in vascular biology.
Tools to Study Mechanoredox Biology
There are unique challenges in studying mechano-redox regulation of protein interactions due to the fast and reversible nature of force and redox reactions. Nevertheless, emerging tools and techniques have provided options for observing components of mechanical and redox processes (Fig. 5). These tools provide methods for visualizing changes in the redox environment, analyzing the redox state of proteins, and measuring specific effects of redox changes on protein function that occur under the effect of shear or traction forces.
Redox probes and microfluidics to visualize mechano-redox dynamics
The incorporation of redox probes in a cellular environment can report on changes in the redox environment. The ROS probes exhibit low levels of fluorescence but can react with ROS to form a highly fluorescent molecule. Traditional ROS probes include the non-fluorescent dye dihydrorhodamine 123, which reacts with peroxides and superoxides to form rhodamine 123. Rhodamine fluoresces at excitation/emission (exc/emm) wavelengths of 507/529 nm. Dihydrorhodamine 123 and proprietary CellROX have been used in flow cytometry to measure ROS production by neutrophils and monocytes/macrophages on exposure to bacterial peptides and chemokines (60).
Thiol-based probes work in a similar manner, exhibiting low levels of fluorescence when oxidized but can react with thiol-reducing molecules to produce fluorescence. Di-Eosin glutathione (GSH) is a molecule with two fluorescent eosin moieties covalently bonded to oxidized GSH. The close proximity of the two eosin moieties causes auto-quenching. When the GSH becomes reduced, for example, by PDI, the eosin groups are separated and emit fluorescence at exc/emm 530/550 nm (124).
This probe has been used to measure the reductase activity of thiol oxidoreductases or cell surface oxidoreductase activity (117). Other thiol-reactive probes, which use the same principle and are commercially available, include boron-dipyrromethene probe BODIPY-FL-L-cysteine. BODIPY-FL-L-cysteine can fluorescently label disulfides in proteins that are cleaved by temperature (11) and mechanical force (80).
Although the probes just mentioned are useful for detecting the production of ROS and disulfide-reducing agents, modifications of these probes are permanent. Hence, the emitted fluorescence only reflects the production of oxidizing and reducing molecules at a particular timepoint. Newer reversible dyes have been developed to reflect the dynamic redox environment. The compound 2,2′-diselanediyldibenzoic acid (FSeSeF) is an example of a reversible redox probe that has been used to identify changes in the redox environment of HeLa cells (94). With the increasing range of fluorescent redox probes available, together with microfluidic technology, real-time visualization of mechano-redox changes under shear can be studied.
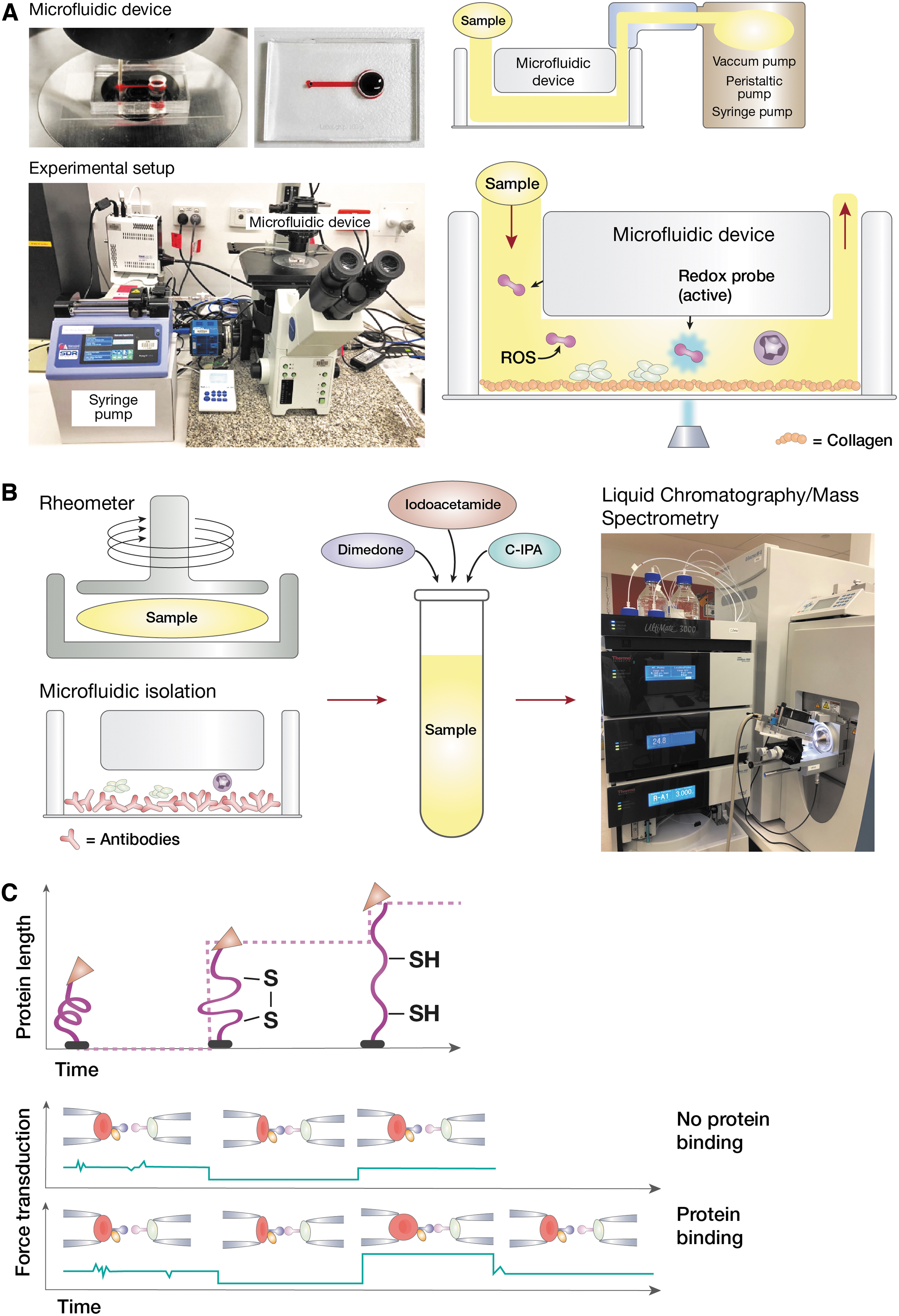
Microfluidics involve the perfusion of samples through sub-millimeter channels at controlled flow rates by using a pump. These flow rates can be adjusted to reflect different levels of shear, thus providing a method to control the mechanical forces applied to cells (Fig. 5A). Early forms of microfluidics involved coating straight channels with proteins of interest and flowing cells across. Recent developments in microfabrication and patterning techniques have provided new avenues for the study of mechano-redox biology.
Novel microfluidic designs can be achieved by patterning silicon molds to reflect different disease pathologies, such as straight channels (laminar flow) and stenosed devices (turbulent flow). In addition, methods such as injection molding are available for mass production of these devices, allowing high-throughput experiments to be performed (128). Apart from the design of the channel, different cellular surfaces can be used in the channel; that is, endothelial cells or leukocytes (HL-60 or THP-1 cells) that express the integrins of interest.
The microfluidic system has been used to study thiol oxidoreductase activity under shear in real time. For this, a platelet antibody (anti-β3 integrin) was linked to a C-terminal azido disulfide-linked peptide construct with a quenched reporter. Fluorescence was produced after reduction of the disulfide at the site of the peptide attachment to the platelet surface.
Human blood perfused over collagen at a shear rate of 100 s−1 showed an increase in disulfide reductase activity for the first 200 s of platelet thrombus formation over 500 s, suggesting that vascular thiol oxidoreductases play a role in the primary adhesion of platelets to collagen, but not secondary aggregation (178). Thus, a combination of microfluidics with fluorescent redox sensors allows for the visualization of mechano-redox interactions.
Microfluidic devices have also been employed to identify proteomic changes in biological samples (31, 52). By coating the surface of the microfluidic channel with antibodies, corresponding cells can be captured and isolated from blood. For example, microfluidic devices coated with anti-CD66b antibody have been used to capture and isolate neutrophils from blood under defined shear force for proteomic analysis (Fig. 5B) (79). Analysis of redox modifications of proteins can be achieved by enriching proteins of interest through immunoprecipitation.
Mass spectrometry and shear systems to quantify redox modifications in proteins
Advancement of mass spectrometry methods has allowed for precise detection of post-translational modifications of proteins (141, 151), including modifications of cysteines. Cysteine contains a redox-sensitive thiol group (-SH) that can protonate to become a highly reactive thiolate ion (S−). The thiolate group can form covalent bonds with other cysteines, lysines within peptides, or with amine groups to form products such as S-glutathione or S-sulfenymides. Redox modifications of the thiol group include: the conjugation with NO in S-nitrosylation and with GSH in S-glutathionylation. In addition, cysteine thiol can oxidize to a variety of oxoforms such as sulfenic, sulfinic, or sulfonic acid (29).
Integrin αIIbβ3 contains a total of 74 cysteines, forming 37 disulfide bonds. Extensive mutagenesis of C > S or C > A has been performed to determine the function of disulfide bonds in integrin activation and fibrinogen binding. The disruption of disulfide bonds by C > A mutations in the four epidermal growth factor-like domains of αIIbβ3 changes the conformation of αIIbβ3 to active open (Fig. 1A). The cleavage of disulfide bonds in αIIbβ3 by redox agents, such as GSH, enhances integrin activation whereas NO inhibits integrin activation (105, 106).
A major breakthrough in the detection of cysteine redox modifications was the development of cysteine-reactive probes (143, 152) in parallel with increased sensitivity and accuracy of mass spectrometry to sub-fentamole quantities of peptides and resolution down to 0.5–1 parts per million (ppm) (149). The nucleophilic nature of the cysteine thiolate enables selective labeling of reactive cysteines by using iodoacetic acid or iodoacetamide.
By incorporating cleavable tags to these chemical alkylators, proteins containing reactive cysteines can be isolated. Redox-modified cysteines can be further enriched with the use of chemical probes or antibodies that are specific to unique cysteine oxoforms. For instance, DYn-2 [4-(pent-4-yn-1-yl)cyclohexane-1,3-dione] selectively reacts with cysteine sulfenic acid (168) whereas DiaAlk [1-(tert-Butyl) 2-(2-methyl-4-(prop-2-yn-1-yloxy)butan-2-yl) (E)-diazene-1,2-dicarboxylate] reacts with cysteine sulfinic acid (2). This array of chemical probes has revealed the complexity of the cysteome in cells for the first time.
To study the effect of shear in mediating disulfide redox modifications in integrins, methods are introduced to expose biological samples to shear. One such method is the cone and plate rheometer/viscometer. Rheometers have been used to introduce controlled levels of shear to analyze platelet integrin function (40, 113, 115). After subjecting cells or biological fluids to shear, shear-induced protein modifications can be analyzed by mass spectrometry (Fig. 5B). In addition, by incubating the proteins with cysteine alkylators during the application of shear, the effect of shear on the redox state of protein disulfide bonds can be quantified (14, 80). A description of our approach to study shear-redox modifications in proteins is given next:
Our method to quantitate the redox state of disulfide bonds in a protein is named differential cysteine labeling. It uses a pair of isotopic cysteine alkylators, N-12C-iodo-phenylacetamide (12C-IPA) and N-13C-iodo-phenylacetamide (13C-IPA) (27). The probe 12C-IPA is added to a platelet suspension (or purified integrins), and the suspension is subjected to shear (i.e., on cone-plate) (Fig. 5B). Platelets or integrins are incubated with 12C-IPA in the presence or absence of shear for 30–60 min in the dark. Platelets are then lysed, and the protein of interest (i.e., αIIbβ3) is immunoprecipitated. Immunoprecipitated proteins are resolved on gel electrophoresis; the integrin band is excised, destained, dried, and incubated with 40 mM dithiothreitol to fully reduce all disulfide bonds in the protein. The fully reduced protein is then alkylated with 13C-IPA.
The gel slice is washed, dried, and deglycosylated before digestion of the integrin with chymotrypsin and trypsin. Peptides are eluted from the slices with formic acid and acetonitrile. Liquid chromatography, mass spectrometry, and data analysis are performed, as described (27). Cysteines labeled with 12C-IPA were unpaired (reduced disulfide bonds) in the original condition of the protein, whereas cysteines labeled with 13C-IPA were paired (oxidized disulfide bonds) in the original protein.
The redox condition of the protein's cysteines is calculated from the relative ion abundance of peptides labeled with 12C-IPA or 13C-IPA. To calculate the ion abundance of peptides, extracted ion chromatograms are generated by using the XCalibur Qual Browser software (v2.1.0; Thermo Scientific). The area is calculated by using the automated peak detection function built into the software (27). By quantifying the ratio of 12C labeled cysteine to 13C labeled cysteine of a particular disulfide bond, we can calculate the ratio of reduced to oxidized state of that disulfide bond at the time of exposure to mechanical force (shear).
Using differential cysteine labeling, we successfully mapped the redox state of 24 of the 28 β3 integrin disulfide bonds in purified human αIIbβ3 or on platelet surface αIIbβ3 occurring in disulfide bonds of αIIbβ3 and fibrinogen under shear force (14, 116). Using a rheometer to shear human plasma at 10,000 s−1 for 5 min combined with the 12C-IPA labeling, Butera et al. measured the reduction of 13 disulfide bonds in fibrinogen.
Combining cysteine-specific probes with shear systems is an effective experimental system to identify mechano-redox alterations in integrins and other proteins.
DFS to characterize the mechano-redox control of protein interaction
The DFS techniques such as atomic force microscopy and BFP have been employed to examine the effect of force and redox changes on protein function (102). The DFS analyzes receptor–ligand interactions and measures binding kinetics under various tensile forces (71). All DFS methods utilize a force transducer to probe molecular binding in force spectroscopy (23, 176).
We have utilized BFP to measure the mechano-redox control of integrin αIIbβ3 binding to fibrinogen (71). This is our approach.
The BFP utilizes two micropipettes, with one aspirating a biotinylated human red blood cell (RBC) with a glass bead to serve as a force transducer, termed “probe” (Fig. 5C). The bead is attached on the RBC apex via biotin-streptavidin (SA) interaction. The other micropipette aspirates a second bead, termed “target.” The probe and target beads are, respectively, coupled with purified human fibrinogen and αIIbβ3 with maleimide-PEG3500-NHS in carbonate/bicarbonate buffer (pH 8.5).
The two probes come in contact with a buffer to which we add the redox reactive enzyme (i.e., ERp5). The force spectroscopy traces are obtained by measuring the RBC-bead deflection from the probe beads edge tracking. Bond formation/dissociation and force application are enabled and monitored in controlled BFP touch cycles (∼2.5 s each). In each cycle, the αIIbβ3-bearing target bead is driven to approach and contact the fibrinogen-probe bead with a 20-pN compressive force for a certain contact time (0.2 s) that allows for bond formation. The target is then retracted at a constant speed (3.3 μm/s) for bond detection.
During the retraction phase, a “bond” event is signified by a tensile force. Conversely, no tensile force indicates a “no-bond” event. For the adhesion frequency assay, “bond” and “no-bond” events are enumerated to calculate an adhesion frequency in 50 repeated cycles for each probe–target pair (Fig. 5C) (71 –73). Using BFP, we have previously shown that in the presence of soluble ERp5, the bond lifetimes of fibrinogen–αIIbβ3 decrease under all tensile forces tested, displaying a slip-bond behavior (116).
The BFP has demonstrated that the cleavage of the disulfide bond in the vWF-A2 domain decreases the lifetime of the vWF–GPIbα bond under force, but it still retains its catch-bond behavior (15). Thus, DFS analyses can be used to study the effect of redox changes on force-dependent receptor–ligand interaction, which promises unprecedented molecular insight into the effect of mechano-redox changes in integrin function in thrombosis and inflammation.
Altered Mechanoredox Conditions in Thromboinflammation and Integrin Function
In thrombo-inflammation there is a varying degree of thrombosis, inflammation, mechanical perturbation, and redox imbalance (Fig. 6). These components of thrombo-inflammation can adversely affect integrin function. Conversely, integrin function contributes to the development of thrombo-inflammation.
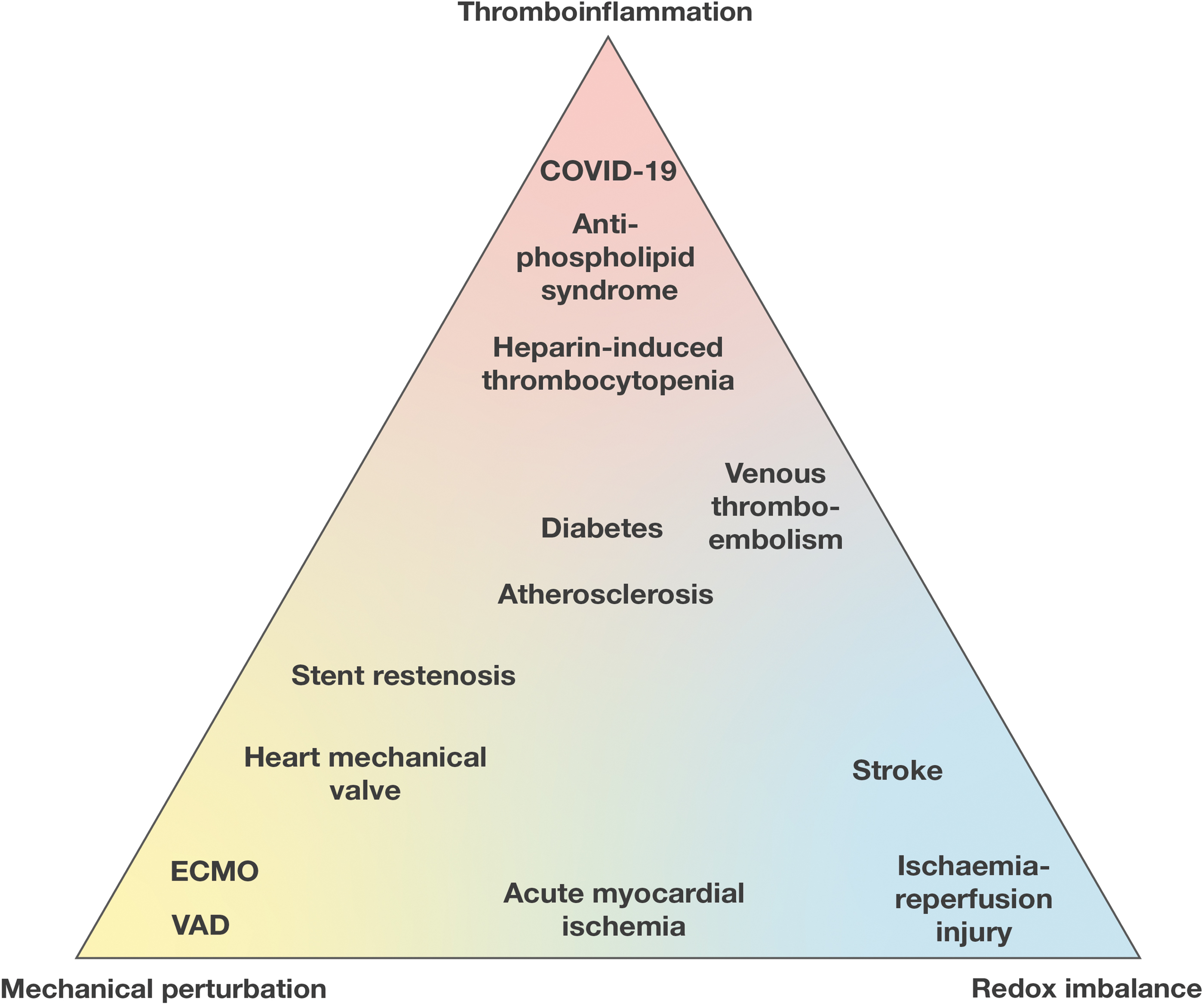
Diabetes and atherosclerosis sit at the center of thromboinflammatory conditions, with contribution of both mechanical and redox imbalance. Hyperglycemia alters the redox environment of cells through both ROS- and thiol-dependent pathways. Under normal circumstances, ROS is enzymatically sequestered by superoxide dismutase and catalase. However, hyperglycemia and hyperlipidemia saturate the activity of these enzymes, leading to the accumulation of ROS, which promotes oxidative stress and damage (150).
Platelets and leukocytes from patients with diabetes are more adhesive compared with patients without diabetes, which is enhanced by the upregulation of surface integrins allbb3 and aMb2, respectively (49, 121). Diabetic platelets also display increased disulfide reductase activity, which may contribute to their thrombotic tendency (123). Atherosclerotic lesions lead to increased mechanical shear of the blood flowing through stenotic lesions. Diabetic platelets are more prone to shear-dependent activation and thrombosis. This is attributed to the increased adhesiveness of the αIIbβ3 integrin from compression forces in the flowing blood (74).
A recent thromboinflammatory challenge is SARS-Cov-2 infection. Hottz et al. reported that patients with severe COVID19 had increased circulating platelet-monocyte aggregates that were inhibited by the αIIbβ3 inhibitor abciximab (69). Interestingly, SARS-Cov-2 contains an RGD motif that may bind to integrins to facilitate adhesion (18).
Some conditions have greater contribution of mechanically induced thromboinflammation; extracorporeal membrane oxygenation (ECMO) is a prominent example. ECMO circuits bypass the lungs in respiratory failure or bypass the lungs and heart in cardiopulmonary failure. Blood is circulated by a pump through an oxygenator to the patient. High shear rates develop by the function of the pump and the flowing of the blood through the tubing, which may activate circulating platelets (36). Fibrinogen is adsorbed on the surfaces of the circuit and promotes integrin-mediated adhesion of platelets and leukocytes (36). ECMO has been shown to upregulate integrin αMβ2 expression on circulating leukocytes (75). In addition, hemolysis in the circuit leads to increased production of ROS (65).
Conditions with a greater contribution of redox-induced thromboinflammation include ischemia/reperfusion (I/R) injury. I/R injury occurs after restoration of vascular supply to an organ, that is, brain or heart, after a critical period of arterial occlusion. After the reinstitution of oxygenated blood to the ischemic tissue, there is a rapid burst of ROS that is mediated by xanthine oxidase, NOX, and mitochondria (55). Ischemia increases the activity of inducible NO synthase, which leads to the production of peroxynitrite, one of the main reactive oxide species leading to both necrosis and apoptosis. The concomitant increase in the production of inflammatory cytokines leads to increased activation of β2 integrins (164, 165) and increased permeability of the vascular system. The inhibition of β2 protects from I/R injury in humans and rats (134, 143).
Mechanical and redox perturbations are key to the development of a variety of cardiovascular pathologies (Fig. 6). The contribution of mechano-redox control of integrins in disease conditions remains an area of investigation.
Conclusion: Future Perspectives
Integrins are fundamental adhesion receptors during thrombosis and inflammation. Although mechanical and redox regulation of integrins have been identified separately in thrombosis and inflammation, the combined mechano-redox control of integrin function has been less studied.
Moving forward, it is expected that emerging tools will be further refined to study the spatiotemporal details of integrin function within their redox and mechanical environment.
There is the potential for inhibitors of ROS and thiol oxidoreductases to be developed as therapeutics to prevent unwanted mechano-redox alterations of vascular integrins.
The effect of disease states on redox and mechanical forces in the circulation, and how they influence integrin function, is an open avenue for further research.
Footnotes
Acknowledgment
The authors would like to thank Ms. Betty Loi from the Heart Research Institute Australia for preparation of the figures in this article.
Authors' Contributions
A.D. wrote the article and drafted the figures, J.C. wrote and revised the article, L.A.J. wrote and revised the article, and F.H.P. wrote and revised the article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This study was funded by the Sydney Cardiovascular Fellowship, University of Sydney, Heart Research Institute (to F.H.P), Ministry of Health New South Wales Cardiovascular Early-Mid Researcher Grant (to F.H.P) and by a National Heart Foundation Future Leader fellowship 105863 (to L.A.J.).