
Abstract
Significance:
Limited oxygen availability (hypoxia) commonly occurs in a range of physiological and pathophysiological conditions, including embryonic development, physical exercise, inflammation, and ischemia. It is thus vital for cells and tissues to monitor their local oxygen availability to be able to adjust in case the oxygen supply is decreased. The cellular oxygen sensor factor inhibiting hypoxia-inducible factor (FIH) is the only known asparagine hydroxylase with hypoxia sensitivity. FIH uniquely combines oxygen and peroxide sensitivity, serving as an oxygen and oxidant sensor.
Recent Advances:
FIH was first discovered in the hypoxia-inducible factor (HIF) pathway as a modulator of HIF transactivation activity. Several other FIH substrates have now been identified outside the HIF pathway. Moreover, FIH enzymatic activity is highly promiscuous and not limited to asparagine hydroxylation. This includes the FIH-mediated catalysis of an oxygen-dependent stable (likely covalent) bond formation between FIH and selected substrate proteins (called oxomers [oxygen-dependent stable protein oligomers]).
Critical Issues:
The (patho-)physiological function of FIH is only beginning to be understood and appears to be complex. Selective pharmacologic inhibition of FIH over other oxygen sensors is possible, opening new avenues for therapeutic targeting of hypoxia-associated diseases, increasing the interest in its (patho-)physiological relevance.
Future Directions:
The contribution of FIH enzymatic activity to disease development and progression should be analyzed in more detail, including the assessment of underlying molecular mechanisms and relevant FIH substrate proteins. Also, the molecular mechanism(s) involved in the physiological functions of FIH remain(s) to be determined. Furthermore, the therapeutic potential of recently developed FIH-selective pharmacologic inhibitors will need detailed assessment. Antioxid. Redox Signal. 37, 913–935.
Introduction
Vertebrates utilize molecular oxygen (O2) as prime substrate for energy metabolism, rendering them vulnerable to disturbances in oxygen supply. A decreased oxygen availability relative to the demand (hypoxia) frequently occurs in numerous physiological and pathophysiological conditions, including development, physical activity, inflammation, ischemia, and cancer. Cellular oxygen sensing is therefore central for the survival of vertebrates, enabling cells and tissues to survive by adaptation to hypoxia. Cellular oxygen sensors are characterized by an essential requirement of O2 for their catalytic activity combined with a low oxygen affinity within the physiologically relevant range of oxygen partial pressure.
The first identified oxygen sensors, prolyl-4-hydroxylase domain (PHD) proteins 1–3 and the asparagine hydroxylase factor inhibiting hypoxia-inducible factor [FIH; originally called FIH-1 (103), but no other hypoxia-inducible factor (HIF)-α asparagine hydroxylase has been identified since the discovery of FIH, which is why it is more often referred to as FIH by now], are members of the 2-oxoglutarate (2-OG)-dependent dioxygenase superfamily that comprises 60–70 different enzymes (58, 64). PHDs and FIH catalyze the hydroxylation of prolyl or asparaginyl residues of target proteins, using O2 and 2-OG as cosubstrates and Fe(II) and a reductive agent (such as ascorbate) as cofactors (145, 148). The PHDs and FIH confer hypoxia sensitivity to the transcriptional activator HIF (149).
Several members of the histone lysine demethylases (KDM), also members of the 2-OG-dependent oxygenase superfamily, have recently been proposed as additional oxygen sensors (Fig. 1), including KDM4A, KDM4E, KDM5A, and KDM6A (2, 6, 43, 97, 140). Remarkably, all hitherto analyzed 2-OG superfamily members directly catalyze substrate hydroxylation in higher organisms. Interestingly, Jumonji C-domain containing protein (JMJD) 5 and JMJD7 are bifunctional enzymes, containing an additional peptidase activity (91, 92, 150). Histone and DNA demethylation by KDMs and ten-eleven-translocation 5-methylcytosine dioxygenases (TETs) are catalyzed by hydroxylation of the methylated lysine or cytosine residue, respectively, followed by release of formaldehyde (128).
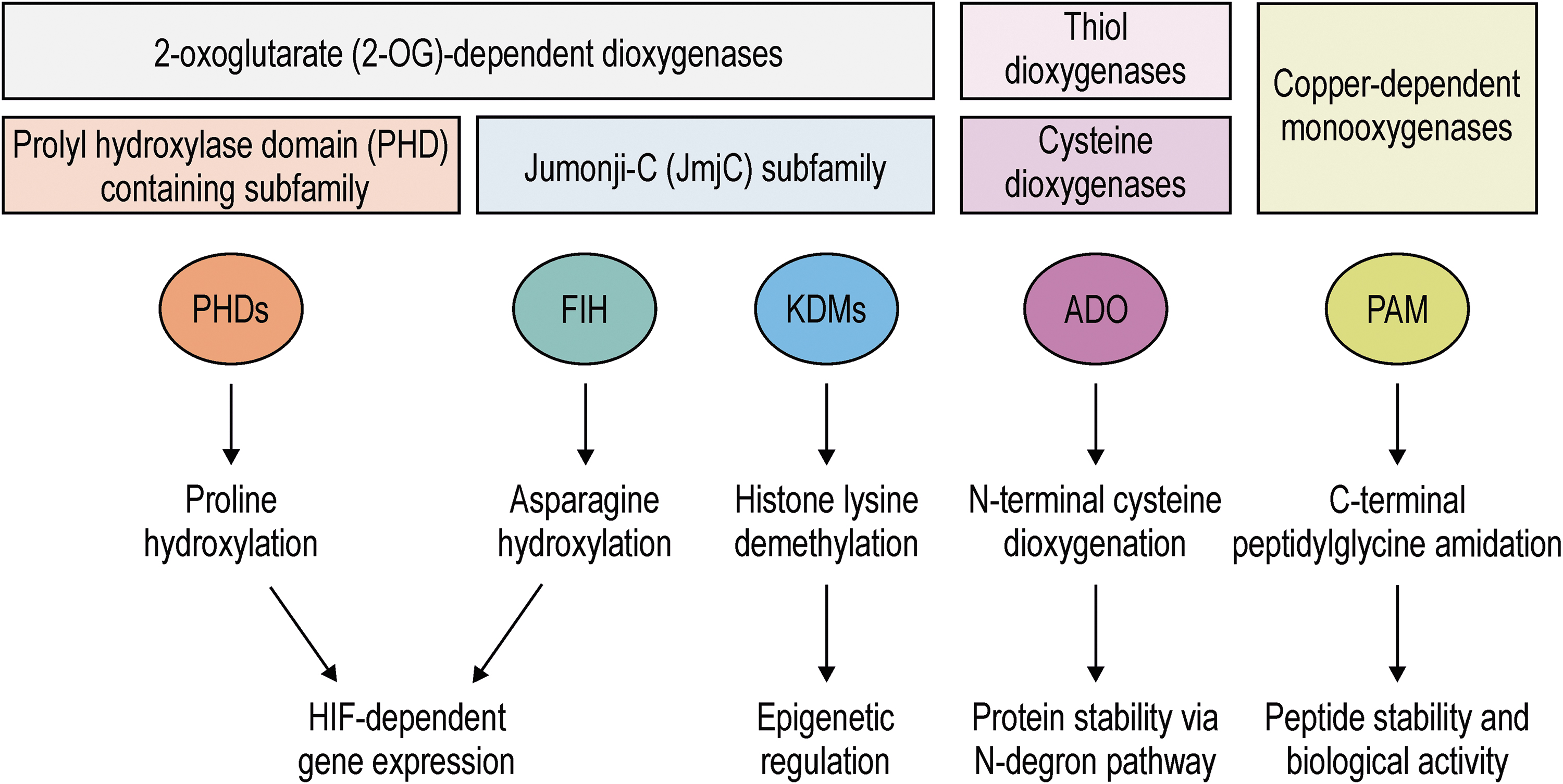
Another recently identified possible cellular oxygen sensor is the peptidylglycine α-amidating monooxygenase (PAM) (Fig. 1), which belongs to the family of copper-dependent monooxygenases, catalyzing the C-terminal amidation of peptides in neuroendocrine cells, regulating peptidergic pathways in an oxygen-dependent manner (154). The enzyme cysteamine (2-aminoethanethiol) dioxygenase (ADO) regulates the N-degron pathway by N-terminal cysteine dioxygenation in an oxygen-dependent manner as another likely oxygen sensor (Fig. 1) (107). ADO belongs to the thiol and cysteine dioxygenases and its activity regulates, for example, the stability of the cytokine interleukin-32 (107). However, the relevance for mammalian (patho)physiology of all of the suspected additional oxygen sensors remains to be investigated.
FIH is the only known asparagine hydroxylase among these sensors (Fig. 1). FIH is more closely related to the Jumonji C (JmjC) subfamily of 2-OG-dependent dioxygenases than to the PHDs (105). The suggested JmjC oxygen sensors KDM4A, 4E, 5A, and 6A target aminomethylated lysyl residues for hydroxylation and not asparagine (58). In addition, as described below, FIH is also a peroxide sensor (108), may target the amino acids aspartate (184), histidine (183), and tryptophan (66) for hydroxylation, and forms oxygen-dependent (likely) covalent protein complexes in a highly hypoxia-sensitive manner (127, 161). Therefore, FIH represents a unique oxygen sensor, which is the focus of the remainder of this review.
FIH in the HIF Pathway
HIF consists of an α and a β subunit, which together form the active transcription factor. A single HIF-β subunit interacts with one out of three HIF-α isoforms, HIF-1α, HIF-2α, and HIF-3α. The half-life of HIF-α subunits (especially of HIF-1α and HIF-2α) is regulated in an oxygen-dependent manner by prolyl-4-hydroxylation through the PHDs. The two hydroxylated prolyl residues are recognized by the E3 ligase adaptor the von Hippel–Lindau (VHL) protein, leading to HIF-α poly-ubiquitination and subsequent proteasomal degradation (Fig. 2) (31, 144).
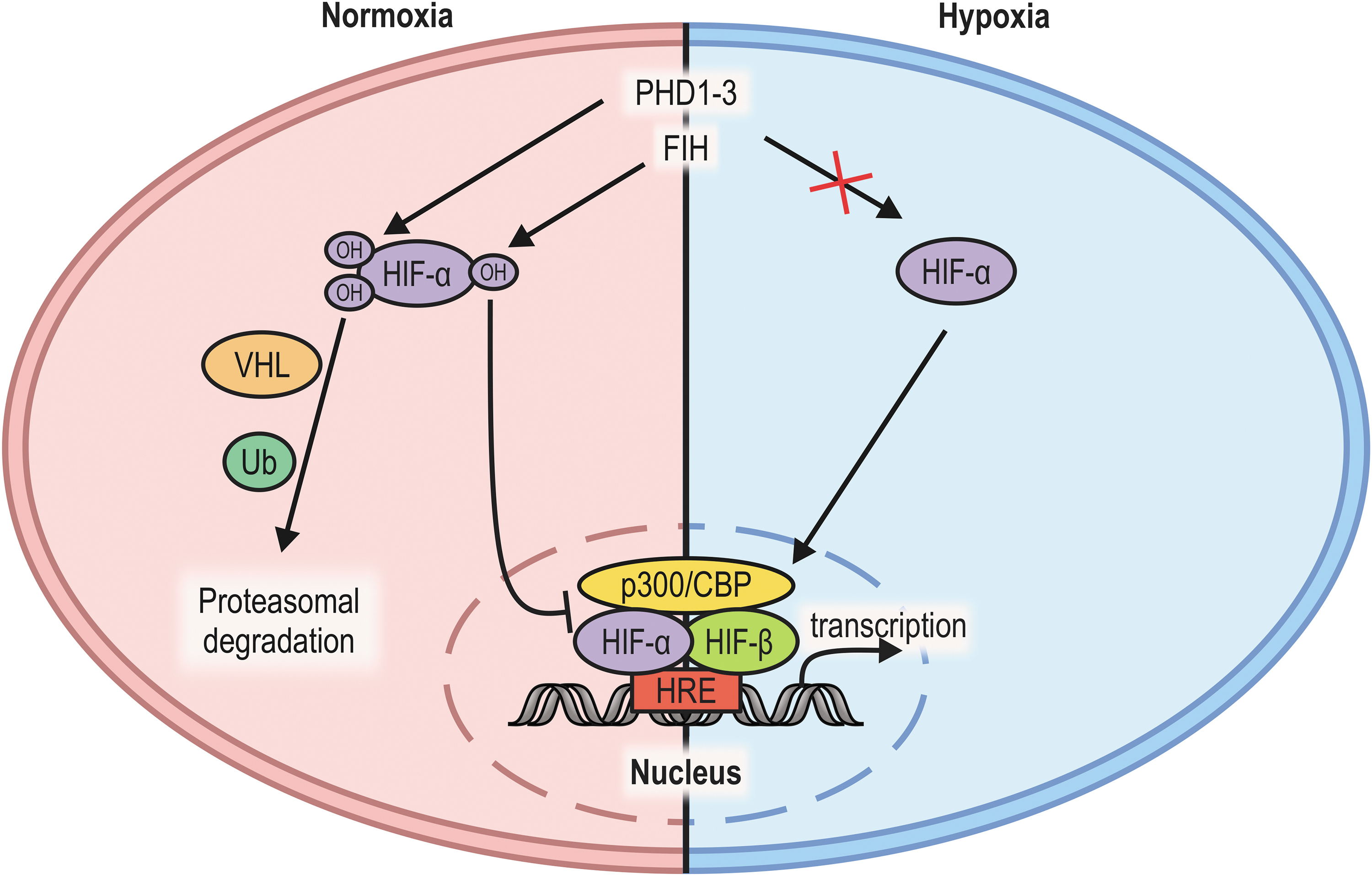
FIH has first been identified as an interaction partner of HIF-1α that reduces HIF-1-dependent reporter gene activity (103). Only later it was demonstrated that FIH modifies HIF-1α by hydroxylation as well: HIF-1α contains an N-terminal transactivation domain (N-TAD) and a C-terminal transactivation domain (C-TAD), the latter of which has been demonstrated to be hydroxylated on a conserved asparagine residue (Asn-803) (48, 81, 141). This hydroxylation prevents the interaction between HIF-1 and its transcriptional coactivators, the histone acetyl transferases p300 and CBP, regulating HIF-α C-TAD transactivation activity (22, 28, 36, 48, 80, 81, 111). FIH plays a key role in the regulation of HIF target gene expression (159).
Further analyses of HIF-dependent gene expression revealed that FIH catalytic activity affects a specific subset of HIF target genes (8, 24, 122), because FIH only regulates one of the two HIF transactivation domains and HIF-mediated enhancement of gene expression involves the N-TAD and/or the C-TAD in a gene-specific manner. A decrease of PHD activity alone leads thus to a different HIF target gene expression (increased N-TAD activity, while the C-TAD is still inhibited by FIH) compared with a selective downregulation of FIH activity (increase of C-TAD activity alone) or a decrease in both PHD and FIH enzymatic activity (increase of both N-TAD and C-TAD activity) (8, 24, 122).
In addition, FIH hydroxylates HIF-1α more efficiently than HIF-2α (3, 75), contributing to the selectivity of the FIH-dependent regulation of HIF-mediated transcription. Interestingly, FIH was also reported to interact with VHL, which is recruited to HIF-α subunits following HIF-α proline hydroxylation (83, 87, 103). VHL may thus modulate FIH activity toward HIF-α, potentially linking PHD- and FIH-dependent HIF-α regulation. The actual contribution of the FIH:VHL interaction to the FIH-mediated regulation of HIF transactivation activity, however, remains unclear.
In summary, FIH is a key regulator of HIF transactivation activity with differential effects on HIF-α subunits and HIF target gene expression.
FIH Catalytic Activity
The protein structure and enzymatic activity of FIH have been investigated in detail and have recently been expertly reviewed (181). In brief, the active site of FIH contains a conserved two-histidine, one-carboxylate motif for the coordination of an Fe(II) ion, and mutation of His-199 or Asp-201 inactivates the enzyme (23). FIH dimerization is essential for FIH activity, which is mediated by Leu-340 and Ile-344 (79).
Studies on various 2-OG-dependent dioxygenases indicated a common enzymatic mechanism that is shared by the HIF hydroxylases (46, 114). 2-OG binds to the active site of the enzyme before binding of the prime substrate (such as Asn-803 of human HIF-1α), and release of a water molecule (181). The binding of 2-OG and the substrate is reversible and likely triggers binding of O2 to Fe(II) in the active center. The subsequent oxidative decarboxylation of 2-OG leads to the production of succinate and CO2. The concomitantly formed highly reactive ferryl intermediate [Fe(VI) = O] then oxidizes a C-H bond in the prime substrate, which represents the actual hydroxylation step. Following the release of the hydroxylated product, succinate is replaced by water (181).
Investigations of the affinity of FIH for O2 determined its K M value as 90 ± 20 μM (27, 75, 164), which is below the PHD K M value of 229 ± 60 μM (27, 50). This indicates that FIH is enzymatically active under lower oxygen partial pressure than the PHDs, but it has to be taken into account that these measurements were obtained from purified proteins and with HIF-α peptides as substrates. It was further shown that the activity of all four HIF hydroxylases is strongly dependent on substrate length in vitro, resulting in higher substrate conversion rates and lower K M values with longer substrates (27, 75, 76, 176).
Analyses of HIF-1α prolyl and asparaginyl hydroxylation in cells using specific antibodies further supported that PHD-dependent HIF-1α hydroxylation is more sensitive to hypoxia than FIH-dependent HIF-1α hydroxylation (168). It has previously been suggested that FIH-dependent asparagine hydroxylation is irreversible (156), but recently it was proposed that asparagine hydroxylation can be reversed, indicating the existence of a yet uncharacterized dehydroxylase (132).
2-OG-dependent dioxygenases demonstrate a remarkable catalytic flexibility that is among the highest of all enzyme families (15). Therefore, it is probably not too surprising that FIH catalytic activity is not restricted to asparagine hydroxylation. FIH is capable of catalyzing the hydroxylation of aspartate (184) and histidine (183) in cellulo. The range of the FIH activity was subsequently extended to the catalysis of the oxidation of neutral, polar, hydrophilic, hydrophobic, acidic, and basic amino acid side chains in peptides using recombinantly expressed FIH proteins, resulting not only in the formation of hydroxylated amino acids but potentially also in β-oxo-histidine and formylglycine/enol or cyclic modifications of serine (185). It is currently unclear if these additional reactions are also catalyzed by FIH in cells.
Recently, the FIH-dependent hydroxylation of tryptophan was reported to occur in the protein N-alpha-acetyltransferase 10 (NAA10) in cellulo (66). Moreover, we previously showed that FIH catalytic activity can also lead to the formation of a (likely) covalent bond between FIH itself and specific target proteins in cells (see section “Oxomer [Oxygen-Dependent Stable Protein Oligomer] Formation”) (127). These findings demonstrate that FIH activity is highly promiscuous and indicate that its substrate pool might be considerably larger than expected based on its currently established targets. However, it remains unclear which factors determine the reaction that is catalyzed by FIH in cells and to what extent each of the reported modifications contributes to the (patho-)physiological function of FIH.
Regulation of FIH
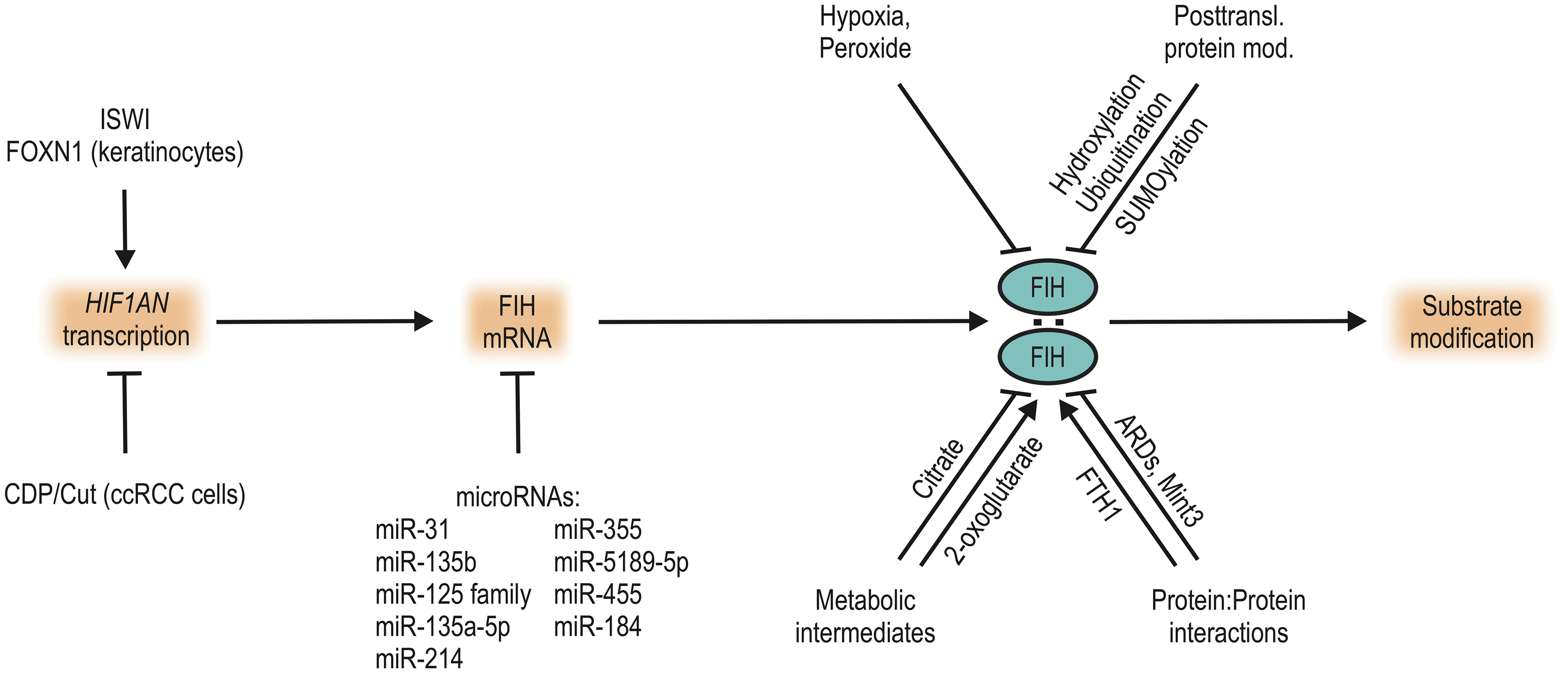
Elucidating the (patho-)physiological regulation of FIH is crucial for understanding its functional relevance. As described above, FIH activity is directly dependent on the availability of its cosubstrate O2. It is also possible to pharmacologically inhibit FIH by sequestration of iron or competition for 2-OG binding (8, 161); for a recent expert review see Wu et al. (181). Within cells, a range of mechanisms potentially affects FIH-mediated hydroxylation by regulating FIH protein levels, enzymatic activity, and/or substrate targeting (Fig. 3).
Transcription
In contrast to the HIF prolyl hydroxylases PHD2 and PHD3 (158), the gene encoding human FIH (HIF1AN) is not an HIF target gene and it is not regulated by hypoxia in mammals according to current knowledge. In renal cell carcinoma, HIF1AN gene expression is regulated by the Cut-like homeodomain protein (CDP/Cut), which binds to a specific binding site in the HIF1AN promoter and represses HIF1AN transcription (86). CDP/Cut binding to the HIF1AN promoter is facilitated by phosphorylation of CDP/Cut through the protein kinase C (PKC) ζ (86). RNA polymerase II recruitment to the HIF1AN promoter is facilitated by the chromatin remodeling enzyme imitation switch (ISWI), enhancing HIF1AN expression (112).
In keratinocytes, the transcription factor forkhead box protein N1 (FOXN1) was reported to affect HIF1AN expression (100), but the underlying mechanism remains unclear. Overall, little is known about the regulation of HIF1AN expression and—to our knowledge—no external factor that alters HIF1AN expression has been identified to date, although FIH messenger RNA (mRNA) and protein levels have repeatedly been reported to be increased in diverse diseases and/or conditions (see section “FIH in Organisms” and Table 2).
microRNAs
Various microRNAs (miRNAs) regulate FIH on the mRNA level. To the best of our knowledge, other noncoding RNAs have only been indirectly associated with the regulation of FIH to date and are therefore not further discussed.
microRNA-31 (miR-31) regulates FIH mRNA in head and neck squamous cell carcinoma (HNSCC) (90), keratinocytes (124), corneal epithelial cells (123), limbal (corneal epithelial) stem cells (95), keloid (193), lung cancer cell lines (198), nonsmall cell lung cancer (NSCLC) (45), and (when applied in exosomes as a therapeutic) in diabetic wounds (54). miR-31 reduces FIH mRNA (and thus the FIH protein) levels by targeting its 3′-untranslated region (3′-UTR) (45, 90).
miR-31-mediated reduction of FIH has been associated with favouring HNSCC development (90), promoting keratinocyte differentiation (124), increasing glycogen synthesis in corneal epithelial cells (123), reducing stemness in limbal stem cells (95), decreasing apoptosis while enhancing cell proliferation in keloid tissue (193), contributing to the Warburg effect in lung cancer cell lines (198), supporting gefitinib (an inhibitor of the epidermal growth factor receptor) resistance in NSCLC (45), and with promoting diabetic wound healing (54) in an HIF-dependent and/or HIF-independent manner.
In bone microvascular endothelial cells (BMECs), miR-135b decreases FIH mRNA by targeting its 3′-UTR, which was associated with increased angiogenesis and BMEC proliferation (104). miR-135b was also associated with decreasing FIH mRNA levels in HNSCC, correlating with increased tumor growth (194). In thyroid cancer cells, miR-18a-5p negatively regulates FIH mRNA, which was associated with the promotion of tumor growth and invasion (93).
The miR-125 family (178), miR-135a-5p (82, 189), miR-214 (172), and miR-355 (180), negatively regulate FIH mRNA in the murine and rat heart and/or in cardiomyocytes. miRNA-mediated reduction of FIH mRNA was protective against cardiac injury (82, 172, 180).
In rat renal fibroblasts, miRNA-184 was reported to decrease FIH mRNA, which was linked to increased profibrotic gene expression (9). In mesangial cells, miR-5189-5p-mediated decrease of FIH mRNA was associated with (among others) the regulation of 5′ adenosine monophosphate (AMP)-activated protein kinase α1 (AMPKα) phosphorylation (10). In brown adipocytes, miR-455 can downregulate FIH levels, affecting differentiation, phosphorylation of AMPKα, and mitochondrial biogenesis (192).
In summary, numerous miRNAs have been reported to regulate FIH mRNA, but it remains unclear whether the FIH mRNA 3′-UTR can be targeted by several miRNAs simultaneously and/or if at least some of these miRNAs have cell-type-specific functions. Moreover, the associated functional downstream effects are often rather mild and a clear mechanistic link to FIH activity is missing. Interpretation of miRNA effects on FIH function is thus in need of some caution.
Protein:protein interactions
FIH enzymatic activity is effectively regulated by interaction with other proteins. Most importantly, FIH acts as a homodimer (23, 28, 83) and disruption of this interaction by point mutation of Leu-340 to Arg-340 inhibits FIH enzymatic activity (79). Developing pharmacologic agents that interfere with FIH homodimerization may present an opportunity for FIH-selective inhibition, because PHD1–3 act as monomers.
As outlined in detail in the section “FIH Substrates Outside the HIF Pathway”, FIH interacts with many ankyrin repeat domain (ARD)-containing proteins (19). Interestingly, the affinity of FIH for ARDs is even higher than its affinity for HIF-1α (19, 176). Therefore, ARD-containing proteins are believed to sequester FIH, reducing the FIH-dependent regulation of HIF-1α and hence increasing the HIF-mediated response to hypoxia (94, 142, 151, 175). Intriguingly, this mechanism can also be recruited by viruses to co-opt the cellular hypoxia adaptive response (11).
Interaction of FIH with Mint3 (also called amyloid-beta A4 precursor protein-binding family A member 3; APBA3), a protein interacting with the Alzheimer β-amyloid precursor protein, prevents HIF-1α Asn-803 hydroxylation (138). The interaction of FIH with Mint3 occurs via the Mint3 amino acids 78–88 (167). In macrophages, Mint3 scavenges FIH to the perinuclear region and increases HIF target gene expression (138). The Mint3-mediated regulation of FIH may also occur in mouse embryonic fibroblasts (MEFs) and in human breast cancer cells, epidermoid carcinoma cells (117), and pancreatic cancer cells (65). In nucleus pulposus cells, a regulation of HIF target gene expression by Mint3:FIH interaction was not observed, indicating to some degree a cell-type specificity (49).
Opposite to Mint3, interaction of ferritin heavy chain (FTH1) with FIH increases FIH-dependent HIF-1α N803 hydroxylation and represses HIF target gene expression (63). FTH1 is a subunit of ferritin, which plays an important role in iron metabolism. Thus, these findings indicate a link between iron metabolism and FIH enzymatic activity. However, HIF-2α rather than HIF-1α appears to be the main HIF-α isoform involved in iron metabolism and it remains to be determined whether FTH1-dependent FIH regulation affects HIF-2 transcriptional activity under conditions of altered iron status.
The p53 tumor suppressor E3 ubiquitin ligase murine double-minute type 2 (Mdm2; also called Hdm2 for the human orthologue) competes with FIH for binding of the HIF-1α C-TAD and thus regulates HIF transactivation activity, potentially playing a role in tumor adaptation to hypoxia (84).
In summary, the FIH activity is regulated by protein:protein interactions in a complex manner. These interactions can be protein specific or widespread and domain, but not protein, specific (ARDs). The exact level of contribution to FIH function and a possible cell-type specificity remain to be assessed.
Posttranslational modifications
FIH autohydroxylates itself at Trp-296 in the absence of a prime substrate such as HIF-α in vitro (13, 14). This autohydroxylation can lead to oxidation of the active site iron and the formation of an Fe(III)-O-Trp species, rendering FIH inactive and possibly preventing the uncoupled formation of reactive oxygen species (ROS) (13, 14).
It is unclear if the FIH autohydroxylation also occurs in cells. Recombinantly expressed FIH from bacteria was used for these analyses, which autohydroxylated itself in an in vitro FIH activity assay (14). However, FIH is active in bacteria (101, 127, 160), which raises the question why the utilized FIH was not already autohydroxylated at the beginning of the experiment. One possible explanation is that bacterial proteins serve as FIH substrates, preventing FIH autohydroxylation in bacterial cells. More analyses are required to determine the physiological relevance of this autohydroxylation.
Several E3 ubiquitin ligases have been identified to target FIH (39). FIH protein levels are regulated by the activity of Siah-1, which ubiquitinates FIH and thereby targets it for proteasomal degradation (37). The ARD-containing proteins and E3 ubiquitin ligases mind bomb (mib) and mib2 interact with FIH (157, 170) and ubiquitinate it (170). However, the outcome of this ubiquitination of FIH remains unclear. Alongside ubiquitin, ubiquitin-like proteins are also covalently attached to FIH. The most prominent of these is the small ubiquitin-like modifier (SUMO). FIH is differentially SUMOylated at diverse stages of placental development, regulating FIH proteasomal degradation (139).
Phosphorylation of HIF-1α Thr-796 prevents hydroxylation of Asn-803 by FIH and thus enhances HIF transactivation activity, because the phosphorylation likely disrupts hydrophobic interactions between FIH and HIF-1α, thereby enhancing HIF transactivation activity (38, 79). In addition, protein kinase A (PKA) phosphorylates HIF-1α on Thr-63 and Ser-692, which enhances HIF transactivation activity, but without disturbing the interaction between FIH and HIF-1α (4). The exact mechanism of action remains to be elucidated.
Overall, posttranslational modifications of FIH appear to be scarce, and common modifications such as protein phosphorylation have not been reported to date. This may be the result of a lack of investigation of this specific area. Taking the vast amount of available proteomics data into account though, it appears more likely that such modifications simply do not occur, highlighting the importance of the regulation of FIH activity by substrate availability (such as O2).
Metabolic intermediates
Reductive agents such as ascorbate or glutathione serve as a cofactor for HIF hydroxylases by keeping active site iron in the reduced state (35, 119, 120). The citric acid cycle intermediate 2-OG serves as a cosubstrate, and succinate (also a citric acid cycle intermediate) is a coproduct of the catalyzed protein hydroxylation. Analyzing the effect of other citric acid cycle intermediates on hydroxylase activity, some differences were identified between the PHDs and FIH (47, 77). Purified PHD activity was inhibited by succinate, fumarate, oxaloacetate, and to a certain extent also by pyruvate, lactate, and malate, whereas FIH was only inhibited by citrate (47, 77).
These results indicate that FIH is less sensitive to changes in intermediates of the citric acid cycle than the PHDs, which may have implications especially in tumor cells with a genetic dysregulation of these metabolic intermediates.
FIH and Oxidant Stress
ROS, mainly produced by the electron transport chain in mitochondria, have been suggested to be increased in hypoxia and to regulate HIF activity through inhibition of the HIF hydroxylases (169, 174). However, these findings are highly controversial, and other investigations reported that ROS production is decreased under hypoxic conditions or that ROS do not play a role in the regulation of HIF-α (16, 52).
Analysis of a possible functional interaction between hypoxia and oxidants revealed that there was a striking difference in the sensitivity of PHDs and FIH to hydrogen peroxide (108). FIH-dependent hydroxylation of HIF-1α and Rabankyrin 5 was inhibited by peroxide treatment in cellulo, whereas HIF-1α prolyl hydroxylation was not regulated at the same peroxide concentrations (108). HIF target gene expression was regulated accordingly and it was thus concluded that FIH serves as a cellular peroxide sensor (108). Iron chelators could protect against peroxide-mediated FIH inactivation, indicating that Fenton chemistry contributed to the FIH inhibition. The exact mode of inhibition remains to be elucidated, especially because reconstitution of FIH cofactors (including iron) could not reestablish FIH activity following peroxide treatment (108).
These results indicate that hypoxia and oxidant stress can functionally interact via regulation of FIH activity. However, the contribution of peroxide sensitivity to (patho-)physiological FIH functions in organisms is unclear.
FIH utilizes O2 for protein hydroxylation, for which oxygen has to be transiently converted into a highly reactive ferryl [Fe(IV) = O] intermediate. This mechanism could potentially lead to the release of ROS, especially if uncoupled from substrate hydroxylation. However, FIH apparently does not activate O2 in the absence of a substrate and no FIH-dependent ROS production could be detected (136).
In summary, these findings imply that hypoxia and oxidant stress can functionally interact via the regulation of FIH enzymatic activity. FIH activity in turn does not contribute to oxidant stress within cells, but FIH acts as an oxygen and peroxide sensor regulating HIF transactivation activity.
FIH Substrates Outside the HIF Pathway
A key question for the understanding of the cellular and organismal response to hypoxia is whether the activity of the oxygen sensing hydroxylases is limited to the modification and regulation of HIF-α proteins. A functional relevance of HIF-independent FIH signaling has been implied by the phenotypic analysis of whole-body HIF1AN knockout mice (195). The observed metabolic phenotype (see section “FIH in Organisms” and Figure 5) could not readily be explained by aberrant HIF activation, as these mice lacked classical signs of enhanced HIF activity such as increased angiogenesis, erythropoiesis, or glycolysis (195).
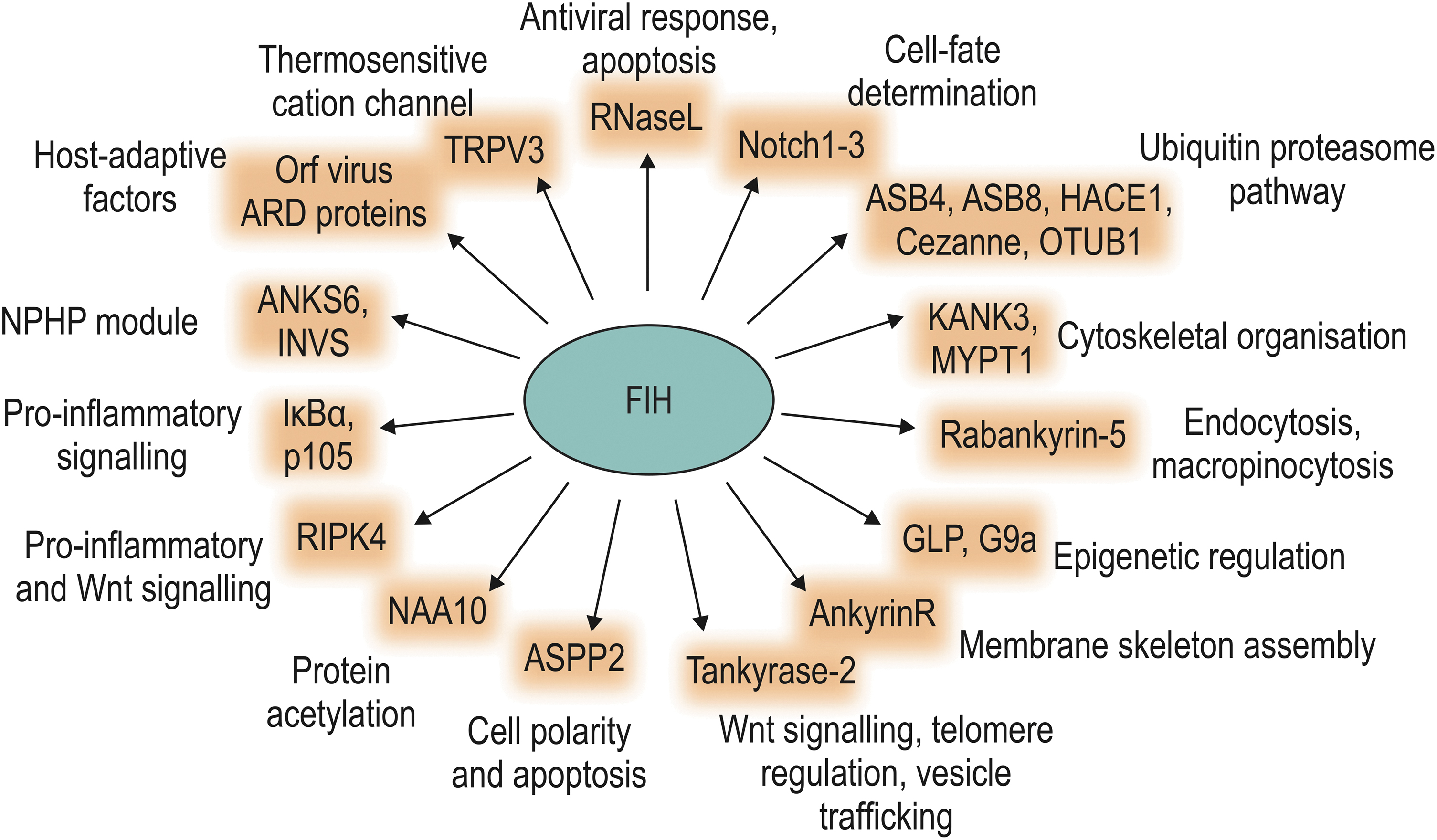
ARD-containing proteins were the first FIH substrates that were described outside the HIF pathway (17, 19, 20, 33, 89, 196). To date, several proteomic approaches have been performed to characterize the FIH interactome and to identify putative novel FIH substrate proteins, confirming that many ARD-containing proteins are FIH interaction partners and/or substrates, but also indicating that FIH interaction and/or activity are not restricted to ARD-containing proteins outside the HIF pathway (18, 68, 133, 146). Indeed, also non-ARD-containing proteins have been reported to be hydroxylated by FIH. This section summarizes the current knowledge about FIH substrates outside the HIF pathway (Table 1 and Fig. 4).
Bona Fide and Potential FIH Substrates Outside the HIF Pathway
FIH-dependent hydroxylation was demonstrated for full-length proteins or peptides in cellulo or in vitro. Interaction with FIH was confirmed by Co-IP from cultured cells or by pull-down of recProt. For some in vitro substrates, interaction with FIH was not determined in cellulo. FIH substrates are referred to as bona fide, if it was demonstrated that FIH enzymatic activity leads to oxidation of a specific amino acid of the investigated full-length protein in cellulo. Identification as bona fide substrate does not imply that the hydroxylation was linked to a mechanistic effect, which is not referred to in this table. Non-ARD proteins are given in bold.
Recently, it was reported that NAA10 is not hydroxylated by FIH in several human cell lines (131).
ANKS6, ankyrin repeat and sterile alpha motif domain containing 6; ARD, ankyrin repeat domain; ASB, ankyrin repeat and SOCS box protein; ASPP2, apoptosis-stimulating p53-binding protein; Co-IP, co-immunoprecipitation; FIH, factor inhibiting hypoxia-inducible factor; GLP, G9a-like protein; HACE1, HECT domain and ankyrin repeat-containing E3 ubiquitin protein ligase 1; IκBα, inhibitor of NF-κB α; INVS, inversin; KANK3, KN motif and ankyrin repeat domain-containing protein 3; MYPT1, myosin phosphatase target subunit 1; NAA10, N-alpha-acetyltransferase 10; n.d., not determined; NPHP, nephronophthisis; OTUB1, ovarian tumor domain containing ubiquitin aldehyde binding protein 1; recProt, recombinantly expressed protein; RIPK4, receptor-interacting serine/threonine-protein kinase 4; TRPV3, transient receptor potential vanilloid 3.
ARD-containing FIH substrates
In 2006, two proteins of the NF-κB signaling pathway were identified as FIH substrates (17). FIH hydroxylates inhibitor of NF-κB α (IκBα) and p105 at conserved asparagine residues located in ARDs (17), but it remained unclear if these hydroxylations have any functional consequence (17, 25). ARDs serve as a module for protein:protein interactions and represent one of the most commonly occurring protein motifs (116). One ankyrin repeat (AR) consists of 33 amino acids and is repeated between 1 and 33 times within a protein (116). Due to the widespread distribution and high conservation of ARDs, it was speculated that a number of other ARD-containing proteins might be substrates for FIH-mediated hydroxylation.
This was further supported by the finding that peptide fragments of several other ARD-containing proteins serve as FIH substrates in vitro (17). Subsequently, ankyrin repeat and SOCS box protein (ASB) 4 and Notch were found to be hydroxylated by FIH (20, 33, 176, 196).
Large-scale identification of additional ARD-containing FIH interactors was achieved by bioinformatic and proteomic approaches, such as a substrate trapping technique by which FIH interacting proteins were trapped within the active site of FIH in the presence of the hydroxylase inhibitor (HI) dimethyloxalylglycine (DMOG), which prevents substrate turnover and release (18, 133). This led to the discovery of a number of FIH interactors that cluster in distinct pathways, including NF-κB signaling or the protein ubiquitination pathway (133).
To date, FIH was found to hydroxylate more than 25 different proteins within their ARDs in vitro and a subset of these proteins were confirmed as endogenous FIH substrates or FIH interactors in cellulo. Some of the identified FIH targets were also hydroxylated by FIH at residues other than asparagine, including histidine (183), aspartate (184), or tryptophan (66).
The majority of FIH-mediated ARD hydroxylations have not been linked to a distinct molecular mechanism with functional outcome. However, there are examples highlighting the relevance of this posttranslational modification as a direct signaling component. For several proteins, it was shown that asparagine hydroxylation in the ARD stabilizes the ankyrin fold (44, 70), which can affect protein:protein interactions (184). FIH-dependent hydroxylation of ankyrin repeat and sterile alpha motif domain containing 6 (ANKS6) and inversin (INVS) alters protein interactions within a signaling module that is involved in kidney development and in the development of nephronophthisis (51).
The activity of the ion channel transient receptor potential vanilloid 3 (TRPV3) is inhibited by FIH-mediated hydroxylation, altering the TRPV3-mediated current, which may have implications for peripheral nociception and/or thermosensation (69). Hydroxylation of the receptor-interacting serine/threonine-protein kinase 4 (RIPK4) stimulates RIPK4 activity with functional consequences for the Wnt signaling pathway (133).
FIH also hydroxylates the histone methyltransferases G9a and G9a-like protein (GLP), which leads to their inhibition (67). In hypoxia, both enzymes were activated, leading to an increased invasiveness of ovarian tumors due to loss of metastasis suppressor gene expression (67). Finally, HECT domain and ankyrin repeat-containing E3 ubiquitin protein ligase 1 (HACE1) hydroxylation regulates the interaction of HACE1 with Rac1 (73). In hypoxia, the interaction of HACE1 and Rac1 is increased, enhancing Rac1 ubiquitination and proteasomal degradation, affecting breast cancer metastasis formation (73). Taken together, these studies show that FIH-dependent hydroxylation of ARDs can have distinct functional consequences.
Based on sequence alignments of HIF and ARD substrates, consensus sequences for recognition and hydroxylation by FIH have been suggested (19, 177, 184), which can be summarized as LXXXXX[D/E]VN (41). This fairly degenerated consensus sequence might serve for the identification of additional FIH substrates. However, it has been shown that some ARs fulfil the FIH target criteria based on this motif, but these proteins were not efficiently hydroxylated by FIH, whereas some bona fide target proteins lack some of the apparently conserved residues (177).
This indicates that other factors might be involved in target protein recognition, such as stability of the substrate protein structure or accessibility of the target site. Similar to the HIF C-TAD, which is unfolded in the absence of an interactor such as p300 or FIH (28, 36), also ARD proteins were shown to bind FIH with an induced fit that affects both FIH and ARD protein structure (20), indicating that structural flexibility might be an important determinant of the interaction of FIH with its substrates.
Other FIH substrates
Besides HIF-1α and HIF-2α, only a few proteins without ARDs have been reported to be hydroxylated by FIH. As described above, in the absence of a protein substrate, FIH can autohydroxylate itself to inhibit its own activity and to prevent substrate-uncoupled O2 activation, which would otherwise lead to ROS production (13, 14).
We previously identified ovarian tumor domain containing ubiquitin aldehyde binding protein 1 (OTUB1), a deubiquitinating enzyme, as an interaction partner of FIH (146). Our subsequent study revealed that OTUB1 is a bona fide FIH substrate, resulting in hydroxylation of Asn-22 (147). This hydroxylation strongly regulated the OTUB1 interactome and thus its substrate selection, affecting cellular energy metabolism (147). Constitutive global Otub1 deletion in mice is lethal due to asphyxiation after birth, caused by increased lung cell proliferation (135). Interestingly, induced global Otub1 deletion in adult mice led to hyperventilation (135), which has also been observed in Hif1an knockout mice (195). Moreover, induced global deletion of Otub1 causes a metabolic phenotype (134a) that is virtually identical to the phenotype reported for Hif1an knockout mice (195). Together, these results indicate that OTUB1 is a physiologically relevant target protein of FIH.
NAA10 (alternatively termed arrest-defective 1, ARD1) is the catalytic subunit of the N-terminal acetyltransferase NatA, which had previously been suggested to acetylate HIF-1α (61). NAA10 is hydroxylated by FIH at residue Trp-38 (66). This is the first report about the hydroxylation of a tryptophan residue by FIH. NAA10 hydroxylation enlarges a gate at the catalytic pocket, permitting NAA10 lysyl-acetyltransferase activity, which is thus regulated in an oxygen-dependent manner (66). However, it was recently reported that no evidence was found for Trp-38 hydroxylation of NAA10 in several human cell lines or for a physical or functional interaction between FIH and NAA10 (131).
A further identified non-ARD FIH target protein is another deubiquitinase, the protein Cezanne (also called OTUD7B). Cezanne is modified on Asn-35 within its N-terminal ubiquitin associated (UBA)-like domain and the Asn-35 hydroxylation inhibits ubiquitin binding (101). Thus, similar to the effect of OTUB1 hydroxylation, FIH-dependent hydroxylation of Cezanne also regulates substrate binding/targeting.
It is intriguing that two separate deubiquitinases have been identified as FIH substrates and that both proteins are hydroxylated in an ubiquitin-binding domain. Both enzymes belong to the ovarian tumor (OTU) protease family (39), which comprises 16 members (113). This raises the question whether also other OTU proteins are targeted by FIH and if (asparagine) hydroxylation is a more common mechanism for the regulation of protein:ubiquitin interaction.
The description of FIH substrates that do not belong to the family of ARD proteins expands the pool of potential FIH substrates. This is further supported by the analysis of the FIH interactome following substrate trapping (133). Of the 192 identified FIH interactors that represent potential substrate proteins, 41 were ARD proteins (133). This not only highlights the relevance of ARDs as FIH targets, but also indicates the presence of a considerable group of non-ARD FIH interactors. Whether these FIH interactors also serve as FIH substrates awaits further experimental clarification.
Overall, FIH clearly plays important roles also outside the HIF pathway, affecting a multitude of different targets, signaling pathways, and biological functions. However, for most of these substrates, it remains to be determined under which physiological or pathophysiological conditions the FIH-mediated regulation becomes relevant and how this multitude of signals act in concert with each other.
Oxomer (Oxygen-Dependent Stable Protein Oligomer) Formation
Mounting evidence suggests that FIH not only hydroxylates asparagine residues of proteins but also catalyzes other reactions, including the hydroxylation of aspartate, histidine, and tryptophan residues as well as the potential formation of β-oxo-histidine and formylglycine/enol or cyclic modifications of serine. We recently reported that FIH and OTUB1 form a stable denaturing condition-resistant protein/protein complex (FIH-OTUB1) catalyzed by FIH (127).
This FIH-OTUB1 “oxomer”—defined as an oxygen-dependent stable protein oligomer—is likely connected via a covalent amide bond (127). The formation of the oxomer occurrs cotranslationally, consists of two FIH and one OTUB1 protein in native conditions (FIH2-OTUB1), regulates OTUB1 enzymatic activity, and has an even higher hypoxia sensitivity than the PHD-mediated stabilization of HIF-1α and HIF-2α (127). This exquisite hypoxia sensitivity is in stark contrast to the known relatively low hypoxia sensitivity of FIH-dependent HIF-α hydroxylation (168). Hence, the oxygen- and FIH-dependent formation of an oxomer with OTUB1 may serve as an alternative mechanism for hypoxia sensing and adaptation.
Of note, the formation of this complex can be used as a readout for FIH activity in cellulo, which we recently used to characterize the PHD/FIH selectivity of all (at that time) available pharmacologic HIs (161). Interestingly, we identified twelve additional putative stable protein complexes formed by FIH with various proteins (127), indicating that oxomer formation by FIH is not exclusive for OTUB1.
Previously, a comparable SDS-PAGE-resistant stable protein complex had been reported between the 2-OG-dependent oxygenase superfamily member 2-OG and Fe(II)-dependent oxygenase domain containing protein 1 (OGFOD1) and the small ribosomal protein S23 (RPS23) (155). OGFOD1 is a prolyl-3-hydroxylase that hydroxylates RPS23 on proline 62, regulating general protein synthesis (53, 155). The formation of the oxomer was dependent on OGFOD1 enzymatic activity (155). However, the nature of the interaction, the oxygen sensitivity, and a possible functional consequence of OGFOD1-RPS23 complex formation are currently unclear.
Interestingly, an RPS23 point mutation was recently linked to a ribosomopathy in humans. Arg67Lys mutation impairs OGFOD1-dependent RPS23 hydroxylation and stable complex formation (121), indicating a possible relevance of oxomer formation in human disease.
In summary, the activity-dependent formation of (likely) covalent complexes between FIH and substrate proteins is a previously unknown catalytic activity of FIH that may represent a novel oxygen sensing mechanism for the adaptation of cells to hypoxia. Intriguingly, the formation of oxomers is likely neither restricted to FIH nor to the targeted amino acid asparagine.
FIH in Organisms
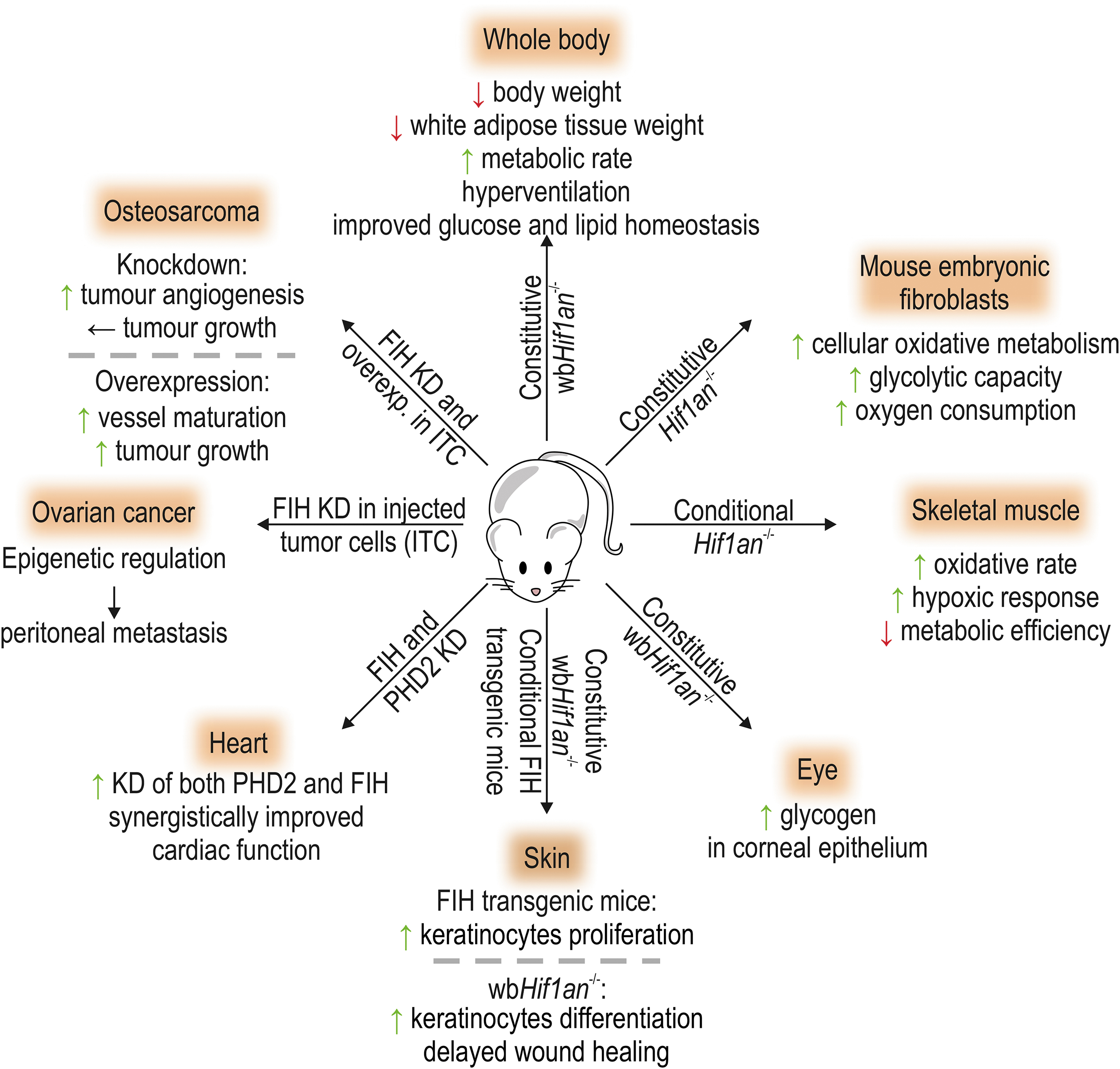
FIH expression was selectively preserved during evolution and can be found in primitive organisms, such as sea anemone and coral, and also in amphibians, fish, birds, reptiles, and mammals (41, 42, 166). In mammals, FIH is ubiquitously expressed with the highest tissue expression levels in the skeletal muscle (152, 195). FIH is thus an important oxygen sensor in most parts of the animal kingdom, but the variability of its conservation is unique in comparison with PHD1–3. FIH expression can also be changed in certain diseases, which is summarized in Table 2. In this section, we highlight the current knowledge about FIH in organisms. Figure 5 displays the present knowledge about the role of FIH in vivo, based on targeted alterations of FIH expression levels in mice.
FIH Messenger RNA and Protein Levels in Different Diseases or Conditions
miR-31, microRNA-31; mRNA, messenger RNA.
Energy metabolism
Constitutive global Hif1an knockout mice present with a reduced body weight gain during aging, a decreased white adipose tissue weight, hyperventilation, an elevated metabolic rate, as well as an increased insulin sensitivity (195). Moreover, these mice were resistant to high-fat diet-induced weight gain and hepatic steatosis. Neuron-specific Hif1an deletion recapitulated some of the metabolic phenotypes, whereas liver-specific Hif1an deletion did not influence the analyzed metabolic parameters (195). Gene expression analysis in brown adipose tissue indicated that Hif1an deletion enhanced thermogenesis.
In agreement with these results, miRNA-dependent negative regulation of FIH was linked to brown adipocyte differentiation and thermogenesis in mice (98, 192). MEFs lacking Hif1an showed an increase in cellular oxidative metabolism and glycolytic capacity combined with increased oxygen consumption. Metabolic adjustments in response to hypoxia were accelerated in the absence of Hif1an (152). Selective Hif1an deletion in the skeletal muscle promoted the oxidative rate and hypoxic response, whereas it decreased the metabolic efficiency (152).
FIH expression was increased in the corneal epithelium in murine diabetic models (123). In both corneal epithelial keratinocytes and limbal epithelial cells, FIH regulated glycogen metabolism (123, 126), and global Hif1an deletion increased glycogen levels in the limbal epithelium (126).
In elite athletes and following a 6-week bicycle training of moderately active individuals, FIH mRNA and protein levels were significantly increased in the skeletal muscle (vastus lateralis) compared with moderately active individuals or with the levels before training, respectively (88). This suggests that FIH serves the adaptation of the skeletal muscle to exercise (which generally includes hypoxia). In genome-wide association studies (GWAS), the HIF1AN gene locus was associated with body mass index (BMI) alterations in humans (96) and pigs (197), and with changes in the food conversion ratio in pigs (137). Polymorphisms in the HIF1AN gene locus were also linked to higher palmitoleic acid plasma levels in humans, which in turn is associated with type 2 diabetes and coronary heart disease (179).
In summary, FIH plays a key role in the regulation of cellular and whole-body energy metabolism in both mice and humans and FIH may be a novel therapeutic target for the treatment of metabolically driven diseases, such as obesity and type 2 diabetes.
High-altitude adaptation
GWAS in various human populations implicated polymorphisms of the HIF1AN gene locus in altitude adaptation in Tibetans (153), and in lowland and moderate-altitude Eurasian populations (62). The mechanistic effect of the observed polymorphisms remains to be elucidated. No association was found between acute mountain sickness and polymorphisms in the HIF1AN 3′-UTR or common HIF1AN haplotypes (191). It remains therefore unclear if polymorphisms of the HIF1AN gene locus play relevant roles for the adaptation to high altitude, and mechanistic analyses of the effect of the identified polymorphisms on FIH protein will need to be performed.
Heart
Using a minicircle-mediated knockdown of FIH and PHD2 following left anterior descending coronary artery ligation in mice, the combinatorial knockdown of both PHD2 and FIH in the myocardium led to a significantly greater improvement than each single knockdown (55). Studies analyzing miRNA-dependent FIH regulation support that a decrease in FIH levels is protective against cardiac tissue injury in mice and rats (172, 178, 180). Overall, these results suggest that inhibition of FIH may be cardioprotective.
Skin
FIH expression is increased in conditions of abnormal epithelial differentiation, including human patients with psoriasis, basal cell and squamous cell carcinomas, and also in the corneal epithelium of patients with diabetic keratopathies (124). Global ablation of Hif1an increased the differentiation of keratinocytes in the limbal epithelium (124) and delayed wound healing (125). Conditional FIH overexpression in keratinocytes increased keratinocyte proliferation (68). Therefore, FIH plays an important role in the response of the skin to injury and disease.
Placenta
HIF1AN mRNA is expressed in the placenta, its mRNA and protein levels increase with advancing gestation and peak at 11–12 weeks of gestation (57, 139). FIH mRNA levels were also increased in placentas from women who conceived, gestated, and delivered at high altitude in comparison with placentas from women who had stayed at sea level (190). However, FIH protein levels were decreased in high-altitude placentas (190). In early-onset preeclampsia, FIH mRNA and protein levels were found to be reduced (134, 139). Increased SUMOylation of FIH was suggested to lead to enhanced FIH protein degradation in preeclampsia (139).
The relevance of alterations of FIH protein levels for human placental and/or embryonic development remains unclear. Of note, mice with global Hif1an deletion propagate normally (195), demonstrating that FIH does not play a major role in murine placental or embryonic development. Whether murine global Hif1an deletion affects gestation under hypoxic conditions has not been analyzed to date.
Kidney
FIH was reported to be expressed in distal tubules and podocytes within the kidney (143). During experimental glomerulonephritis in rats, FIH mRNA in glomeruli gradually decreased with disease progression, which was associated with enhanced HIF target gene expression (143). FIH enzymatic activity was also associated with renal development and the development of nephronophthisis, and knockdown of FIH in Xenopus led to edema and tubular shortening (51). In a mouse model of chronic cyclosporin A-induced renal injury, FIH mRNA levels were increased, but HIF target gene expression was maintained (30).
In summary, FIH mRNA levels change during renal injury and FIH may mechanistically contribute to the outcome of kidney disease. However, this is only beginning to be understood and more analyses are necessary to elucidate the apparently complex function of FIH within the kidney and in renal diseases.
Cancer
Antitumor cytotoxic T cells play an important role in the defensive mechanism of the immune system against cancer. Ectopic HIF-2α expression in CD8+ T cells promoted their antitumor properties (171). Mutation of the FIH-targeted hydroxylation site in HIF-2α further enhanced their effectiveness as antitumor T cells, demonstrating the importance of FIH-dependent HIF-2α regulation in tumor immunity (171). These results further show that FIH is an important functional regulator of HIF-2α in CD8+ T cells, although it preferentially hydroxylates HIF-1α, highlighting the importance of assessing the function of FIH in the HIF pathway in a cell-type-specific manner in vivo.
In a murine osteosarcoma model, FIH knockdown (in the injected cancer cells) increased tumor angiogenesis, but had no effect on tumor development or progression (78). FIH overexpression in turn improved vessel maturation and stimulated tumor growth (78). These effects were likely caused by FIH-dependent regulation of platelet-derived growth factor (PDGF)-C (78). In ovarian cancer, FIH hydroxylates the histone lysine methyltransferases G9a and GLP, thereby indirectly exerting an epigenetic regulation (67). Analyses using murine xenograft models (with tumor cells that were modified in their G9a and FIH expression or expressed G9a containing a point mutation in its hydroxylation site) showed that FIH-dependent hydroxylation of G9a affected peritoneal metastasis formation (67).
Several different types of cancer have been analyzed for changes in FIH mRNA and/or protein levels, which are summarized in Table 2. GWAS investigated potential associations between polymorphisms of the HIF1AN gene locus and cancer. The single-nucleotide polymorphism (SNP) rs17094222 in the HIF1AN gene locus showed no association with breast cancer risk alone (199). In combination with a BMI of ≥24, however, heterozygosity for the SNP increased the risk of developing breast cancer (199). HIF1AN SNPs were not associated with the risk of developing overall or advanced prostate cancer (59). In clear cell renal cell carcinomas (RCCs), papillary RCCs, and oncocytomas, no somatic mutation was found in FIH (115).
Thus, no direct association between the HIF1AN locus and cancer exists to date, but functional studies indicate that FIH may nonetheless be an important modulator of (tumor and antitumor) cell behavior, affecting tumor development and metastasis formation.
FIH functions in fish
Several studies have been performed to analyze the levels and the role of FIH in various fish species. In zebrafish (Danio rerio), knockdown of FIH increased angiogenesis, whereas augmented FIH expression decreased it (157). This phenotype was suggested to be caused by altered HIF-dependent expression of vascular endothelial growth factor-a (VEGF-A) (157). Another investigation of the role of FIH in zebrafish found that CRISPR/Cas9-mediated Hif1an deletion increased the hypoxia tolerance, HIF target gene expression, and decreased the number of apoptotic cells in the brain after hypoxic exposure (5). Overexpression of FIH decreased HIF-mediated gene expression (5). The highest endogenous FIH expression was observed within the liver (5).
In the bighead carp (Aristichthys nobilis), the highest level of FIH mRNA was found in muscle (32). Following hypoxia, FIH mRNA was increased in the gill, liver, spleen, and hypothalamus (32). Following hypoxic exposure, FIH mRNA was also reported to be increased in the Asian sea bass (Lates calcarifer) in the gill, spleen, and heart (187), in the Nile tilapia (Oreochro mis niloticus), in the gill, spleen, heart, and brain (85), and in the developing tropical gar Atractosteus tropicus (106).
The observed phenotypes for Hif1an knockdown/knockout in (zebra)fish differ from the observations made in Hif1an knockout mice. In fish, the FIH-dependent regulation of HIF transactivation activity appears to be FIH's primary function. This may be due to a differential evolutionary conservation of FIH target sites, as, for example, the OTUB1 hydroxylation site is only evolutionarily conserved in mammals, but is greatly altered in fish (147).
In summary, FIH plays a key role for the regulation of energy metabolism. Interestingly, FIH expression is altered under many different disease conditions, indicating that FIH is relevant in these diseases. However, the role of FIH in diseases is poorly understood and has not been largely investigated yet. Nonetheless, the available in vivo data about the tissue protective effects of FIH ablation justify preclinical research on pharmacologic FIH antagonists.
Therapeutic Potential of Pharmacologic FIH Inhibition
In December 2018, FG-4592 (roxadustat) became the first HIF HI to be approved for the treatment of Chinese patients with renal anemia (26). Several further HIs have been approved in China and Japan since (40). In addition, HIs were shown to be protective in animal models of several other diseases, ranging from inflammatory bowel disease over ischemia/reperfusion injury to kidney inflammation and fibrosis, and even cancer (7, 21, 29, 102, 109). However, the HI protective effect is scarcely understood and likely not only due to the regulation of HIF activity (29, 146, 162, 165). Furthermore, the HIF hydroxylase selectivity of these compounds is largely unclear (7, 161), delaying the translation of the preclinical findings into clinical trials.
A recently developed method by us allowed first insights into the HI selectivity toward the PHDs and FIH in cellulo (161), but it cannot yet distinguish between the different PHD isoforms. Hence, a better understanding of HI-regulated proteins and signaling pathways as well as of the pharmacologic selectivity is urgently needed. Pharmacologic inhibition of FIH has recently been expertly reviewed (181) and is only briefly summarized here.
It is possible to selectively inhibit FIH over the PHDs, as demonstrated with the compound N-oxalyl-D-phenylalanine (NOFD) (110) and its dimethyl derivative (DM-NOFD), which penetrates cell membranes (8, 161).
Recently, the (to our knowledge) first use of NOFD was reported in animals. Repeated intraperitoneal injection of NOFD conferred radioprotection to the gastrointestinal tract, which was suggested to be caused by increased HIF-1 transactivation activity (186). Analyzing the effect of 2-OG derivatives on FIH, 10 compounds were found to enhance FIH activity, whereas 17 compounds selectively inhibited FIH (118). These results highlight that there is a large potential for the development of selective compounds that specifically regulate FIH activity. Moreover, FIH activity may thus be regulated by natural 2-OG derivatives in cellulo, which should be further explored in the future.
The important role of FIH in the regulation of energy metabolism and the indications for its functions in other diseases combined with the feasibility of selectively inhibiting FIH demonstrate a great therapeutic potential, which needs to be characterized and harnessed in the future. Current inhibitors that are used in the clinics for the treatment of renal anemia and that target the HIF hydroxylases are to the best of our knowledge PHD selective (and are thus called PHD inhibitors or PHIs) (161, 188). These inhibitors have proven to be highly efficient for the activation of erythropoietin (Epo) expression.
So far, no major side effects have been identified that would prevent their clinical use, but PHIs are not long enough in the clinics to allow sufficient conclusions (40). Epo is apparently not an FIH target gene (195), and thus, PHIs are effective and sufficient for the treatment of renal anemia.
Preclinical studies have shown that HIs may also be novel treatment options for other hypoxia-associated diseases, including ischemia/reperfusion injury, (chronic) inflammation, fibrosis, and metabolic diseases. However, for the treatment of such diseases, it would not be favorable to induce Epo expression and red blood cell production. FIH inhibiting compounds allow the increase of (selective) HIF target gene expression while omitting Epo, which may be a safer option for future therapies of such diseases compared with the PHIs that are currently on the market.
Conclusions
The cellular oxygen and peroxide sensor FIH is a highly promiscuous enzyme catalyzing not only the asparagine hydroxylation of HIF-α subunits, but also posttranslational modifications of other targets outside the HIF pathway. The regulation of FIH enzymatic activity occurs mainly through the availability of its substrates, particularly of O2, with contribution of alterations of its protein levels via miRNA-mediated regulation of its mRNA and ubiquitination-mediated proteasomal degradation, respectively. The most prominent physiological function of FIH is the regulation of metabolic homeostasis, but FIH plays also a role in altitude adaptation and in the damage response of the skin with further functions currently emerging. FIH expression is differentially regulated in various types of cancer, but the relevance of the observed alterations remains largely unclear.
In general, the pathophysiological function of this unique oxygen sensor is rather poorly understood. Nonetheless, the existing data demonstrate that FIH is a likely novel therapeutic target in metabolically driven diseases and it has been demonstrated that FIH-selective pharmacologic inhibition is feasible. Expanding our knowledge on molecular mechanisms underlying known FIH functions, on the contribution of FIH to pathologies, and on extending the pharmacologic substances targeting FIH, appear to be the prime tasks of research on this intriguing enzyme in the near future.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
There is no funding agency that needs to be acknowledged.