
Abstract
Significance:
Autophagy is critical to cellular homeostasis. Emergence of the concept of regulated necrosis, such as necroptosis, ferroptosis, pyroptosis, and mitochondrial membrane-permeability transition (MPT)-derived necrosis, has revolutionized the research into necrosis. Both altered autophagy and regulated necrosis contribute to major human diseases. Recent studies reveal an intricate interplay between autophagy and regulated necrosis. Understanding the interplay at the molecular level will provide new insights into the pathophysiology of related diseases.
Recent Advances:
Among the three forms of autophagy, macroautophagy is better studied for its crosstalk with regulated necrosis. Macroautophagy seemingly can either antagonize or promote regulated necrosis, depending upon the form of regulated necrosis, the type of cells or stimuli, and other cellular contexts. This review will critically analyze recent advances in the molecular mechanisms governing the intricate dialogues between macroautophagy and main forms of regulated necrosis.
Critical Issues:
The dual roles of autophagy, either pro-survival or pro-death characteristics, intricate the mechanistic relationship between autophagy and regulated necrosis at molecular level in various pathological conditions. Meanwhile, key components of regulated necrosis are also involved in the regulation of autophagy, which further complicates the interrelationship.
Future Directions:
Resolving the controversies over causation between altered autophagy and a specific form of regulated necrosis requires approaches that are more definitive, where rigorous evaluation of autophagic flux and the development of more reliable and specific methods to quantify each form of necrosis will be essential. The relationship between chaperone-mediated autophagy or microautophagy and regulated necrosis remains largely unstudied. Antioxid. Redox Signal. 38, 550–580.
Introduction
Macro-autophagy (hereafter referred to autophagy) is a highly conserved process that targets the cargos of autophagosome for degradation by lysosomes. It is involved in a wide range of physiological and pathological processes, such as development, immunity, infection, and aging (Glick et al, 2010). Autophagy occurs constitutively at basal level and increases significantly under stress conditions. On starvation, autophagy is responsible for recycling portion of cytoplasm to provide energy and metabolites for the cell.
Meanwhile, it also ameliorates cellular stress by removing aberrant protein aggregates and damaged organelles, which makes autophagy a highly selective quality-control mechanism (Parzych and Klionsky, 2014). Thus, autophagy is primarily a cytoprotective mechanism and prevents cell death (Shimizu et al, 2014). However, autophagy also contributes to cell death. The role of autophagy in cell death can be defined as two major forms: (1) autophagy-mediated cell death, where autophagy triggers apoptosis or necrosis; (2) autophagy-dependent cell death recently refers to autosis (Denton and Kumar, 2019).
Morphologically, cell death is classified into apoptosis and necrosis. A cell undergoing apoptosis in tissues keeps its membrane integrity. Even at the late stage when the apoptotic cell breaks into smaller pieces, each of these pieces known as apoptotic bodies is enclosed within cellular membranes. Tissue phagocytes eventually uptake the apoptotic bodies and degrade them (Del Re et al, 2019). Thus, the intracellular content of apoptotic cells does not come into direct contact with the extracellular space; as such, apoptosis does not trigger inflammation.
A cell undergoing necrosis, however, loses its cell membrane integrity and swells, leading to plasma membrane rupture and the leakage of intracellular components to the extracellular space and thereby triggering in inflammatory responses (Lewno et al, 2021). Necrosis was previously assumed as a form of cell death that is incidental or accidental and not regulated, which is distinct from apoptosis that orderly engages caspases activation and can be intervened at various stages. In contrast to necrosis, apoptosis presents morphological features, including cell shrinkage, nuclear fragmentation, and chromatin condensation (Proskuryakov et al, 2003; Su et al, 2015; Tonnus et al, 2021).
Recent advances in cell biology overturned the long-standing paradigm in necrosis and unveiled that at least some of the necrosis are reversibly programmed molecular events, referred to as regulated necrosis. There are many forms of regulated necrosis, such as necroptosis, ferroptosis, pyroptosis, and mitochondrial membrane-permeability transition (MPT)-dependent necrosis. Among them, necroptosis is the earliest one described.
Tumor necrosis factor alpha (TNFα) via TNF receptor 1 (TNFR1) usually causes apoptosis. However, in caspase 8 deficient or suppressed cells (e.g., L929 cells), TNFα treatment did not induce apoptosis but triggered a unique type of regulated necrosis that was coined as necroptosis (Degterev et al, 2005). Subsequent studies revealed that necroptosis can be blocked by a small molecular compound necrostatin-1 (Nec-1) that turned out to be a specific inhibitor of the kinase activity of receptor interacting protein kinases 1 (RIPK1) (Degterev et al, 2005).
More recent studies revealed that RIPK3-mediated phosphorylation of mixed lineage kinase domain like psudokinase (MLKL) and resultant oligomerization of the phosphorylated MLKL at the cell membrane are central to the necroptotic pathway (Wang et al, 2014) and, in some circumstances, RIPK1 is not required for necroptosis. For example, the activation of Toll-like receptor 3 (TLR3) and TLR4 respectively by poly(I:C) and lipopolysaccharide (LPS) induced macrophages to undergo necroptosis through engaging RIPK3 by an adaptor protein Toll/interleukin-(IL)-1 receptor domain-containing adapter inducing interferon (IFN)-β (TRIF) (He et al, 2011). Nevertheless, RIPK1 remains a key player in the canonical necroptotic pathway (i.e., RIPK1-RIPK3-MLKL pathway) triggered by TNFα (Wu et al, 2021a; Xiao et al, 2020).
Ferroptosis is an iron-dependent and reactive oxygen species (ROS)-reliant cell death, which was coined in 2012 by Stockwell's group. They characterized this distinct form of regulated necrosis when they conducted a phenotypic screen for small molecules that were selectively lethal to tumor cells overexpressing oncogenic mutation of HRAS (Dixon et al, 2012). Three core hallmarks of ferroptosis are defined, including (1) oxidation of polyunsaturated fatty acid (PUFA)—containing phospholipids (PLs), (2) redox-active iron, and (3) deficiency of lipid peroxide repair (Dixon and Stockwell, 2019).
PUFA oxidation is essential for the execution of ferroptosis by forming structured lipid pores on the plasma membrane. Free iron or iron-containing lipoxygenase enzymes mediate oxidization of PUFAs, leading to the lipid peroxidation. Lipid hydroperoxidase, glutathione peroxidase 4 (GPX4) maintains the endogenous redox balance by eliminating the iron-catalyzed PL-PUFA oxidation; hence, GPX4 deficiency leads to ferroptosis.
Pyroptosis is a more recently identified necrotic and inflammatory cell death, triggered by inflammasomes (Fang et al, 2020). It is stimulated by a range of microbial infections and non-infectious stimuli (Bergsbaken et al, 2009). Pyroptosis shares some morphological characteristics with apoptosis, such as DNA damage, chromatin condensation, and caspase activation but has its own features. First, pyroptosis-related DNA damage does not require the participation of caspase-activated DNase (CAD) and the nucleus keeps its integrity (Bergsbaken et al, 2009). Second, pyroptosis features with rapid plasma-membrane rupture mediated by caspase-activated gasdermins (GSDMs, the executer) (Ding et al, 2016; Liu et al, 2016c). Moreover, pyroptotic activation of caspases does not induce poly(ADP)-ribose polymerase (PARP) cleavage and associated ATP depletion (Ha and Snyder, 1999).
The MPT-driven necrosis is characteristic of necrotic morphology and proinflammatory effects, which are triggered by severe perturbations of the intracellular redox level or cytosolic homeostasis of Ca2+ (Galluzzi et al, 2018; Izzo et al, 2016). MPT is precipitated by the sudden loss of impermeability to small solutes of the inner mitochondrial membrane (IMM), which is mediated by a supramolecular complex, permeability transition pore complex (PTPC) (Galluzzi et al, 2018; Izzo et al, 2016). PTPC assembles between the IMM and outer mitochondrial membrane (OMM) whose opening triggers the MPT-driven necrosis (Bonora et al, 2015; Vanden Berghe et al, 2014).
Cyclophilin D (CYPD; also known as peptidylprolyl isomerase F) is critically significant for MPT induction (Baines et al, 2005; Schinzel et al, 2005). Genetic or chemical inhibition of CYPD prevents MPT-driven necrosis (Baines et al, 2005; Kwong and Molkentin, 2015; Mukherjee et al, 2016).
Coexistence of autophagy and cell death was widely revealed long time ago, but studies to delineate the mechanistic relationship between autophagy and regulated necrosis have just begun to emerge. The dual roles of autophagy also intricate its relationship with regulated necrosis. In this review, we analyze the crosstalk between autophagy and regulated necrosis and illustrate the mechanisms and associated factors connecting these important processes.
Autophagy
Autophagy is an evolutionarily conserved catabolic process, occurring at a basal level in all cells. In response to the stimuli, autophagy is initiated through recruiting autophagy related gene (ATG) proteins to phagophore assembly site (PAS), following nucleation of the isolation membrane to form phagophores. Then, the phagophore elongates by recruiting isolation membrane and forms a double-membraned vesicle, termed an autophagosome. During the process of expansion, autophagosome engulfs cytoplasmic materials. After fusion with the lysosome, autophagosome matures as autolysosome and concludes with degradation of intra-vesicular products (Dikic and Elazar, 2018; Levy et al, 2017) (Fig. 1).
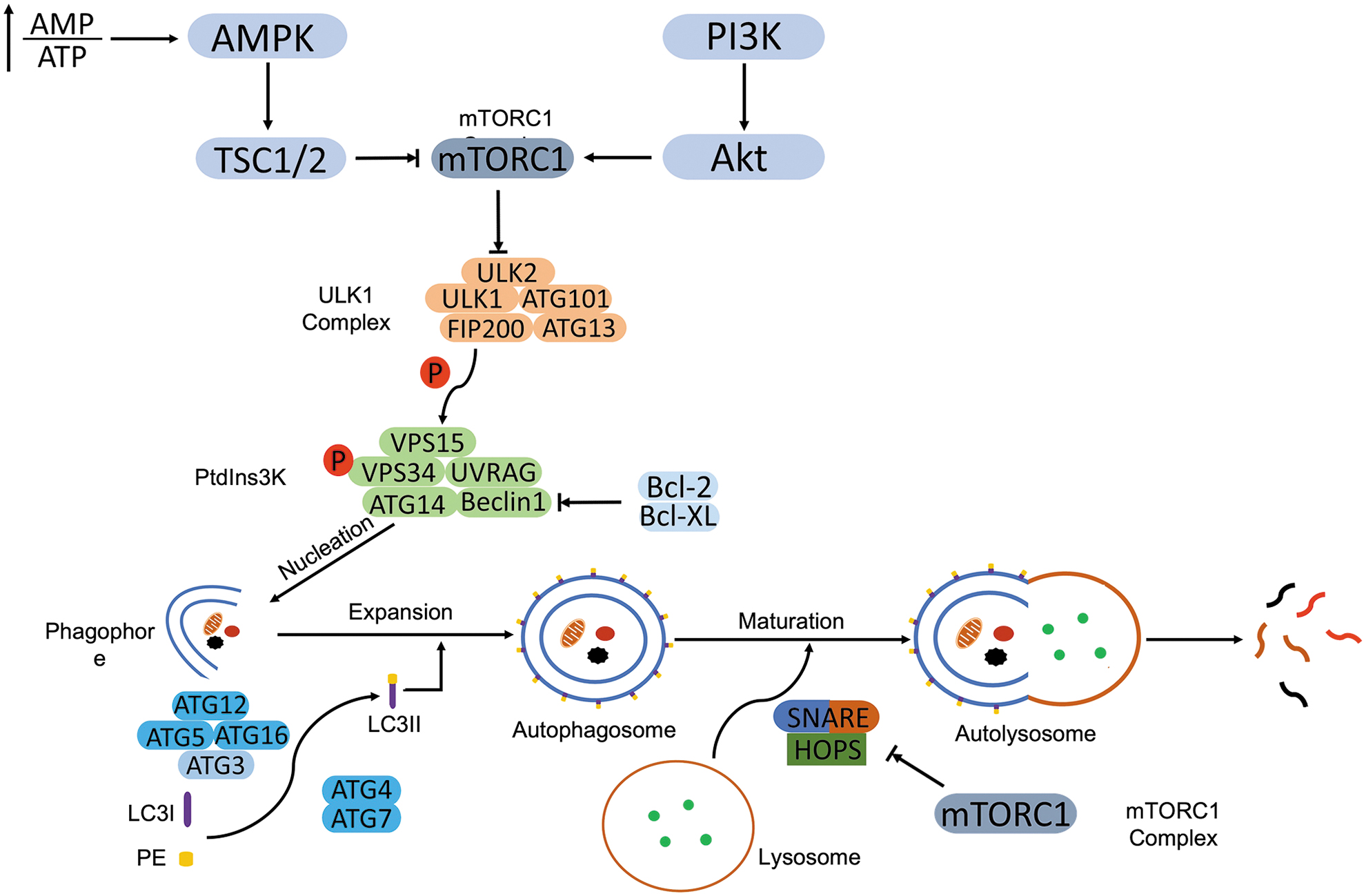
Initiation of autophagy starts from the activation of the Unc-51 like autophagy activating kinase 1 (ULK1) complex, which consists of ULK1, ULK2, ATG13, 200 kDa FAK family kinase-interacting protein/RB1 inducible coiled-coil 1 (FIP200/RB1CC1), and ATG101 (Mercer et al, 2009). The ULK1 complex phosphorylates the class III PI3K complex (Ptdlns3K) to facilitate the formation of the phagophore. PtdIns3K is composed of vacuolar protein sorting 15 (VPS15)/PIK3 regulatory subunit 4 (PIK3R4), VPS34/PIK3 catalytic subunit type 3 (PIK3C3), ATG14, p63/UV radiation resistance-associated gene protein (UVRAG), and Beclin-1 (Rashid et al, 2015), the scaffold protein for the PtdIns3K proteins (Liang et al, 1999). B cell lymphoma-2 (Bcl-2) or Bcl-XL counteracts the autophagy initiation by directly binding to Beclin-1 through BH3 domain and restricts the construction of PtdIns3K (Liang et al, 1998).
Phagophore expansion requires the involvement of ubiquitin-like (UBL) proteins, ATG12, ATG5, and ATG16. The ATG12-ATG5-ATG16 complex mediates the recruitment and conversion of microtubule-associated protein 1 light chain 3 (LC3) from LC3-I to its lipidated form, LC3-II (marker of autophagy) (Hanada et al, 2007). The cysteine protease activity of ATG4 promotes the cleavage of nascent LC3, following conjugation with phosphatidylethanolamine (PE) and subsequent incorporation into the phagophore membrane with the assistance of ATG7 and ATG3 (Glick et al, 2010; Ohsumi, 2001).
ATG4 also negatively regulates the expansion of phagophore by deconjugating LC3 from PE (Scherz-Shouval et al, 2019). Both activities of ATG4 are needed for the orchestration of autophagy progression. Fusion of autophagosome with lysosomes indicates the maturation of the autophagosome, though the detailed mechanism of this dynamic process is still elusive. Microtubules and motor proteins are required to transport autophagosomes to lysosomes (Olsvik et al, 2015), and the formation of autolysosome is executed by a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex.
The SNARE complex contains syntaxin 17 (Stx17) and synaptosomal-associated protein 29 (SNAP29) on the autophagosome and vesicle associated membrane protein 8 (VAMP8) on the lysosome (Diao et al, 2015; Itakura et al, 2012). The homotypic fusion and protein sorting (HOPS) complex holds the two vesicles together and facilitates the SNARE-mediated fusion (Jiang et al, 2014; Takats et al, 2014). Finally, the sequestered cargo in the autophagosome is hydrolyzed by lysosomal hydrolases and recycled for metabolic pathways.
The identification of autophagy receptors remodifies the autophagy from a nonselective process to a selective process. Through different autophagy receptors, autophagy recognizes and degrades distinct cargoes such as protein aggregates (aggrephagy) and mitochondria (mitophagy) (Deng et al, 2017). For example, p62/sequestosome 1 (SQSTM1) recognizes ubiquitin or polyubiquitin chains and delivers polyubiquitinated cargoes to autophagy for degradation (Myeku and Figueiredo-Pereira, 2011).
Mechanistic target of rapamycin complex 1 (mTORC1) is a master regulator of autophagy. In normal nutrient status, mTORC1 directly interacts with ATG13 and ULK1, and it suppresses their activation through phosphorylation. On amino acid deprivation, inactive mTORC1 dissociates ULK1, which autophosphorylates to self-activate, followed by phosphorylation of ATG13 and FIP200 to initiate autophagy (Hosokawa et al, 2009; Jung et al, 2009). Besides a role in the initiation of autophagy, mTORC1 is also involved in the regulation of autophagosomal maturation.
Kim et al (2015b) demonstrated that recruitment of the HOPS complex to the autophagosome was mediated by UVRAG, which is negatively regulated by mTORC1. mTORC1 also suppresses a key regulator of autophagy, transcription factor EB (TFEB), that facilitates the expression of genes involved in lysosomal biogenesis and lipid catabolism on starvation (Settembre et al, 2013).
Glucose deprivation-induced autophagy partially depends on the inactivation of mTORC1. Serine/threonine-protein kinase 11 (STK11)/liver kinase B1 (LKB1) and 5′ AMP-activated protein kinase (AMPK) sense the glucose starvation-caused decline of ATP/AMP ratio and activate autophagy through indirect inhibition of mTORC1 (Gurumurthy et al, 2010). LKB1-mediated AMPK activation (through phosphorylation) phosphorylates tuberous sclerosis 2 (TSC2) whose activation inhibits mTORC1 through phosphorylation (Tripathi et al, 2013).
In addition, AMPK-induced autophagy can also bypass mTORC1 through directly phosphorylating ULK1, VPS34, and Beclin-1 (Kim et al, 2011). mTORC1 is also a downstream target of the phosphatidylinositol 3 kinase (PI3K) and kinase AKT, also known as protein kinase B (AKT) pathway. Activation of the PI3K/AKT/mTORC1 pathway inhibits autophagy (Heras-Sandoval et al, 2014).
Regulated Necrosis
Necroptosis
Multiple stimuli have been identified that are capable of activating necroptosis, including members of the TNFR superfamily (Degterev et al, 2005; Moujalled et al, 2013), pattern recognition receptors (PRRs) (Amarante-Mendes et al, 2018), T cell receptors (TCRs) (Osborn et al, 2010), multiple chemotherapeutic drugs (Kim et al, 2017; Lalaoui et al, 2015), and environmental stresses such as hypoxia (Huang et al, 2013). Since TNFα/TNFR1-induced necroptosis is the most intensively investigated model, we illustrate the molecular events of necroptosis activation through this model (Fig. 2). TNFα ligation triggers a conformational change of TNFR1 trimers, which recruit related proteins and form a membrane-bound multimeric protein complex, known as complex I.
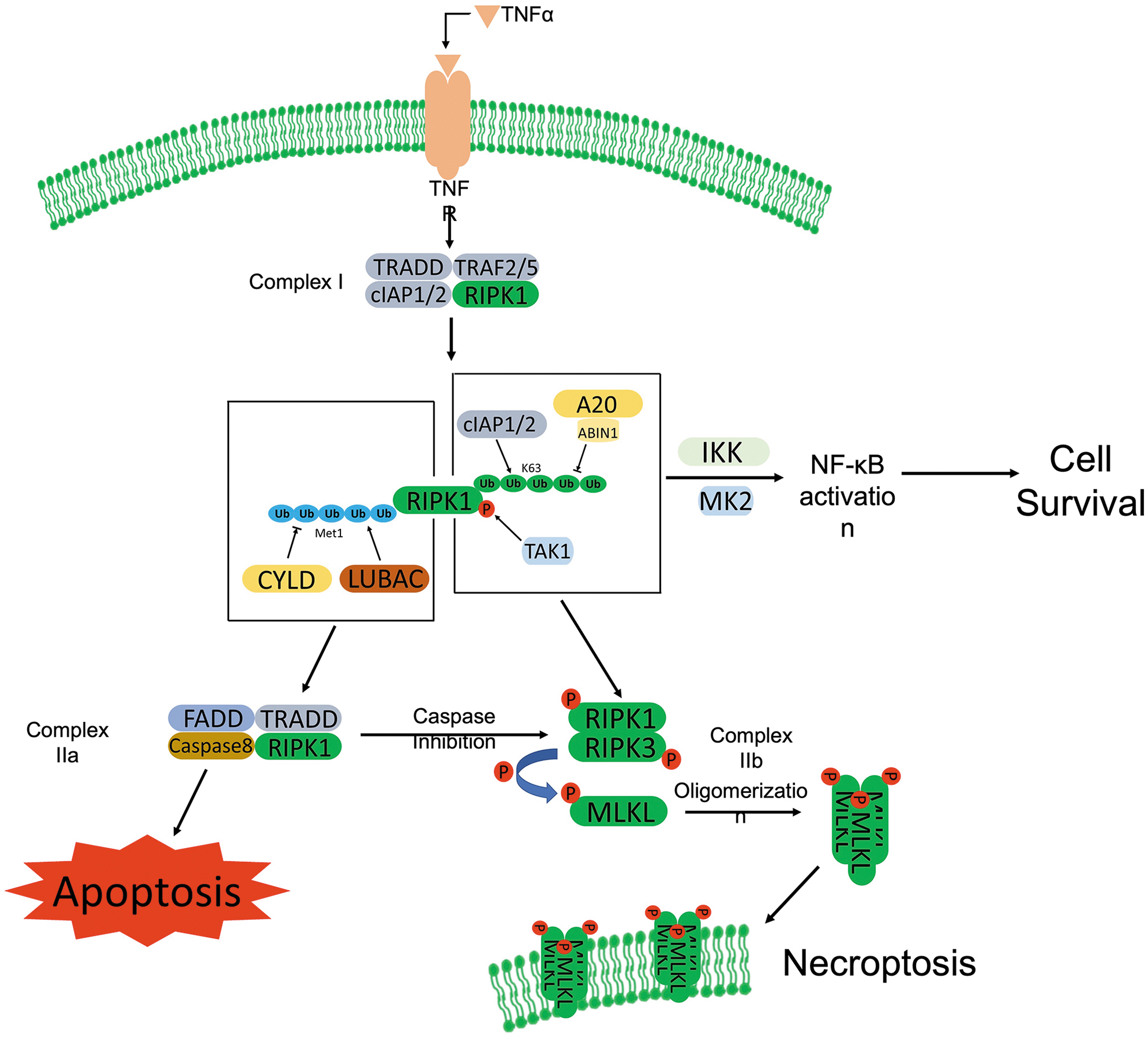
The complex I consists of RIPK1, TNFR-associated death domain (TRADD), cellular inhibitor of apoptosis protein 1 (cIAP1), cIAP2, TNF receptor-associated factor 2 (TRAF2), and TRAF5 (Bertrand et al, 2008; Micheau and Tschopp, 2003; Vandenabeele et al, 2010). As the crucial regulator of necroptosis, RIPK1 is inactivated via cIAP1/2-mediated K63-linked-ubiquitination (Bertrand et al, 2008), which inhibits the necroptosis, but promotes survival through the activation of nuclear factor kappa light chain enhancer of activated B cell (NF-κB) pathway (Fulda, 2013).
The subsequent internalization of TNFα/TNFR1 promotes de-ubiquitination of RIPK1 catalyzed by the de-ubiquitinase, cylindromatosis (CYLD) (Trompouki et al, 2003). The de-ubiquitinated RIPK1 interacts with caspase-8, TRADD, and FAS-associated death domain protein (FADD) to form complex IIa, which triggers apoptosis (Shan et al, 2018). Activated caspase-8 not only initiates pro-apoptotic caspase activation cascade, but it also cleaves RIPK1 and RIPK3 to inhibit necroptosis (Kaiser et al, 2011; Newton et al, 2019).
Inhibition of caspase-8 blocks the cleavage of RIPK1 and RIPK3, which promotes the formation of complex IIb with recruitment of RIPK3 to RIPK1. RIPK1 is activated through autophosphorylation (Degterev et al, 2005) and interacts with RIPK3 through RIP homotypic interaction motifs (RHIMs), activating RIPK3 through phosphorylation (Cho et al, 2009b). The activated RIPK1 and RIPK3 form a heterodimer, named necrosome, which phosphorylates the executer, MLKL (Linkermann and Green, 2014). MLKL oligomerizes and translocates to the plasma membrane to form pores, leading to loss of membrane integrity and thereby necrosis (Murphy et al, 2013).
The RIPK1-RIPK3-MLKL pathway
RIPK1 is a protein of 671 amino acids, containing an N-terminal kinase domain, an intermediate domain, and a C-terminal death domain (Ofengeim and Yuan, 2013). Activation of RIPK1 is important in regulated cell death. Ubiquitination status regulates the activation of RIPK1 (Zhang et al, 2019b). RIPK1 is inactivated by cIAP1/2-mediated K63-linked ubiquitination (Feoktistova et al, 2011), and the latter recruits the TGFβ activating kinase 1 binding protein (TAB)/TGFβ activating kinase 1 (TAK1) complex, as well as the IκB kinase (IKK) complex to activate NF-κB signaling and promote cell survival (Ea et al, 2006; Li et al, 2006). In addition, the K63 ubiquitination also recruits the linear ubiquitination assembly complex (LUBAC) (Draber et al, 2015) and deubiquitinating enzyme, CYLD to RIPK1 (Kovalenko et al, 2003).
CYLD deubiquitylates LUBAC-formed Met1 (M1)-linear ubiquitin chains, but it does not affect the K63-linked ubiquitin chains. Through ABIN-1, a ubiquitin-binding protein, the M1-ubiquitin linear chain further recruits ubiquitin-editing enzyme, A20, deubiquitinating K63-linear chain of RIPK1 (Dziedzic et al, 2018). The ratio of K63/M1 ubiquitination of RIPK1 determines cell fate. Enhanced K63 ubiquitination promotes RIPK1-dependent necroptosis, whereas a higher M1 ubiquitination protects against apoptosis and necroptosis after TNFα stimulation (Draber et al, 2015; Dziedzic et al, 2018). Moreover, A20 also acts as an E3 ligase to ubiquitinate RIPK1 with K48-linked chains, which promotes RIPK1 degradation through the proteasome (Wertz et al, 2004).
Phosphorylation status also determines the function of RIPK1 in the regulated cell death. K63-ubiquitination of RIPK1 is required for the phosphorylation of RIPK1 (Geng et al, 2017). Autophosphorylation of RIPK1 at Ser166 activates RIPK1, and it enhances the kinase activity of RIPK1, promoting the kinase-dependent apoptosis or necroptosis (Laurien et al, 2020). TAK1 transiently phosphorylates RIPK1 at Ser321 of the intermediate domain on TNFα stimulation. The phosphorylation status of RIPK1 at Ser321 determines the cell fate. Blocking the phosphorylation of RIPK1 at Ser321 enhances the interaction of RIPK1 with FADD and promotes RIPK1 dependent apoptosis.
Transient phosphorylation of RIPK1 at Ser321 facilitates RIPK1-independent apoptosis. Sustained phosphorylation of RIPK1 promotes activation of RIPK3 and necroptosis (Geng et al, 2017). Mitogen-activated protein kinase (MAPK) pathways also play an important role in regulating RIPK1. Besides TAK1, MAPK-activated protein kinase 2 (MK2) also directly phosphorylates RIPK1 at Ser321 in response to pro-inflammatory stimuli and infection. p38 MAPK/MK2 mediated RIPK1 phosphorylation inhibits RIPK1 autophosphorylation and suppresses RIPK1-dependent apoptosis and necroptosis (Menon et al, 2017). In addition, IKK kinase complex interacts with RIPKs and phosphorylates RIPK1, which inhibits RIPK1-dependent cell death and promotes the activation of NF-κB signaling and associated cell survival (Dondelinger et al, 2015).
Ubiquitination at K5 of RIPK3 is required for RIPK3 to form the necrosome with RIPK1. A20 as a deubiquitinase is also involved in removing poly-ubiquitin chains from RIPK3, which restricts necroptosis through inhibiting necrosome formation (Onizawa et al, 2015). The expression of RIPK3 is negatively regulated by carboxyl terminus of heat shock protein 70 (HSP70)-interacting protein (CHIP). CHIP-mediated ubiquitination promoted RIPK3 degradation through lysosomes in response to TNFα (Seo et al, 2016). Meanwhile, CHIP was also involved in the negative regulation of RIPK1 expression (Seo et al, 2016), suggesting a regulatory role of CHIP in necrosome formation.
MLKL contains an N-terminal four helix bundle domain and a C-terminal pseudo-kinase domain, which are connected by a flexible brace (Flores-Romero et al, 2020). MLKL requires RIPK3-mediated phosphorylation at the Thr357 and Ser358 sites to form oligomers and to perforate the plasma membrane (Wang et al, 2014).
RIPK3-CamKII-MPT pathway
In addition to being a critical linkage in the RIPK1/RIPK3/MLKL-mediated necroptotic pathway, RIPK3 was also shown to induce myocardial necroptosis through activating Ca2+ and calmodulin-dependent protein kinase II (CamKII), independently of RIPK1 and MLKL, under an ischemia/reperfusion (I/R) condition. CamKII is a multifunctional protein kinase that couples disease stress to mitochondrial injury through triggering MPT pore opening in I/R induced myocardial injury (Joiner et al, 2012). Zhang et al (2016) revealed that RIPK3-mediated direct phosphorylation at Thr287 and indirect oxidation at Met281/282 equally contributed to the activation of CamKII in RIP3K-CaMKII-dependent myocardial necroptosis.
Here, activated CamKII triggered the opening of MPT pores and led to the I/R induced cardiac cell death signaling cascade (Zhang et al, 2016). Dynamin related protein 1 (DRP1) was demonstrated to be a downstream factor that mediated CamKII dependent MPT pore opening. Xu et al (2016) showed that chronic β-adrenergic receptor (β-AR) stimulation promoted CamKII-mediated phosphorylation of DRP1 at Ser 616, subsequently facilitating MPT pore opening. However, Wu et al (2021a) reported that subsequently two daily doses of β-AR agonist isoproterenol (85 mg/kg/day) cause massive cardiomyocyte necroptosis through activation of the RIPK1-RIPK3-MLKL pathway.
Ferroptosis
GPX4 dependent ferroptosis pathway
GPX4, a selenoprotein, is the major enzyme that catalyzes the reduction of PL and cholesterol hydroperoxides to their corresponding alcohols (Jiang et al, 2021). Glutathione (GSH), as a potent reductant, provides the required two electrons during the process and promotes GPX4-mediated reduction of phospholipid hydroperoxides (PLOOHs) (Jiang et al, 2021; Pierzynowska et al, 2021). The oxidized GSH is reduced by glutathione-disulfide reductase with the electrons from NAPDH/H+ (Jiang et al, 2021; Lei et al, 2019; Pierzynowska et al, 2021).
To synthesize GSH, cysteine, glutamate, and glycine are needed (Lei et al, 2019). The uptake of cysteine is mediated by the cysteine/glutamate antiporter system xc -, which can be inhibited by erastin, an indirect inhibitor of GPX4 (Dixon et al, 2012). Decreased cysteine level detains GSH biosynthesis and subsequently inactivates GPX4. Another compound, RAS-selective lethal 3 (RSL3), directly binds the active site of GPX4 driven by selenocysteine and inhibits GPX4 activity (Yang et al, 2016). Dysfunction of GPX4 accumulates PLOOHs and lipid-free radicals.
The iron-dependent Fenton chain reaction rapidly amplifies PLOOHs and initiates ferroptosis (Conrad and Pratt, 2019). The Fenton reaction involves both ferrous (Fe2+) and ferric ions (Fe3+), which react with PLOOHs to generate free radicals, PLO• and PLOO•, respectively, leading to propagation of PLOOH production. Increased PLOOHs generate a myriad of secondary products, including breakdown products of lipid peroxides and oxidized proteins, ultimately rupturing organelle and cell membrane (Jiang et al, 2021).
GPX4 independent ferroptosis pathway
In addition to the cysteine-GSH-GPX4 axis, other mechanisms that facilitate ferroptosis are also reported. Ferroptosis suppressor protein 1 (FSP1) protects cells against GPX4 deletion-induced ferroptosis. Myristoylation recruits FSP1 to the plasma membrane where it reduces coenzyme Q10 (CoQ10, also known as biquinone) via NAD(P)H. The reduced CoQ10 traps lipid peroxyl radicals and halts the propagation of lipid peroxides (Bersuker et al, 2019; Doll et al, 2019). Through FSP1-CoQ10-NAD(P)H pathway, FSP1 suppresses ferroptosis. Thus, the inhibition of FSP1 sensitizes cells to ferroptosis.
Besides FSP1, GTP cyclohydrolase 1 (GCH1) was reported to protect against ferroptosis via its metabolic products tetrahydrobiopterin (BH4) and dihydrobiopterin (BH2) (Kraft et al, 2020). BH4/BH2 led to lipid remodeling by preventing oxidative degradation of the two PUFA tails of PLs, which acted as a direct radical-trapping antioxidant and promoted CoQ10 synthesis (Kraft et al, 2020; Soula et al, 2020).
Pyroptosis
Canonical pathway
The activation of the canonical pyroptotic pathway initiates from the assembly of the inflammasome that contains three components: sensor, adaptor, and pro-caspase-1 (Yu et al, 2021). The sensors are cytosolic PRRs that are capable of recognizing pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (Yu et al, 2021). The PRRs include Nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3), NLR family caspase activation and recruitment domain (CARD) containing 4 (NLRC4), NLR family pyrin domain-containing 1B (NLRP1B), and absent in melanoma 2 (AIM2) (Zheng et al, 2021).
Pathogenic stimuli (such as anthrax lethal toxin and muramyl dipeptide) promote the recruitment of pro-caspase-1 to the PRRs with the coordination of adaptor protein, apoptosis-associated speck-like protein containing a CARD (ASC) (Broz and Dixit, 2016; Lamkanfi and Dixit, 2012; Latz et al, 2013; Strowig et al, 2012). The recruited pro-caspase-1 is activated through autocleavage, which subsequently hydrolyzes the executor protein, gasdermin D (GSDMD) into C-terminus of GSDMD (C-GSDMD) and N-terminus of GSDMD (N-GSDMD) (Kesavardhana et al, 2020; Rathinam and Fitzgerald, 2016; Shi et al, 2015).
N-GSDMD perforates the plasma membrane and forms pores, leading to pyroptosis and release of pro-inflammatory cytokines (He et al, 2015; Rathinam and Fitzgerald, 2016; Sborgi et al, 2016). In addition, activated caspase-1 generates mature IL-1β and IL-18 through cleaving their precursors; the IL-1β and IL-18 are then released via the pores (Rathinam and Fitzgerald, 2016).
Non-canonical pathway
In the non-canonical pyroptosis pathway, the LPS derived from gram-negative bacteria directly binds to caspase-4/5/11 through the N-terminal of CARD and activates the caspases. Activated caspase-4/5/11 cleave GSDMD (Kayagaki et al, 2015; Shi et al, 2015). The generated N-GSDMDs oligomerize and form pores on the plasma membrane. Meanwhile, the activated inflammatory caspases mediate maturation of IL-1β and IL-18 through the NLRP3/caspase-1 pathway (Shi et al, 2017; Zheng et al, 2021).
Recently, apoptosis-related caspases are also identified to be involved in the cleavage of GSDM family members to induce pyroptosis. Chemotherapeutic drugs promoted caspase-3-dependent cleavage of gasdermin E (GSDME) and induced pyroptosis by N-terminal of GSDME-formed pores in tumor cells (Wang et al, 2017). Caspase-8 was also reported to cleave GSDMD and trigger pyroptosis during Yersinia infection. Yersinia effector protein, YopJ inhibited TAK1, which promoted caspase-8-dependent cleavage of GSDMD, thereby inducing pyroptosis and GSDMD-processing-mediated NLRP3 inflammasome-dependent release of IL-1β (Orning et al, 2018; Sarhan et al, 2018).
MPT-driven necrosis
MPT-driven necrosis is triggered by PTPC mediated-MPT pore opening. PTPC is a highly dynamic entity that physically and functionally involves proteins from the mitochondrial matrix, IMM and OMM, intermembrane space, and cytosol (Galluzzi et al, 2018; Izzo et al, 2016). Although under intense investigation, the mechanism and regulation of PTPC action are still elusive. Currently, CYPD is the only validated protein that is required for MPT induction (Galluzzi et al, 2018; Izzo et al, 2016). Inhibition of CYPD protects cells from Ca2+-overload and oxidative stress-induced MPT-driven necrosis (Baines et al, 2005; Clarke et al, 2002).
Interplay Between Autophagy and Necroptosis
Autophagy co-exists with necroptosis
Co-existence of autophagy and necroptosis was observed in various studies. TNFα induced necroptosis and autophagy in L929 cells, which was augmented by the pan-caspase inhibitor, zVAD ( Ye et al, 2013; Ye et al, 2011). Pretreatment of Nec-1 completely inhibited necroptosis and autophagy, suggesting that autophagy might be a downstream consequence of TNFα-induced necroptosis (Ye et al, 2011). zVAD alone also induced necroptosis and autophagy in L929 cells (Chen et al, 2011). Activated estrogen receptor β (ERβ) bound to Sp1 on phosphatase and tensin homolog (PTEN) promoter, restoring PTEN transcription that suppressed by Sp1 and inhibiting PI3K/AKT pathway, which led to autophagy and necroptosis in human seminoma cells, TCAM2 (Guido et al, 2012).
Saturated free fatty acid, palmitic acid (PA)-induced lipotoxicity promoted autophagy and necroptosis in endothelial cells. PA activated RIPK3 and CYLD to induce necroptosis and increased the level of Ca2+ to trigger autophagy (Khan et al, 2012). Macrophage necroptosis induced by sitosterol, a plant sterol, was also accompanied by autophagy (Bao et al, 2006).
Multiple chemotherapeutic regiments induce necroptosis and autophagy in cancer cells. Shikonin induced necroptosis and autophagy in various cancer cells (Lin et al, 2018). Inhibition of autophagy did not affect the shikonin-induced necroptosis, suggesting that autophagosome formation might be a downstream sequence of necroptosis in shikonin-treated cells. However, Han et al (2007) and Kim et al (2017) recently found that autophagy prevented necroptotic cell death in shikonin-treated lung cancer cells.
The anti-bacterial substance taurolidine exhibited anti-neoplastic effects with a mixed types of programmed cell death, including upregulation of necroptosis and autophagy (Stendel et al, 2009). Pentagalloylglucose increased autophagy and necroptosis in prostate cancer cells (Hu et al, 2009).
Autophagy regulates necroptosis
The role of autophagy in necroptosis is controversial. On one hand, autophagy prevents necroptosis and protects cells against necroptotic cell death in breast carcinoma cells, MCF7 (Farkas et al, 2011). On the other hand, autophagy promotes necroptosis and inhibition of autophagy attenuates necroptotic cell death in endothelial cells (Khan et al, 2012). Wu et al even demonstrated that inhibition of autophagy at an early stage protected cells against zVAD-induced necroptotic cell death in L929 cells.
However, disruption of the fusion of autophagosome with lysosomes or autophagosome maturation sensitized cells to zVAD-induced necroptotic cell death in the same cells (Wu et al, 2021b). Here, we discuss the dual roles and potential mechanisms of autophagy in the regulation of necroptosis (Fig. 3).

Autophagy antagonizes necroptosis
Many studies have demonstrated that autophagy protects cells from necroptotic cell death and acts as a negative regulator of necroptosis caused by multiple stresses. Starvation-induced autophagy protected cells from both apoptosis and necroptosis in MCF7 cells (Farkas et al, 2011). Inhibition of autophagy by Ulk1 deletion enhanced the starvation-induced necroptotic cell death, which was rescued by Nec-1 (Farkas et al, 2011). Genetic and pharmacological inhibition of autophagy increased TNFα-induced necroptosis in L929 and intestinal epithelial cells (Matsuzawa-Ishimoto et al, 2017; Ye et al, 2013; Ye et al, 2011).
Dysregulation of autophagosome fusion with the lysosome or of the late stage of autophagy enhanced TRAIL-induced necroptosis in murine prostate cells, suggesting a protective effect of basal autophagy on necroptosis (Goodall et al, 2016). Autophagy defect caused by Atg7 inactivation in pancreatic acinar cells led to pancreatitis through enhancing the RIPK3 expression and necroptosis. Dual inhibition of necroptosis and autophagy promoted apoptosis in pancreatic acinar cells (Zhou et al, 2017b). Impairment in myocardial autophagosome maturation is a major and early defect in mice with cardiomyocyte-restricted knockout of the constitutive photomorphogenesis 9 (COP9) signalosome subunit 8 (Cops8) (Su et al, 2011a). Massive cardiomyocyte necroptosis mediated by the RIPK1-RIPK3 pathway occurred in these mice after the autophagic impairment became discernible (Su et al, 2011b; Xiao et al, 2020).
Autophagy degrades pro-necroptotic factors
Autophagy antagonizes necroptosis through autophagic degradation of pro-necroptotic regulatory factors. Xie et al (2020) observed that autophagy degraded RIPK3 and MLKL, preventing necroptosis in amino acid starvation cells. The key components of necroptosis, RHIM harboring proteins, were also degraded through autophagy (Lim et al, 2019). Active/oligomerized, but not monomeric forms of RHIM proteins, such as TRIF, RIPK1, and RIPK3, were degraded through autophagy in bone marrow-derived macrophages (BMDMs) (Lim et al, 2019).
Another RHIM protein, ZBP1 was also degraded through autophagy in BMDMs. Deletion of ZBP1 in autophagy-deficient BMDMs promoted LPS/zVAD-induced and TRIF-mediated necroptosis (Lim et al, 2019), indicating that accumulation of ZBP1 protects against necroptosis in autophagy-deficient macrophages.
Additional mechanisms mediating the anti-necroptotic effect of autophagy
Autophagy was also shown to prevent necroptosis through blocking the RIPK3-mediated MLKL phosphorylation during the process of human cytomegalovirus (HCMV) infection. Inhibition of autophagy restored RIPK3-dependent phosphorylation of MLKL and led to necroptotic cell death in infected monocytes (Altman et al, 2020).
Li et al demonstrated that I/R injury-induced myocardial necroptosis was exaggerated in aged mice due to the autophagy insufficiency. During the process, the accumulated p62 interacted with RIPK1 and RIPK3, promoting the necrosome formation and thereby necroptosis in cardiomyocytes. Metformin treatment, which is known to activate autophagy, disrupted the p62-RIPK1-RIPK3 complexes and repressed I/R-induced necroptosis (Li et al, 2020a).
Beclin-1 might negatively regulate necroptosis through repressing the oligomerization of MLKL, preventing the perforation of plasma membrane. Through the coiled-coil domain, Beclin-1 interacted with phospho-MLKL and incorporated into necrosome to inhibit necroptosis. Deletion of Beclin-1 promoted the RIPK3-MLKL-dependent necroptosis in human colorectal carcinoma cells and multiple mouse-originated cells (Seo et al, 2020).
The ULK1 complex is central to the initiation of autophagy, which is composed by ULK1, ATG13, FIP200, and ATG101 (He and Klionsky, 2009). Inhibition of ULK1 compromised autophagy initiation, which promoted necroptotic cell death in MCF7 cells (Farkas et al, 2011). ULK1 also seems to be directly involved in the regulation of necroptosis. Wu et al (2020a) demonstrated that ULK1 interacted with RIPK1 and phosphorylated RIPK1 at Ser357, which disrupted the necrosome formation and inhibited TNFα-induced necroptosis in murine embryonic fibroblasts (MEFs).
Autophagy promotes necroptosis
The promotion of necroptosis by autophagy is less reported. Bonapace et al (2010) demonstrated that combined treatment of rapamycin and dexamethasone led to autophagy and cell death with characteristics of necroptosis in acute lymphoblastic leukemia cells. PA induced necroptosis accompanied with autophagy in endothelial cells, and the autophagy increased the fatty acid mediated necroptosis-related cell death (Khan et al, 2012). Further, the inhibition of autophagy abolished Chal-24-induced necroptotic cell death in different cancer cells (He et al, 2014).
Autophagy may promote necroptosis through degradation of anti-necroptotic regulatory factors. He et al (2014) revealed that autophagy degraded cIAP1/2, the E3 ligases that block RIPK1 from forming complex IIa with FADD and caspase-8, thereby inhibiting necroptosis in cancer cells. Autophagy-dependent degradation of caspse-8 may promote the necroptosis. Using human colon cancer cells, Hou et al (2010) revealed that caspase-8 was sequestered by autophagosomes and degraded by lysosomes, which may initiate necroptosis through blocking the cleavage of RIPK1.
Autophagosomes provide docking sites for the assembly of necrosomes. Autophagosomal membrane protein ATG5 interacted with FADD, RIPK1, and RIPK3 in rhabdomyosarcoma cells (Basit et al, 2013). Atg5 knockdown completely abolished the interaction of RIPK1 with FADD and RIPK3, concurrent with severely impaired autophagosome formation (Basit et al, 2013). Goodall et al (2016) also showed that autophagy at the expansion stage, but not nucleation or maturation stages, promoted necroptosis through providing a scaffold for necroptosis signaling in TRAIL-induced necroptosis in murine prostate cells.
RIPK1 was associated with proteins involved in early-to-mid stages of autophagy, including ATG5/ATG12, ATG7, and p62. Among these proteins, ATG5 was required for TRAIL-induced interaction of RIPK1-p62-phospho-MLKL, and p62 recruited RIPK1 to the autophagy machinery that resulted in activation of necroptosis in murine prostate cells (Goodall et al, 2016).
Necroptosis regulates autophagy
Multiple lines of evidence are emerging to support a dichotomy for necroptosis-mediated regulation of autophagy. On the one hand, activation of the necroptotic pathway was shown to promote autophagy; on the other hand, there were also reports supporting an inhibitory effect of necroptosis on autophagy (Fig. 4). This dichotomy is perhaps governed by differences in cell types and in stressors.
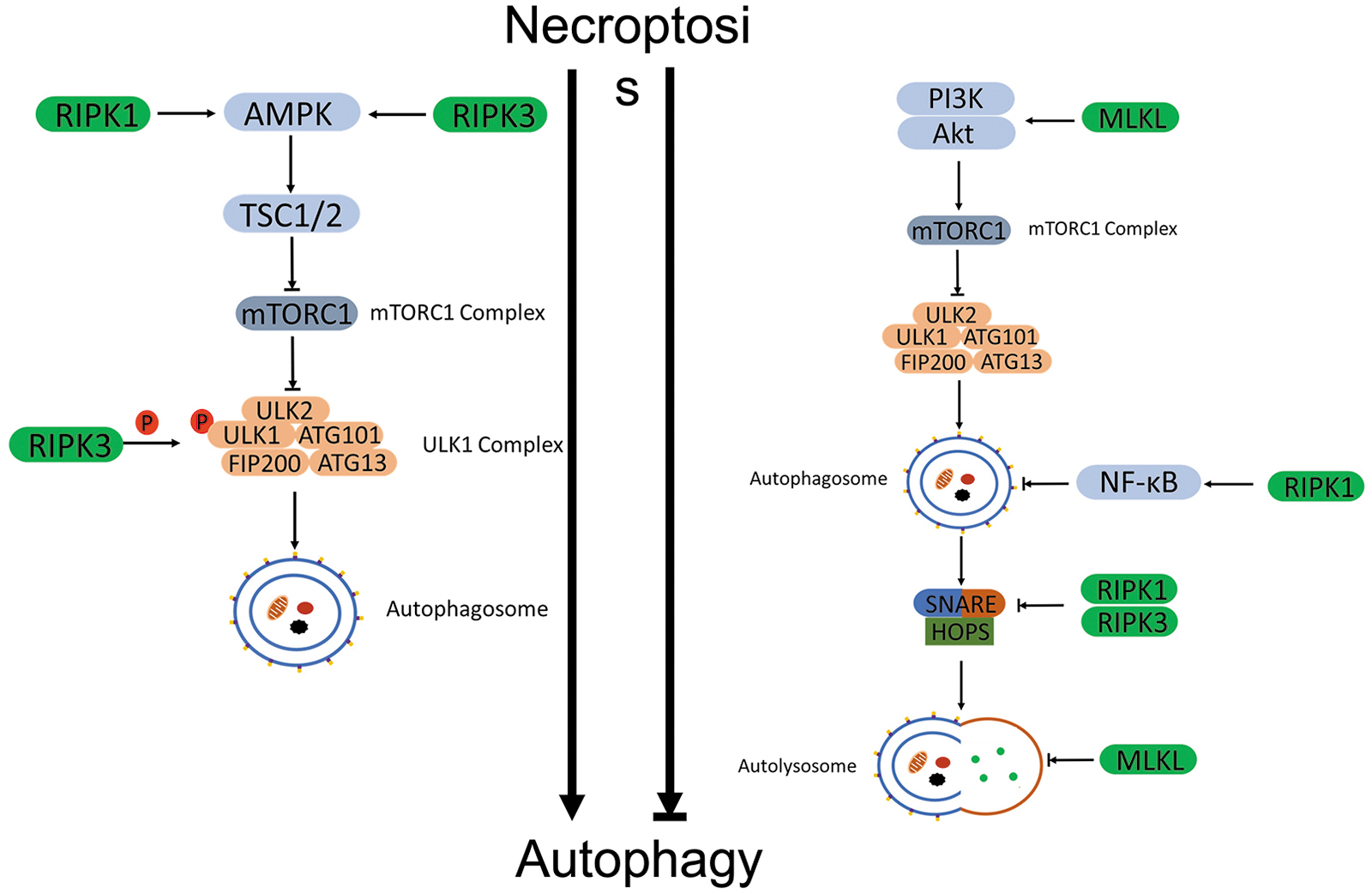
Necroptosis promotes autophagy
Nec-1 inhibited autophagy, indicating that necroptosis may promote autophagy (Rosenbaum et al, 2010). Ni et al (2018) revealed that RIPK1 participated in activation of the autophagic-lysosomal pathway in the ischemia-induced neuronal and astrocytic cell death. Knockdown of RIPK1 decreased autophagy by showing reduced LC3II. Kikuchi et al (2012) reported that induction of autophagy and autophagic cell death by deletion of caspase-8 required RIPK1 and RIPK3 in mouse T-lymphoma cells. Inhibition of either RIPK1 or RIPK3 completely prevented caspase-8 knockdown from inducing autophagic cell death.
RIPK1 also seems to participate in AMPK-mediated mTORC1 inhibition during energetic stress. RIPK1 promoted the interaction between AMPK and TSC2 and facilitated TSC2 phosphorylation at Ser1387 by AMPK. The activated TSC2 further phosphorylated mTORC1 and initiated the autophagy in MEFs (Najafov et al, 2021). Wu et al (2021b) observed that TNFα-induced necroptosis initiated the early events of autophagy by activating AMPK and subsequently ULK1 and Beclin-1 in L929 cells. RIPK3 directly interacted with AMPK and phosphorylated the catalytic α-subunit of AMPK, PRKAA1, at Thr183, thereby activating AMPK (Wu et al, 2021b).
Inhibition of RIPK3 abolished the activation of AMPK, the phosphorylation of ULK1 and Beclin-1, and associated autophagosome formation (Wu et al, 2021b). In MEFs, Torii et al (2020) also revealed that RIPK3 phosphorylated ULK1 at Ser746 and initiated the alternative autophagy that is an ATG5 independent, Golgi membrane derived, and genotoxic stress-induced autophagy type. In addition, CamKII-mediated phosphorylation of MLKL activated MLKL and facilitated autophagic flux during short-term starvation, accompanied by decreases of LC3-II and p62 in multiple mouse and human cells (Zhan et al, 2022).
Necroptosis inhibits autophagy
Liu et al (2020b) demonstrated that RIPK1 inhibited autophagy through the activation of NF-κB signaling in neurons. Deletion of RIPK1 promoted autophagy via inhibition of NF-κB signaling; conversely, RIPK1 overexpression activated NF-κB signaling and abolished autophagy (Liu et al, 2020b). In addition, necroptosis was also found to inhibit autophagic flux via dysregulating SNARE-mediated autophagosome-lysosome fusion. Wu et al found that necroptosis destabilized SNARE complexes. Inhibition of either RIPK1 or RIPK3 strengthened the interactions of SNARE complexes in L929 cells (Wu et al, 2021b).
MLKL displays multiple roles in the suppression of autophagy. Frank et al (2019) revealed that MLKL inhibited the fusion of autophagosome and lysosome or autophagic flux through disrupting the integrity of autolysosome membranes in mouse dermal fibroblasts. The ability of membrane translocation of MLKL was required in this process. The mutant MLKL that was unable to translocate failed to inhibit autophagic flux (Frank et al, 2019). The MLKL-mediated autophagic inhibition was also observed in western diet-induced non-alcoholic fatty liver disease (NAFLD). Western diet activated MLKL; then, the translocation of the latter to the autophagosome and plasma membrane blocked the autophagy and initiated necroptosis in hepatocytes (Wu and Nagy, 2020).
Interestingly, the sequence of MLKL translocation was first to the autophagosomes and then to the plasma membrane in hepatocytes (Wu et al, 2020b). Long noncoding RNA, lncRNAs-FA2H-2 downregulated MLKL expression through binding to the promoter of MLKL, which restored the autophagy in macrophages (Guo et al, 2019). In addition, MLKL activated PI3K/AKT/mTORC1 signaling, contributing to the inhibition of autophagy in an RIPK3 independent manner (Guo et al, 2019).
Factors simultaneously regulate autophagy and necroptosis
MAPK pathway
Ye et al (2011) reported that p38 was involved in TNFα-induced autophagy and necroptosis. A reduced phosphorylation of p38 (p-p38) and downregulated NF-κB signaling were observed in TNFα-induced necroptosis and autophagy in L929 cells. Further downregulation of p-p38 and NF-κB signaling potentiated TNFα-induced necroptosis and autophagy. Nec-1 reversed the p-p38 and NF-κB, and it attenuated TNFα-induced necroptosis and autophagy (Ye et al, 2011). Activation of c-Jun N-terminal kinase (JNK) and extracellular signal regulated kinase (ERK) signaling involved in Chal-24-induced autophagy and necroptosis in cancer cells.
Suppression of JNK or ERK effectively inhibited Chal-24-induced autophagy and necroptosis (He et al, 2014). Activated JNK phosphorylated Bcl-2 and Bcl-xL, releasing Beclin-1 from both Bcl-2/Beclin-1 and Bcl-xL/Beclin-1 complexes. The released Beclin-1 initiated autophagy (He et al, 2014). Meanwhile, active ERK phosphorylated cIAP1, which triggered autophagic degradation of cIAP1 and promoted necroptosis (He et al, 2014).
Death-associated protein kinase 1 (DAPK1) seems to be involved in the regulation of autophagy and necroptosis. DAPK1 inhibited necroptosis through direct interaction with p38 MAPK, which activated p38 MAPK/MK2 signaling. The activated MK2 phosphorylated RIPK1 at Ser321, which inhibited the complex IIa formation and subsequent RIPK1-RIPK3-MLKL dependent necroptosis in colon cancer cells (Wu et al, 2020c).
FAS-associated death domain protein
It was reported that FADD repressed necroptosis and autophagy in T cells (Bell et al, 2008). FADD interlaced with necroptotic factors, caspase-8 and RIPK1, and autophagic factors ATG5, ATG12, and ATG16L. The resultant complex tethered on the pre-autophagosomal membrane and activated caspase-8, which suppressed autophagic signaling and RIPK1-dependent necroptosis (Bell et al, 2008). Inhibition of autophagosome formation completely rescued the necroptotic death in FADD-deficient cells. In contrast, Osborn et al observed that inhibition of autophagosome formation was unable to rescue the necroptotic death in FADD knockdown T cells. Their results suggested that autophagy was the consequence of necroptosis, not the cause (Osborn et al, 2010).
Caspases
TNFα increased the activation of caspase-6 by promoting procaspase-6 cleavage. However, the inhibition of caspase-6 had no effect on the TNFα-induced necroptosis, but it increased TNFα-induced autophagy. Inhibition of either autophagy or necroptosis increased the activation of caspase-6. Deletion of caspase-6 attenuated autophagic inhibition-enhanced necroptosis in L929 cells, suggesting that caspase-6 was required for the antagonized effects of autophagy to necroptosis (Ye et al, 2013). This was also consistent with the evidence that ATG proteins were targeted for cleavage by caspase-6 in Hela cells (Cho et al, 2009a).
Caspase-8 suppresses necroptosis and autophagy. Caspase-8 activation leads to the proteolytic inactivation of RIPK1, thereby preventing the induction of necroptosis. Inhibition of caspase-8 also prevented autophagic signaling in T cells (Bell et al, 2008). A similar observation was confirmed by Kikuchi et al. Their results indicated that knockdown of caspase-8 promoted RIPK1 and RIPK3 dependent necrotic cell death, concurring with ROS accumulation, caspase activation, and autophagosome formation in T cells (Kikuchi et al, 2012).
Song et al also showed that the reduction of pro-caspase-8 enhanced the induction of autophagy in leukemia cells by tetrahydrobenzimidazole derivative TMQ0153. However, autophagy was not involved in the degradation of pro-caspase-8. Inhibition of autophagy did not significantly rescue the TMQ0153-induced pro-caspase-8 reduction in leukemia cells (Song et al, 2020).
Cellular FADD-like IL-1β-converting enzyme-inhibitory protein
Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP) is a major anti-apoptotic factor. c-FLIP forms heterodimers with pro-caspase 8 and inhibits its activation. Previous reports revealed that the long splice variant of c-FLIP (c-FLIPL) repressed necroptosis by interfering the recruitment of RIPK1 to the complex II in response to Fas or TLR3 in keratinocytes (Feoktistova et al, 2011; Geserick et al, 2009). Deletion of c-FLIPL sensitized hepatocytes to TNFα-induced RIPK1/RIPK3-dependent necroptosis (Oberst et al, 2011).
In addition, Lee et al (2009) showed that c-FLIP suppressed autophagy by preventing ATG3 from binding and processing LC3 in virus-infected lymphoma cells. He and He (2013) revealed that c-FLIPL repressed necroptosis and autophagy simultaneously. They showed that c-FLIPL-deficient T cells exhibited enhanced necroptosis and autophagy.
mTORC1 signaling pathway
Inhibition of mTORC1 increases the autophagy and, sometimes, necroptosis in cells. Wang et al demonstrated that cigarette smoke extract activated TSC2, inhibited mTORC1, and induced autophagy and necroptosis in bronchial epithelial cells. Depletion of mTORC1 augmented the induction of autophagy and necroptosis by cigarette smoke extract (Wang et al, 2018a). Simultaneous inhibition of mTORC1 and of the fusion of autophagosome with lysosome triggered RIPKs-dependent necroptosis in cancer cells (Bray et al, 2012).
mTORC1 inhibition elevated oxidative metabolism and ROS production through blocking glycolytic metabolism. The metabolic disorder promoted fragmentation of mitochondria, which activated mitophagy to remove fragmented mitochondria and ROS. Once the mitophagy was blocked, the accumulated ROS promoted necroptotic cell death (Bray et al, 2012). However, Abe et al also revealed that mTORC1 inhibition suppressed necroptosis in cardiomyocytes.
They found that inhibition of mTORC1 downregulated TNFα/zVAD-induced RIPK1 autophosphorylation at Ser166, but it upregulated the inhibitory phosphorylation at Ser320 site. Both Ser166 dephosphorylation and Ser320 phosphorylation in RIPK1 were required for the mTORC1 inhibition to prevent necroptotic cell death (Abe et al, 2019).
Hyperactivation of mTORC1 was also involved in western diet-induced necroptosis and suppression of autophagy in intestinal epithelium cells (Xie et al, 2020). Western diet enhanced phosphorylation of AKT and GSK3α/β, which induced hyperactivation of mTORC1 through inhibiting the TSC1/TSC2 complexes (Xie et al, 2020). Activated mTORC1 phosphorylated RIPK3 and MLKL, initiating the necroptosis. Meanwhile, hyperactivation of mTORC1 downregulated autophagy and blocked autophagic degradation of RIPK3, which further potentiated necroptosis (Xie et al, 2020).
A20, a negative regulator of necroptosis, was involved in the activation of autophagy through the inhibition of mTORC1 in CD4 T cells after TCR stimulation. Matsuzawa et al reported that A20 directly interacted with mTORC1, removed the polyubiquitination of mTORC1, restrained the mTORC1 activity, and induced autophagy. A20 deficient cells displayed enhanced polyubiquitination and activity of mTORC1 (Matsuzawa et al, 2015).
ROS-related signaling pathway
ROS contributes to the crosstalk between necroptosis and autophagy. TNFα-induced mitochondrial dysfunction, and ROS production upregulated necroptosis and autophagy in L929 cells (Ye et al, 2012). RIPK1 was shown to directly participate in the TNFα-induced mitochondrial dysfunction and ROS production. Inhibition of RIPK1 significantly reduced TNFα-induced total ROS production, necroptosis, and autophagy (Ye et al, 2012).
S. Typhimurium infection-induced type I interferons (IFN-I) signaling activation damaged mitochondria and increased ROS production, resulting in enhanced autophagy and necroptosis in bacteria-infected macrophages (Hos et al, 2017). The enhanced autophagy promoted the degradation of autophagy receptor p62, which was supposed to activate nuclear factor erythroid 2-related factor 2 (Nrf2) through impairing the interaction between Nrf2 and Kelch-like ech-associated protein 1 (Keap1). Meanwhile, the upregulated RIPK3 promoted the expression of the mitochondrial phosphatase phosphoglycerate mutase (PGAM5), which sequestered Nrf2 in the cytosol (Hos et al, 2017). Both effects attenuated antioxidant effects of Nrf2 and further sensitized the cells to the ROS-mediated necroptosis.
In NAFLD, Mohammed et al showed that the deletion of antioxidant enzyme, Cu/Zn superoxide dismutase (SOD1) induced oxidative stress. The stress upregulated necroptosis, but it downregulated autophagy in the NAFLD mouse model (Mohammed et al, 2021). Tetrahydrobenzimidazole, TMQ0153, disrupted the mitochondrial bioenergetics and enhanced ROS level, which activated autophagy and necroptosis in cancer cells. Inhibition of autophagy augmented the TMQ0153 induced necroptosis (Song et al, 2020). Increased ROS level and MPT pore opening contributed to mitochondrial oxidative phosphorylation (OXPHOS) dysregulation, inducing ATG5 mediated-autophagy and necroptosis in melanoma cells (Basit et al, 2017).
JNK and ERK signaling pathways are involved in ROS production and associated autophagy. Chen et al (2011) showed that zVAD-induced ROS production and autophagy were attenuated by JNK or ERK inhibitors in L929 cells. c-Src was the upstream factor of zVAD-induced activation of JNK and ERK (Chen et al, 2011).
Endoplasmic reticulum stress and unfolded protein response
Endoplasmic reticulum (ER) is responsible for the folding of approximately one-third of cellular proteins. The ER stress triggers the activation of unfolded protein response (UPR) through pathways mediated by protein kinase RNA-activated (PKR)-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) (Almanza et al, 2019). The ER stress also contributes to the initiation of necroptosis and autophagy.
The ER stress induces necroptotic cell death. For instance, Saveljeva et al revealed that ER stress induced RIPK1-RIPK3-MLKL dependent necroptosis through TNFR1 signaling. Inhibition of necroptosis through RIPK1 repression switched ER stress-induced necroptosis to apoptosis in L929 cells (Saveljeva et al, 2015). The ER stress-triggered activation of the RIPK1-RIPK3 pathway was also observed in the cardiomyocyte lipotoxicity and hypertrophy induced by PA. Suppression of ER stress downregulated transcription of RIPK1 and RIPK3, whereas inhibition of necroptosis also decreased ER stress marker, glucose regulated protein 78 (GRP78), and calreticulin in cardiomyocytes (Zhao et al, 2016).
Co-expression of GRP78 and phospho-MLKL was observed in spinal cord injury-derived microglia/macrophages, which also indicated a link between necroptosis and ER stress. The subsequent studies revealed that RIPK3 and MLKL localized on the ER of necroptotic microglia/macrophages, whereas inhibition of ER stress attenuated necroptosis (Fan et al, 2015).
The detailed signaling mechanism between ER stress and necroptosis is not fully understood. One of the UPR branches, PERK signaling may link the ER stress with necroptosis. Tian et al (2020) showed that phosphorylation of eukaryotic translation initiation factor 2 subunit α (eIF2α) downregulated RIPK3 and mitigated ER stress-induced necroptotic cell death in hepatocytes (Tian et al, 2020). In addition, C/EBP homologous protein (CHOP) activation was found to lead to RIPK1 dependent necroptosis, concurrent with ROS production in lung cancer cells. Removal of ROS rescued the cells from CHOP activation and caused necroptotic cell death (Ma et al, 2016). Caspase-8 negatively regulated ER stress-induced necroptosis.
Kishino et al demonstrated an inverse correlation between caspase-8 and RIPK1 in auditory cells. Deletion of caspase-8 enhanced ER stress-induced RIPK1 and necroptosis (Kishino et al, 2019).
The connections between ER stress and autophagy were extensively investigated, and all three branches of UPR were all involved in the regulation of autophagy. The activation of autophagy by ER stress was first described in 2006, which indicated a survival function of autophagy to counteract ER stress-induced toxicity in neuroblastoma cells (Ogata et al, 2006). For the IRE1 signaling pathway, activation of JNK1 by IRE1 kinase activity induced autophagy through disrupting the interaction between Beclin-1 and Bcl-2 in MEFs (Wei et al, 2008), or upregulating the transcription of Beclin-1 by JUN/c-Jun in cancer cells (Li et al, 2009).
The spliced X-box–binding protein 1 (XBP1) generated by endoribonuclease activity of IRE1 upregulated Beclin-1 by direct binding to the promoter of Beclin-1 in endothelial cells (Margariti et al, 2013). For the PERK signaling pathway, phospho-eIF2α enhanced the expression of ATG12 in C2C5 cells (Kouroku et al, 2007); PERK-mediated activation of ATF4 and CHOP was responsible for the upregulation of a dozen autophagy genes, including MAP1LC3, Beclin-1, ATG3, ATG12, p62, neighbor of BRCA1 gene 1 (NBR1), ATG7, ATG10, GABARAP, and ATG5 in MEFs and human tumor cells (B'Chir et al, 2013; Rouschop et al, 2010).
Therefore, PERK-eIF2α-ATF4-CHOP appeared to be the most important UPR branch in autophagy regulation due to the extensive involvement in autophagy signaling. For ATF6 signaling, ATF6 downregulated AKT to initiate autophagy in human choriocarcinoma cells (Yung et al, 2011). In addition, ATF6 was also involved in DAPK1 dependent autophagy initiation in the bacteria-infected mouse model (Kalvakolanu and Gade, 2012).
Although there is a lack of direct support that ER stress affects necroptosis and autophagy concurrently, it has been confirmed that ER stress triggers necroptosis and autophagy respectively in various studies (Rashid et al, 2015; Zhu et al, 2018). PERK signaling might be the possible candidate in the ER stress-mediated crosstalk between necroptosis and autophagy.
Stimulator of IFN response cGAMP interactor 1 signaling pathway
Stimulator of Interferon Response cGAMP Interactor 1 (STING1) is a conserved transmembrane protein that mediates cyclic GMP-AMP synthase (cGAS) induced innate immune response (Sun et al, 2013). Activation of STING1 mediates autophagy and necroptosis through sensing the abnormal cytosolic nucleic acids. For autophagy activation, Watson et al (2012) demonstrated that mycobacterium tuberculosis infection initiated the STING1-dependent, ubiquitin-mediated selective autophagy to restrain the infection.
The c-di-AMP (a bacterial second messenger) induced STING1-dependent autophagy via mTORC1 inactivation in macrophages during gram-positive bacterial infection (Moretti et al, 2017). Besides microbial infection, abnormal Aβ accumulation also suppressed autophagy and mitophagy through downregulated cGAS-STING1-TBK1 (TANK-binding kinase 1) signaling in cardiomyocytes, which was correlated with cardiac dysfunction in Alzheimer's disease patients and the mouse model (Wang et al, 2020a). For necroptosis induction, Luo et al (2020) showed that the leakage of damaged nuclear and mitochondrial DNA to the cytosol activated STING1 signaling and triggered the necroptotic cell death in smooth muscle cells.
In addition, Chen et al (2018) also elucidated that the proapoptotic factor, PUMA triggered the cytosolic release of mitochondrial DNA and induced STING1 dependent necroptosis in colon cancer cells.
Crosstalk Between Mitophagy and Regulated Necrosis
Mitochondria are essentially involved in energy production and metabolites synthesis as well as programmed cell death (Galluzzi et al, 2012). To maintain the diverse and complex functions of mitochondria, different mechanisms of mitochondrial quality control have evolved to antagonize the dysfunction of mitochondria caused by various molecular damages. These mechanisms contain systems that prevent molecular damage (ROS scavenging), restore damaged components (repair and refold), replace the damaged components (degradation and new synthesis), promote mitochondrial dynamics (fission and fusion), and eliminate dysfunctional mitochondria (mitophagy) (Fischer et al, 2012).
Mitophagy is characterized as the process of selective removal of mitochondria via autophagy. It involves the crosstalk between mitochondria and autophagy. Dysregulation of mitochondrial OXPHOS stimulates MPT pore opening, Δψ depolarization, and activation of phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1). These changes lead to the initiation of mitophagy, whereas the inhibition of mitophagy increases ROS levels and stimulates necroptotic cell death (Basit et al, 2017). There is an intricate crosstalk between mitophagy and regulated necrosis (Fig. 5).
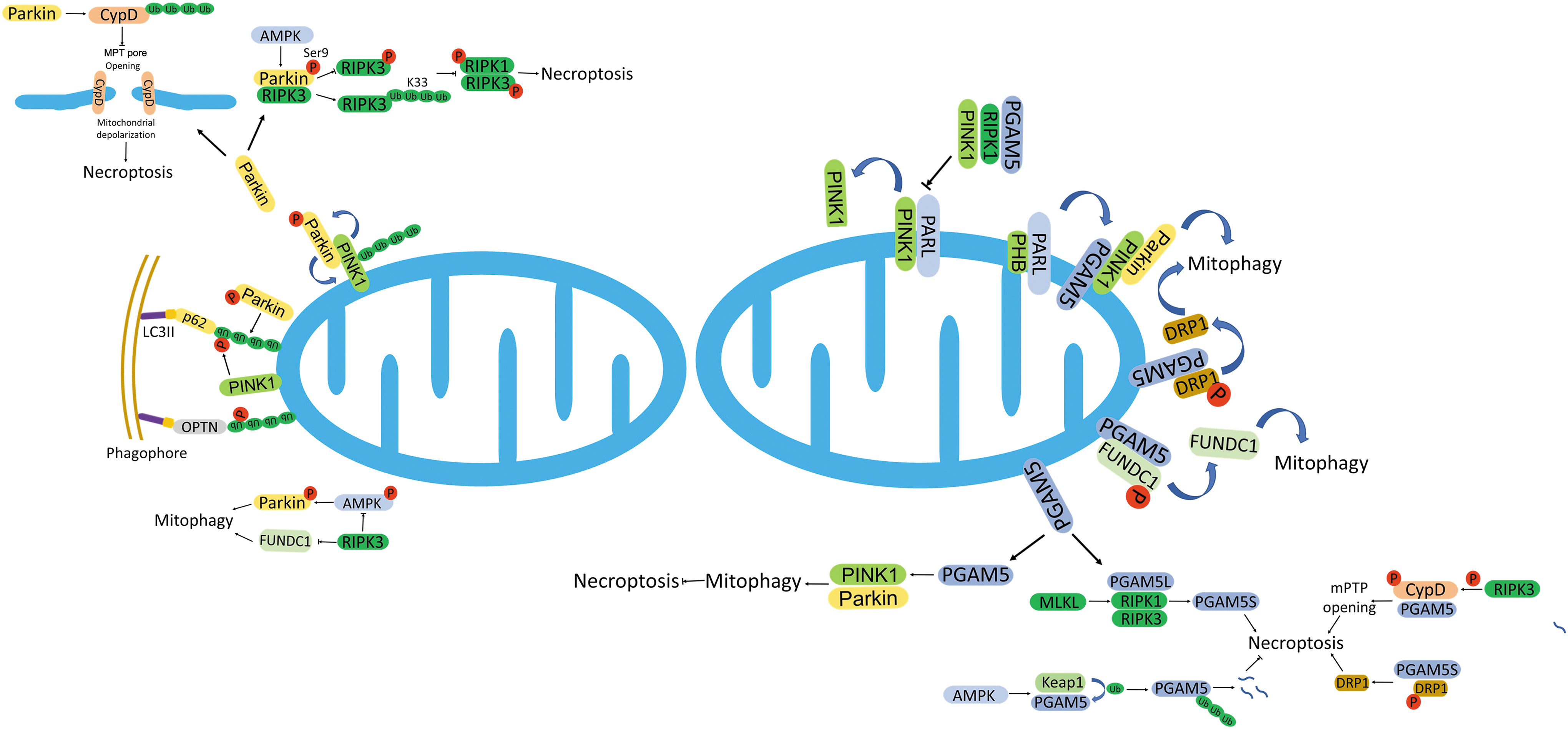
Mitophagy
PINK1/Parkin-mediated mitophagy
Healthy mitochondria import the transported-PINK1 from OMM to IMM via the cooperation of translocases of outer membrane (TOM) and translocases of inner membrane (TIM) (Liang and Gustafsson, 2020). The resident protease, PINK/PGAM5-associated rhomboid-like (PARL) cleaves PINK1 and then the resultant PINK fragments are retro-translocated to the cytosol and degraded by the proteasome. PINK1 senses the depolarized mitochondrial membrane (Liang and Gustafsson, 2020). Loss of mitochondrial membrane potential stabilizes PINK1 on the OMM whose accumulation recruits E3 ligase, Parkin (Pickles et al, 2018).
PINK1-dependent phosphorylation triggers the Parkin's E3 ligase activity. Meanwhile, PINK1-mediated phosphorylation of ubiquitin also potentiates the bondage and activation of Parkin, which forms a positive feedback loop to amplify the mitophagy signal. Parkin further catalyzes the formation of poly-phospho-ubiquitin chain with several OMM proteins, including voltage dependent anion channel 1 (VDAC1), Mitofusin 2 (MFN2), TBK1, and Mitochondrial Rho GTPase (MIRO) (Chourasia et al, 2015). These signals are recognized by mitophagy cargo receptors, such as p62 and optineurin, which facilitate the degradation of mitochondria through autophagy (Palikaras et al, 2018).
In addition to Parkin, other ubiquitin E3 ligases were also identified in the regulation of mitophagy, such as Glycoprotein 78 (Gp78), SMAD specific E3 ubiquitin protein ligase 1 (SMURF1), Siah E3 Ubiquitin Protein Ligase 1 (SIAH1), mitochondrial E3 ubiquitin protein ligase 1 (MUL1), and Ariadne RBR E3 ubiquitin protein ligase 1 (ARIH1) (Palikaras et al, 2018). For instance, Gp78, a critical E3 ligase in ER-associated degradation, promotes depolarization-induced mitophagy, which is Mfn1-dependent (Fu et al, 2013). Similar to Parkin, these recruited E3 ligases also generate ubiquitin chains that facilitate the autophagy adaptors-mediated mitophagy (Palikaras et al, 2018).
PINK1-dependent phosphorylation significantly contributes to mitophagy. Phosphorylation of ubiquitin by PINK1 antagonizes the hydrolysis mediated by deubiquitinases, such as USP30, which potentiates the Parkin-mediated mitophagy (Bingol et al, 2014; Harper et al, 2018). Besides Parkin, PINK1 also phosphorylates MFN2, which serves as a docking site for Parkin to ubiquitinate various OMM proteins (Chen and Dorn, 2013). The AAA+-ATPase ATAD3A facilitates the import and processing of PINK1 and suppresses PINK1-dependent mitophagy. Defective ATAD3A increases mitophagy of both damaged and healthy mitochondria (Jin et al, 2018).
Mitochondrial proteins-mediated mitophagy
In addition to ubiquitin-dependent mitophagy, mitophagy receptors also can mediate the autophagosome-dependent degradation of dysfunctional mitochondria. Generally, mitophagy receptors directly interact with LC3 and GABARAP through their LC3-interacting region (LIR) motifs and initiate mitophagy (Palikaras et al, 2018). In mammalian cells, the OMM proteins, Bcl-2-like protein 13 (Bcl-L-13), and FK506-binding protein 8 (FKBP8) directly bind to LC3 through LIR motifs, initiating Parkin-independent mitophagy (Bhujabal et al, 2017; Murakawa et al, 2015).
The OMM protein NIP3-like protein X (NIX) plays a critical role in programmed mitophagy during cell differentiation (Sandoval et al, 2008). Phosphorylation of NIX LIR domain enhances its interaction with LC3 and resultant mitophagy (Rogov et al, 2017). Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (Bnip3) is a pro-apoptotic BH3-only protein, which also mediates mitophagy as a mitophagy receptor (Quinsay et al, 2010). Both NIX and Bnip3 share a similar working mechanism, that is, recruitment of Parkin.
As a substrate of Parkin, NIX in its ubiquitinated form recruits NBR1 to the mitochondria, initiating mitophagy (Gao et al, 2015). Similarly, the deletion of Parkin also attenuated Bnip3-mediated mitophagy (Lee et al, 2011).
FUN14 domain-containing protein 1 (FUNDC1) is another OMM mitophagy receptor that promotes mitophagy during hypoxia (Liu et al, 2012). The phosphatase PGAM5 and kinase CK2 coordinate to regulate the activation of FUNDC1 and its interaction with LC3 via modulating the phosphorylation status of FUNDC1 (Chen et al, 2014). Besides CK2, FUNDC1 can also be recruited and phosphorylated by ULK1, which enhances the mitophagy through strengthening the interaction between FUNDC1 and LC3 (Wu et al, 2014b).
Prohibitin 2 (PHB2) and cardiolipin are IMM proteins that facilitate mitophagy as mitophagy receptors. The OMM dissipation externalizes PHB2 and cardiolipin, which then interact with LC3 and trigger mitophagy (Chu et al, 2013; Wei et al, 2017). PHB2 is essential in Parkin-mediated mitophagy. PHB2 knockdown blocks Parkin-mediated mitophagy (Wei et al, 2017). Meanwhile, PHB2 was also reported to form a complex with LC3 and p62, which facilitates the phagophore generation near damaged mitochondria (Xiao et al, 2018).
Mitophagy facilitates necroptosis
Similar to autophagy, key players of mitophagy also vividly participate in the regulation of necroptosis. Accumulated evidence supports that mitophagy contributes to necroptosis. The inhibition of mitophagy protected against the necroptosis of pulmonary epithelial cells induced by cigarette smoke (Mizumura et al, 2014). In addition, mitophagy was associated with enhanced ROS, which led to the activation of the necroptotic pathway in melanoma cells (Basit et al, 2017).
The mitochondria and mitochondrial ROS production proved to be downstream effectors of RIPK3-mediated necroptosis in L929 and Hela cells (Vanlangenakker et al, 2011; Wang et al, 2012). However, the observation was challenged by that deletion of mitochondria via mitophagy did not compromise necroptosis in other cell types, such as murine endothelial or fibroblastic cells. Tait et al (2013) reported that mitophagy-dependent mitochondrial elimination did prevent necroptosis-associated ROS production, but it did not attenuate the induction of necroptosis by either death receptor or dimerization of active RIPK3. Hence, there seems to be an intricate crosstalk between mitophagy and necroptosis (Fig. 5).
Parkin-dependent regulation of necroptosis
As a positive regulator in the mitophagy process, the role of Parkin in necroptosis is controversial. On the one hand, Parkin seems to exert a negative regulation on necroptosis. Knockdown of Parkin increased RIPK3 and MLKL-dependent necroptosis in response to necroptotic stimuli. Parkin directly bound to the N-terminal of RIPK3 through its IBR-R2 domain. The interaction disrupted the phosphorylation of RIPK3 but facilitated the K33-linked ubiquitination of RIPK3, which blocked the necrosome formation (Lee et al, 2019). During the process, AMPK was required to activate Parkin through phosphorylation at the Ser9 residue of Parkin. Either AMPK depletion or mutation of Ser9 site compromised Parkin-RIPK3 pathway during necroptosis (Lee et al, 2019).
Sun et al (2019) demonstrated that Parkin was downregulated in response to severe oxidative stress in cardiomyocytes, which coincided with the enhanced mitochondrial MPT pore opening and related necroptosis. Further studies revealed that Parkin catalyzed K63-linked polyubiquitination of CYPD, the essential regulator of MPT pore opening, and blocked its activity, which inhibited the necroptosis through diminishing the MPT pore opening (Sun et al, 2019).
Clinical observation also revealed an inverse relationship between Parkin and RIPK3 dependent necroptosis in breast cancer. Low RIPK3, but high Parkin expression was characterized with more aggressive and metastatic clinical features in breast cancer (Won et al, 2021). On the other hand, Parkin was also found to promote necroptosis. Parkin knockdown prevented necroptotic cell death in zVAD-treated microglial cells accompanied with an enhanced ubiquitination of RIPK1 (Dionisio et al, 2019). Thus, it is indicated that Parkin may dually regulate necroptosis through modulating RIPK1 and RIPK3 separately.
PGAM5 mediated interplay between mitophagy and necroptosis
PGAM5 belongs to the PGAM family that catalyzes the conversion of 3-phosphoglycate (3-PG) to 2-phosphoglycate (2-PG) and shares a conserved domain, PGAM domain (Cheng et al, 2021). However, PGAM5 is mutase deficient because its PGAM domain is poorly conserved. Two isoforms of PGAM5 (PGAM5-L and PGAM5-S) exist, due to the alternative splicing of PGAM5 transcript (Cheng et al, 2021). Recent studies have shown that PGAM5 is involved in the regulation of both mitochondrial dynamics and programmed cell death.
PGAM5 promotes the clearance of unhealthy or damaged mitochondria to maintain mitochondrial homeostasis through mitophagy. Generally, the deletion of PGAM5 accumulated damaged mitochondria and elevated intracellular ROS level (Lu et al, 2014). The reduced mitochondrial clearance indicates that PGAM5 is required in the process of mitophagy. Through stabilization of PINK1, PGAM5 promotes PINK1/Parkin-mediated mitophagy. It was reported that PGAM5 directly bound with PINK1 in Drosophila (Imai et al, 2010). Both PINK1 and PGAM5 in Hek293T cells were substrates of PARL. Their cleavage by PARL depends on the health status of mitochondria; PINK cleavage occurs in healthy mitochondria and PGAM5 cleavage in unhealthy mitochondria. (Sekine et al, 2012).
Under mitochondrial dysfunction in MEFs, PGAM5 interacted with PINK1, which prevented its cleavage by PARL in the IMM (Lu et al, 2014). Meanwhile, PGAM5 enabled the translocation of PINK1 from IMM to OMM, which initiated PINK1/Parkin-mediated mitophagy (Lu et al, 2014). PHB binds to PGAM5 in healthy mitochondria. However, PHB tended to interact with PARL, which blocked the cleavage of PGAM5 on mitochondria depolarization in Hela cells (Yan et al, 2020). The released PGAM5-L retained PINK1 at the OMM and initiated mitophagy (Yan et al, 2020).
DRP1 was reported to be involved in the recruitment of PINK1 caused by PGAM5. PGAM5 positively regulated DRP1 activity through dephosphorylation, and DRP1 knockdown inhibited mitophagy in human neuroblastoma cells (Park et al, 2018). The function and localization of PGAM5 are regulated by Stx17. Stx17 deficiency led to aggregation of PGAM5 in mitochondria and halted the dephosphorylation of DRP1 by disrupting their proximity in Hek293T cells (Sugo et al, 2018).
Mitochondrial-localized PGAM5 also dephosphorylated and activated FUNDC1, the mitophagy receptor, on mitophagic stimuli in Hela cells (Chen et al, 2014). The dephosphorylation of FUNDC1 promoted its interaction with LC3, which induced mitophagy (Chen et al, 2014). During this process, Stx17 facilitated the association of cleaved PGAM5 (by PARL) with FUNDC1 in Hek293T cells (Sugo et al, 2018). However, Bcl-2L1 counteracted this interaction and inhibited the dephosphorylation of FUNDC1 and associated mitophagy in Hela cells (Wu et al, 2014a).
On the other hand, PGAM5 also positions centrally in the execution of necroptosis. Necroptosis-inducing condition facilitated PGAM5 to form a protein complex with RIPK1/RIPK3 or necrosome, which phosphorylated PGAM5 and increased its phosphatase activity. Knockdown of PGAM5 attenuated necroptosis, and both variants of PGAM5 were involved in the necroptotic execution in L929 cells (Wang et al, 2012). MLKL mediated the proper interaction between PGAM5-S and necrosome, but not affecting PGAM5-L's binding to RIPK1/RIPK3.
This indicates that PGAM5-S is the downstream effector of complex RIPK1/RIPK3/MLKL/PGAM5-L (Wang et al, 2012). RIPK3 upregulated PGAM5 expression and phosphorylated CYPD in endothelial cells (Zhou et al, 2018). The activated PGAM5-CYPD signaling promoted necroptosis by augmenting the MPT pore opening and thereby damaged mitochondria in endothelial cells and prolactinoma cells (Zhang et al, 2019a; Zhou et al, 2018). AMPK downregulated PGAM5 expression through Keap1-mediated ubiquitination and degradation of PGAM5. Loss of AMPK upregulated PGAM5 level and potentiated necroptosis in cardiomyocytes (Wang et al, 2018b).
DRP1 is a substrate of PGAM5 that is required for necroptotic execution. The activation of DRP1 needs dephosphorylation at amino acid residue Ser637 (Wang et al, 2012). PGAM5-S directly recruited DRP1 to mitochondria, dephosphorylating DRP1 and activating its GTPase activity (Wang et al, 2012). As a mediator, PGAM5 connects necrosome with DRP1 during necroptosis.
However, the central position of PGAM5 is being challenged. Moriwaki et al (2016) have demonstrated that PGAM5 is dispensable for necroptosis caused by either necroptotic inducers or development in the PGAM5-deficient mouse model. A similar observation by Safferthal et al (2017) also revealed that Smac mimetics-triggered necroptosis did not require the participation of PGAM5 in acute myeloid leukemia cells. PGAM5 was even shown to prevent necroptosis. PGAM5 deficiency exacerbated necroptosis in response to various necroptotic stimuli in mouse models (Lu et al, 2016). Through stabilizing PINK1, PGAM5 promoted PINK1-dependent mitophagy and ameliorated necroptosis by preventing the accumulation of abnormal mitochondria and related overproduction of ROS (Lu et al, 2016).
The RIPK1-RIPK3 axis
The crosstalk between mitophagy and necroptosis appears to be bidirectional. Necroptotic factors are also involved in the regulation of mitophagy. RIPK1 has been shown to promote mitophagy. RIPK1 was responsible for the necroptotic cell death induced by extracellular matrix (ECM)-detachment. During ECM-detachment, RIPK1-mediated cell death was independent of the classical effectors, RIPK3 and MLKL, but dependent on PGAM5/PINK-mediated mitophagy.
RIPK1 formed a multi-protein complex with PGAM5 and PINK1, which stabilized PINK1 and prevented PARL-mediated proteolytic cleavage. The intact PINK1 induced mitophagy. The mitophagy contributed to ROS production that triggered the cell death in ECM-detached tumor cells (Hawk et al, 2018).
Another key player of necroptosis, RIPK3 is demonstrated to antagonize mitophagy. Through direct interaction, RIPK3 blocked dephosphorylation of FUNDC1 so as to inactivate FUNDC1 and FUNDC1-mediated mitophagy in I/R injured cardiomyocytes and cardiac microvascular endothelial cells (Zhou et al, 2017a). The impaired mitophagy contributed to the accumulation of mitochondrial fragments and mitochondria-induced apoptosis (Zhou et al, 2017a). RIPK3 also inhibited Parkin-mediated mitophagy in hypoxic cardiomyocytes.
Hypoxia promoted MPT pore opening and related necroptosis, but alleviated mitophagy, which were reversed by either deletion of RIPK3 or overexpression of Parkin. The regulatory effect of RIPK3 on Parkin was mediated by AMPK. RIPK3 suppressed the phosphorylation of AMPK, which sequentially downregulated the phosphorylation of Parkin. The inactivated-Parkin diminished the mitophagy in hypoxia-stressed cardiomyocytes (Zhu et al, 2021a).
Crosstalk Between Autophagy and Other Types of Regulated Necrosis
Autophagy promotes ferroptosis
As a novel form of regulated cell death, ferroptosis is distinct in morphology, biochemistry, and genetics from other types of necrosis. It is implicated in a variety of physiological and pathological processes. The identification that critical modulators in ferroptosis also exhibit the ability to regulate autophagy has revealed the connections between the two processes.
For example, mitochondrial aldehyde dehydrogenase (ALDH2) has been shown to suppress myocardial autophagy under various pathological conditions (Pang et al, 2019; Zhang et al, 2017), and more recently, Zhu et al (2022) reported that transgenic overexpression of ALDH2 attenuated cardiac dysfunction in an AD mouse model via suppression of acyl-CoA synthetase long chain family member 4 (ACSL4)-dependent ferroptosis, indicative of promotion of ferroptosis by autophagy in the heart. Accumulated confident data show that autophagy contributes to ferroptosis (Fig. 6). Li et al (2021b) observed that core components of autophagy and ferroptosis were widely co-elevated in different types of cancer cells when they were exposed to erastin and RSL3.
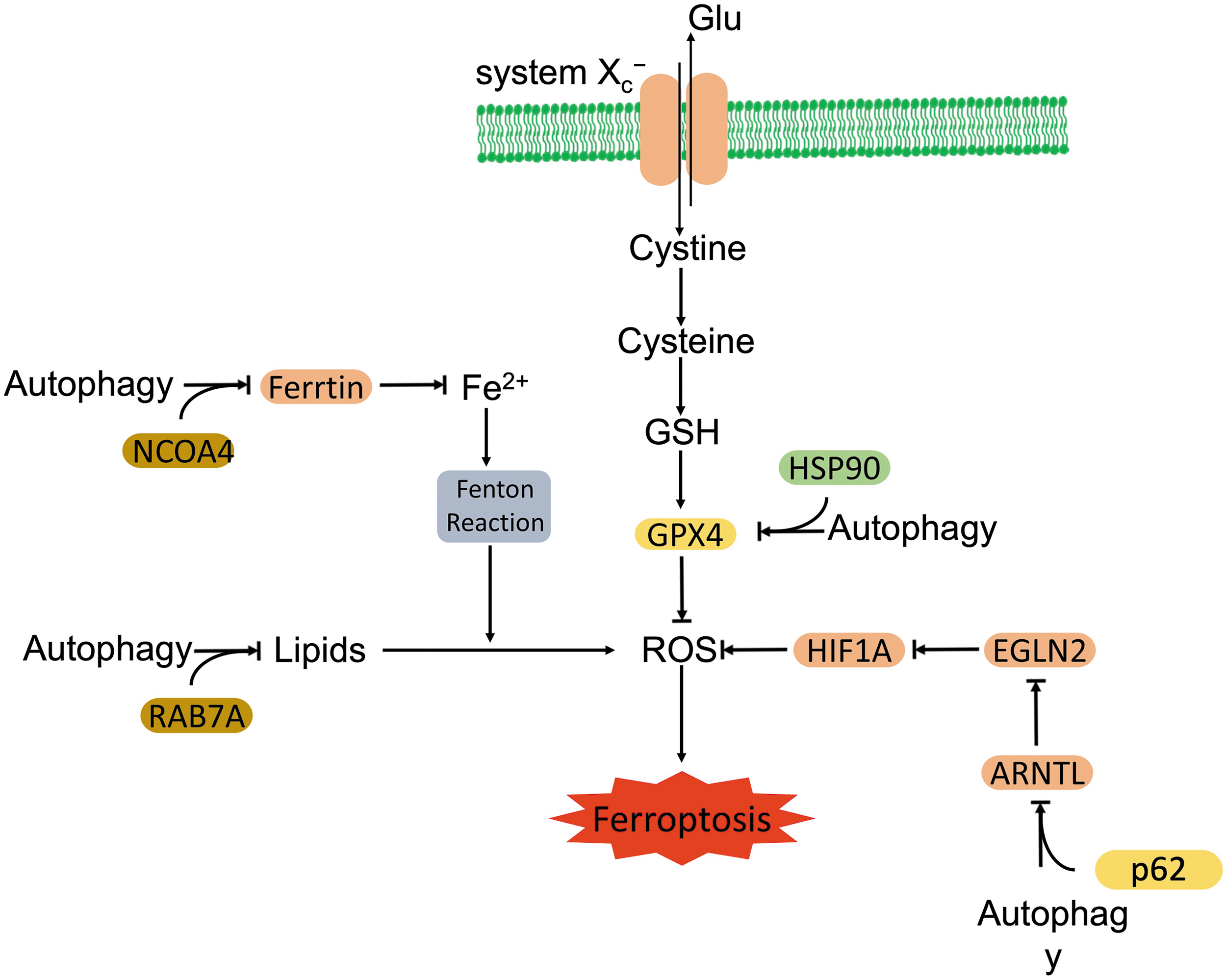
The cancer cells that exhibited a higher autophagic activity on ferroptotic stimuli were also more sensitive to ferroptotic cell death (Li et al, 2021b). Increased autophagy was observed in various cells in response to ferroptotic activators, such as erastin and RSL3 (Gao et al, 2016; Li et al, 2021b). Moreover, pharmacological inhibition of lysosomes prevented erastin- and RSL3-induced ferroptosis, and it attenuated resultant ferroptotic ROS burst in human fibrosarcoma cells (Torii et al, 2016). Concurrently, autophagy deficiency diminished ferroptosis-related lipid peroxidation and intracellular iron level in fibroblasts (Park and Chung, 2019).
More than stimulating ROS, ferroptosis associated factors also modulate autophagy via other mechanisms, including regulating the stability of autophagic proteins, Beclin-1 and ATG16L1. Shen et al demonstrated that ferroptotic inducers increased the N6-methyladenosine (m6A) modification of Beclin-1 messenger RNA (mRNA) through the upregulation of methylase methyltransferase 4 (METTL4), but downregulation of demethylase fat mass and obesity-associated (FTO). Stabilized Beclin-1 activated autophagy and promoted ferroptosis in hepatic stellate cells (Shen et al, 2021).
RNA-binding protein, ELAV like RNA binding protein 1 (ELAVL1), which was required to activate autophagy and promote ferroptosis in hepatic stellate cells, also stabilized Beclin-1 via binding to the AU-rich elements within the 3′-untranslated region (3′-UTR) of Beclin-1 mRNA (Zhang et al, 2018). Ubiquitin specific protease 11 (USP11) deubiquitinated and stabilized Beclin-1, thereby facilitating ferroptosis in spinal neurons after spinal cord I/R injury (Rong et al, 2022). However, poly(rC)-binding protein 1 (PCBP1), a cytosolic iron chaperone, repressed ferritinophagy-mediated ferroptosis. PCBP1 downregulated autophagosome generation via promoting Beclin-1 mRNA decay due to binding to 3′-UTR of Beclin-1 mRNA, whereas it concurrently attenuated lipid peroxidation by decreasing LOX15 expression in head and neck cancer cells (Lee et al, 2022).
Another key factor, ATG16L1 was stabilized by F-box and WD repeat domain containing 7 (FBXW7)-dependent ZFP36 ring finger protein (ZFP36) degradation in hepatic stellate cells (Zhang et al, 2020b). On exposure to ferroptotic inducers, the ubiquitin ligase FBXW7 ubiquitinated ZFP36 and degraded ZFP36 through the ubiquitin-proteasome system (UPS), which abolished ZFP36-mediated ATG16L1 mRNA decay (Zhang et al, 2020b).
Beyond stabilizing autophagy related proteins, ferroptotic inducers also activate autophagy via inhibiting the mTORC1 pathway. Han et al (2020) found that ferroptotic inducer treatment upregulated Sirtuin 3 in trophoblasts, which activated the AMPK-mTORC1 pathway and triggered autophagy.
On the other hand, autophagy is identified to boost ferroptosis. To facilitate ferroptosis, autophagy promotes the degradation of key factors that prevent ferroptosis. These factors are actively involved in iron metabolism (e.g., ferritinophagy), circadian clock cycle (clockophagy), lipid metabolism (lipophagy), and other biological processes.
Ferritins are iron storage proteins that are critical for the regulation of intracellular iron levels. Three forms of ferritin are known: ferritin heavy chain (FTH), ferritin light chain (FTL), and mitochondrial ferritin (FTMT) (Arosio and Levi, 2002; Arosio et al, 2017). Ferritinophagy is the process of autophagic degradation of ferritin, which releases labile iron and activates ferroptosis (Ajoolabady et al, 2021). Erastin-triggered ROS activated ferritinophagy and promoted ferroptosis in fibroblasts (Park and Chung, 2019).
Genetic screening identified the autophagic cargo receptor, nuclear receptor coactivator 4 (NCOA4) as a positive regulator of ferroptosis (Gao et al, 2016). NCOA4 recognized, bound, and trafficked ferritins to the lysosome where the ferritins were degraded (Quiles Del Rey and Mancias, 2019). ATG5 and ATG7, coordinated with NCOA4, initiated selective autophagy that specifically degraded ferritin in fibroblasts and cancer cells when they were exposed to erastin (Gao et al, 2016; Hou et al, 2016). The LPS treatment increased cellular NCOA4 expression and triggered ferritinophagy. Released iron activated siderofexin and then transported cytoplasmic iron into mitochondria, which promoted mitochondrial ROS production and subsequent ferroptosis (Li et al, 2020c).
Inhibition of NCOA4-mediated ferritinophagy antagonized ferroptosis in various cells (Fang et al, 2021; Fuhrmann et al, 2020; Gao et al, 2016; Hou et al, 2016). Interestingly, it seems that the contribution of NCOA4-mediated ferritinophagy to the execution of ferroptosis depends on the types of ferroptotic inducers. Gryzik et al (2021) demonstrated that NCOA4-mediated ferritinophagy promoted erastin-induced ferroptosis, but it delayed RSL3-induced ferroptosis in Hela cells.
Released iron negatively regulates the intracellular level of NCOA4 in a feedback mechanism. Excess cellular iron promoted HERC2 (a ubiquitin E3 ligase) dependent ubiquitination and degradation of NCOA4 through the UPS in Hek293T and cancer cells (Mancias et al, 2015). In contrast, the deubiquitinase, USP14 deubiquitinated NCOA4 and promoted ferritinophagy-dependent ferritin degradation in neurons under spinal cord I/R injury (Li et al, 2021a). Coatomer protein complex subunit zeta 1 (COPZ1) was another regulator that downregulated NCOA4 level in glioblastoma cells, whose knockdown increased NCOA4 and promoted ferritinophagy (Zhang et al, 2021).
Hypoxia inducible factor 1α (HIF1α), on the other hand, was reported to show dual roles in the regulation of ferritinophagy. In one scenario, hypoxia triggered HIF1α activation upregulated NCOA4 and promoted ferritinophagy in dental pulp stem cells (Yang et al, 2021a). However, HIF1α also downregulated ferritinophagy in osteoclasts followed by receptor activator of NF-κB ligand (RANKL) stimulation under hypoxia (Ni et al, 2021). In addition to ferritinophagy, autophagy also promoted iron-derived ferroptosis via degradation of ferroportin-1, an intracellular iron exporter. Autophagic receptor, p62 was responsible for recognizing and executing autophagic degradation of ferroportin-1 on erastin treatment (Li et al, 2021b).
GPX4, as a key regulator of ferroptosis, is universally identified to be degraded in response to various ferroptosis activators. Autophagy also contributes to the degradation of GPX4. Fin56-induced autophagy promoted the degradation of GPX4 and ferritin, whereas inhibition of autophagy dampened Fin56-induced lipid peroxidation and ferroptosis (Sun et al, 2021b). Rapamycin or RSL3 was shown to inactivate mTORC1, which led to autophagy-dependent GPX4 degradation in human pancreatic cancer cells.
Moreover, GPX4 inhibited autophagy-dependent ferroptosis induced by rapamycin or RSL3 (Liu et al, 2021b). In addition, acid sphingomyelinase (ASM)-related redox amplification also facilitated ferroptotic inducers-induced autophagic degradation of GPX4, and it promoted ferroptosis in cancer cells (Thayyullathil et al, 2021). Chaperone-mediated autophagy (CMA) is a type of selective autophagy that specifically delivers certain cytosolic proteins to the lysosome for degradation with coordination of chaperones. GPX4 was reported to be degraded via CMA in an HSP90-dependent manner, followed by ferroptosis activation.
Ferroptotic stimuli increased the interactions of GPX4 with HSP90 and heat shock cognate protein 70 (HSC70). HSC70 recognized GPX4 through a KEFRQ-like motif and coordinated lysosome-associated membrane glycoprotein 2a (LAMP-2a) that promoted CMA-mediated degradation of GPX4 and facilitated ferroptosis in hippocampal cells (Wu et al, 2019). Further, HSC70-dependent CMA also promoted erastin-induced GPX4 degradation in breast cancer cells (Wu et al, 2019). However, GRP78, an ER stress-associated chaperone, was reported to protect against ferroptosis via preventing erastin-induced GPX4 degradation in pancreatic cancer cells (Zhu et al, 2017).
Aryl hydrocarbon receptor nuclear translocator-like (ARNTL) is the key circadian clock transcriptional factor that regulates circadian clock related genes by binding to their E-box motifs of promoters. ARNTL was identified to upregulate HIF1α through downregulated egl nine homolog 2 (EGLN2), an ARNTL target gene in RSL3-induced ferroptosis in different cancer cells (Yang et al, 2019b). Enhanced HIF1α upregulated fatty acid binding proteins (FABPs) to promote fatty acid uptake and lipid storage, which prevented ferroptosis (Yang et al, 2019b).
In response to ferroptotic stimuli, autophagy (clockophagy) selectively degraded ARNTL through cargo receptor p62. Loss of ARNTL activated EGLN2 and inactivated HIF1α, which promoted ferroptosis via by the initiation of lipid peroxidation (Yang et al, 2019b).
Lipid droplets are complex organelles whose digestion releases free fatty acid and fuels the peroxidation of lipids (Bai et al, 2019). Autophagy or lipophagy was reported to promote the lysosomal decomposition of lipid droplets. Bai et al (2019) demonstrated that RAB7A-dependent lipophagy promoted lipid droplet degradation and facilitated lipid peroxidation-mediated ferroptosis in hepatocytes.
Recently, Chen et al (2022) identified a novel autophagic receptor, hippocalcin like 1 (HPCAL1) that selectively degraded cadherin 2 (CDH2), a protein that increased ferroptotic susceptibility by reducing membrane tension and favoring lipid peroxidation. The autophagic degradation of CDH2 required protein kinase C theta (PRKCQ)-mediated HPCAL1 phosphorylation in pancreatic cells (Chen et al, 2022).
Besides degradation, autophagy also promotes ferroptosis via other mechanisms. Key regulator of autophagy, Beclin-1 promoted ferroptosis through directly blocking system Xc − activity via binding to its core component, solute carrier family 7 member 11 (SLC7A11) in cancer cells (Kang et al, 2018; Song et al, 2018). AMPK-mediated phosphorylation of Beclin-1 facilitated Beclin1-SLC7A11 complex formation and subsequent lipid peroxidation in cancer cells (Kang et al, 2018; Song et al, 2018).
Moreover, autophagy sensitized cells to erastin-induced ferroptotic cell death via upregulating VDAC3 post-transcriptionally. Autophagy downregulated FBXW7, which ubiquitinated VDAC3 and promoted its degradation through the UPS (Zhu et al, 2021b). Ablation of FUNDC1-dependent mitophagy significantly attenuated paraquat-induced myocardial ferroptosis and cardiomyocyte contractile malfunction (Peng et al, 2022).
In contrast, evidence that autophagy inhibits ferroptosis is also reported. Buccarelli et al (2018) reported that quinacrine-mediated autophagy inhibition augmented the temozolomide-induced ferroptosis, which was alleviated by iron chelator or ferrostatin-1 in glioblastoma cells. Epigallocatechin-3-gallate alleviated doxorubicin-induced ferroptosis in the myocardium by upregulating AMPK-dependent adaptive autophagy, which increased energy supply, and maintained mitochondrial function (He et al, 2021). Further studies also revealed that HSC70 mediated CMA promoted the degradation of ACSL4 via lysosomes, which removed the accumulation of lethal lipid species and inhibited ferroptosis in retinal pigment epithelial cells (Liu et al, 2022).
One more thing that needs to be mentioned is the autophagy-dependent ferroptosis-mediated intracellular content release. Dai et al demonstrated that autophagy-dependent ferroptotic cell death facilitated oxidative stress-induced mutant KRAS (G12D) release from pancreatic cancer cells to an extracellular microenvironment, which stimulated a macrophage-derived pancreatic adenocarcinoma growth. During the process, the inhibition of autophagy abolished KRAS G12D release, whereas ferroptosis inhibitors blocked autophagy, ferroptosis, and KRAS G12D release (Dai et al, 2020).
Meanwhile, high mobility group box protein 1 (HMGB1), as a DAMPs, was released by ferroptotic cells in an autophagy-dependent manner. Autophagy-mediated histone deacetylase (HDAC) inhibition promoted HMGB1 acetylation and the subsequent release. HMGB1 was recognized by advanced glycation end-product receptor (AGER), which mediated HMGB1 mediated inflammation in macrophage in response to ferroptotic cells (Wen et al, 2019).
Autophagy counteracts pyroptosis
Inflammasome activation not only triggers pyroptosis but also accompanies autophagy (Byrne et al, 2013; Shi et al, 2012). Induced autophagy negatively regulates pyroptosis (Fig. 7). Pharmacological or genetic inhibition of autophagy enhanced pyroptosis and associated production of inflammatory cytokines (Pu et al, 2017; Saitoh et al, 2008; Suzuki et al, 2007), whereas autophagy induction conferred resistance to pyroptotic cell death (Suzuki and Nunez, 2008).
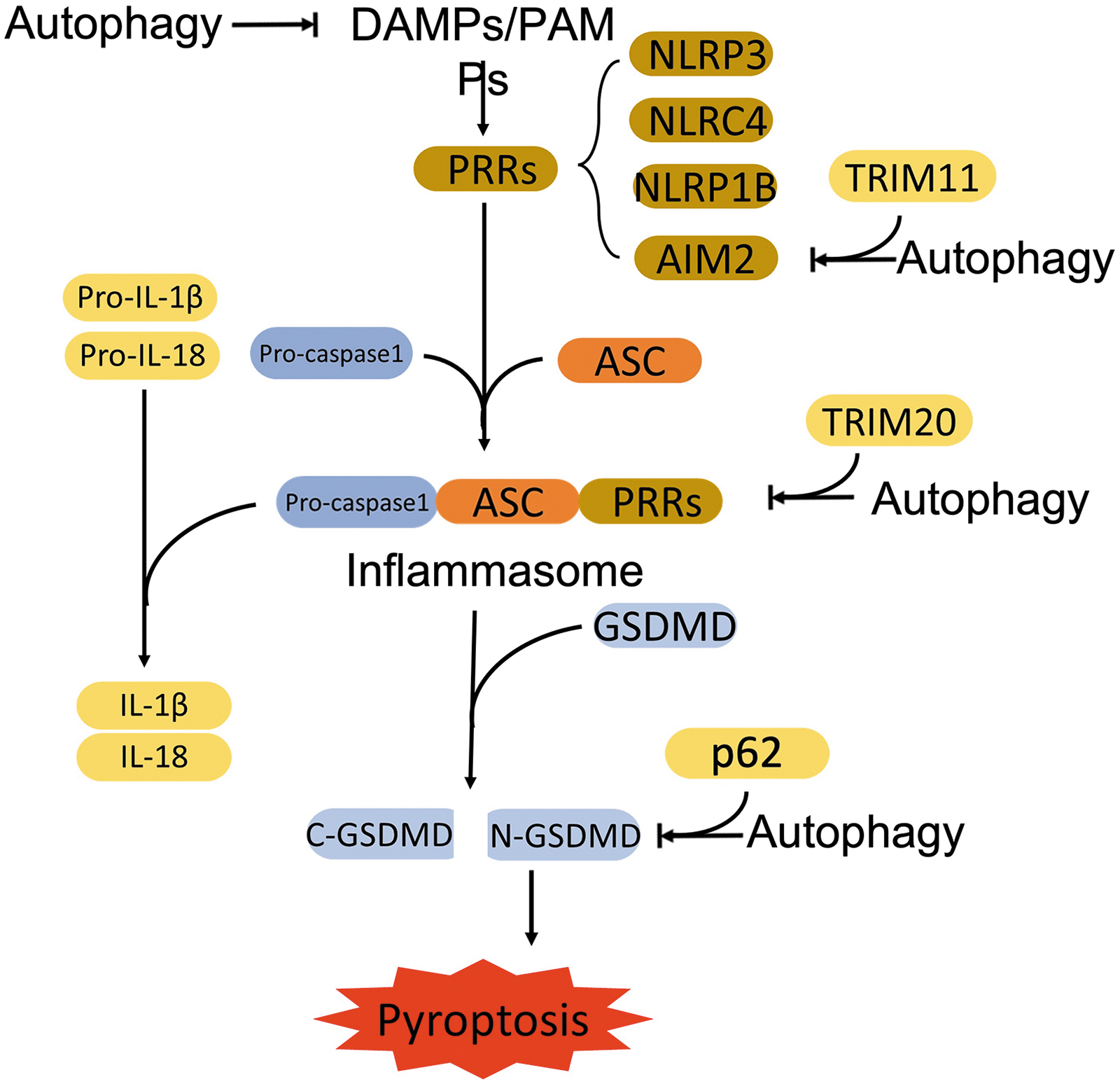
Mechanistically, the anti-pyroptotic roles of autophagy can be summarized mainly as the elimination of DAMPs and PAMPs, and the removal of key components of inflammasome. DAMPs and PAMPs are recognized by PRRs to initiate canonical pyroptosis. Nakahira et al (2011) showed that autophagy suppressed LPS/ATP-caused NALP3-dependent release of mitochondrial DNA, which inhibited pyroptosis and the secretion of IL-1β and IL-18 in macrophages. Similar results were also observed in microglia-mediated neurotoxicity.
Quercetin treatment promoted mitophagy to eliminate damaged mitochondria-associated ROS accumulation, which alleviated LPS-induced NLRP3 inflammasome activation and pyroptosis in neurons, and protected against microglial toxicity (Han et al, 2021). Increased ROS (sensed by NLRP3) also stimulated autophagy and, in turn, inhibited pyroptosis in Leydig cells and microglia (Kim et al, 2015a; Li et al, 2019).
Further, autophagy facilitates the clearance of essential components of inflammasome. Magupalli et al (2020) demonstrated that inflammasome was degraded by autophagosome by showing a co-localization of ASC and LC3II at the microtubule-organizing center (MTOC) after nigericin stimulation. Exogenous DNA-induced inflammasomes ubiquitination recruited autophagic receptor p62 and facilitated autophagic degradation of AIM2-based inflammasome (Shi et al, 2012). Tripartite motif 11 (TRIM11) served as a regulator that promoted p62-mediated autophagic degradation of AIM2 in macrophages (Liu et al, 2016a).
TRIM20 also promoted autophagic degradation of inflammasome components, NLRP3, NLRP1, and pro–caspase-1 in immune cells, including myelomonocytes and macrophages (Kimura et al, 2015). Accumulation of p62 due to autophagy flux impairment activated Nrf2, which, in turn, activated pyroptosis in macrophage on oxidized low-density lipoprotein treatment (Liu et al, 2021a). Besides the removal of inflammasome components, autophagy was also reported to downregulate the executors of pyroptosis, GSDMs.
Autophagy induction alleviated pyroptosis via the downregulation of cleaved GSDND level (Wang et al, 2020b; Zhuo et al, 2020). Liao et al (2021) further revealed that/p62 mediated autophagic degradation of N-GSDMD in LPS-treated nucleus pulposus cells. Meanwhile, enhanced autophagy also inhibited GSDME-mediated pyroptosis in melanoma cells (Yu et al, 2019).
In addition, autophagy also regulated the secretion of mature cytokines. Harris et al (2011) demonstrated that autophagy sequestered pro-IL-1β and facilitated its degradation by the lysosome in macrophages.
Not limited to the inhibition of the canonical pyroptotic pathway, autophagy is also identified to suppress the non-canonical pyroptosis pathway. It was found that Beclin-1 antagonized caspase-3-GSDME axis-mediated pyroptosis independent of its autophagic roles in hematopoietic stem cells (Yang et al, 2020). In human cardiac microvascular endothelial cells, overexpression of Beclin-1 suppressed caspase-4 activity and GSDMD expression caused by I/R, which alleviated pyroptosis and IL-1β levels (Sun et al, 2021a).
GTPases dependent lysis of vacuole released gram-negative bacteria whose LPS activated caspase-11 and non-canonical pyroptosis. The released bacteria also promoted galectin-8-mediated autophagy, which removed the pathogens and reduced caspase-11 dependent pyroptosis in macrophages (Meunier et al, 2014).
Some other factors are also found to mediate the antagonistic consequence of autophagy to pyroptosis. Enhanced expression of vascular endothelial growth factor receptor 3 (VEGFR3) and its ligand VEGFC was induced on Salmonella typhimurium infection in macrophages, whose activation inhibited caspase-1-dependent inflammasome formation and associated pyroptosis, but agonized AMPK dependent autophagy to eliminate bacteria (Ma et al, 2022). The ubiquitin ligase, TRAF3 inactivated autophagy via proteasomal degradation of ULK1 with the coordination of TRAF2 and cIAP1, which promoted LPS/nigericin-induced pyroptosis by activating inflammasomes in macrophages (Shen et al, 2020).
TRK-fused gene (TFG) rescued pyroptotic cell death by stabilizing ULK1 via blocking TRAF3-mediated ubiquitination and proteasomal degradation, which promoted autophagy and removed intracellular ROS on the same treatment (Shi et al, 2022). Recently, immunity-related GTPases M clade (IRGM) family proteins were identified as novel mediators between autophagy and canonical or non-canonical pyroptosis. Increased IRGM, stimulated by pyroptotic stimuli, interacted with NLRP3 and ASC, blocking their oligomerization and promoting their selective autophagic degradation, which inactivated NLRP3 inflammasomes and inhibited canonical pyroptosis (Mehto et al, 2019).
Meanwhile, it was also reported that IRGM inactivated caspase-11-dependent inflammatory response, whereas it activated autophagy via direct interaction with ATG8 (Eren et al, 2020; Finethy et al, 2020; Nath et al, 2021).
The counteracted role of autophagy to pyroptosis is also challenged by several reports. For example, benzo[a]pyrene-induced pyroptosis was not potentiated by autophagy inhibition in human liver cells (Yuan et al, 2017). Dupont et al (2011) and Karmakar et al (2020) also found that IL-1β was secreted via an autophagy-dependent mechanism in BMDMs and neutrophils. The cause for this discrepancy is unclear but may be attributable to the difference in pathological conditions and cell types.
Crosstalk between MPT and autophagy
Increase of ROS resulting from MPT could trigger autophagy. During the process, CYPD, as the key regulator of MPT induction, promoted starvation-induced autophagy. Inhibition of CYPD preserved mitochondrial membrane potential and blocked MPT-induced autophagy in cardiac cells (Carreira et al, 2010). In addition, CYPD-enhanced autophagy acted as a pro-survival pathway in response to oxidative stress and in aging Podospora anserina (Kramer et al, 2016).
Conclusions and Perspectives
Compared with what is known about the interrelationship between autophagy and other types of regulated necrosis, the crosstalk between autophagy and necroptosis seems to be more complicated, which may be simply due to the fact that it has been investigated more in depth and breadth. It appears that autophagy can promote, suppress, or independently co-exist with necroptosis under certain circumstances. Degterev et al described that autophagy and necroptosis were independently regulated. Autophagy was not involved in the necroptosis-executed cell death, but a cellular response to necroptosis (Degterev et al, 2005).
However, seemingly more evidence suggests that these two processes are interwoven together. In terms of a temporal relationship between autophagy and necroptosis in the signaling process of necroptosis, autophagy has been shown to position either upstream or downstream of necroptosis. For instance, Ye et al demonstrated that Nec-1 completely blocked the autophagy during the process of TNFα-induced necroptosis, which suggested that autophagy might be a downstream consequence of necroptosis (Ye et al, 2011).
On the other hand, autophagosomes were found to provide a platform for the assembling of necrosome, indicating an upstream role of autophagy in necroptotic signaling (Goodall et al, 2016). So, it is very possible that the hierarchical relationship between autophagy and necroptosis is etiology and cellular circumstance dependent. It should be pointed out that many earlier studies relied solely on the inhibition of RIPK1 kinase activity to suppress necroptosis but it is now clear that RIPK1 has many other roles than just mediating the necroptotic pathway. As a result, the conclusions of some of the studies may need to be re-evaluated, which may help clarify the controversy.
The dual roles of autophagy, both cell-protective and cell-destructive, also may determine its opposing contributions to necroptosis, either counteracting or promoting necroptotic cell death. Autophagy is predominately known for its ability to maintain cellular homeostasis, which prevents cell death in response to different cellular stresses. For example, autophagy was activated to promote cell survival when cells underwent amino acid starvation (Thomas et al, 2018). This protective mechanism of autophagy was applied to amino acid starvation-induced necroptosis, where autophagy suppressed necroptosis by degradation of RIPK3 and MLKL (Xie et al, 2020).
It was widely observed that the inhibition of autophagy deteriorated necroptotic cell death (Farkas et al, 2011; Goodall et al, 2016), which supports a protective role for autophagy against necroptosis. On the opposite side, autophagy is also identified to exert cell death that is termed “autophagic cell death” (Denton and Kumar, 2019). However, in our discussion, autophagic cell death does not refer to autosis, but autophagy-mediated necroptosis. Autophagy promoted necroptosis by removal inhibitory molecules of necroptosis (He et al, 2014) and provided a platform for the necrosome (Goodall et al, 2016).
Inhibition of autophagy impeded necroptotic cell death (He et al, 2014). The dual functions of autophagy may also contribute to the controversial interplay between autophagy and necroptosis.
The key components of necroptosis are also involved in the regulation of autophagy. RIPK1 promoted autophagy through activating the upstream regulators of mTORC1, AMPK (Najafov et al, 2021). RIPK1-deficient cells were vulnerable to energetic stress, indicative of an important role of RIPK1 in the cellular response to low energy crisis (Najafov et al, 2021). Through activation of nutrient sensor, RIPK1 triggered autophagy. In addition, it was also reported that RIPK3 directly interacted with AMPK and initiated autophagy (Wu et al, 2021b).
Both RIPK1 and RIPK3 mainly enhance autophagy at the early stage. However, RIPK1 and RIPK3 are also involved in the downregulation of autophagy through the dysregulation of autophagosome maturation (Wu et al, 2021b). MLKL is the key factor that suppresses autophagy. MLKL displays multiple roles in the suppression of autophagy, including damaging the integrity of autophagosomes (Frank et al, 2019) and inhibiting autophagy initiation (Guo et al, 2019). The necroptotic regulation of autophagy suggests that the interplay between autophagy and necroptosis is bi-directional.
Both autophagy and necroptosis are implicated in multiple human diseases, such as cardiovascular diseases and cancer. In a normal heart, the basic level of autophagy is responsible for recycling cytoplasmic components but altered in response to I/R stress or other cardiovascular diseases. For example, enhanced autophagy was observed in human failing hearts with idiopathic dilated cardiomyopathy (Kostin et al, 2003). Upregulation of autophagy protects cardiomyocytes. Cardiac-specific autophagy deficiency caused cardiac dysfunction accompanied by abnormal protein accumulation after transverse aortic constriction (TAC) (Nakai et al, 2007).
Necroptosis also exerts an important effect on cardiovascular disease. Abnormal induction of necroptosis led to adverse remodeling and heart failure (Li et al, 2014). The disrupted relationship between autophagy and necroptosis contributed to cardiovascular diseases. Zhang et al (2020a) demonstrated that autophagy dysregulation led to activation of necroptosis which caused loss of cardiomyocytes, adverse ventricular remodeling, and heart failure.
Restoration of autophagic flux protected cardiomyocytes and ameliorated necroptotic cell death after I/R injury (Qiu et al, 2021). The results confirmed the protective and anti-necroptotic effect of autophagy in cardiovascular diseases. However, Liu et al (2016b) revealed that the inhibition of autophagy attenuated necroptosis in cardiomyocytes. HSP70 downregulated necroptosis through suppressing autophagy (Liu et al, 2016b). Here, it suggests that the destructive role of autophagy in necroptosis caused cell death of cardiomyocytes. It is important to clarify the crosstalk between autophagy and necroptosis systematically as it is expected to improve our understanding of the causes of cardiovascular diseases and its treatment.
Likewise, both autophagy and necroptosis also exhibit dual roles in cancer. For instance, autophagy served as either a resisting or a promoting mechanism to death receptor agonist-induced apoptosis. The opposing effects appear to depend on the degradation of either pro-apoptotic or anti-apoptotic molecules by autophagy, respectively (Gump et al, 2014; Thorburn et al, 2014). Necroptosis may serve as an alternative cell death mode when cancer cells acquire apoptotic resistance (Gong et al, 2019), preventing tumor development. However, necroptosis-associated DAMPs trigger inflammatory responses that promote cancer metastasis and immunosuppression (Seifert et al, 2016; Strilic et al, 2016).
Inhibition of autophagy potentiated chemotherapeutics-induced necroptosis (Han et al, 2007; Kim et al, 2017). Meanwhile, induction of autophagy overwhelmed the acquired-apoptotic resistance of anti-cancer regiment by enhancing necroptosis (Bonapace et al, 2010). So, proper modulation of the interplay between autophagy and necroptosis is expected to improve the efficacy of cancer therapy.
The intricate crosstalk between autophagy and necroptosis may also be stimuli and cell-type dependent. For instance, different stimuli-triggered autophagy selectively degraded different factors that either antagonize or promote necroptosis. Starvation-triggered autophagy removed RHIM proteins, which protected cancer cells from necroptosis (Lim et al, 2019); however, anticancer chemotherapy-mediated autophagy degraded cIAPs to facilitate necroptosis in cancer cells (He et al, 2014).
Meanwhile, autophagy also displayed opposing effects in response to same stimuli-induced necroptosis in different cells. PA-mediated lipotoxicity induced necroptosis, which was attenuated by autophagy in pheochromocytoma cells (Montero et al, 2020) but was aggravated by autophagy in endothelial cells (Khan et al, 2012). Wu et al (2021b) even reported TNFα-induced necroptosis differentially affected the different stages of autophagy, promoting the autophagy initiation but disrupting autophagosome maturation in L929 cells.
The variation in the relationship between autophagy and necroptosis originated from different stimuli, and cell types may partially explain the intricacy of the interplay between autophagy and necroptosis. Appropriate manipulation of the variation may help to treat different diseases more precisely.
In comparison to the crosstalk between autophagy and necroptosis, the interplay between autophagy and ferroptosis or pyroptosis seems to be much less complicated. Ferroptosis is considered as a type of autophagy-dependent cell death (Liu et al, 2020a; Zhou et al, 2020). Classical ferroptotic activators, such as erastin and RSL3, increase autophagy flux in various cells. Meanwhile, ferroptosis is associated with autophagy activation and autophagy promotes the ferroptotic cell death process (Gao et al, 2016). Components of autophagy machinery contribute to ferroptosis.
For instance, knockout of ATG3 and ATG13, two key factors that function at different stages of autophagy, significantly downregulated the sensitivity of MEFs to erastin or cystine starvation-induced ferroptosis (Gao et al, 2016). In addition, Beclin-1 directly inhibited system Xc − to facilitate ferroptosis. Further, selective autophagy, such as NCOA4-facilitated ferritinophagy (Hou et al, 2016), HSP90-associated CMA (Wu et al, 2019), RAB7A-dependent lipophagy (Bai et al, 2019), and p62-mediated clockophagy (Yang et al, 2019b), can also promote ferroptosis via selective degradation of key players involved in the ferroptotic process.
Recent studies have revealed the important role of ferroptosis in the occurrence and development of many diseases such as cancer and I/R injury (Li et al, 2020b). The delineation of the interplay between autophagy and ferroptosis inspires exploration for novel therapies for these diseases.
Studies reported so far have mainly revealed that autophagy negatively modulates pyroptosis and minimize the harmful effect of pyroptosis. Pyroptosis is regarded as a part of innate immunity, which also activates adaptive immunity. The released IL-1β and IL-18 not only recruit neutrophils and macrophages, but also stimulate CD4+ T cells and promote B cell proliferation and related antibody production (Van Gorp et al, 2019). Autophagy antagonizes pyroptosis through elimination of its causes (DAMPs and PAMPs) and its indispensable components.
An improved understanding of the relationship between autophagy and pyroptosis will also likely lead to novel therapeutic strategies to manage pyroptosis-associated diseases. For example, chemicals were identified to inhibit LPS-stimulated inflammasome activation and cytokine release through induction of autophagy for sepsis (Ge et al, 2019; Yang et al, 2021b; Yang et al, 2019a). Further studies are required to unveil the complete picture of autophagic regulation of pyroptosis.
In summary, the interplay between autophagy and regulated necrosis involves both promotion and suppression. The influence between these two processes is not unidirectional but bidirectional, and it is conditional. The wide involvements of autophagy and necroptosis in physiological and pathological processes make their interplay extremely important. Investigation of the crosstalk benefits the further understanding of the maintenance of cellular homeostasis and pathogenesis.
Footnotes
Authors' Contributions
L.Z.: Conceptualization (equal); writing—original draft (lead); writing—review and editing (supporting). T.C.: Writing—review and editing (supporting). X.W.: Conceptualization (equal); writing—original draft (supporting); writing—review and editing (lead).
Author Disclosure Statement
The authors declare that there is no conflict of interest.
Funding Information
X.W.'s research was supported in part by NIH grants HL72166, HL085629, HL153614, and AG072510 and American Heart Association grant 20TPA35490091. L.Z.'s research is supported in part by Jilin Agricultural University Startup Grant 0205/201020785.