
Abstract
Significance:
Translation is an essential cellular process, and diverse signaling pathways have evolved to deal with problems arising during translation. Erroneous stalls and unresolved ribosome collisions are implicated in many pathologies, including neurodegeneration and metabolic dysregulation.
Recent Advances:
Many proteins involved in detection and clearance of stalled and collided ribosomes have been identified and studied in detail. Ribosome profiling techniques have revealed extensive and nonprogrammed ribosome stalling and leaky translation into the 3′ untranslated regions of mRNAs. Impairment of protein synthesis has been linked to aging in yeast and mice.
Critical Issues:
Ribosomes act as sensors of cellular states, but the molecular mechanisms, as well as physiological relevance, remain understudied. Most of our current knowledge stems from work in yeast and simple multicellular organisms such as
Future Directions:
A better insight into the physiological roles of ribosome-surveillance pathways and their crosstalk could lead to an improved understanding of human pathologies and aging. Antioxid. Redox Signal. 39, 336–350.
Introduction
Protein translation occurs constantly in all living cells, and needs to be tightly controlled to ensure homeostasis and survival on a cellular as well as organismal level. A diverse range of ribosome-surveillance pathways recognize and respond to physiological and pathological obstructions to translation.
The major response systems are as follows: (i) ribosomal subunit splitting, where activating signal cointegrator (ASC) proteins ASCC1, ASCC2, ASCC3, and TRIP4 dissociate stalled ribosomes into 40S and 60S subunits; (ii) ribosome quality control (RQC), where the protein nuclear export mediator factor (NEMF) binds to the 60S subunit with an attached peptidyl-tRNA, and the E3 ligase Listerin (LTN1) ubiquitinates the nascent polypeptide targeting it for degradation by the proteasome; (iii) the ribosomal branch of the integrated stress response (ISR), which is mounted by the GCN2 kinase to repress bulk translation; and (iv) the ribotoxic stress response (RSR), which through the MAP3 kinase ZAKα regulates inflammation and cell death in response to severe stress (Table 1). Together, these pathways act to protect the cell from the detrimental consequences of compromised translation.
Key Ribosome-Surveillance Factors
ASC, activating signal cointegrator; CAT, C-terminal alanine and threonine; ISR, integrated stress response; NEMF, nuclear export mediator factor; RSR, ribotoxic stress response.
During the last few years, many factors involved in ribosome-surveillance pathways have been identified and studied in various organisms. Several of these pathways are conserved throughout evolution and have as well been identified in prokaryotes. In this review, we summarize recent findings, focusing on translation stress responses in a physiological context, and their links to pathologies in mice and humans. For more molecular aspects of these pathways, we refer to recent reviews (Filbeck et al., 2022; Howard and Frost, 2021; Kim and Zaher, 2022; Meydan and Guydosh, 2021; Vind et al., 2020a; Yip, 2021).
Sources of Translation Stress
Many different perturbations from both endogenous and exogenous sources cause ribosomal impairment, resulting in compromised translation (Fig. 1). Exogenous sources of translational stress have been widely studied, and include ultraviolet (UV) irradiation, small-molecule translation inhibitors (such as the antibiotic anisomycin or cycloheximide), ribotoxins (such as the castor bean toxin ricin or the bacterial toxin Shiga), and the introduction of damaged or difficult-to-translate reporter plasmids to cells. Other examples of exogenous sources are translation stress due to high proliferation in cancer cells, or viruses that can modify and exploit the host translation machinery, which will be further discussed in the chapter on immune signaling.
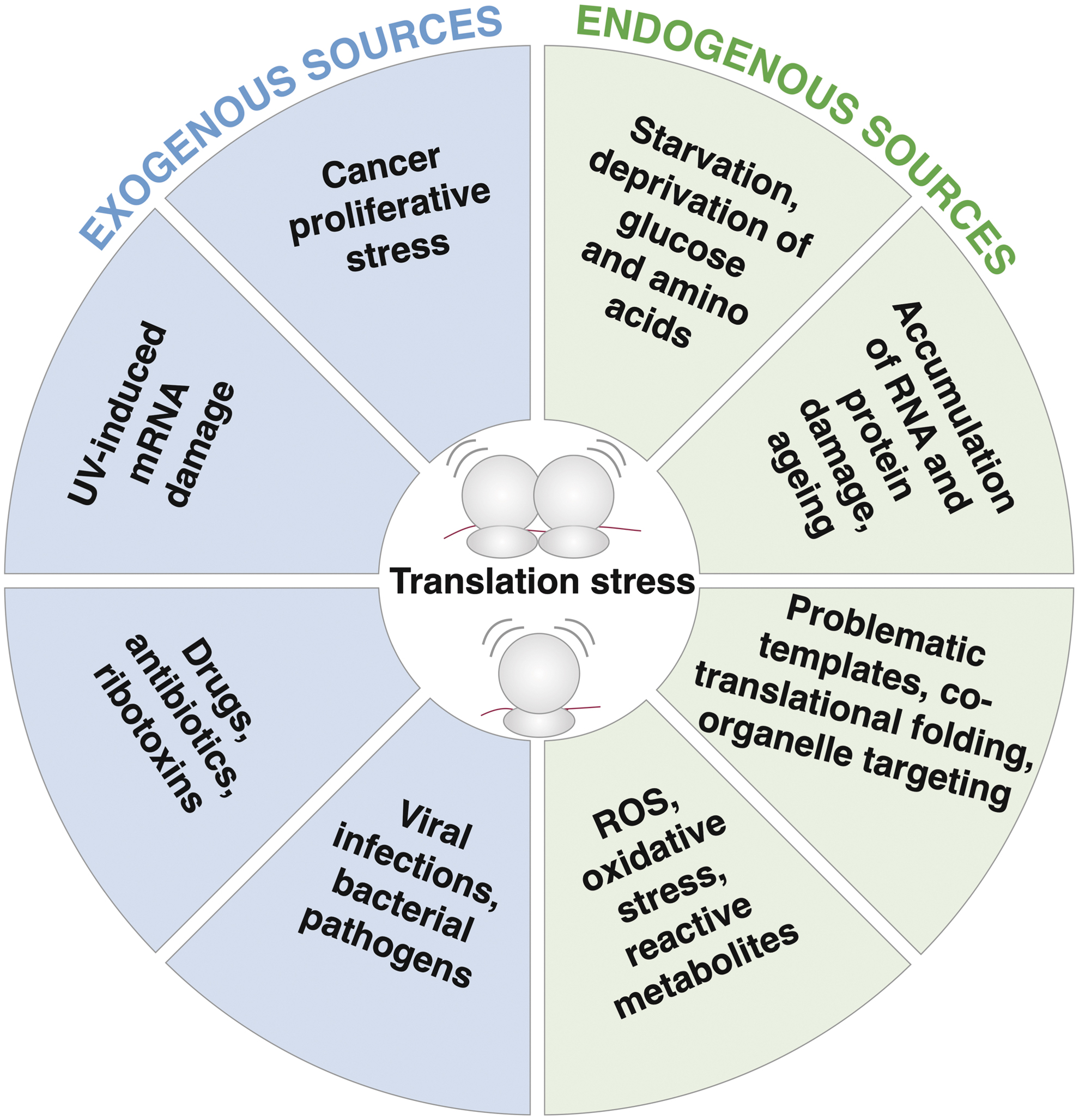
Endogenous sources of translation stress are less well studied as they are often more challenging to control and manipulate. They comprise shortage of nutrients leading to unloaded tRNAs, inherently problematic RNA sequences, or oxidative stress as a metabolic by-product.
Recently, RNA sequences enriched for stalled and collided ribosomes revealed abundant translation problems on endogenous mRNA substrates (Arpat et al., 2020; Han et al., 2020; Meydan and Guydosh, 2020). A failure to decode stop codons can cause translation of the 3′ untranslated region, and premature cleavage and polyadenylation of mRNA lead to ribosome stalling. This is especially evident in the context of long stretches of adenine-rich sequences and translation of proline tracts (Guydosh and Green, 2017; Han et al., 2020). Wobble base pairing between tRNA and mRNA (Stadler and Fire, 2011), reduced levels of aminoacyl-tRNAs (such as during amino acid shortage), and chemical modification of tRNAs can add to translation stress (Gu et al., 2014).
Oxidative agents can cause nucleotide damage to mRNA, and adducts such as 8-hydroxyguanine can lead to ribosome stalling (Simms et al., 2014). Increased levels of 8-hydroxyguanine damage to mRNA and rRNA were found in brain tissue from patients with neuropsychiatric disorders (Che et al., 2010).
Ribosome stalling also occurs when proteins are cotranslationally targeted to the endoplasmic reticula or mitochondria. Specialized RQC pathways are dealing with those types of translation stalls (Joazeiro, 2019).
Ribosome-Surveillance Pathways
The ribosome-surveillance pathways are important mechanisms employed by the cell to detect and respond to obstructed translation. During elongation, the ribosome may encounter obstacles or lack the required amino acid, which will cause it to pause (Buskirk and Green, 2017). If the ribosome is more permanently stuck, this stalling can lead to ribosome collision if an elongating ribosome runs into the stalled one (Box 1). These so-called “disomes” constitute both a substrate for faithful resolution and a molecular signal for the presence of severe translation stress. The interface between collided ribosomes is generally considered a recruitment platform for the sensor components of ribosome-surveillance pathways (Fig. 2).
Ribosome pausing, stalling and collision

Prevention of translation initiation on problematic mRNA templates
One of the early responders to collided ribosomes is EDF1, which recruits GIGYF2 that together with 4EHP represses translation locally to prevent reinitiation on problematic mRNAs (Hickey et al., 2020; Sinha et al., 2020). Recruitment of EDF1 is dependent on RACK1, a protein component of the 40S subunit, highlighting the central placement of RACK1 on the interface of collided ribosomes.
In parallel, activation of the ISR via the GCN2 kinase reduces the risk of further ribosome collision by globally inhibiting new rounds of mRNA translation initiation (Darnell et al., 2018; Stoneley et al., 2022; Wu et al., 2020). Here, GCN2 phosphorylates eIF2α, which in turn inhibits eIF2B. This is prohibitory to initiation of cap-dependent translation but does not impair a subset of transcripts that are required for ISR-induced translational reprogramming, such as the transcription factor ATF4 (Wek et al., 2006).
Disassembly of stalled and collided ribosomes
Another sensor of ribosome collision is the E3 ubiquitin ligase ZNF598 (Juszkiewicz et al., 2020a), which ubiquitinates RPS20/uS10 and RPS10/eS10. These ubiquitination events facilitate the splitting of the leading 80S ribosome by the ASC-1 complex composed of ASCC1–3 and TRIP4 (Juszkiewicz and Hegde, 2017; Juszkiewicz et al., 2020b; Stoneley et al., 2022). Despite ZNF598 playing an important role in recognizing collided ribosomes, very few RQC proteins directly depend on ZNF598 for recruitment to disomes (Sinha et al., 2020), highlighting a gap in our understanding of the role(s) of 40S subunit ubiquitination.
Once the leading ribosome is removed, trailing ribosomes can resume elongation and only become targets of degradation if they themselves stall and cause a collision (Juszkiewicz et al., 2020b). Stalled ribosomes with empty decoding centers are not disassembled by the ASC-1 complex but can be recognized by the HBS1L–Pelota complex. HBS1L hydrolyzes GTP and dissociates from the ribosome, allowing Pelota to bind and recruit the ATPase ABCE1, which then separates the ribosomal subunits (Juszkiewicz et al., 2020b).
ZNF598 is not the only E3 ligase recognizing stuck ribosomes. Mono-ubiquitination by the E3 ligase RNF10 on the 40S proteins RPS2/uS5 and RPS3/uS3 has been observed on blocked initiating ribosomes as well as on stalled elongating ribosomes (Garshott et al., 2021; Garzia et al., 2021), contrary to ZNF598, which only recognizes collided ribosomes. Stalled preinitiation complexes are ubiquitinated by RNF10, leading to 40S degradation, while ribosomes already engaged in translation undergo resolution, and the nascent chain is degraded by the proteasome (Garshott et al., 2021).
While the functional consequences of ubiquitination of the different 40S proteins by ZNF598 and RNF10 are still rather unclear, the ubiquitin code appears to be involved in (i) distinguishing physiologically paused from pathologically stalled ribosomes, (ii) disassembling compromised ribosomes, and (iii) acting as a signal for lysosomal or proteasomal degradation of the 40S subunit (An and Harper, 2020; Garshott et al., 2021; Meyer et al., 2020).
Degradation of the nascent peptide chain
After disassembly of the terminally stalled ribosome, the RQC system ensures degradation of the incomplete protein product. The obstructed 60S subunit is recognized by NEMF that recruits the E3 ubiquitin ligase LTN1, which ubiquitinates the lysine residues on the nascent chain, thereby marking it for proteasomal degradation. NEMF can facilitate ubiquitin-mediated proteolysis by adding C-terminal alanine and threonine (CAT) tails to stalled polypeptides, pushing the chain out of the exit tunnel and exposing lysine residues otherwise buried inside the ribosome (reviewed in Filbeck et al., 2022).
This process is evolutionarily conserved, and the bacterial NEMF homolog RqcH supports Ltn1/Listerin in degrading aberrant nascent polypeptides by adding C-terminal polyalanine tails (Crowe-McAuliffe, 2021; Filbeck et al., 2021; Lytvynenko et al., 2019). Tail composition differs between organisms, with mammalian C-terminal tails mainly being composed of alanine (Udagawa et al., 2021). In the absence of LTN1 or lysine exposure, the alanine tails are recognized as a degron by Pirh2 or CRL2KLHDC10 E3 ubiquitin ligases, ensuring nascent chain ubiquitination and degradation (Thrun et al., 2021).
In addition, CAT-tailed peptides are more prone to aggregation, which may be a contingency plan to sequester aberrant protein products and trigger a process called aggrephagy (autophagy-mediated degradation of protein aggregates) (Lamark and Johansen, 2012). This must be tightly regulated as excess aggregation can lead to cytotoxicity and disrupts neuronal morphogenesis (Udagawa et al., 2021). Once the incomplete protein has been released from the tRNA by ANKZF1, VCP delivers it to the proteasome for degradation, and the 60S subunit is free to be recycled (Verma et al., 2013; Yip et al., 2019).
Signaling from stalled and collided ribosomes
In contrast to local-acting ribosome splitting and subsequent RQC, the ISR and the RSR are two pathways with a broader reach: the ISR induces translation reprogramming to adapt or restore cellular homeostasis, and the RSR is a mitogen-activated protein kinase cascade directing multiple biological processes. The RSR is initiated by the mitogen-activated protein kinase kinase kinase ZAKα in response to stalled or collided ribosomes (Vind et al., 2020b; Wu et al., 2020) leading to activation of the mitogen-activated protein kinases p38 and JNK, which are both master regulators of many cellular functions including proliferation, inflammation, apoptosis, and differentiation (Wagner and Nebreda, 2009).
A possible interplay between ISR and RSR has recently become apparent under certain conditions (Snieckute et al., 2022; Stoneley et al., 2022; Wu et al., 2020). Indirect impairment of translation, such as arginine deprivation, causes ribosomal pausing and GCN2 activation (Darnell et al., 2018). Under these conditions, inhibition of GCN2 (or downstream steps of the ISR) promotes disome formation, likely resulting from a failure to block new initiation on the affected mRNA template (Darnell et al., 2018; Stoneley et al., 2022). In addition, translation inhibition by the antiviral protein Viperin leads to GCN2 activation in a ZAK-dependent manner (Hsu et al., 2022).
The presence of both stalled and collided ribosomes is sensed by specific ribosome-binding proteins. These factors initiate the activation of pathways for separation of the ribosomal subunits, degradation of the nascent polypeptide, local inhibition of new translation initiation and cell fate signaling (Fig. 3). In the next sections, we will discuss the signaling pathways in response to translational stress on a cellular level.

Cellular Signaling Downstream of Impaired Ribosomes
The rapid speed of protein synthesis allows the ribosome to pick up on immediate changes in the cellular environment. Stalling and collision of ribosomes can thus be considered “molecular flags,” notifying the cell of its current state. Depending on the nature, duration and severity of the stress, the ribosome-surveillance responses will try to mitigate the consequences of compromised translation by influencing different biological processes such as immune signaling, cell proliferation, autophagy, and cell death.
Viral signaling
There seems to be a connection between ribosome collisions and viral infection as depletion of collision disassembling (ZNF598 and ASCC3) or scaffolding factors (RACK1) results in interferon-stimulated gene (ISG) expression (DiGiuseppe et al., 2018; Long et al., 2014; Wan et al., 2021), which inhibits viral replication (Fig. 4). Unresolved ribosome collisions are most likely sensed by the cGAS–STING pathway, triggering the innate immune system (Wan et al., 2021).
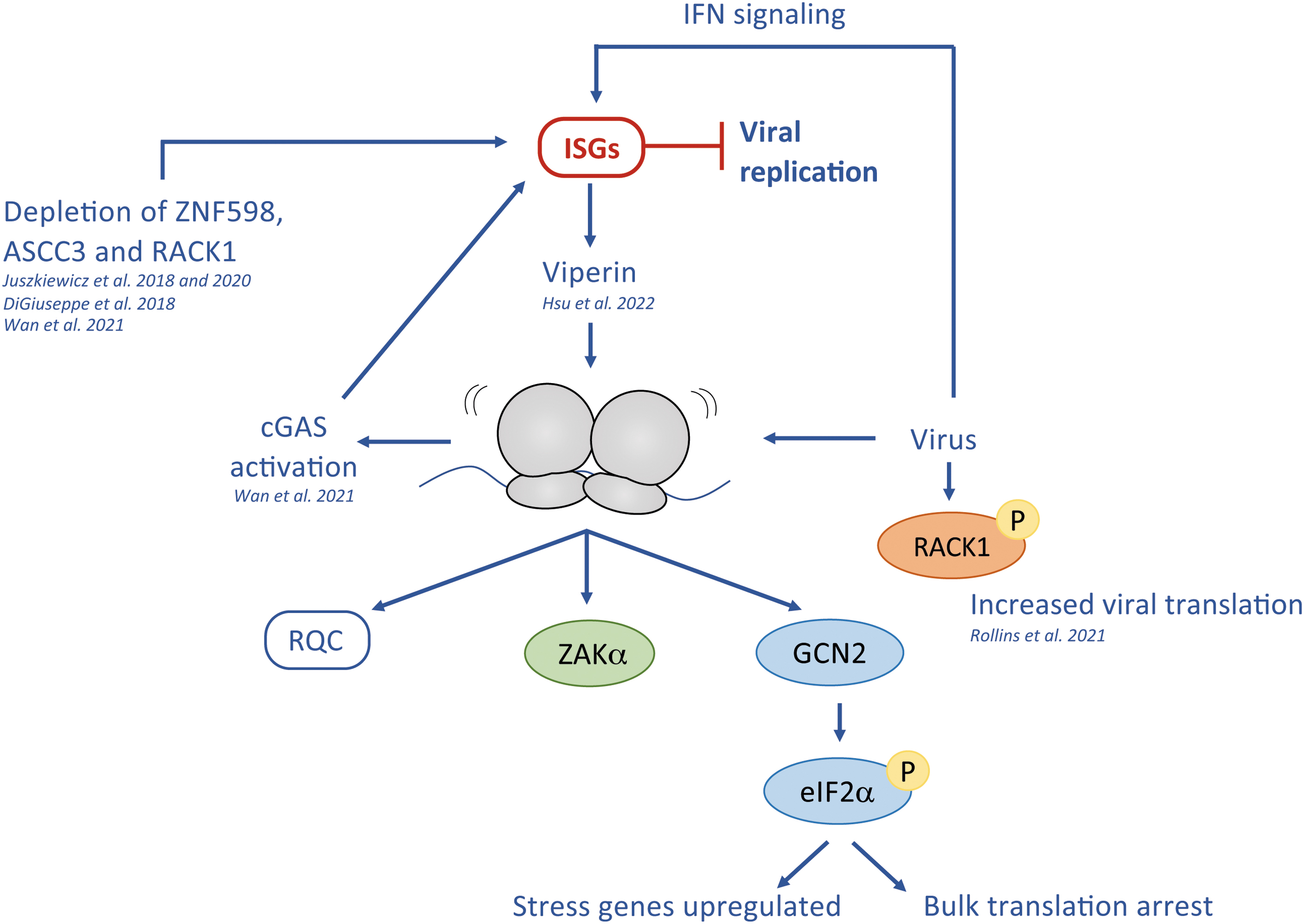
In support of this, depletion of either NEMF (a downstream RQC factor), or the main splitting factors for stalled ribosomes, HBS1L and Pelota, does not seem to affect ISG expression (Wan et al., 2021), suggesting that ribosome collision itself constitutes the relevant signal. This mechanism appears potentially conserved in evolution, since the bacterial STING/cGAS system is also a key player in phage defense (Morehouse et al., 2020). In addition, a recent study showed that ISGs induce Viperin, a virus-inhibitory enzyme. The product of Viperin induces ribosome collisions that are sensed by GCN2 and ZAKα, reducing overall translation, which interferes with the replication of the RNA-virus Zika (Hsu et al., 2022).
The importance of Viperins is further underlined by their evolutionary conservation, as prokaryotic Viperins protect bacteria against phage infection (Bernheim et al., 2021). In parallel to ISGs induction via RQC, the RQC contributes to efficient MHC-1 presentation possibly facilitating early viral detection (Trentini et al., 2020).
Many RNA viruses induce 40S remodeling via their complex internal ribosome entry sites to favor translation of the viral RNA (reviewed in Johnson et al., 2017). In a similar manner, phosphorylation in the RACK1 loop broadens the ribosome's translation capacity by supporting eIF4A-independent translation initiation (Rollins et al., 2021). This noncanonical mode of translation can be used by DNA viruses such as poxvirus to ensure optimal translation of its viral proteome (Rollins et al., 2021).
This process depends on both RACK1 (Park and Walsh, 2022) and ZNF598 and its RPS20/uS10 ubiquitination (DiGiuseppe et al., 2018), as genetic deletion of ZNF598 or mutation of RPS20/uS10 preventing its ubiquitination results in global reduction of viral protein synthesis of vaccinia virus (Sundaramoorthy et al., 2021). In addition to RQC, viral pathogens activate and regulate the ISR to facilitate their replication (recently reviewed in Wu et al., 2022).
Inflammatory signaling
Besides a role in viral propagation, ribosomal stress responses have been implicated in inflammatory responses. Activation of the RSR leads to upregulation of proinflammatory cytokines such as interleukin (IL)-8 (Jandhyala et al., 2008; Vind et al., 2020b), and to NLRP1 inflammasome activation and subsequent IL-1β secretion in response to UV-B in primary human keratinocytes (Robinson et al., 2022). Considering the possible crosstalk between ZAKα and GCN2 (Hsu et al., 2022; Wu et al., 2020), it is interesting to note that activation of GCN2 in macrophages promotes the expression of anti-inflammatory IL-10 and TGF-β (Ravishankar et al., 2015), and inhibits the production of the proinflammatory cytokine IL-1β (Battu et al., 2018).
Several reports have also proposed a function for GCN2 in T-helper cell proliferation and differentiation (Brenk et al., 2009; Munn et al., 2005; Sundrud et al., 2009). RSR and ISR may thus serve opposing functions in the context of immune signaling as GCN2 generally has an anti-inflammatory role, whereas ZAKα is involved in proinflammatory cytokine production.
Proliferation, autophagy, and senescence
Immune signaling is not the only cellular process affected by ribosome impairment. The different ribosome-surveillance pathways can also trigger changes in the cell cycle, and induce autophagy, senescence, or transcriptional reprogramming depending on the type of cell and translational problem.
It was recently shown that ASCC3 helps distinguish between resolvable collisions, such as the ones caused by anisomycin, and unresolvable ribosome collisions caused by UV-B or 4-nitroquinoline 1-oxide. In ASCC3-depleted cells, anisomycin treatment caused a ZAKα/p38-mediated G2 arrest due to ribosome collision, whereas unresolvable ribosome collision incurred after UV-B resulted in a more extended cell cycle arrest (Stoneley et al., 2022). This suggests that mechanisms exist that respond dynamically and that can modulate cell fate depending on the severity and persistence of the initial perturbation.
Activation of the ISR via GCN2 leads to upregulation of many autophagy genes in response to short-term leucine deprivation both in cultured cells and in mouse liver (Maurin et al., 2022). Accordingly, GCN2 deletion results in reduced autophagy and stress granule formation upon induction by Halofuginone (Battu et al., 2018), while
While protein synthesis and the activity of the nutrient-sensing mammalian target of rapamycin (mTOR) kinase are important at the onset of senescence, translation is globally reduced in fully senescent cells except for senescence-associated secretory phenotype (SASP) factors. Interestingly, mTOR supports senescence-associated expression of MAPKAPK2, the effector kinase downstream of ZAKα and p38. MAPKAPK2 is a negative regulator of the RNA-binding protein ZFP36L1 that mediates degradation of SASP-encoding mRNAs. This mechanism thus allows for mTOR-dependent translation of SASP factors in senescence (Anerillas et al., 2020; Herranz et al., 2015).
In skin keratinocytes, UV stress can also cause senescence. UV damage activates GCN2, which in turn phosphorylates eIF2α, leading to translational inhibition in senescent skin cells (Payea et al., 2021; Wu et al., 2020).
Interestingly, ribosomes also act as sensors of translation stress in intestinal stem cells, where cellular changes mediated by ZAKα allow cells to adapt to the changing environment and adopt a fetal-like identity (Silva et al., 2022).
Cell death
Persistent ribosome stalling and collision can overwhelm the cellular stress response pathways and lead to cell death. Caspase-dependent apoptosis can be induced by the ZAKα–JNK pathway as seen after prolonged treatment of cells with doxorubicin (Sauter et al., 2010), anisomycin (Wu et al., 2020), or high doses of UV-B (Vin et al., 2014). Apoptosis can also be triggered by CAT-tailed nascent chain aggregation, which results from excessive expression and accumulation of nonstop protein products (Udagawa et al., 2021).
CAT-tail–mediated apoptosis is likely distinct from the above, as ZAKα binds directly to ribosomes for activation and CAT-tail–mediated apoptosis is only initiated after ribosome splitting. ZAKα is also associated with pyroptosis, a type of cell death characterized by caspase-1 activation and IL-1β secretion. Unlike classic apoptosis, which seems to be induced via JNK, pyroptosis is linked to the p38 prong of the RSR (Robinson et al., 2022).
Although ISR signaling mainly operates to transiently inhibit cap-dependent translation, some stress conditions may induce p-eIF2α/ATF4–mediated apoptosis (Ishimura et al., 2016). The contribution of the RSR, CATylation, and ISR pathways to apoptotic signaling varies among cell lines and stress agents (Stoneley et al., 2022; Udagawa et al., 2021), suggesting cell-type and tissue-specific susceptibility to impaired translation and proteotoxicity.
Physiological and Pathological Consequences of Ribosome Stalling and Collision
The cellular importance of ribosome-surveillance pathways has become evident in the past decade, but we have barely scratched the surface of their physiological function in complex organisms. In the following sections, we will review our current knowledge of the links between ribosomal impairments and neurodegeneration, inflammation, metabolic diseases, cancer, and aging (Table 2).
Physiological Implications of Ribosome-Surveillance Factors in Different Pathologies
IL, interleukin; KO, knockout; UV, ultraviolet.
Neurodegenerative disorders
Dysregulation of ribosome-surveillance pathways is tightly coupled to neurodegenerative disorders. Both in mice and in humans, mutation of ribosome disassembling factors, RQC factors, and ISR factors has been implicated in neurological dysfunction. Interestingly, the RSR sensor ZAKα is not expressed in the brain (Liu et al., 2000), indicating that it does not play a role in neuronal homeostasis. The importance of functional RQC for neuronal health is evident from the embryonic lethality of LTN1 knockout (KO) or RING domain-mutated mice (Chu et al., 2009), and in line with this, knockdown of LTN1 (but not NEMF, ZNF598, or ASCC3) impaired neurite outgrowth in vitro (Udagawa et al., 2021).
Moreover, while a hypomorphic LTN1 allele bypasses embryonic lethality in mice, they suffer from an amyotrophic lateral sclerosis (ALS)-like disease (Chu et al., 2009). LTN1 and ZNF598 were also shown to suppress atypical translation of the pathogenic poly(GR) protein from the C9ORF72 locus, a genetic contributor to the pathogenesis of ALS and frontotemporal dementia (Park et al., 2021). In addition, Park et al. (2021) observed that RQC function is impaired in C9-ALS patient-derived neurons.
Mutations in RQC factor NEMF have been associated with juvenile neuromuscular disease (Martin et al., 2020) and inherited neurological disorders (Ahmed et al., 2021). Mechanistically, these mutations have been linked to NEMFs CAT-tailing ability, which normally supports protein degradation via RQC. However, if the RQC fails to remove CAT-tailed products, they will accumulate and aggregate, which is detrimental to proteostasis (Howard and Frost, 2021). Indeed, there is a correlation between the mutation-associated degree of impairment of CAT tailing and phenotype severity in mice (Martin et al., 2020).
Aberrant CAT tailing has also been implicated in the development of Alzheimer's disease, where translation of amyloid precursor protein APP.C99 leads to ribosome stalling and collision and CAT tailing of APP.C99 peptides (Rimal et al., 2021). These CAT-tailed APP.C99 products can act as a seed for amyloid-β aggregation, which is the main component of amyloid plaques.
Consistent with the idea that unresolved stalling and collision may contribute to Alzheimer's disease pathogenesis, brain tissue from Alzheimer's disease patients also shows an enrichment of ZNF598, NEMF, and ANKZF1 at the core of the amyloid plaque (Rimal et al., 2021). Finally, mice that are heterozygous for the ZNF598 interactor GIGYF2 show neurodegeneration and motor dysfunction in adulthood (Giovannone et al., 2009).
Biallelic variants of ASCC3 were recently shown to underlie a neuromuscular syndrome in 11 individuals (Nair et al., 2021), and a homozygous mutation has been reported in 1 family with intellectual disability (Najmabadi et al., 2011). However, it is not yet known if these patients' disabilities are linked to the established functions of ASCC3 in the DNA damage response or ribosome splitting.
While a full KO is embryonically lethal, a hypomorphic mutation of the ribosome rescue factor HBS1L in mice and humans leads to developmental cerebellar defects (O'Connell et al., 2019; Terrey et al., 2021). Embryonic fibroblasts from KO mice show specific pause sites that are found mostly in protein coding sequences. Loss of HBS1L also leads to activation of mTOR signaling, although this might be an indirect effect of the defective quality control pathway (Terrey et al., 2021).
In humans, mutations in GTPBP2, a binding partner of Pelota, are linked to the development of Jaberi-Elahi syndrome. This newly classified neurodegenerative disorder is characterized by dystonia, motor and sensory neuropathy, ataxia, and cognitive dysfunction (Jaberi et al., 2016). In support of this, mice with GTPBP2 mutations coupled with a central nervous system-specific tRNA mutation suffer from age-dependent neurodegeneration, and exhibit increased ribosome stalling in neuronal tissue (Ishimura et al., 2014). The increased stalling activates GCN2, and like RQC, the ISR appears neuroprotective, since loss of GCN2 in the GTPBP2-mutated mice causes increased ataxia, tremors, and locomotor deficits (Ishimura et al., 2016).
While ISR and the GCN2 kinase mostly have protective effects, some data indicate a role for the phospho-eIF2α/ATF4 axis in neuronal cell death, suggesting a differentiated response depending on the intensity and duration of stress conditions (Han et al., 2013; Lange et al., 2008). This is also seen in mice where dominant mutations in at least six aminoacyl-tRNA synthetases cause peripheral neuropathy. These mutations activate the ISR via GCN2, and inhibition or genetic deletion of GCN2 improved the pathophysiology of the mice (Spaulding et al., 2021).
Chronic activation of ISR in the mutant mice suggests an increased level of stalled and collided ribosomes, and it is tempting to speculate that this overwhelms the ribosome-surveillance pathways consequently leading to increased neurodegeneration.
Other neurodegenerative disorders have been linked to ribosome stalling and RQC problems as well: In a Drosophila model of Parkinson's disease, a link between mitochondrial RQC and mitochondrial dysfunction was suggested (Wu et al., 2019), while in Huntington's disease CAG repeats in the Htt gene negatively impact translation and cause ribosome collisions in a cellular as well as a mouse model (Eshraghi et al., 2021).
RQC failure results in stalled and collided ribosomes that cannot be rescued. This leads to aberrant and aggregated protein products, accumulation of unresolved 60S subunits, and organelle dysfunction. All of these contribute to proteotoxicity and proteome imbalance, which are well-known hallmarks of neurodegenerative disorders. Interestingly, the viral protein Nsp1 was recently shown to offer neuroprotection due to its ability to manipulate the RQC pathway in Drosophila models of Alzheimer's disease, Parkinson's disease, and ALS (Wang et al., 2022).
Immune system and inflammation
Viral infections are accompanied by a substantial reprogramming of the translation machinery, in general suppressing translation of host factors and favoring extensive translation of viral proteins. Such changes may be accompanied by deregulation of the translation process itself and lead to ribosome collision. Early-acting RQC factors suppress innate immune signaling as they allow for disassembly of collided ribosomes. Such structures would otherwise be sensed as danger signals by the cGAS system resulting in ISG induction (Wan et al., 2021).
In general, ISGs function to inhibit viral replication, and all the ribosome-surveillance pathways have been implicated in viral propagation and immune signaling in a cellular context. Future work in animal models will be required to elucidate the biological importance of these stress responses in viral infections.
At an organismal level, both RSR and ISR have been associated with inflammatory signaling in various disease models. The RSR and its upstream kinase ZAKα trigger production of the proinflammatory cytokine IL-8 in response to ricin and Shiga toxins, and ZAK inhibitors might alleviate the illnesses and pathogenesis caused by ricin and Shiga toxin-producing bacteria (Jandhyala et al., 2008).
Recently, ZAKα was also shown to be responsible for inflammasome induction after UV-B irradiation of human primary keratinocytes. Interestingly, ZAKα phosphorylates a linker region in the immune sensor NLRP1, which is present in humans but not in mice. This suggests differences in the inflammatory response to UV damage, which should be considered for potential future treatments of inflammatory disorders (Robinson et al., 2022).
In humans, the highest expression of GCN2 is found in the colon, and these levels are increased in patients with ulcerative colitis and Crohn's disease compared with healthy controls (Ravindran et al., 2016). GCN2 KO mice do not present with obvious gut deficiencies, suggesting that GCN2 is not required for differentiation or proliferation of intestinal cells. However, GCN2 KO mice are more sensitive to dextran sodium sulfate-induced colitis and exhibit greater weight loss, inflammation, TH17 responses, and colon shortening compared with wild-type littermates (Ravindran et al., 2016). These exacerbated effects were suggested to be caused by reduced autophagy in antigen presenting cells, leading to increased production of reactive oxygen species (ROS) and inflammasome activation (Ravindran et al., 2016).
As GCN2 inhibition in amino acid starved cells leads to increased levels of ribosome collisions (Stoneley et al., 2022) and the RSR initiates proinflammatory signaling and inflammation upon collisions, it would be interesting to examine a possible contribution of the RSR in the observed GCN2 KO phenotype (Jandhyala et al., 2008; Vind et al., 2020b). Also, in an experimental autoimmune encephalomyelitis mouse model mimicking multiple sclerosis, deletion of GCN2 was characterized by increased central nervous system inflammation, presence of TH17 cells, and increased IL-17 and interferon-γ (Orsini et al., 2014).
In accordance with this, a GCN2 agonist protected mice from nephritic kidney damage by inducing autophagy and suppressing proinflammatory cytokine production (Chaudhary et al., 2015).
Metabolic dysregulation
The ISR-inducing kinase GCN2 has been shown to contribute to various aspects of metabolic regulation upon dietary challenges. GCN2 KO mice are hypersensitive to dietary leucine starvation correlating with an inability to phosphorylate eIF2α and induce the ISR in the liver (Anthony et al., 2004). Upon prolonged amino acid deprivation, mice normally repress lipid synthesis in the liver, improve hepatic insulin sensitivity, and mobilize adipose tissue lipid deposits. However, leucine-starved GCN2 KO mice increase expression of lipogenesis genes resulting in liver steatosis (Guo and Cavener, 2007), cannot suppress diet-induced mTOR activity, and do not improve insulin sensitivity (Xiao et al., 2011).
Moreover, leucine deprivation causes a rapid weight reduction, lower adiposity, and browning of white adipose tissue. Similarly, in mouse amygdalar PKC-δ neurons, GCN2 deletion blocked leucine deprivation-induced white adipose tissue browning and PKC-δ neuron activation, which could be rescued by overexpression of the downstream ISR-factor ATF4 (Yuan et al., 2020). We have recently shown that, unlike GCN2 KO mice, ZAK KO mice present with more browning of white adipose tissue, reduced steatosis, and improved glucose tolerance (Snieckute et al., 2022). These opposing phenotypes seen in GCN2 and ZAK KO mice in response to leucine-deficient diet underline a likely interplay between the ISR and RSR pathways.
A single nucleotide polymorphism in the GCN2 gene has been linked to type 2 diabetes mellitus in genome-wide association studies of the Japanese population (Kou et al., 2013; Yasuda et al., 2008), underscoring the role of GCN2 in metabolic regulation. In a follow-up study, carriers of the GCN2 risk allele showed reduced insulin secretion but no change in insulin sensitivity, when compared with controls (Kanno et al., 2020). When fed a high-fat diet, whole-body GCN2 KO and pancreatic β cell-specific GCN2 KO mice exhibited reduced glucose tolerance, insulin secretion, and reduced pancreatic β cell mass (Kanno et al., 2020).
A likely explanation is that WT mice increase proinsulin translation in pancreatic β cells when on a high-fat diet, potentially depleting the amino acid pool, leading to ribosome stalling and GCN2 activation. This leads to the induction of ATF4 and subsequent mTOR inhibition, which is required to maintain healthy pancreatic β cell mass. Thus, without GCN2, mice experience exacerbated metabolic decline (Kanno et al., 2020).
The ribosomal branch of the ISR is important for metabolic adaption during under and overfed conditions, likely due to diet-induced ribosome stalling/collisions as seen in cell culture systems (Darnell et al., 2018). Intriguingly, GCN2 is also activated in brain cells in response to amino acid deficiency and imbalanced nutrition, linking the ISR to behavioral adaption to malnutrition and starvation (Hao et al., 2005; Maurin et al., 2022).
The RSR appears to contribute to metabolic adaption in response to translation stress as well. In intestinal organoids and mouse models, ribosomes and ZAKα can act as sensors for nutrient availability by reprogramming intestinal stem cell identity to a more fetal-like phenotype. This is followed by the metabolic switch from oxidative phosphorylation to glycolysis. This change is not mediated through the typical ZAKα-p38/JNK axis, but instead involves an alternative ZAKα–SRC–YAP signaling pathway (Silva et al., 2022).
Cancer
Tumor microenvironments are often characterized by hypoxia, oxidative stress, and nutrient starvation—stress conditions that lead to ISR-mediated global translational inhibition that cancer cells need to cope with (Leprivier et al., 2015). To adapt, cancer cells undergo translational reprogramming, where eIF2α favors translation of specific transcripts. The ISR target gene ATF4 is often upregulated in cancer, and has been shown to increase migration of breast cancer cells and metastasis of squamous cell carcinoma (Nagelkerke et al., 2013; Zhu et al., 2014).
In some colorectal cancers, the proto-oncogene MYC mediates an increase in global protein synthesis depleting the cellular amino acid and nucleotide pools, and ultimately causing cell death. However, depletion of energy resources activates the GCN2–eIF2α axis that suppresses translation, and thus preserves resources and prevents apoptosis. Inhibition of GCN2 in tumor organoids suppresses tumor growth and has the potential in targeted therapy of tumors with deregulated MYC expression (Schmidt et al., 2019).
The RSR is often upregulated in cancers, too. Transcriptome analysis comparing cancer tissue with adjacent healthy tissue showed that ZAKα expression is highly upregulated in gastric, colorectal, urinary, and breast cancer (Adler et al., 2014; Liu et al., 2000; Rey et al., 2016). In addition, overexpression of ZAKα negatively correlates with cancer survival, especially in breast cancer patients (Li et al., 2018; Tang et al., 2019).
Intriguingly, the risk of developing cutaneous squamous cell carcinoma, a type of UV-induced skin cancer, is increased in melanoma patients treated with the BRAF inhibitor sorafenib. Sorafenib displays a strong off-target effect against ZAK and has been reported to suppress UV-induced apoptosis in keratinocytes in a ZAK-dependent manner (Vin et al., 2014).
In support of this, UV-B–induced ribosome collisions are resistant to the ASC-1 complex, and unresolved stalled ribosomes are recognized by the RSR inducing a G2 cell cycle arrest (Stoneley et al., 2022). ZAKα also mediates UV-B–induced pyroptosis in human skin keratinocytes by activating the NLRP1 inflammasome (Robinson et al., 2022). These connections offer a potential explanation for the skin cancer-promoting effects of sorafenib and related BRAF inhibitors.
Aging
Reducing protein synthesis promotes longevity. A classic example is attenuation of the mTOR pathway, which extends life span in yeast, nematodes, fruit flies, and mice (Bjedov and Rallis, 2020; Papadopoli et al., 2019). While it was originally believed that calorie restriction was necessary for life span extension, it is now clear that limiting essential amino acids is sufficient to extend life span in many organisms. Like mTOR, GCN2 responds to amino acid restriction, although indirectly, and mediates global translation inhibition and induction of stress response genes. Together with mTOR signaling, GCN2 and the ISR thus directly impact organismal longevity, fitness, and stress resistance (Gallinetti et al., 2013; Rousakis et al., 2013).
Aging increases the level of ribosomal pausing, resulting in more ribosome collisions in yeast and worms (Stein et al., 2022). Consequently, the RQC is overwhelmed, causing polypeptides to accumulate and aggregate, which contributes to aging-associated proteome imbalance (Stein et al., 2022). These connections have yet to be observed in mammals, but it has been shown that translation elongation rates are decreased by ∼20% in livers from old mice (Gerashchenko et al., 2021). This might be due to exacerbated ribosome pausing, considering that liver is the tissue with the highest elongation rate (Gerashchenko et al., 2021).
Further links between translation fidelity and aging were provided by a recent study showing premature aging phenotypes in mice harboring a heterozygous mutation of the ribosomal protein RPS9/uS4 (Shcherbakov, 2022).
Future Perspectives and Therapeutic Potential
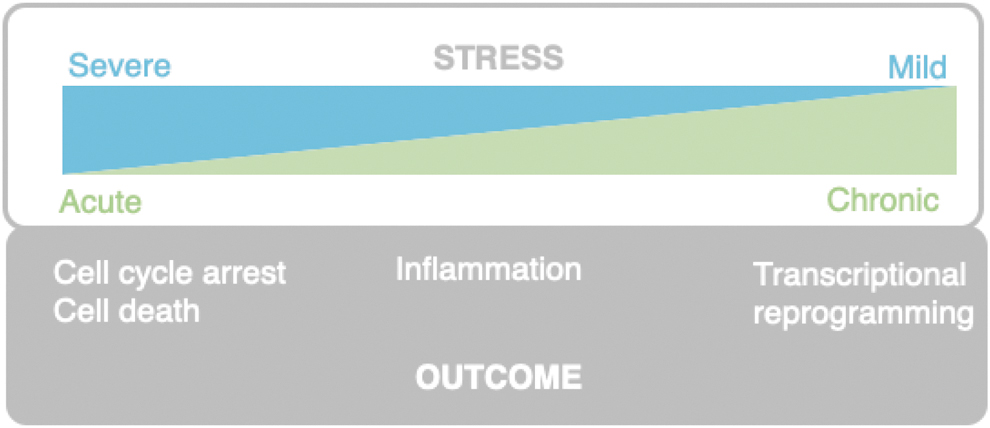
Physiological and pathological consequences of compromised translation are rapidly emerging, but much of our mechanistic insight is limited to model systems with reduced complexity such as yeast and human cell lines. We have described many pathologies associated with defective ribosome surveillance, yet few direct links between ribosome stalling or collision and disease stages have been made. From the current literature, chronic but mild ribosomal stress levels have been shown to snowball into pathological features such as accelerated aging, metabolic dysregulation, and neurodegeneration (Giovannone et al., 2009; Shcherbakov, 2022; Xiao et al., 2011).
On the contrary, more acute and severe stress such as UV exposure or doxorubicin activate the RSR and rapidly cause cell cycle arrest, or even cell death as illustrated in Figure 5 (Sauter et al., 2010; Stoneley et al., 2022). As the ribosome-surveillance pathways are implicated in many different cellular responses such as autophagy, senescence, immune signaling, or apoptosis, future research will be needed to uncover how cells decide on the appropriate biological outcome.
One interesting question that will require more research is how a cell distinguishes between paused and permanently stalled ribosomes, and between aberrant and natural ribosome pausing. Is there a measure of time involved and if so, how does it work? Is it regulated by a balance of ubiquitination and deubiquitination of ribosomal proteins? The ubiquitination of 40S ribosomal proteins has been studied diligently after different stresses (Garshott et al., 2021; Garshott et al., 2020), and determining additional post-translation modifications induced by translation stress agents of ribosomal proteins and their interactors will continue to be an important avenue for future research as well as the functional outcomes of these modifications.
We know, for example, that the E3 ubiquitin ligase RNF10 ubiquitinates RPS2/uS5 and RPS3/uS3 upon inhibition of translation initiation (Garshott et al., 2021; Garzia et al., 2021), while ZNF598 ubiquitinates RPS20/uS10 and RPS10/eS10 upon ribosome collision (Juszkiewicz and Hegde, 2017; Sundaramoorthy et al., 2017). ZNF598-dependent RPS10/eS10 ubiquitination happens on Lys 138 and Lys 139, whereas the E3 ligase MKRN1 preferentially ubiquitinates RPS10/eS10 on Lys53 and Lys107, and the outcomes of these different ubiquitination reactions remain disputed.
To advance our understanding of the physiological roles of the ribosomal-surveillance machinery, research will have to focus more on endogenous sources of translation stress, such as ROS. ROS can damage mRNA, rRNA, and tRNA as well as proteins involved in translation, and it remains unclear how cells detect the different damaged molecules and coordinate their repair or removal. It would also be interesting to better understand when and in what order the different ribosome-surveillance factors interact with the ribosome. Ribosome affinity purification techniques could be used to compare ribosome interactors in response to different kinds of stress such as ROS and starvation (Simsek et al., 2017).
While it is apparent that severe and prolonged translational aberration leads to activation of the ZAKα–p38/JNK pathway and subsequent cell death, there is probably no clear cell type-independent threshold for the induction of cell death. Translational stress tolerance might vary a great deal between cell lines or tissues, probably depending on the type and severity of stress they are routinely exposed to, for example, UV irradiation in the skin.
The structurally diverse perturbations may also account for the different downstream signaling events, and while structures of RACK1, ZNF598, and EDF1 bound to ribosomes have been solved (Juszkiewicz et al., 2018; Rabl et al., 2011; Sinha et al., 2020), these are lacking for other factors. Especially, cryo-EM structures of collided as well as individual ribosomes in complex with ZAKα and GCN2 will be key to further our understanding of how ribosomes are recognized by these kinases. It remains unclear if the different ribosome-surveillance factors can bind the same ribosome simultaneously, and to what extent binding hierarchies are determined by protein abundance and/or stoichiometry.
Many aspects of translational stress are present during tumor formation, including ROS, nutrition deprivation, proteotoxicity, and a high level of proliferation, making cancer a potential target for RQC, ISR, and RSR medical intervention. As mentioned above, the ISR is often activated in cancers, manifested as eIF2α phosphorylation and ATF4 expression. The role of this pathway appears to be complex and highly context dependent, and both ISR-activating and ISR-inhibiting drugs are being developed and tested clinically (Licari et al., 2021).
ISR activation can, for example, be achieved by drugs that activate the ISR kinases HRI and PKR, or by drugs such as Halofuginone that impair prolyl tRNA-synthetase and thus mimic amino acid starvation to activate GCN2 (Keller et al., 2012). Since tumors differ in their amino acid metabolism, it would be attractive to employ ribosome profiling to identify tumor-specific vulnerabilities in amino acid availability and to exploit this knowledge for individualized therapy (Loayza-Puch et al., 2016).
Salubrinal, an inhibitor of the eIF2α phosphatase GADD34, has also shown therapeutic potential as an anticancer agent in preclinical studies (Licari et al., 2021). Direct inhibition of ISR kinases may also be of therapeutic value. GCN2 inhibitors have potential both in cancer treatment (Kato et al., 2020) and in antiviral therapy (Jiang et al., 2017; Torres et al., 2021), but more work is needed to develop highly potent and selective drugs (Licari et al., 2021).
A recent screen for drugs targeting FBXW7-mutated tumors that are multidrug resistant found that ISR-activating drugs are very toxic to these tumors. Since the E3 ubiquitin ligase FBXW7 is among the most mutated tumor suppressors, this finding could help understand and develop drugs for otherwise therapy-resistant cancers (Sanchez-Burgos et al., 2022).
ZAK inhibition might be a potential approach for a subset of cancers, and ZAKα is most likely off-targeted by anticancer drugs such as nilotinib, ponatinib, or sorafenib with unknown therapeutic consequences (Mathea et al., 2016). In addition, the ZAK inhibitor M443 sensitized medulloblastoma brain tumors to the effects of radiation (Markowitz et al., 2016). Unwanted side-effects of cancer treatment could also be caused by ZAKα activation, for example, the cardiac inflammation associated with doxorubicin therapy, for which ZAK inhibition could prove beneficial (Wong et al., 2013).
In addition to small-molecule inhibitors of ZAK and GCN2, E3 ligases are druggable cancer targets, and more focus could be put on targeting E3 ligases involved in ribosomal signaling.
Using the knowledge gained from cancer research, there is a clear potential in targeting ribosome-surveillance factors in drug development in other disease areas. As most of our knowledge about the ribosome-surveillance apparatus originates from work with bacteria, yeast, and human cancer cell lines, advancing our understanding of their mechanistic, cellular, and physiological functions will hopefully allow us to harness them for medical interventions.
Concluding Remarks
Ribosomes are emerging as sensors of cellular fitness (Kim and Zaher, 2022), and much of the ongoing research focuses on improving our mechanistic understanding of the different pathways responding to translation stress. In an organismal setting, defects in ribosome surveillance have been linked to inflammation, immunity imbalance, neurodegeneration, and other disease aspects. Future work should focus on individual organs as well as whole organisms. Finally, as several ribosome-surveillance and rescue factors are potential drug targets, their therapeutic potential should be investigated within areas such as metabolic disease, neurological disorders, inflammation, and cancer.
Footnotes
Authors' Contributions
A.C.V., G.S., M.B., and S.B.-J. designed, wrote, and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This project has received funding from the European Research Council (ERC) under the European Horizon 2020 research and innovation programme (Grant Agreement No. 863911-PHYRIST).