
Abstract
Significance:
A cell plays its roles throughout its life span, even during its demise. Regulated cell death (RCD) is one of the key topics in modern biomedical studies. It is considered the main approach for removing stressed and/or damaged cells. Research during the past two decades revealed more roles of RCD, such as coordinating tissue development and driving compensatory proliferation during tissue repair.
Recent Advances:
Compensatory proliferation, initially identified in primitive organisms during the regeneration of lost tissue, is an evolutionarily conserved process that also functions in mammals. Among various types of RCD, apoptosis is considered the top candidate to induce compensatory proliferation in damaged tissue.
Critical Issues:
The roles of apoptosis in the recovery of nonregenerative tissue are still vague. The roles of other types of RCD, such as necroptosis and ferroptosis, have not been well characterized in the context of tissue regeneration.
Future Directions:
In this review article, we attempt to summarize the recent insights on the role of RCD in tissue repair. We focus on apoptosis, with expansion to ferroptosis and necroptosis, in primitive organisms with significant regenerative capacity as well as common mammalian research models. After gathering hints from regenerative tissue, in the second half of the review, we take a notoriously nonregenerative tissue, the myocardium, as an example to discuss the role of RCD in terminally differentiated quiescent cells. Antioxid. Redox Signal. 39, 1053–1069.
Introduction
Tissue regeneration is common among species. Extreme cases include Hydra and planarians, which can survive bisection (King and Newmark, 2012). Drosophila, a common genetic model, has highly regenerative imaginal discs in the larval stage (Bergantinos et al., 2010). Among vertebrates, zebrafish can fully regenerate fins and heart (Nakatani et al., 2007; Poss et al., 2002). The axolotl and newts regrow their appendages (Murawala et al., 2012). By contrast, adult mammals have limited regenerative capacity during adulthood with some exception such as skin, liver, and bones. Mammalian liver can efficiently restore 70% of its mass (Taub, 2004). However, other vital organs, such as heart, repair their wounds with scars, which compromise tissue functions (Sousounis et al., 2014).
The process of tissue regeneration normally includes acute injury response, blastema formation, differentiation, and remodeling, with discrepancies among different tissue types (Fig. 1) (Gurtner et al., 2008). These stages can be overlapping. A blastema is composed of progenitor cells, which can be resident stem cells or dedifferentiated cells located near the wound (Fig. 1). Cell division within the blastema is considered the main driving force of tissue regeneration (Brockes and Kumar, 2008). Interestingly, the formation of blastema is concomitant with histolysis in the injury border zone (Weiss and Rosenbaum, 1967). However, the role of cell death has not been appreciated in tissue repair until recent years. Researchers proposed the concept of “altruistic cell death,” which induces proliferation of adjacent cells (Kondo, 1988). This phenomenon resembles the evolutionary advantage of the bee colony to have self-sacrificing worker bees. Later studies showed that signals derived from apoptotic cells participate in cell proliferation in the wound, a phenomenon termed apoptosis-induced compensatory proliferation (Fig. 1) (Bergmann and Steller, 2010).

In Drosophila larval imaginal discs, genetic and chemical manipulations of caspases allow cells to stay in an apoptotic status but remains “undead” (Hay et al., 1994). This model was used to show that the c-Jun N-terminal kinase (JNK) activation and release of growth factors by the dying cells regulate compensatory proliferation (Morata et al., 2011; Worley et al., 2012). Similar observation was made in Hydra, zebra fish, and mice, supporting that apoptotic signaling is necessary for triggering cellular remodeling and regeneration (Bergmann and Steller, 2010).
In this study, we systematically review the current knowledge on the roles of regulated cell death (RCD) during tissue regeneration. We discuss recent progress in our understanding of how apoptosis can trigger compensatory proliferation. We also discuss the evidence that supports the involvement of necroptosis and ferroptosis in tissue regeneration. By summarizing the knowledge gained from regenerative tissue, we share our perspective on the role of RCD in the repairing of the nonregenerative mammalian myocardium.
Regulated Cell Death Under Physiological Context
In 1972, apoptosis was defined as a type of RCD that demonstrates morphological differences from necrotic cells (Kerr et al., 1972). After decades of effort, our knowledge of RCD has been significantly enriched. Based on morphological characteristics, molecular mechanisms, functions, and pathophysiological context, cell death was classified by the Nomenclature Committee on Cell Death (NCCD) into a dozen of categories, including apoptosis, necroptosis, ferroptosis, and pyroptosis (Galluzzi et al., 2018). The molecular mechanisms that initiate and/or propagate each type of RCD show interconnectivity. Meanwhile, each RCD exhibits distinguishable morphological characters and immunomodulatory features varying from proinflammatory to anti-inflammatory (Fig. 2). However, not every type of RCD has been studied in the context of tissue regeneration.
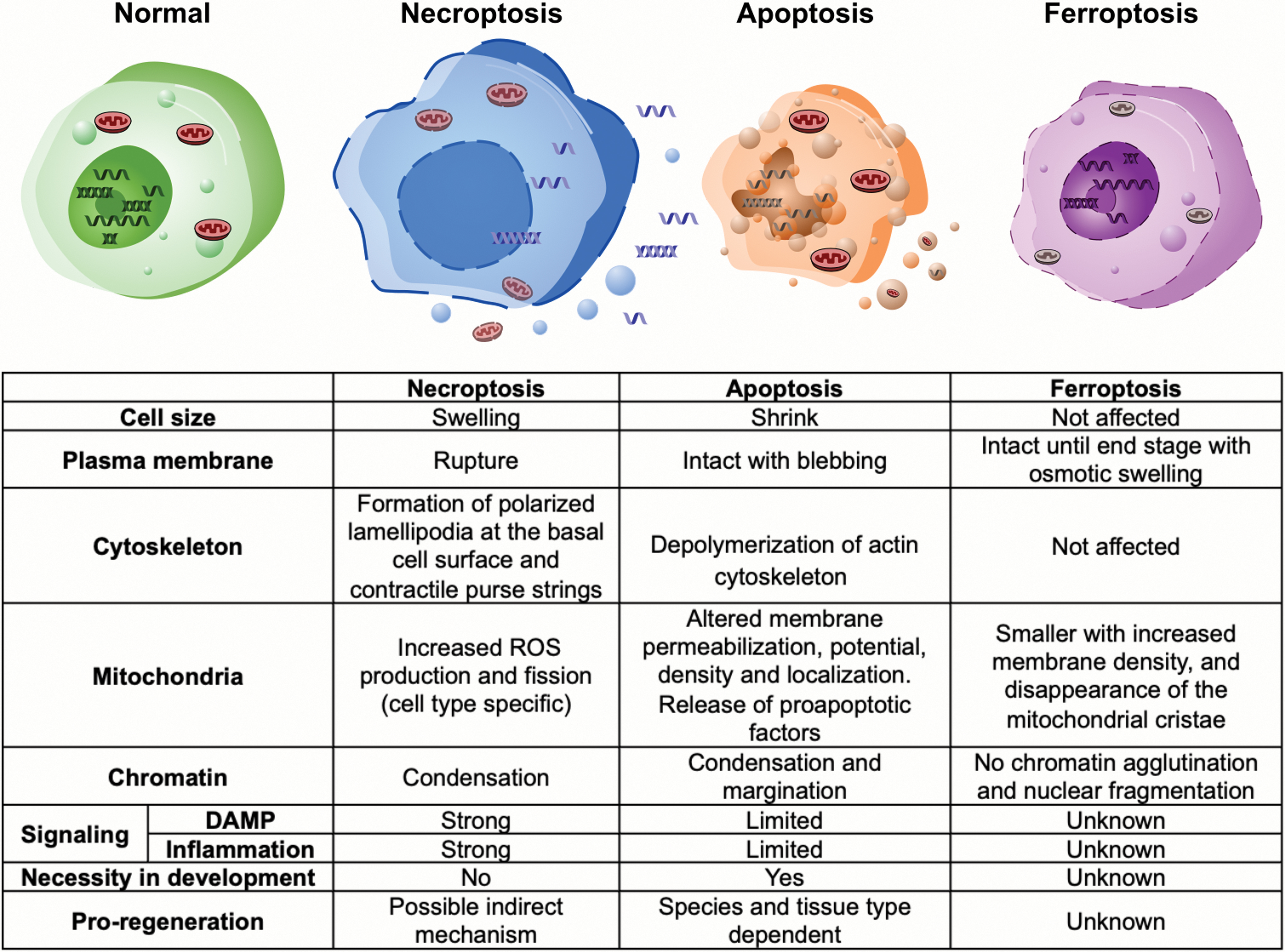
Apoptosis, the founding member of the RCD family, features shrunken cells with condensed cytoplasm and organelles, and breakdown of cytoskeleton (Kothakota et al., 1997). Chromatin also progressively condenses (pyknosis) and fragments (karyorrhexis) (Saraste and Pulkki, 2000). Following karyorrhexis, plasma membrane appears blebbing and is separated into membranous apoptotic bodies containing compacted organelles (Saraste and Pulkki, 2000). Apoptosis causes low inflammatory reaction due to limited exposure of intracellular components and efficient phagocytosis of cellular debris (Fig. 2) (Savill and Fadok, 2000).
Two distinct apoptotic pathways, the intrinsic and the extrinsic, have been described (Igney and Krammer, 2002). In response to stimuli such as DNA damage, the intrinsic mitochondrial pathway is propagated by Bcl-2 family proteins (Roos et al., 2016), which stimulates the release of cytochrome c from mitochondria that binds to Apaf-1 to form a heptameric complex called an apoptosome (Bao and Shi, 2007; Czabotar et al., 2014). The apoptosome recruits procaspase 9 for its catalytic activation (Bao and Shi, 2007). The active caspase 9 activates caspase 3, which cleaves over 1000 cellular substrates to complete apoptosis (Parrish et al., 2013). On the contrary, the extrinsic pathway is triggered by death factors of tumor necrosis factor (TNF) receptor gene superfamily, such as TNF-α and Fas ligand (FasL) (Locksley et al., 2001; Nagata, 1997; Strasser et al., 2009), which bind to specific death receptors TNFR1 and Fas, respectively (Locksley et al., 2001).
The death domain of Fas recruits Fas-associated protein with death domain (FADD), procaspase 8, etc., to form a death-inducing signaling complex (DISC). DISC can recruit caspase 8 to activate downstream caspase for substrate cleavage and execution of apoptosis (Lavrik and Krammer, 2012; Nagata, 1997).
During tissue development, various structures with transient functions are removed by RCD when they are no longer required. One well-known example is the formation of digits in vertebrates, during which apoptosis eliminates the interdigital webs (Lindsten et al., 2000). RCD by apoptosis contributes to the patterning and development of virtually all adult structures in the fly, including legs, wings, eyes, genitalia, digestive system, and the nervous system (Fuchs and Steller, 2011). In vertebrates, physiological RCD occurs in differentiating inner cell mass of blastocysts and lasts till adulthood for maintaining tissue homeostasis (Hardy et al., 1989). Interestingly, inactivation of mouse cell death genes only causes minor developmental defects, likely due to the redundancy within the caspase family and the dual apoptotic pathways (Lindsten and Thompson, 2006). However, block of apoptosis itself can lead to developmental abnormalities (Thompson, 1995).
Coordinated cell division and RCD rates maintain an appropriate cell population in developing organs with normal functions. In some organs, cells are overproduced and subsequently removed by RCD. For example, nearly 80% of human oocytes are removed by RCD before birth, and more than half of neurons in developing mammalian central nerve system are eliminated (Barres and Raff, 1999; Reynaud and Driancourt, 2000). It is likely that cells are selected meticulously for survival or undergoing RCD based on the presence of genetic mutation and structural defects occurred during development. This evolutionary advantage guarantees a high-quality pool of vital cell types.
In other scenarios, tissue primordia release limited amounts of survival signals to control the size of cell population. In Drosophila, cell competition contributes to the establishment of homeostatic cell variations for a certain type of tissue, “Loser” cells are eliminated by apoptosis (de la Cova et al., 2004). These observations showed a physiological role of RCD during development as a cell quality checker in coordination with cell division and differentiation.
The Concept of Regenerative Cell Death
Rapid and robust cell division occurs in regenerative tissue such as the epithelium and liver after injury where the roles of cell death are often overlooked. Most concepts treat cell death as a major obstacle of tissue regeneration. However, it is believed that activated developmental signaling can repair injured mature tissue. Since RCD is essential for the development of many tissue types, a compelling question would be if it can coordinate with other developmental signaling to contribute to tissue regeneration.
The concept of compensatory proliferation was initially described in planaria, Hydra and Drosophila, and was later supported by evidence from Xenopus and mouse models. It suggests that stressed cells undergoing apoptosis can stimulate the proliferation of neighbor cells (Perez-Garijo et al., 2004; Ryoo et al., 2004). In planarians, apoptotic genes, such as DjClg3, are involved in head and tail regeneration (Hwang et al., 2004). In the tail of Xenopus tadpole, intensive apoptosis occurs in the nascent regeneration bud soon after amputation and is critical for initiating regeneration (Tseng et al., 2007). During Hydra head regeneration after midgastric bisection, a rapid onset of apoptosis leads to secretion of Wnt3 from the dying cells (Chera et al., 2009). Wnt3 then activates β-catenin in surrounding cells to induce proliferation (Galliot and Chera, 2010).
The compensatory proliferation is critical for Hydra head regeneration, which can be blocked by a caspase inhibitor, but restored by the exogenous Wnt3 protein (Chera et al., 2009). In Drosophila, cell death in the wing imaginal discs caused by lineage-restricted ricin expression triggered cell proliferation in the neighboring compartment, likely by emitting a diffusible mitogenic signal (Milan et al., 1997). Apoptosis is scarce in normal wing imaginal discs but can be rapidly induced by radiation and heat shock. The rapid repopulation of wing imaginal discs allows them to develop into normal adult tissue even after losing 60% of cells (Haynie and Bryant, 1977). The efficient clearance of apoptotic bodies used to cause a technical difficulty for experimental examination.
To prolong the regenerative effect of apoptotic cells, the potent caspase inhibitor baculovirus P35 was used to block the execution of apoptosis and maintain cells in the apoptotic state. Because P35 specifically inhibits effector caspases, the initiator caspase death regulator Nedd2-like caspase (Dronc) can still perform nonapoptotic functions. This model granted the researchers a comparatively longer time window to study the mechanisms underlying compensatory proliferation. P35 and Dronc collaborate to induce nonautonomous cell proliferation and stimulate blastema formation, likely through upregulation of the Wingless (Wg) and Decapentaplegic (Dpp) pathways (Huh et al., 2004; Perez-Garijo et al., 2004; Ryoo et al., 2004; Wells et al., 2006). Alternatively, in Drosophila eye tissue, compensatory proliferation occurs through activation of the Hedgehog (Hh) signaling pathway by effector caspases, drICE and Dcp-1 (Fan and Bergmann, 2008). These findings suggest different mechanisms of compensatory proliferation preferred by different tissue types.
Studies in mice further support a link between apoptosis and regeneration. Mice lacking either caspase-3 or -7 have impaired skin wound healing and liver regeneration. Interestingly, a downstream target of these caspases is prostaglandin E2 (PGE2), known to work in tandem with the Wnt pathway to promote stem cell maintenance and tissue regeneration (Fig. 3) (Goessling et al., 2009; Li et al., 2010). Numerous aspects of regeneration can be dependent on the apoptotic signaling that includes morphogenesis, inflammation, and tissue regrowth (Bergmann and Steller, 2010). These critical steps are coregulated by a group of mitogens, including Wnt, transforming growth factor β (TGFβ), EGF, and Hh, all of which can be released from dying cells (Fogarty and Bergmann, 2017). In vertebrate tissue regeneration, Wnt signaling plays critical but sometimes opposing roles. For example, zebra fish fin regeneration is promoted by Wnt8, but suppressed by Wnt5b (Stoick-Cooper et al., 2007).

In Drosophila, contradictory results were reported regarding the necessities of Wg in apoptosis-induced proliferation (Perez-Garijo et al., 2009; Smith-Bolton et al., 2009). It is likely that the function of Wg pathway varies by the tissue type, injury type, as well as the severity of cellular stress. Apoptosis and compensatory proliferation allow tissues to eliminate damaged cells and replace them with the progeny of healthy neighbors. When designing therapeutic methods using apoptosis inhibitors, it is necessary to consider the timing and strength of treatment to avoid disrupting the onset of compensatory proliferation.
Necrotic Cell Death in Tissue Regeneration
It is generally agreed that cell death with preserved plasma membrane integrity will have a lower amount of, but more controllable, death-related signaling. Different from apoptosis, necrosis was considered an unregulated accidental cell death (Fig. 2). Cells undergoing necrosis swell, causing plasma membrane rupture and release of cytoplasmic and nuclear components (Srinivasula et al., 2000). Multiple factors derived from necrotic cells might influence the behavior of surviving tissues, although further studies are needed to identify their roles in regeneration (Venereau et al., 2015). In humans, necrosis can occur to all cell types due to microenvironmental stress such as ischemic injury (Konstantinidis et al., 2012; Yuan, 2009). Drosophila wing discs can regenerate following necrosis but do so by inducing apoptosis in cells away from the injury site. This necrosis-induced apoptosis is required for localized proliferation and efficient regeneration (Klemm et al., 2021).
Apoptosis and necrosis may occur simultaneously, and in some cases can be induced by the same stimulus at different dosages. For instance, hydrogen peroxide can induce apoptosis at a low concentration and necrosis at a high dosage (Saito et al., 2006). Similar to the necessity of an intrinsic apoptotic pathway during embryogenesis, lack of caspase-8 or its adaptor FADD causes early embryonic lethality, a phenotype dependent on receptor interacting protein kinase-3 (RIPK3) and the necroptotic effector mixed lineage kinase domain-like protein (MLKL) (Ke et al., 2018; Weinlich et al., 2017). Necroptosis is a regulatory form of necrosis (Weinlich et al., 2017). Considered more controllable than necrosis, necroptosis is triggered by microenvironmental disturbance, which activates pathogen recognition receptors (PRRs) such as Z-DNA binding protein 1 (ZBP1), toll-like receptor 3 (TLR3), TLR4, and death receptors including Fas and TNFR1 (Kaiser et al., 2013; Vercammen et al., 1998).
Active receptors will further activate downstream necroptotic regulators including RIPK1. RIPK1 activates RIPK3 (Li et al., 2012), which then phosphorylates MLKL, leading to the formation of MLKL homotrimers, which can disrupt cell membrane integrity (Murphy et al., 2013; Sun et al., 2012). Remodeling of cytoskeleton in necroptotic cells is different from apoptotic cells and shows controllable patterns (Fig. 2) (Shkarina et al., 2022). Increased reactive oxygen species (ROS) production and fission of mitochondria were described, however, whether mitochondria play a role during necroptosis is still under debate (Tait et al., 2013; Wang et al., 2012).
As a key necroptotic regulator, RIPK1 can activate nuclear factor kappa light chain enhancer of activated B cells (NF-κB), resulting in production of cytokines and chemokines within the cell, later released into the extracellular space (Yatim et al., 2015). These evidences suggest that necroptosis may play roles during tissue repair. In mouse cell culture, dimerization of an N-terminal fragment of MLKL that is unlikely to interact with other proteins causes plasma membrane damage and induces the expression of the chemokines CXCL1 and CXCL10 (Gong et al., 2017b). This suggests that the cell membrane damage might itself initiate a transcriptomic shift in response to stress (Gong et al., 2017a). The resulted altered chemokine level in the extracellular matrix (ECM) will likely affect immune response and inflammation, both of which are crucial for tissue regeneration.
In another example, studies using peripheral blood mononuclear cells (PBMCs) showed that high-dose γ-irradiation induces both apoptosis and necroptosis, and that the proangiogenic capacity of PBMC secretome requires the onset of necroptosis, but not apoptosis (Simader et al., 2019). The authors further identified TNF receptor superfamily member 1B, activated by TNF-α, as the main driver of necroptosis in response to γ-irradiation in PBMCs (Simader et al., 2019). The regulatory pathways of apoptosis and necroptosis could intertwine, and the roles of cell death during tissue regeneration expand beyond apoptosis (Fig. 3).
Regenerative Signaling Induced in and Derived from Dying Cells
In a certain species, different organ and tissue types show different capacities and patterns of regeneration after injury/disease. In mammals, skin and liver can fully restore both tissue structure and function after injury, while heart muscle, limb appendages heal wounds with scarring tissue (Brockes and Kumar, 2008; Tanaka and Reddien, 2011). These observations suggest that injury activates distinct signaling cascades in tissue with different regenerative capacities, leading to cell dedifferentiation, proliferation, or scarring. It is also likely that each tissue/cell type has a preferred form of cell death. Therefore, a profound investigation on injury-induced signals in each model animal and tissue type would provide valuable insight into the regenerative programs required for a full structural and functional restoration.
JNK signaling
In Drosophila, JNK signaling plays a critical role in compensatory proliferation and wound healing, either by directly answering to the caspase Dronc or act independently of apoptosis (Perez-Garijo et al., 2009; Ryoo et al., 2004). Compensatory proliferation requires mitogenic signals secreted by dying cells, including Wg (Wnt ortholog) and Dpp (TGFβ ortholog) (Ryoo et al., 2004). These factors are essential during early development and can stimulate tissue regeneration in vertebrates and insects (Bergmann and Steller, 2010). Importantly, active JNK signaling induces transcription of Wg in apoptotic cells (Fig. 3) (Bergantinos et al., 2010; Ryoo et al., 2004; Smith-Bolton et al., 2009). Furthermore in Drosophila, JNK and p38 coregulate Foxo and AP-1-related factor ATF2 to control stress resistance and antioxidant gene expression (Panayidou and Apidianakis, 2013).
In mammals, JNK-dependent AP-1 and p38 MAPK act in nonparenchymal liver cells to induce TNF and IL-6, both required for liver regeneration (Seki et al., 2012). Interestingly, NF-κB activation protects hepatocytes from TNF-induced apoptosis, meanwhile, allows them to respond to proliferative signals generated through TNFR1 activity (Maeda et al., 2003). Some of NF-κB's protective effects are mediated through GADD45β, which inhibits prolonged JNK activation (Papa et al., 2008). In mouse hepatocytes, when the NF-κB pathway was inhibited, treatment with the liver carcinogen led to JNK activation and increased compensatory proliferation in the surviving hepatocytes. In addition, mice lacking JNK had noticeable decreases in cyclin D, vascular endothelial growth factor (VEGF), and cell proliferation (Sakurai et al., 2006). The coordinated action of JNK and NF-κB pathways maintains a balanced regenerative activity in injured tissue to facilitate regeneration and avoid carcinogenesis.
Tissue regeneration driven by damage-associated molecular patterns
Cells communicate through direct and/or indirect molecule exchange, and the communicative pattern changes dramatically under stress. Dying cells release a group of heterogeneous molecules called damage-associated molecular patterns (DAMPs), to regulate sterile inflammation (Fig. 3) (Zitvogel et al., 2010). The DAMP molecules have a vital biological function under physiological circumstance and will not be accessed by immune cells (Zitvogel et al., 2010). A diverse group of molecules have been reported to act as DAMPs during tissue damage and regeneration, including nuclear factors (Tian et al., 2007), DNA (Muruve et al., 2008), adenosine triphosphate (ATP) (Elliott et al., 2009). Necrotic cells are a major source of DAMPs, largely due to the compromised cell membrane integrity (Zitvogel et al., 2010).
On the contrary, the plasma membrane remains comparatively intact throughout apoptosis, therefore limiting the release of DAMPs (Li et al., 2003). DAMPs can be either pro- or anti-inflammatory, a dual-directional mechanism to keep inflammatory response under control (Chen et al., 2009). DAMPs bind to cell surface receptors (normally PRRs) to recruit immune cells for cleaning cell debris and activate compensatory proliferation in either healthy neighboring cells or local progenitors (Venereau et al., 2015; Zong et al., 2013). Immune cells are not the only cell population responding to DAMP signaling. Endothelial cells and fibroblasts also express various immune receptors and can be activated by DAMPs during injury or disease (Gong et al., 2020). The source of DAMPs is not limited to cells undergoing terminal steps of RCD, but also can be cells under stress (Venereau et al., 2015). This provides a fast stress-responding mechanism for injured tissue.
Well-studied DAMP molecules, such as ATP and high-mobility group box 1 (HMGB1), are involved in tissue regeneration. HMGB1 and ATP can be passively released by necrotic cells or actively secreted by stressed cells independent of the ER pathway (Pandolfi et al., 2016).
HMGB1, one of the first DAMPs identified, is upregulated during wound healing and ischemic injury (Biscetti et al., 2010). As a redox-sensitive molecule, HMGB1 binds to CXCL12 to recruit leukocytes in a reduced microenvironment, and when oxidized, activates the TLR4 to regulate downstream chemokine and cytokine release (Janko et al., 2014; Venereau et al., 2013; Venereau et al., 2012). HMGB1 can be released by both apoptotic and necroptotic cells to recruit innate immune cells and promote their secretion of cytokines and chemokines (Galluzzi et al., 2017; Yatim et al., 2015; Zhang et al., 2012). During tissue repair, HMGB1 promotes proliferation and differentiation of tissue-specific progenitors in fibrotic and angiogenic events (Palumbo et al., 2004; Ranzato et al., 2009; Zhang et al., 2012).
The regenerative properties of HMGB1 have been studied in multiple organs. It promotes locomotor recovery and axonal formation in zebra fish spinal cord (Fang et al., 2014). In skin wound, topical application of recombinant HMGB1 accelerated wound healing (Straino et al., 2008). In mature mammalian myocardial tissue, commonly recognized as nonregenerative, overexpression of HMGB1 can be beneficial after a heart attack (Kitahara et al., 2008). Furthermore, HMGB1 level increases in skeletal muscle cells after ischemia/reperfusion (I/R) to enhance vascularization and myofiber restoration (De Mori et al., 2007).
Downstream cytokines and chemokines of DAMP signaling deliver a stoichiometric and pleiotropic effect on repairing and deterioration of damaged tissue. This process could be highly dynamic, even chaotic. The degree of the cytokine activity in turn is an important determinant of the regenerative outcome. Severe traumatic injury could lower the level of circulating cytokines known to have tissue protective and repairing functions, such as IL-9, IL-22, and IL17E/25 (Cai et al., 2021; Namas et al., 2016). Meanwhile, IL-33 may initiate early detrimental immune responses in acute lung injury (Xu et al., 2017).
The spatiotemporal regulation and interaction of these factors in different types of injury and disease have been attracting a significant amount of effort. For example, HMGB1 induces the production of TNF-α, which can either promote apoptosis through the death receptors or initiate a cytoprotective effect by activating NF-κB and stress-activated protein kinase/JNK (Nian et al., 2004; Venereau et al., 2012).
ATP, on the contrary, can be actively released via vesicles and connexin hemichannels by apoptotic cells in a caspase-dependent mechanism (Fig. 3) (Chekeni et al., 2010; Eltzschig et al., 2008). During angiogenesis, ATP binds to the P2Y receptor to promote the migration of vascular smooth muscle cells and the proliferation of endothelial cells and fibroblasts (Albert et al., 1997; Jin et al., 2014; Yu et al., 2008). During regeneration, extracellular ATP stimulates cell proliferation through MAPK activation (Lin et al., 2007; Tu et al., 2000). ATP release also participates in the “find me” and “eat me” signal, attracting phagocytes to clear cellular debris (Elliott et al., 2009; Ravichandran, 2010). In acute kidney injury, such signals induce the proliferation of neighboring tubular cells to promote wound healing (Nakagawa et al., 2014). In UV-irradiated keratinocytes, ATPs are released and participate in the DNA repair (MacLeod et al., 2014).
After hepatectomy, ATP is released from hepatocytes and Kupffer cells to promote hepatocyte division (Gonzales et al., 2010). ATP function can be complicated by its ability to activate nicotinamide adenine dinucleotide phosphate (NADPH) enzymes to produce H2O2 (Cordeiro and Jacinto, 2013). At high concentration, intracellular H2O2 induces cell death (Kamata et al., 2005).
ROS as regenerative signaling
H2O2 and other ROS members are oxygen-containing reactive molecules that can be naturally generated from intracellular structures such as mitochondria and NAD(P)H oxidase enzymes (Fig. 3). At physiological levels, ROS act as important signaling molecules involved in a variety of physiological activities such as immune response and exercise adaptation (Zuo et al., 2015). Endogenous antioxidants including catalase, superoxide dismutase, glutathione, and glutathione peroxidase (Gpx) are responsible for maintaining ROS at normal levels. However, under specific pathological conditions, antioxidant defenses can be overwhelmed, resulting in cellular oxidative stress, which damages common biomolecules such as DNA, protein, and lipid (Zuo et al., 2015). The cells undergo different RCD pathways when different biomolecules are affected by high ROS levels.
In adult Drosophila midgut, ROS at low levels are microbicidal and promote the replacement of the stressed enterocytes through enteroblast differentiation (Lee et al., 2013). At high levels, ROS cause delamination and death of enterocytes, which release cytokines to induce the proliferation of neighboring intestine stem cells (Buchon et al., 2009). In Xenopus laevis tadpoles, a high ROS level induces Wnt signaling, which promotes cell proliferation and axial growth during later stages of regeneration (Love et al., 2013). In adult zebra fish, caudal fin injury causes a rapid and lasting increase of ROS level to activate JNK and apoptosis, both of which are essential for blastema formation (Gauron et al., 2013).
In mammals, the extracellular ROS level spikes to stress neighboring cells after ischemic or traumatic injury in muscle tissue (Tao et al., 2016). Redox stress is also widely involved in the progression of hepatic injury during I/R and viral infection (He et al., 2006; Korenaga et al., 2005). Contradictorily, ROS protect cirrhotic hepatocytes from TGFβ-induced apoptosis and participate in the beneficial effects of ischemic preconditioning in hepatic and renal I/R (Lee et al., 2012; Song et al., 2012). High ROS levels in mammals can also activate JNK and p38 MAPK, which activate caspases (Kamata et al., 2005; Ravindran et al., 2011).
Among the RCDs, ferroptosis, an iron-dependent form of regulated necrosis mediated by lipid peroxidation, is a unique manifestation of oxidative stress under high ROS levels. The Stockwell group started to use the term ferroptosis in 2012 to describe this erastin (a system xc- inhibitor)-induced RCD (Dixon et al., 2012). In ferroptotic cells, free intracellular Fe2+ combines with peroxides to produce highly reactive hydroxyl radicals and peroxide radicals through Fenton reaction, thus promoting nonenzymatic oxidation of lipids, especially unsaturated fatty acids. The products of lipid peroxidation include the initial lipid hydroperoxides and subsequent reactive aldehydes, which increase during ferroptosis. Endogenously, ferroptosis is suppressed by the system xc--Gpx4 pathway with glutathione as substrate, or coenzyme Q10 system. The repairing of membranous structure damaged by the lipid ROS can also prevent ferroptosis (Tang et al., 2021; Xie et al., 2016).
Therapeutically, ferroptosis can be inhibited by iron chelators and lipophilic radical-trapping antioxidants (Dixon et al., 2012). Thus, when a fine balance between lipid ROS and the scavenging system targeting them is tilted, ferroptosis occurs.
By far, the most reliable morphological identity of a ferroptotic cell is shrunken mitochondria with increased membrane density and decreased or disappeared mitochondrial cristae. The nuclear morphology is normal, with either no or moderately condensed chromatin depending on the cell type. The cell membrane remains undamaged but will be affected through an osmotic mechanism at the end stage (Dixon et al., 2012; Riegman et al., 2020; Yagoda et al., 2007) (Fig. 2). These features theoretically limit the amount of DAMPs released when compared with necrosis, and may provide the cells under oxidative stress an alternative way to control inflammation, stress signaling, and self-repair. Compared with other types of RCDs, ferroptotic cells may induce a unique pattern of inflammatory reactions, which will clean the dead cells and repair damaged tissue in a distinctive way.
However, our knowledge about the molecular mechanisms of ferroptosis and its roles during tissue regeneration is still scarce. In addition, the tissue repair process seems to vary significantly based on the affected cell type (Tang et al., 2021).
Caspase activity in compensatory proliferation
Caspases are cysteine proteases synthesized as inactive zymogens. They gain catalytic activity in response to upstream stimuli and act as key executioners in apoptosis (Thornberry and Lazebnik, 1998). Caspases are categorized into initiators and effectors. Initiator caspases, such as caspase-8, caspase-9, and Drosophila Dronc, possess long prodomains for interacting with upstream apoptotic stimuli (Kumar, 2007). Active initiator caspases cleave and activate effector caspases including caspase-3, caspase-7, Drosophila DrICE, and Dcp-1, which then cleave a broad range of substrates that contribute to the execution of apoptosis (Dix et al., 2008; Kerr et al., 1972).
As briefly mentioned above, specific inhibition of Drosophila effector caspases by P35 in stressed wing and eye imaginal discs leads to hyperplastic growth likely due to persistent mitogenic signaling (Clem et al., 1991). This showed a nonapoptotic function of initiator caspases in compensatory proliferation. Worth noticing, the same genetic strategy will stop compensatory proliferation in more differentiated eye tissue where the effector caspases DrICE and Dcp-1 are also required for regeneration (Fan and Bergmann, 2008). In this case, differentiated photoreceptor neurons emit mitogenic signals for apoptosis-induced proliferation (Fan and Bergmann, 2008). The necessity of caspase during compensatory regeneration has also been shown in other regenerative models. During Xenopus tadpole tail regeneration, inhibition of effector caspase blocks cell proliferation (Tseng et al., 2007).
In a mouse model, irradiated mouse embryonic fibroblasts (MEFs) promote stem cell division when cocultured. Importantly, caspase-3 and caspase-7 are required in MEFs for affecting stem cells, indicating the requirement of full execution of apoptotic program (Li et al., 2010). These reports suggest that apoptotic signaling plays an active role during tissue regeneration. The nature (stemness, status of differentiation, etc.) of the injured tissue may call for the involvement of apoptotic signaling at different degrees and stages.
In tissue/organ where effector caspases are not required for compensatory proliferation, a potential therapeutic strategy would be to inhibit the effector caspase to preserve stressed cells and prolong the beneficial effect of apoptotic-relevant regenerative signaling. However, when activation of effector caspases is required, an important question would be if it is still possible to prevent the apoptotic cells from dying, meanwhile, get the most out of their regenerative potential. As a matter of fact, a phenomenon named “anastasis,” during which cell survives despite effector caspases being activated, has been described (Sun et al., 2017). This allows the investigation of nonapoptotic functions of effector caspases and a potential method for rescuing terminally apoptotic cells.
Worth mentioning, it is also possible for cells to survive p-MLKL action during necroptosis, a cellular event named resuscitation (Gong et al., 2017b). Interestingly, the transcriptomic feature of anastasis and resuscitation shares a certain level of similarity (Gong et al., 2017a). In Drosophila, reports indicate that anastasis could be common during development (Tang et al., 2015). Meanwhile, after patient kidney transplantation, endothelial cells of the donor kidney express p-MLKL without ongoing necroptosis (Gong et al., 2017b). However, both phenomena will need to be validated in mammalian injury/regeneration models.
One intriguing question is that how the cells decide to stay “undead” or progress directly into the terminally steps of RCD until the cell body is destroyed and cleared. The switch between these two decisions might be controlled by the activation level of the caspases or MLKL. In other words, when upstream stimuli are too strong, the room for the cells to keep the death machinery in a controllable scale will be limited. When using in vitro cell culture, cell death is typically executed rapidly due to the strong stimuli researchers treat the cells with. By correlating the stress level to the activity of RCD machinery, one may define the threshold of caspase or MLKL activity that decides the fate of cells.
Going through the literature, we learned that caspase activity is detectable in healthy cells. In fact, controllable levels of caspase activity participate in cell migration (Geisbrecht and Montell, 2004), cytoskeletal rearrangements (Kuranaga et al., 2006), and cell fate decision (De Maria et al., 1999). Together, these observations suggest that lower-than-critical level of caspase activity may allow cells to signal for proliferation and tissue repair. However, what are the downstream factors of controllable caspases during tissue regeneration?
In MEFs, as an example, activated caspase-3 cleaves and activates independent phospholipase A2 (iPLA2), which enhances the secretion of arachidonic acid and PGE2, reported to stimulate stem cell and keratinocyte proliferation (Liou et al., 2007; North et al., 2007). These findings point to PGE2, produced by apoptotic cells, as a link between caspase activation and downstream cellular effects (Fig. 3). Indeed, PGE2 further activates the Wnt/β-catenin pathway in nonapoptotic cells to promote compensatory proliferation in the context of wound healing in mammalian cells (Castellone et al., 2005; Goessling et al., 2009; Li et al., 2010).
RCD in Terminally Differentiated Mammalian Cells
RCD facilitates the elimination of damaged cells and paves the road for regeneration. However, how to take advantage of the to-be-disposed cells varies in different tissue types, which demonstrate a different intrinsic regenerative capacity and preferred type of RCD. In neonatal mouse, as an example, the sound-sensing cochlear hair cells regenerate after cell death caused by diphtheria toxin, but not neomycin (Hu et al., 2016). It turned out that diphtheria-toxin-treated hair cells have higher Wnt pathway activity, compared with the neomycin group (Hu et al., 2016). Regeneration of damaged tissue can vary due to difference in stimuli, the level of regenerative signals, and the type of RCD the stressed cells choose to undergo.
Mammals have limited regenerative ability. Vital organs such as heart are notoriously nonregenerative (Deshmukh et al., 2019). Derived from the primitive heart fields in mesoderm, heart is a muscular organ that drives blood flow in the circulation system. In comparison, another mesoderm-derived striated muscle type, the skeletal muscle, can regenerate after severe damage. This is due to the presence of satellite cells, a group of multipotent progenitors residing in between the sarcolemma and basement membrane (Fig. 4A) (Mauro, 1961). After the loss of adjacent myofiber (skeletal muscle cell), Pax7-positive satellite cells proliferate to produce a pool of MyoD-positive myoblasts, which then proliferate, differentiate, and fuse into bona fide myofibers (Fig. 4B) (Zammit et al., 2006).

It has been reported that an inflammatory response is required for activating the satellite cells, likely initiated by the release of DAMPs and paracrine factors from stressed myofibers after the onset of RCD (Tidball and Wehling-Henricks, 2007). Apoptosis has been well documented in skeletal muscle as a consequence of trauma, genetic defect, and intensive exercise (Andrianjafiniony et al., 2010; Dupont-Versteegden et al., 2006; Smith et al., 2000). The typical apoptotic mechanisms occurring in common somatic cells cannot fully describe the complicated RCD pattern in multinucleated myofibers. RCD can selectively remove an affected nucleus and surrounding cytoplasm in a myofiber, causing skeletal muscle atrophy (Fig. 4C) (Allen et al., 1999; Marzetti et al., 2013). This mode of partial RCD can spare the life of the affected cell, which has the chance to be repaired through protein, lipid synthesis, and anabolism.
Also, the partial RCD pattern likely causes less inflammation, and is faster and energy-efficient than the differentiation of a satellite cell. During this process, the contractility of a myofiber is partially maintained.
On the contrary, heart damages caused by heart disease including myocardial infarction (MI) claim millions of lives worldwide annually (Benjamin et al., 2019). Cardiac and skeletal muscle cells are functionally and anatomically similar, demonstrated as large cell size and enrichment of sarcomeres and mitochondria. However, unlike skeletal muscle, there is a lack of progenitor population in the myocardium (van Berlo et al., 2014). Cardiomyocytes, although large in size compared with most somatic cells, do not have the long fibril structure as seen in myofibers. Moreover, murine cardiomyocytes typically have two diploid nuclei, and human cardiomyocytes demonstrate a dominant karyotype of mononuclear tetraploidy (Gan et al., 2020). Therefore, the partial RCD observed in myofiber is not likely utilized by cardiomyocytes.
After reaching adulthood, cardiomyocyte division is exceedingly rare, either in healthy or injured status (Bergmann et al., 2015; Senyo et al., 2013). Although they possess considerable longevity and resilience, cardiomyocytes are susceptible to ischemic injury typically caused by MI (Tao et al., 2016). Unlike zebra fish and salamander, which display robust heart regeneration at adult stages (Foglia and Poss, 2016), the microenvironment of damaged mammalian myocardium is not optimized for tissue repair. Massive loss of cardiomyocytes after MI will be compensated by fibrotic scarring to preserve contractility that, however, will eventually lead to heart failure (Deshmukh et al., 2019).
Mammalian heart growth during embryonic development is driven by cardiomyocyte division. After birth, cardiac growth switches to hypertrophic pattern, concomitant with karyokinesis in the absence of cytokinesis, and therefore, myocardial tissue volume expands without cell division (Soonpaa et al., 1996). Neonatal mammalian cardiomyocytes have residual proliferative capacity, which fades by juvenile stages (Porrello et al., 2011). Current strategies for treating an infarcted myocardium include genetic manipulation to promote cell cycle reentry of cardiomyocytes (Tzahor and Poss, 2017). Forced regulation of cell cycle genes may reactivate DNA synthesis, however, the occurrence of cytokinesis seems to be less common, unless the key tumor inhibitors such as the Hippo pathway components are removed (Deshmukh et al., 2019; Leach et al., 2017). Compared with cell proliferation, it seems that the roles of RCD in cardiomyocytes have not attracted an equal amount of attention.
Several death-relevant signaling pathways involved in tissue regeneration mentioned above have been shown to play roles in myocardial homeostasis and disease. Physiologically, ROS can be produced by mitochondria, xanthine oxidase, and NADPH oxidase in cardiomyocytes (Zuo et al., 2015). ROS regulate cardiomyocyte contractility by altering ion flux in cardiomyocytes (Kaplan et al., 2003). During disease such as heart failure, an increased amount of ROS can be detected, along with damage in DNA, protein, membranous structure, and organelles (Zhou et al., 2018). Interestingly, ROS can also be stimulated by TNF-α (Meier et al., 1989), which, when overexpressed specifically in cardiomyocyte, increases cytochrome c level in cytoplasm (Haudek et al., 2007). In addition, the proinflammatory effect of HMGB1-RAGE (receptor for advanced glycation end products) axis promotes the onset of inflammatory cardiomyopathy (Bangert et al., 2016).
Conversely, an exogenous supplement of HMGB1 preserves cardiac function and decreases scarring after MI (Pellegrini et al., 2019). The beneficial effects are likely implemented by regulating transient inflammation after MI, which if prolonged, aggravates ischemic damages and promotes adverse remodeling (Bangert et al., 2016). Furthermore, HMGB1 promotes the release of cytokines, chemokines, and angiogenic factors from cardiac fibroblasts (Rossini et al., 2008). Although multiple roles of HMGB1 signaling have been reported in the context of heart injury, it seems that HMGB1 cannot promote the proliferation of cardiomyocytes, but of noncardiomyocyte cells only (Pellegrini et al., 2019).
RCD plays a vital part in the cardiac morphogenesis. The outflow tract (OFT) portion of the myocardium is shortened by apoptosis at specific stages as observed in rodent and human embryos (Goor et al., 1972; Ya et al., 1998). The prevalence of these spatiotemporal apoptosis can reach up to 50% of cardiomyocytes in the OFT area (Watanabe et al., 1998). OFT shortening is necessary for the transition of the embryonic heart from single to dual circulation. Although RCD helps to reduce the regional population of cardiomyocyte during this process, it has not been reported that these apoptotic primitive cardiomyocytes can affect the proliferative events of adjacent myocardium.
Although many cardiomyocytes are lost during a severe heart attack (1 billion for human heart, millions for mouse heart) (Benjamin et al., 2019; Monroe et al., 2019), cell death seems to be inefficient to induce cardiomyocyte proliferation. Meanwhile, noncardiomyocytes such as fibroblast and endothelial cells might proliferate in response to cardiomyocyte death, to contribute to fibrosis and angiogenesis (Das et al., 2019; Fu et al., 2018). Even in the injured neonatal mouse heart, it is not clear if compensatory proliferation contributes to the low percentage of proliferative cardiomyocytes observed (Porrello et al., 2011). These observations raised an interesting question: Can we simply apply our knowledge of compensatory proliferation to cardiomyocytes?
In heart disease models, one would expect a large quantify of apoptotic cardiomyocytes after traumatic or ischemic injury. Although cleaved caspase-3 or TUNEL-positive cardiomyocytes have been observed in animal models of heart attack, the amount is surprisingly low. In a pig model of I/R injury, TUNEL reagent marks roughly 17 cardiomyocytes in every 1 cm2 of area (Weil et al., 2017). Interestingly, the low apoptotic rate is concomitant with cardiac troponin I release, indicating that the cardiomyocytes are indeed under stress (Weil et al., 2017). Similarly, postmortem animal and human biopsies with end-stage heart failure display apoptotic rates ranging between 0.12% and 0.7% (van Empel et al., 2005). As mentioned above, the presence of activated caspases is not necessarily equal to ongoing apoptosis. It has been reported that a sublethal level of caspase activity facilitates cardiomyocyte differentiation from embryonic stem cells (Bulatovic et al., 2015).
The Megeney group also reported an alternative role of caspase 3 as stress-responding signaling during cardiomyocyte hypertrophy (Putinski et al., 2013). In their report, phenylephrine or isoproterenol treatment induces cardiomyocyte enlargement with active cleaved caspase 3; meanwhile, the cardiomyocytes maintain nuclei and cytoplasmic integrity (Putinski et al., 2013). In fact, it is possible that cardiomyocytes have a prolonged apoptotic procedure, which provides enough time for cellular self-repair (Kanoh et al., 1999; Narula et al., 1999). Another way to ask this question would be: Do cardiomyocytes actually undergo apoptosis or do they prefer an alternative type of RCD?
In neonatal mouse heart, we surgically amputated a piece of apical myocardial tissue to establish a traumatic model for studying heart regeneration (Tao et al., 2016). Interestingly, at 4 days after the injury, immunostaining of cleaved caspase 3 only highlighted a low ratio of apoptotic cardiomyocytes (Fig. 5A–C). At this time point, scarring is being built up, and ROS accumulate in the microenvironment of the injury border zone, stressing resident cardiomyocytes (Tao et al., 2016). The rate of apoptotic cardiomyocytes did not increase significantly even when Pitx2, a master regulator of redox balance, was deleted in cardiomyocytes (Fig. 5D–F) (Tao et al., 2016). The scarce presence of apoptotic cardiomyocytes may indicate that apoptosis is not required during neonatal cardiomyocyte proliferation.

Recent studies showed that regional inflammation and macrophage infiltration are required for neonatal mouse heart regeneration (Aurora et al., 2014). This might partially explain the low level of apoptosis in cardiomyocytes since apoptosis is considered an anti-inflammatory form of RCD. The death signal released by apoptotic cells is limited due to well-maintained cell membrane integrity, and the fact that apoptotic cells are rapidly cleared by phagocytes, further shrinks the signaling window (Ravichandran and Lorenz, 2007).
Another hint is provided by the roles of RCD during cardiac valvulogenesis. The heart valves derive from primordial structures called endocardial cushions, formed through endocardial delamination and endothelial-to-mesenchymal transformation at the sites of atrioventricular and ventriculoarterial connections (Tao et al., 2012). Significant levels of apoptosis have been observed in the remodeling endocardial cushions of avian and mammalian heart models (van den Hoff et al., 2000). In later developmental stages, the cushions elongate and stratify into leaflets (for atrioventricular valves) and cusps (for aortic and pulmonary valves). Mature valves are composed of an outer endothelial sheet that surrounds three stratified layers of specialized ECM, interspersed with valvular interstitial cells (Tao et al., 2012). Each ECM layer is organized according to the blood flow to withstand the continual change in hemodynamic flow during the cardiac cycle.
One of the three layers, the fibrosa, is located away from blood flow. This layer is rich in collagen fibers aligned along the circumferential direction of the leaflets/cusps (Lincoln et al., 2006). This arrangement provides tensile strength to the valve during opening, while transmitting forces to promote coaptation of the leaflets/cusps in the closed position (Aldous et al., 2009). Interestingly, we detected apoptotic cells within the cushions right before the valvular remodeling stages begin (Fig. 6A, B). By combining our findings with the current reports that indicate apoptosis promoting fibrosis, one would speculate that, besides reducing the population size of valvular interstitial cells during the valvular remodeling stages, if apoptosis is required for the formation of the fibrosa layer (Fig. 6C, D) (Johnson and DiPietro, 2013).
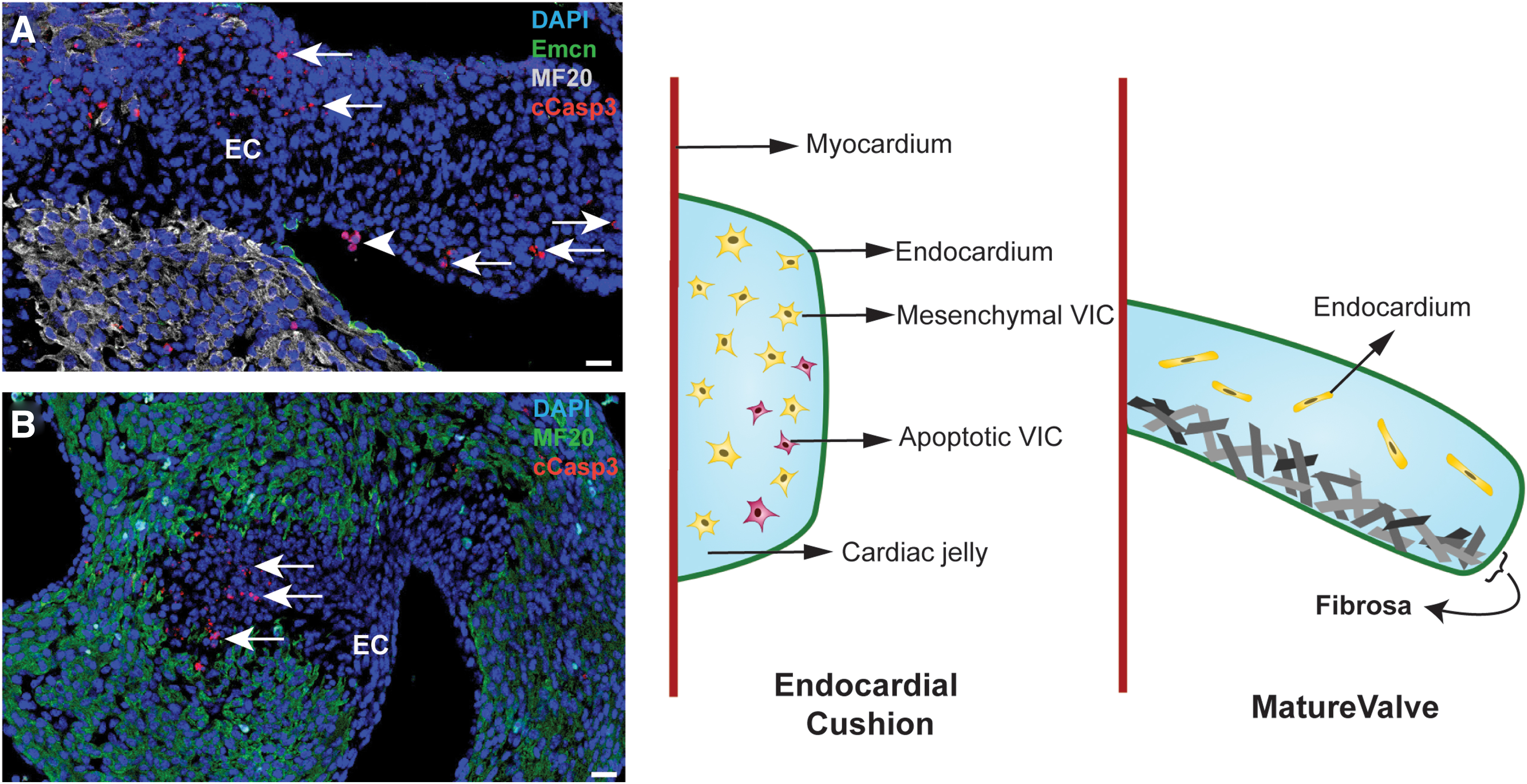
Should this be the case, apoptosis in a cardiomyocyte population during heart injury might be profibrotic, which might cause maladaptive remodeling and impede myocardial regeneration. To avoid this scenario, cardiomyocytes may have evolved a preference for an alternative form of RCD.
As mentioned above, PGE2 released by apoptotic cells downstream of the caspases participates in tissue regeneration. PGH2, the substrate for synthesizing PGE2, was produced by cyclooxygenases 1 and 2 (Cox1 and 2) using arachidonic acid. Cox2 (PTGS2) is also used as a marker for ferroptosis (Tang et al., 2021). The role of Cox2 during ferroptosis is still under debate, it is believed that Cox2 is not a part of the ferroptotic machinery but expressed during ferroptosis (Tang et al., 2021). Combining these findings, one could speculate that the signaling pathway in charge of the regenerative features during apoptosis could be shared with ferroptosis. In general, regulated necrosis including ferroptosis and necroptosis elicits a stronger inflammatory response compared with apoptosis (Davidovich et al., 2014), which could meet the needs of inflammatory signaling by cardiomyocytes (Fig. 2).
However, more direct evidence is needed to assess the inflammatory effect of ferroptosis. In fact, ferroptosis has been implicated in adult cardiomyopathy as a major contributor of cardiomyocyte death (Fang et al., 2019). In cardiomyocytes, Slc7A11 encodes the cysteine–glutamate antiporter to regulate glutathione levels within the cell and the activity of Gpx4, a master inhibitor of ferroptosis (Fang et al., 2020). After MI or trauma, severe redox stress could easily increase the level of lipid ROS, the major cause of ferroptosis, in membranous structures (Fig. 2) (Dixon et al., 2012). However, the roles of ferroptosis in heart regeneration models will need further investigation.
Worth mentioning, although not discussed here, ferroptosis may play major roles in multiple prevalent neural diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease. A majority of the studies focus on ROS, lipid peroxidation, and the susceptibility of neuron to ferroptosis, which has significant contribution to neuron degeneration (Do Van et al., 2016; Hambright et al., 2017; Lee et al., 2011). Similar to cardiomyocytes, neurons are terminally differentiated cells with significant longevity and a low turnover rate. An interesting thought would be that ferroptosis is preferred by highly differentiated cells.
Remaining Questions and Perspectives
RCD during tissue regeneration is a fascinating topic. With hundreds of literatures available, there are still many questions unanswered. One of them is how many waves of RCD occur in tissue after damage. Immediate injury-induced cell death is a direct consequence of damage observed in the leading edge area independently of any regenerative process (Cordeiro and Jacinto, 2013). By contrast, when a later round of cell death is observed, it appears specific to regeneration as observed in planarians and zebra fish. During most studies, researchers would collect samples at a certain time point defined as the early time point, and therefore, the temporal information of tissue regeneration could be neglected. This happened to most of the studies in heart muscle regeneration, as 4 or 7 days postinjury are constantly assumed as an early injury response stage (Porrello et al., 2011; Tao et al., 2016).
The progression of RCD is highly dynamic and the status of a cell undergoing RCD constantly changes. This leads to another question: What do stressed, dying, and end-stage dying cells release into the microenvironment and will they cause different responses in neighboring healthy cells? To answer this question, researchers need to establish a decent model/platform to achieve and maintain each stage of RCD in cells. For example, in cardiomyocyte studies, induced pluripotent stem cell-derived cardiomyocytes can be treated with a different reagent at high to low dosage to mimic stress and dying stages in different types of RCD.
Another important question is that should the RCD be stopped for therapeutic purpose. In mammalian ischemic injury, the shortage of blood and nutrition supply calls for the removal of affected cells to restore homeostasis and reach balance between cell number and available nutrient. Ideally, strategies to eliminate the pathological cause of ischemia, such as angioplasty, are the top choice in clinic. After that, halting of RCD may preserve more somatic cells to minimize tissue damage and facilitate tissue repair. However, in the case when the cause of ischemia cannot be removed immediately, stopping RCD by inhibiting the execution pathway (e.g., caspase) might push the stressed cells toward alternative RCD pathways, or even necrosis. Should this happened, excessive inflammation and lack of RCD-derived signal for compensatory proliferation may cause more harm than good.
Terminally differentiated cells such as cardiomyocytes and neurons do not seem to regenerate through compensatory proliferation. Instead, when cell death occurs among them, it is very likely that they activate resistant mechanism to avoid the final engagement of RCD. One potential advantage of this repairing strategy is to preserve cellular and organ function during tissue repair due to the importance of heart and nerve system. Meanwhile, it would be interesting to find out if it is more time- and/or energy-efficient to repair a cell than to generate a daughter cell. For those nonregenerative cell types, research groups are also interested in where the “point-of-no-return” is for a cell undergoing RCD, and to what extent can a dying cell be “saved”? Should these questions be answered, we may have a better understanding and new perspectives on tissue regeneration in organs considered nonregenerative.
Footnotes
Authors' Contributions
G.T. and S.L. studied the literature, performed the experiments, and prepared the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the grants from the National Institutes of Health (NIH; R01HL148728 to G.T.). G.T. was also partially supported by the National Science Foundation (EPSCoR RII Track-1: MADE in SC OIA-1655740) and Saving Tiny Hearts Society.