
Abstract
Significance:
Dihydrolipoamide dehydrogenase (DLDH) is a flavin-dependent disulfide oxidoreductase. The active form of DLDH is a stable homodimer, and its deficiencies have been linked to numerous metabolic disorders. A better understanding of redox and nonredox features of DLDH may reveal druggable targets for disease interventions or preventions.
Recent Advances:
In this article, the authors review the different roles of DLDH in selected pathological conditions, including its deficiency in humans, its role in stroke and neuroprotection, skin photoaging, Alzheimer's disease, and DLDH as a nondehydrogenating protein, and construction of genetically modified DLDH animal models for further studying the role of DLDH in specific pathological conditions. DLDH is also vulnerable to oxidative modifications in pathological conditions.
Critical Issues:
Novel animal models need to be constructed using gene knockdown techniques to investigate the redox- and nonredox roles of DLDH in related metabolic diseases. Specific small-molecule DLDH inhibitors need to be discovered. The relationship between modifications of specific amino acid residues in DLDH and given pathological conditions is an interesting area that remains to be comprehensively evaluated.
Future Directions:
Cell-specific or tissue-specific knockdown of DLDH creating specific pathological conditions will provide more insights into the mechanisms, whereby DLDH may have therapeutic values under a variety of pathological conditions. Antioxid. Redox Signal. 39, 794–806.
Introduction
Dihydrolipoamide dehydrogenase (DLDH) is a flavin-dependent oxidoreductase catalyzing the oxidation of dihydrolipoamide to lipoamide with concurrent production of NADH at the expense of NAD+ (Fig. 1A) (Argyrou et al., 2002; Carothers et al., 1989; Williams, 1992). DLDH is involved in several key metabolic enzyme complexes, including pyruvate dehydrogenase complex (PDC), α-keto glutarate dehydrogenase complex (α-KGDC), branched chain α-keto acid dehydrogenase complex, and the glycine cleavage system (Fig. 1B) (Vettakkorumakankav and Patel, 1996; Yan et al., 2008).

DLDH can also behave as an NADH-specific diaphorase, whereby it catalyzes the oxidation of NADH with reduction of NAD+ and ubiquinone (Patel and Harris, 1995; Xia et al., 2001). The active form of DLDH, when functioning as a dehydrogenase, is a homodimer with each monomer contributing key amino acid residues to the homodimeric functional activities (Argyrou and Blanchard, 2004; Brautigam et al., 2005; Vaubel et al., 2011). In this article, we overview the role of DLDH in health and disease by discussing selected paradigms.
Our Serendipitous Finding of DLDH
Between 2003 and 2005, the first author of this article was heavily involved in investigating protein carbonylation in aging and disease using both mouse and rat as animal models (Wu et al., 2016; Yan, 2009). A major technique used at the time was two-dimensional gel electrophoresis of oxidized proteins using the specific probe 2,4-dinitrophenyl hydrazine (DNPH) that specifically reacts with protein carbonyl groups to produce 2,4-dinitrophenyl hydrazones, which can be further identified by Western blot assays using anti-DNPH antibodies (Reznick and Packer, 1994; Yan, 2009; Yan et al., 1998; Yan et al., 1997a; Yan et al., 1997b).
Protein carbonyls can also be labeled by aldehyde-reactive probes linked to biotin (Wu et al., 2016). After labeling, the oxidized proteins were then separated by two-dimensional gel electrophoresis, followed by Western blotting detection of the oxidized proteins using anti-DNPH antibodies or streptavidin (if the labeling probe contains a biotin moiety) (Wu et al., 2016; Yan, 2009).
The method revealed numerous proteins that underwent oxidative carbonylation in aging or under pathological conditions, leading to identification of a large number of structural proteins or cytoskeleton proteins that cannot be readily characterized by their functions (Yan and Forster, 2011). This is a bottleneck for 2D proteomics as many of the identified proteins cannot be functionally analyzed within a short time. In fact, many identified carbonylated proteins cannot be readily analyzed if no methods are available at the time of identification. This is particularly true if the identified proteins are cytoskeleton proteins instead of enzymes. Therefore, it is difficult to know if the identified carbonylated proteins have any functional abnormalities due to carbonylation.
To avoid this problem, we tweaked our approach by analyzing functional activity first, and then determining whether any detectable changes in activities could be due to oxidative carbonylation or modifications. At the time of this approach pivot, we wanted to focus on mitochondrial NAD+/NADH-specific oxidoreductases using blue native polyacrylamide gel electrophoresis (BN-PAGE). We modified the original BN-PAGE protocol (Schagger, 1995) by performing nongradient gel electrophoresis to resolve whole mitochondrial proteins instead of only mitochondrial membrane proteins (Yan et al., 2007).
As a result, we observed two gel bands containing NADH dehydrogenating activity when the gel strip was incubated with a buffer containing NADH and the artificial electron acceptor nitroblue tetrazolium (Fig. 2). The upper band is obviously that of mitochondrial complex I (NADH-ubiquinone oxidoreductase), and the lower band was then identified to be that of DLDH (Yan et al., 2007). Since then, the first author of this article (L-J.Y.) at the University of North Texas Health Science Center at Fort Worth has been focusing on studying these two mitochondrial enzymes (Yan and Forster, 2009; Yan et al., 2008).

It should be stressed that if only mitochondrial membrane proteins were analyzed by BN-PAGE, DLDH would have not been found, as DLDH itself is not a membrane protein (Yan et al., 2007). It is also possible that the sample preparation procedure in the original BN-PAGE protocol could lead to loss of DLDH due to dissociation from its parent complexes such as PDC and α-KGDC, both of which are known to associate with mitochondrial membranes (Carothers et al., 1989; Williams, 1992).
Structure and Function of DLDH
Mammalian DLDH monomer has ∼475 amino acid residues with very slight variation from rodents to humans. The molecular weight of the monomer is ∼51 kDa (Carothers et al., 1989; Jentoft et al., 1992). In rat, each DLDH monomer has 10 cysteine residues (Yan et al., 2013a), including cysteine 45 (Cys45) and cysteine 50 (Cys50) at the active center, which transfers electrons from the substrate dihydrolipoamide to NAD+ via flavin adenine dinucleotide (FAD) (Fig. 3A, B). As shown in Figure 3C, each monomer contains four domains: the FAD binding domain, the NAD+ binding domain, the central domain, and the interface domain (Brautigam et al., 2005; Jentoft et al., 1992).

The two Cys residues of the CXXXXC active site interact with the FAD cofactor, while the C-terminal interface domain with the conserved H452XXXXE457 also participates in the catalytic activity of the homodimeric enzyme. As shown in Figure 4, there is a 97.8% similarity and 94.1% identity between rat and human DLDH amino acid sequences (Fig. 4, the left panel).
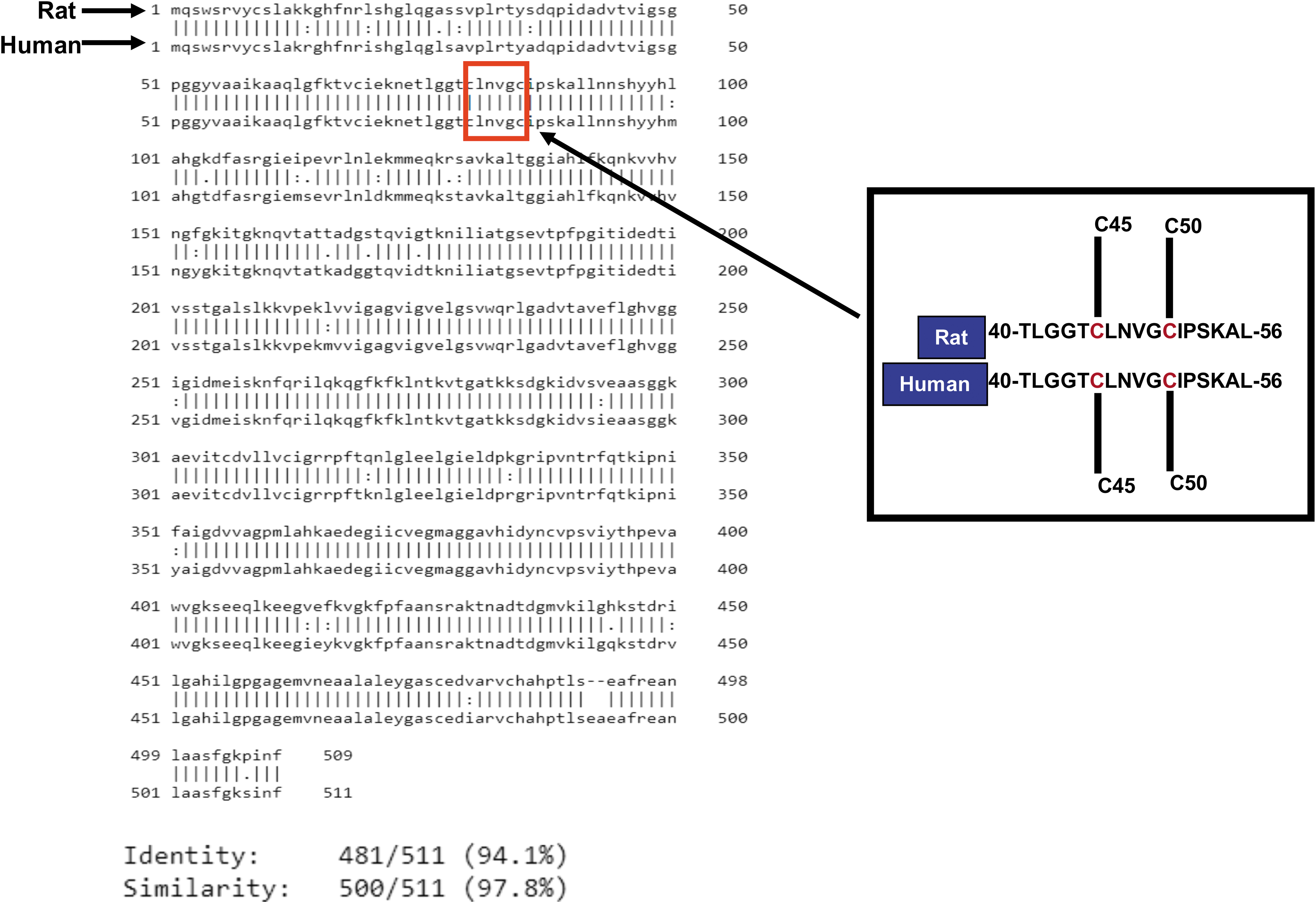
Moreover, the primary peptide sequences containing Cys45 and Cys50 from amino acid residues 40 to 56 in mature form of DLDH are identical (Fig. 4, right panel). These data indicate that DLDH is highly conserved in evolution. Figure 5 shows that electrons from the substrate dihydrolipoamide are transferred to reduce the Cys45–Cys50 disulfide. The electron from reduced Cys50 are transferred to the isoalloxazine ring of FAD and further to NAD+, generating NADH to complete the catalytic reaction and to restore the Cys45–Cys50 disulfide of the enzyme.

As can be seen in Figure 3B, the substrate and the NAD+ molecules are located on different domains of the enzyme. Hence, dihydrolipoamide and NAD+ cannot really “see” each other. Therefore, the enzyme operates like a mini-electron transport system (Fig. 5). Conceivably, disruption of this mini-electron transport system by any means can impair the enzymatic function, leading to accumulation of the upstream metabolic intermediates such as pyruvate or lactate (Broxton et al., 2022).
DLDH Deficiency
Only few cases of DLDH deficiency have been reported in humans (Ambrus, 2019; Hong et al., 1996; Liu et al., 1993; Odievre et al., 2005; Shany et al., 1999; Staretz-Chacham et al., 2021; Szabo et al., 2019). DLDH deficiency may be manifested by several disorders such as maple syrup urine disease (Ambrus, 2019; Chuang et al., 2006; Hengeveld and de Kok, 2002), the early onset of neurological disease, hepatic disease, and myopathic abnormalities (Quinonez and Thoene, 1993). If untreated, the early onset of neurological disease and hepatic disorders caused by DLDH deficiency can lead to death, and myopathic presentation can be reflected by muscle weakness and increased levels of creatinine and lactate (Quinonez and Thoene, 1993).
For the purpose of diagnosis, genetic analysis such as DLD gene sequencing instead of DLDH enzymatic activity measurements is often the primary approach to making sure DLDH deficiency is indeed the cause of the observed metabolic abnormalities (Quinonez and Thoene, 1993). Updated information in reference (Quinonez and Thoene, 1993) such as metabolic abnormalities in DLDH deficiency and levels of certain metabolites representing DLDH deficiency may help design additional animal models for investigating DLDH deficiency and metabolic diseases (Table 1).
Metabolic Abnormalities Associated with Dihydrolipoamide Dehydrogenase Deficiency
This table was reproduced from reference (Quinonez and Thoene, 1993).
DLDH and Ischemic Stroke
During our investigation of the role of DLDH in stroke and neuroprotection in an animal model of middle cerebral artery occlusion, we have found that upon 1 h ischemia and 1 h reperfusion, DLDH activity was nearly completely lost while DLDH protein content did not change when analyzed by BN-PAGE (Fig. 6A, B) (Wu et al., 2017). However, the enzyme's activity showed a quick recovery beyond 2 h's reperfusion after 1 h ischemia of the brain (Fig. 6A). At the end of 24 h reperfusion, the infarction area was much larger than that of control with a full recovery of DLDH activity (Wu et al., 2017).

Such studies indicate that there was a short period loss of DLDH activity after ischemic stroke. It was previously shown that DLDH is a source of reactive oxygen species (ROS) (Bunik and Brand, 2018; Goncalves et al., 2016; Kareyeva et al., 2012; Quinlan et al., 2014), and that slowing down ROS generation during reperfusion can mitigate tissue injury (Jaeschke, 2003; Sehirli et al., 2003). Our results further implied that ROS generation due to the quick recovery of DLDH activity upon reperfusion could be associated with cerebral ischemic injury, and that DLDH underwent reversible modifications during this ischemia–reperfusion process. Therefore, if DLDH's loss of function is prolonged to prevent its ROS production after ischemia reperfusion, the infarction size in the brain after ischemia–reperfusion might be decreased.
This reasoning led to our extensive studies of DLDH preconditioning and postconditioning for neuroprotection against ischemic stroke (Sumien et al., 2020; Wu et al., 2018; Wu et al., 2017). We took the advantage that 5-methoxyindole-2-carboxylic acid (MICA) is a reversible inhibitor of DLDH (Hanson et al., 1969; Haramaki et al., 1997). When MICA was administered to rats either via intraperitoneal injection or via diet, the brain showed mitigated infarction areas upon stroke (Wu et al., 2017). Our studies further elucidated the mechanisms of DLDH inactivation, which was caused by protein sulfenic acid that could be labeled by a sulfenic acid-reactive probe followed by Western blot assays using antibodies recognizing the probe (Wu et al., 2017).
This oxidative modification of cysteine residues by ROS or reactive nitrogen species (RNS) is a reversible process (Yan et al., 2013a; Yan et al., 2012), thus explaining the loss and recovery of DLDH activities observed during cerebral ischemia reperfusion. Nevertheless, the sulfenylated Cys residues are unknown, since the Cys residues in the active site motif could not be identified as sulfenylated using mass spectrometry. Nonetheless, prolonged sulfenation of the enzyme active center's cysteine upon MICA inhibition could be one of the mechanisms underlying neuroprotection of MICA-induced DLDH inhibition.
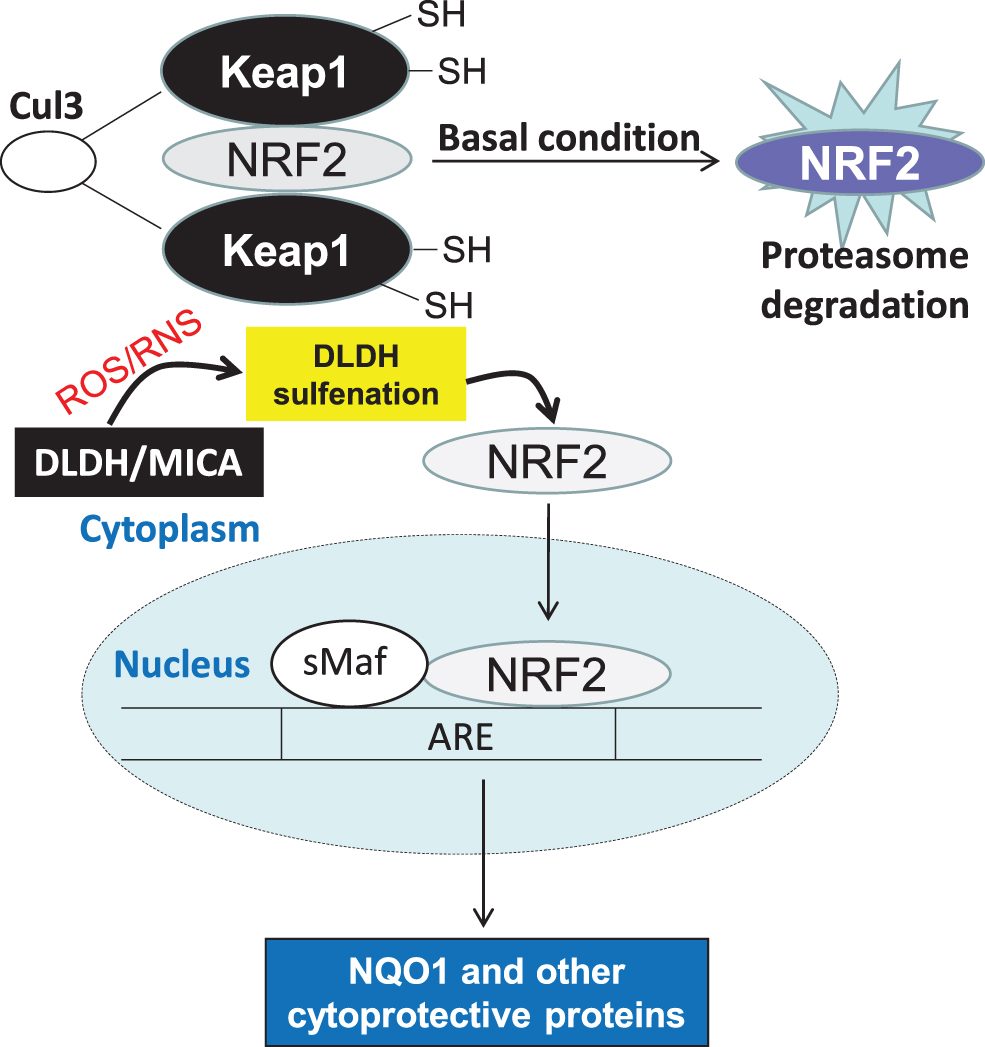
Moreover, we found that DLDH inhibition by MICA involves activation of the nuclear factor-erythroid factor 2-related factor 2 (Nrf2) signaling pathway, leading to upregulation of NAD(P)H quinone dehydrogenase 1 (NQO1) that protected the brain against oxidative damage (Wu et al., 2017). We later found that MICA administered after ischemic stroke is also neuroprotective, and the underlying mechanisms also involve activation of the Nrf2 signaling pathway (Wu et al., 2018). The potential mechanism by which DLDH sulfenation and MICA inhibition activate the Nrf2 signaling pathway is shown in Figure 7. It is likely that DLDH inhibition caused a stress response that triggers Nrf2 translocation to the nucleus.
DLDH and Alzheimer's Disease
It has been reported that DLDH is a risk factor for Alzheimer's disease (AD) in an Ashkenzai Jewish population, and this DLDH genotype of risk only appears to be affecting male population in this ethnic group (Brown et al., 2007; Brown et al., 2004). In Caenorhabditis elegans, suppression of DLDH activity has been shown to be neuroprotective against β amyloid toxicity (Ahmad and Ebert, 2021).
Suppression of DLDH activity in the same C. elegans AD model can also induce human tau protein phosphorylation via elevated whole-body glucose content (Ahmad, 2018). While the results of such studies demonstrate that lowering energy metabolism is neuroprotective in AD (Ahmad and Ebert, 2021), how exactly DLDH is involved in the pathogenesis of AD is not completely understood. In addition, a comprehensive evaluation of the role of DLDH in a variety of rodent models of AD has yet to be performed.
DLDH and Skin Photoaging
In studies focusing on tricarboxylic acid (TCA) cycle enzymes and skin photoaging, it was found that DLDH in α-keto glutarate dehydrogenase in the TCA cycle is the only enzyme whose expression was decreased in the skin upon ultraviolet (UV) irradiation (Moon et al., 2015; Sun et al., 2018). This decline in DLDH protein expression can be corrected by natural antioxidants such as paeonol extracted from Paeonia suffruticosa Andr. (Sun et al., 2018). The increased DLDH expression upon phytochemical stimulation further activates the Nrf2 signaling pathway that leads to increased antioxidant protein expression such as heme oxygenase-1 and NQO1 (Sun et al., 2018). Therefore, DLDH in the TCA cycle could be a major target for antiskin photoaging therapy.
In addition, as recently reported, the role of transketolase in light stress resistance may be mediated by DLDH (Chen et al., 2022) as DLDH expression was downregulated by transketolase knockdown. Furthermore, it has been found that downregulation of DLDH by UVA can inhibit melanoma progression that is mediated by oxidative stress and dysregulated enzyme metabolism (Yumnam et al., 2021).
Nondehydrogenating Functions of DLDH
Enzymatically active DLDH is a stable homodimer (Yan et al., 2007). When the homodimer is unstabilized or disrupted, however, each DLDH monomer can act as a serine protease (Babady et al., 2007; Jeffery, 2011). Such findings demonstrate that DLDH, when existing as a monomer, has a moonlighting function. In addition, DLDH has been shown to possess other nondehydrogenating functions, which include adherence to metal-oxide surfaces (Dayan et al., 2019b), TiO2 binding (Dayan et al., 2017), attenuation of radiation toxicity (Alzahrani and Ebert, 2019; Alzahrani and Ebert, 2018), and ROS production (Starkov, 2008; Starkov et al., 2004; Tahara et al., 2007). It has been suggested that DLDH's DNA binding and ROS production are involved in cellular oxidative damage and cell death (Dayan et al., 2020; Dayan et al., 2019a; Dayan et al., 2019b).
Furthermore, DLDH modified by a small peptide such as arginine-glycine-aspartic acid has been shown to have a variety of applications such as inhibition of tumor growth, enhancement of cancer cell apoptosis, and photodynamic treatment of melanoma cells, acting as a tool for coupling DLDH to cell surface integrins (Fleminger and Dayan, 2021). For detailed DLDH nondehydrogenating functional mechanisms, readers may refer to a recent excellent review article by Fleminger and Dayan (2021).
DLDH Oxidative Modifications
As a redox-sensitive enzyme (Yan et al., 2013a; Yan et al., 2012; Yan et al., 2008), DLDH is susceptible to oxidative modifications by other sources of ROS and RNS (Gutierrez-Correa, 2010; Gutierrez-Correa and Stoppani, 2002; Gutierrez-Correa and Stoppani, 1999; Gutierrez-Correa and Stoppani, 1995; Gutierrez Correa and Stoppani, 1993; Lee et al., 2009). The enzyme can be attacked by both endogenous reactive species generated within mitochondria (Yan et al., 2013a) and those nonmitochondrial sources (Yan et al., 2012). We have found that DLDH can undergo reversible modifications by mitochondrial hydrogen peroxide generated from complex III instead of complex I (Yan et al., 2013a).
In addition, in vitro studies indicate that DLDH is vulnerable to reversible modifications by Angeli's salt via nitroxyl hydride (HNO) modifications of cysteines' sulfur group (Yan et al., 2012). In addition to these reversible modifications, others have reported that DLDH can also undergo irreversible modifications such as carbonylation (Frohnert and Bernlohr, 2013; Hussain et al., 2006) and nitrosylation (Foster and Stamler, 2004).
It should be pointed out that the relationship between oxidation of specific amino acid residues and given pathological conditions remains to be comprehensively investigated. It should also be noted that while most post-translational modifications of DLDH are conceivably deleterious, modifications that are beneficial under particular pathological conditions have also been reported in our investigation of DLDH and stroke neuroprotection, suggesting that DLDH sulfenation was neuroprotective (Wu et al., 2017).
Nonmitochondrial or Extracellular DLDH in Mammalian Cells
There have been scarce reports regarding the existence of nonmitochondrial DLDH in mammalian tissues. We have found incidentally, via mass spectrometry peptide sequencing of a serum protein band separated by BN-PAGE, that DLDH exists in rat serum (Yan et al., 2013b). This isoform of DLDH (if we can name so) is different from mitochondrial DLDH, in that this serum DLDH is vulnerable to experimental procedures, including ammonium sulfate extraction, gel electrophoresis, and column chromatography, as the enzyme activity is lost after these experimental procedures. Enzymatic activity assays also indicate the existence of DLDH in mouse and human serum (Yan et al., 2013b).
Moreover, serum DLDH does not seem to possess diaphorase activities (i.e., NADH oxidation). Our study indicates that unlike mitochondrial DLDH that is highly stable, serum DLDH is labile. In spite of these findings, the function of this serum protein remains presently unknown. In addition, although our results agree with those reported in the 1970s and 1980s that there existed a serum DLDH (Melancon et al., 1980; Pelley and Bradley, 1989; Pelley et al., 1976), that whether a different gene other than the one encoding mitochondrial DLDH encodes serum DLDH also remains to be determined.
Nevertheless, it is known that the DLDH encoding gene can yield three alternatively spliced isoforms of DLDH (
Genetic Manipulation of DLDH for Studies of DLDH Function in Health and Disease
As a critical enzyme involved in several metabolic pathways, knockout of DLDH encoding gene in rodents would be conceivably lethal. In mouse, a global knockout of DLDH indeed yields embryonic lethality, and no viable pups could be obtained (Johnson et al., 1997). Nonetheless, a heterogeneous mouse model of DLDH (Dldh+/− ) with only 50% DLDH expressed when compared with wildtype animals is viable and normal with no apparent disease phenotypes (Johnson et al., 1997). The same DLDH deficient mouse model, however, shows vulnerability to neurotoxic challenges (Klivenyi et al., 2004).
We have found that this DLDH deficient mouse model also exhibited increased cerebral damage upon stroke challenge (Li et al., 2015). What surprised us is that a global DLDH overexpression in a transgenic mouse model is also deleterious as the transgenic mouse brain also shows increased damage after stroke (Li et al., 2015). These results indicate that DLDH overexpression in animals is not beneficial to ischemic challenge, at least in the brain.
One explanation for this is that the overexpressed DLDH cannot be incorporated into the respective DLDH-containing enzyme complexes as other subunits, such as E1 and E2 in the respective enzymes' complexes, are not proportionally overexpressed, leading to free or noncomplexed DLDH intracellularly. This noncomplexed DLDH could be toxic to cells.
Our own studies also demonstrate that either knockout or knock-in of DLDH is harmful to the brain. Therefore, future studies may need to take pathway-specific or cell-specific approaches to understand the role of DLDH under specific pathological conditions. Design of cell-specific or tissue-specific transgenic DLDH animal models may also be used to investigate the role of DLDH under physiological conditions or in any given pathological conditions. For example, such genetic approaches may be used to test whether DLDH itself, when not in its respective enzyme complexes, can serve as a suitable therapeutic target or an antioxidant as has been suggested previously (Ahmad and Ebert, 2021; Chang et al., 2022; Dayan et al., 2019a; Igamberdiev et al., 2004; Kliukiene et al., 1997; Yang et al., 2019).
DLDH Autoantibodies in the Serum of Cancer Patients and Other Diseases
It has been reported that DLDH abnormalities occur in certain cancer cells (Qi and Zhu, 2023), and that DLDH in certain diseases and cancers can induce immune response as anti-DLDH antibodies can be detected in the serum of the patients. For example, primary biliary cirrhosis has been associated with the production of DLDH autoantibodies (Dubel et al., 1999; Yeaman et al., 2000), and DLDH is a major autoantigen in hepatitis C virus infection (Wu et al., 2002).
Evidence has also been presented that in both the sera of ovarian cancer patients and endometrial cancer patients, anti-DLDH antibodies were detected (Yoneyama et al., 2015; Yoneyama et al., 2014). These DLDH antibodies may be a biomarker for such cancers or at least cancer-driven disruption of DLDH involved enzyme complexes. These studies also indicate that free DLDH, not DLDH in each respective enzyme complex, is immunogenic. Nonetheless, how DLDH induces immune responses resulting in the formation of anti-DLDH antibodies remains unclear. Further studies will need to be conducted to screen anti-DLDH antibodies in different patients with different types of cancers to see if this is a universal phenomenon for all types of cancers.
Summary
In this article, after review of DLDH structure and function, we overviewed the role of DLDH in several pathological conditions, including DLDH deficiency and metabolic manifestations in humans, DLDH in ischemic stroke and neuroprotection, and role of DLDH in photo light-induced skin abnormalities. The roles and features of DLDH as a nondehydrogenating protein were also discussed; as well as DLDH autoantibodies in certain diseases and cancers. It should be pointed out that depending on modes of functional manipulation, duration and magnitude of functional modulation, and the physiological or pathological conditions, DLDH may be manipulated for either beneficial (prosurvival) or detrimental (prodeath) purposes via modulation of redox balance and oxidative stress (Fig. 8).
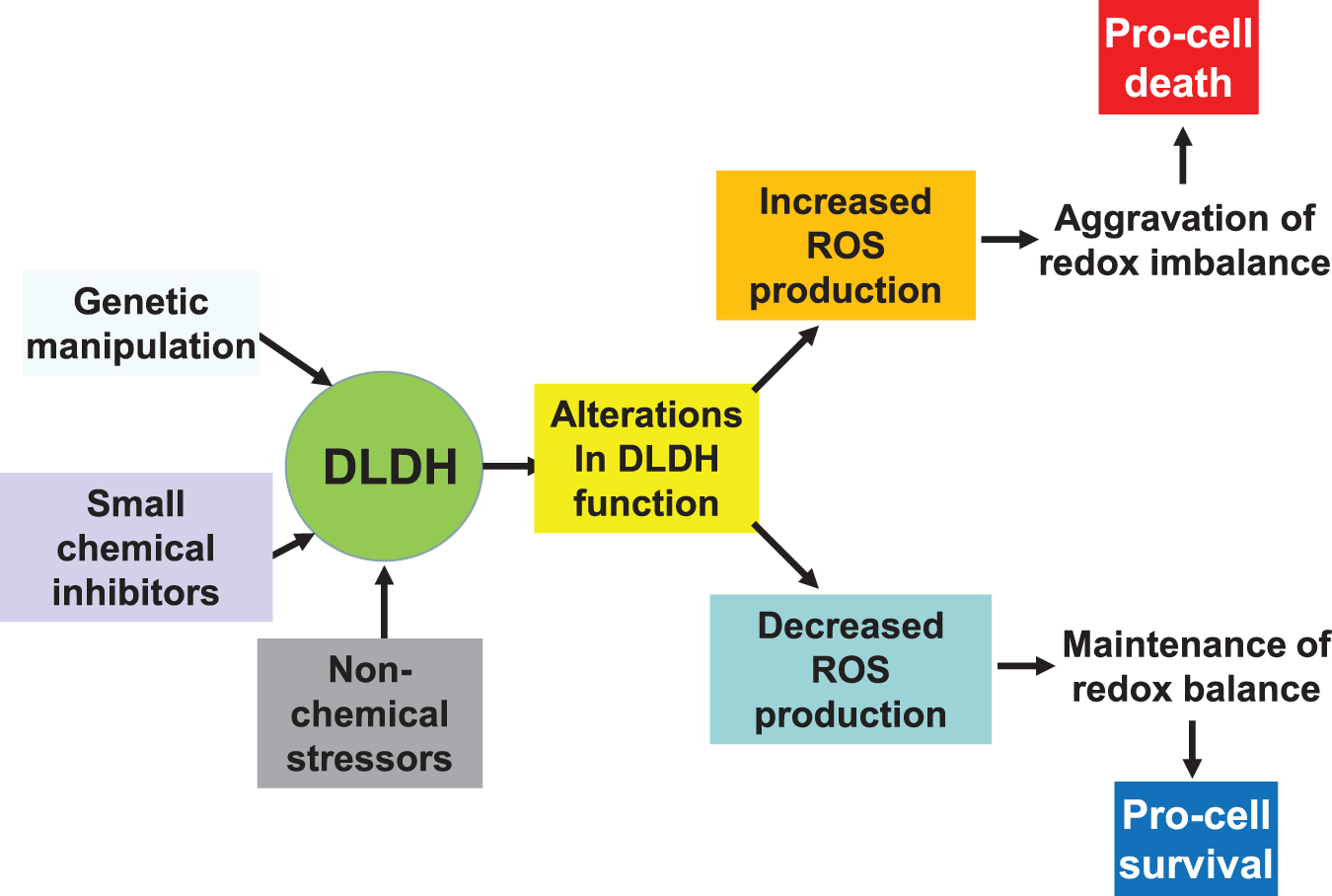
To further explore DLDH as a therapeutic target in a given pathological condition, novel animal models would need to be constructed, which appears to be challenging, given that DLDH is a housekeeping oxidoreductase. In this regard, tissue-specific or cell-specific knockdown of DLDH using small RNA interference techniques (Ahmad, 2018; Broxton et al., 2022) should be a promising approach. Inhibition of DLDH function by DLDH inhibitors such as MICA (Bauman and Pease, 1969; Bauman and Hill, 1968) and valproic acid (Luis et al., 2007) can also offer plausible approaches. Overall, while DLDH structure and physiological function have been well characterized, its roles in numerous pathological conditions such as metabolic disorders, including hypertension, diabetes, and dyslipidemia, are yet to be comprehensively investigated.
Footnotes
Acknowledgments
L-J.Y. would like to thank his postdocs, students, and collaborators who were involved in studies discussed in this article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
L-J.Y. was supported in part by the National Institute of Neurological Disorders and Stroke (R01NS079792) and UNTHSC (seed grants: RI6044, RI10015, and RI10039).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.