
Abstract
Aims:
Studies demonstrated that oxidized fish oil (OFO) promoted oxidative stress and induced mitochondrial dysfunction and lipotoxicity, which attenuated beneficial effects of fish oil supplements in the treatment of nonalcoholic fatty liver disease (NAFLD). The current study was performed on yellow catfish, a good model to study NAFLD, and its hepatocytes to explore whether selenium (Se) could alleviate OFO-induced lipotoxicity via the inhibition of oxidative stress and determine its potential mechanism.
Results:
The analysis of triglycerides content, oxidative stress parameters, and histological and transmission electronic microscopy observation showed that high dietary Se supplementation alleviated OFO-induced lipotoxicity, oxidative stress, and mitochondrial injury and dysfunction. RNA-sequencing and immunoblotting analysis indicated that high dietary Se reduced OFO-induced decline of peroxisome-proliferator-activated receptor alpha (Pparα) and ubiquitin-specific protease 4 (Usp4) protein expression. High Se supplementation also alleviated OFO-induced reduction of thioredoxin reductase 2 (txnrd2) messenger RNA (mRNA) expression level and activity. The txnrd2 knockdown experiments revealed that txnrd2 mediated Se- and oxidized eicosapentaenoic acid (oxEPA)-induced changes of mitochondrial reactive oxygen species (mtROS) and further altered Usp4 mediated-deubiquitination and stabilization of Pparα, which, in turn, modulated mitochondrial fatty acid β-oxidation and metabolism. Mechanistically, Usp4 deubiquitinated Pparα and ubiquitin-proteasome-mediated Pparα degradation contributed to oxidative stress-induced mitochondrial dysfunction.
Innovation:
These findings uncovered a previously unknown mechanism by which Se and OFO interacted to affect lipid metabolism via the Txnrd2-mtROS-Usp4-Pparα pathway, which provides the new target for NAFLD prevention and treatment.
Conclusion:
Se ameliorated OFO-induced lipotoxicity via the inhibition of mitochondrial oxidative stress, remodeling of Usp4-mediated deubiquitination, and stabilization of Pparα. Antioxid. Redox Signal. 40, 433–452.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is becoming the dominant chronic liver disease worldwide (Samuel and Shulman, 2018). Increasing evidence suggested that lipotoxicity was an important initiating event in NAFLD (Mundi et al., 2020; Samuel and Shulman, 2018; Zhang et al., 2021a). As a consequence, mitigation of excess lipotoxicity is considered the most effective approach to prevent the progression of NAFLD.
Innovation
We proposed a mechanistic model underlying selenium (Se), alleviating oxidized fish oil (OFO)-induced lipotoxicity: Se attenuated OFO-induced lipotoxicity and mitochondrial oxidative stress; Se-induced upregulation of thioredoxin reductase 2 (txnrd2) expression alleviated OFO-triggered oxidative stress; and Se reversed oxidative stress-induced decrease of peroxisome-proliferator-activated receptor alpha (Pparα) deubiquitination mediated by ubiquitin-specific protease 4 (Usp4), suppressed Pparα degradation, and restored fatty acid β-oxidation, thereby reducing OFO-induced lipotoxicity (Fig. 1). Our findings suggest that appropriate Se supplementation contributes toward counteracting the adverse effects of fish oil oxidation. It broadens the current knowledge into the role of Pparα ubiquitination in mitochondrial oxidative stress-triggered lipid metabolic disorder and provides the potential new target for the prevention and treatment of nonalcoholic fatty liver disease.

Studies have demonstrated that fish oils, rich in eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), can alleviate mitochondrial dysfunction and stimulate fatty acid β-oxidation, and accordingly exert protective effects on hepatic steatosis (Song et al., 2008). Therefore, the guideline from the American Association for the Study of Liver Disease (AASLD) recommended the omega-3 polyunsaturated fatty acids (n-3 PUFAs) in the treatment of NAFLD patients.
Fish oils are used as supplements widely for adults in the United States, the United Kingdom, and other developed countries (Chalasani et al., 2018; Siscovick et al., 2017). However, fish oils are easily oxidized under ambient conditions because they contain multiple polyunsaturated fatty acids (PUFAs), which produce lipid peroxides (Azizi et al., 2019). Importantly, studies have reported that most commercial fish oils contain lipid peroxides whose amounts often exceed the recommended values (Jackowski et al., 2015).
In addition, our recent work revealed that oxidized fish oil (OFO) intake caused severe lipotoxicity by oxidative stress-induced mitochondrial dysfunction (Zhang et al., 2021a). Thus, it is of utmost importance to seek alternative strategies for counteracting the adverse effects of fish oil oxidation.
Our previous study demonstrated that OFO triggered oxidative stress, which led to mitochondrial dysfunction and reduced fatty acid β-oxidation, and ultimately aggravated lipid deposition (Zhang et al., 2021a). Nonetheless, the exact molecular mechanisms during these processes remain elusive. Peroxisome-proliferator-activated receptor alpha (Pparα), an important transcription factor, exerts critical roles in modulating fatty acid β-oxidation.
Hence, in the present study, one of the aims was to investigate whether Pparα mediated OFO-induced mitochondrial dysfunction and decreased fatty acid β oxidation. Studies point out that Pparα expression and activity are controlled at levels of gene, protein expression, and post-translational modifications (Bougarne et al., 2018). Among these, the regulatory mechanisms of Pparα in gene and protein expression levels are well explored (Bougarne et al., 2018).
However, studies are very scarce on the potential effects of protein post-translational modifications (such as ubiquitination) on Pparα activity. Especially, to the best of our knowledge, the Pparα ubiquitination modification mechanism and its biological significance is still unclear. Notably, oxidative stress is closely associated with protein ubiquitination and protein ubiquitination exhibits an important regulatory role in lipid metabolism (Chen et al., 2021; Sarraf et al., 2013). Collectively, we hypothesize whether the ubiquitination of Pparα is involved in the changes of lipid metabolism induced by selenium (Se) and OFO.
Se is an essential element for vertebrates. Its biological effects are primarily mediated by selenoproteins (Santesmasses et al., 2020; Zhang et al., 2022). Among the selenoproteins, thioredoxin reductases are crucial for the control of redox balance (Sibbing et al., 2011), and the mitochondrial thioredoxin reductase 2 (Txnrd2) is of paramount importance for scavenging reactive oxygen species (ROS) within the mitochondria (Fink et al., 2016; Sibbing et al., 2011).
Thus, considering the key roles of Txnrd2 in antioxidative responses, we hypothesize that Txnrd2 might mediate the Se- and OFO-induced changes of oxidative stress.
Fish exceed 30,000 species and are considered the most diversified category among vertebrates. They also possess conservative metabolic pathways to mammals (Hotamisligil, 2006). In fish, more than 25 selenoproteins were reported (Mariotti et al., 2012). Recently, we identified 28 selenoproteins in yellow catfish Pelteobagrus fulvidraco, a widely distributed teleost fish in some Asian countries, based on its whole genome information and gene cloning (Gong et al., 2018; Xu et al., 2020).
Thus, yellow catfish is an ideal model for investigating the function of selenoproteins. Moreover, yellow catfish has been considered as an appropriate model for studying lipid deposition and development of NAFLD (Lv et al., 2022; Song et al., 2022; Zhao et al., 2020). Hence, using the yellow catfish model, we investigated whether high Se (H-Se) supplementation could attenuate OFO-induced hepatic lipid deposition, oxidative stress, and mitochondrial dysfunction, and the relevant mechanisms were further elucidated.
Results
OFO increased contents of saturated fatty acids and oxylipins but reduced contents of unsaturated fatty acids
Compared with fresh fish oil (FFO), OFO significantly decreased contents of unsaturated fatty acids (UFA; 15.1%–60.0%), including α-linolenic acid (ALA), linoleic acid (LA), EPA, and DHA, but significantly increased saturated fatty acids (SFA) contents (11.7%–15.9%), such as palmitic acid, myristic acid, and stearic acid (Supplementary Fig. S1A and Supplementary Table S1).
Compared with FFO, OFO had higher contents of oxylipins derived from EPA (2.92- to 50.9-fold), DHA (3.76- to 41.4-fold), arachidonic acid (AA; 13.9- to 49.1-fold), LA (3.95- to 35.7-fold), and ALA (6.44- to 37.4-fold), respectively, such as 5-hydroxyeicosapentaenoic acid (HEPE), 15-HEPE, 12-HEPE, 18-HEPE, prostaglandins (PG) D3, PGE3 (from EPA) (Supplementary Fig. S1B and Supplementary Table S2), 4-hydroxydocosahexaenoic acid (HDHA), 14-HDHA, 7-HDHA and 17-HDHA (from DHA), 18-hydroxyeicosatetraenoic acid (HETE), 5-HETE, 15-HETE, 11-HETE, 12-HETE and lipoxin A4 (LXA4; from AA), 9(s)-hydroxy-octadecadienoic acid (HODE) and 13-HODE (from LA), 9(s)-hydroxy-octadecatrienoic acid (HOTrE), and 13(s)-HOTrE (from ALA) (Supplementary Table S2). The oxidation of EPA increased the contents of oxylipins derived from EPA (Supplementary Table S2).
Dietary Se supplementation attenuated OFO-induced lipotoxicity and oxidative stress in the liver of yellow catfish
Next, we investigated the effects of dietary Se and OFO on Se retention, lipid metabolism, and oxidative stress in the liver of yellow catfish. Dietary Se and OFO did not interact to influence hepatic Se content (Supplementary Fig. S2), but they affected hepatic lipid content (Fig. 2). Compared with the control (adequate Se [A-Se]+FFO group), A-Se+OFO diet significantly increased hepatic lipid content (25.7%), based on hematoxylin and eosin (H&E) and oil red O (ORO) staining and the analysis of triglycerides (TGs) content (Fig. 2).
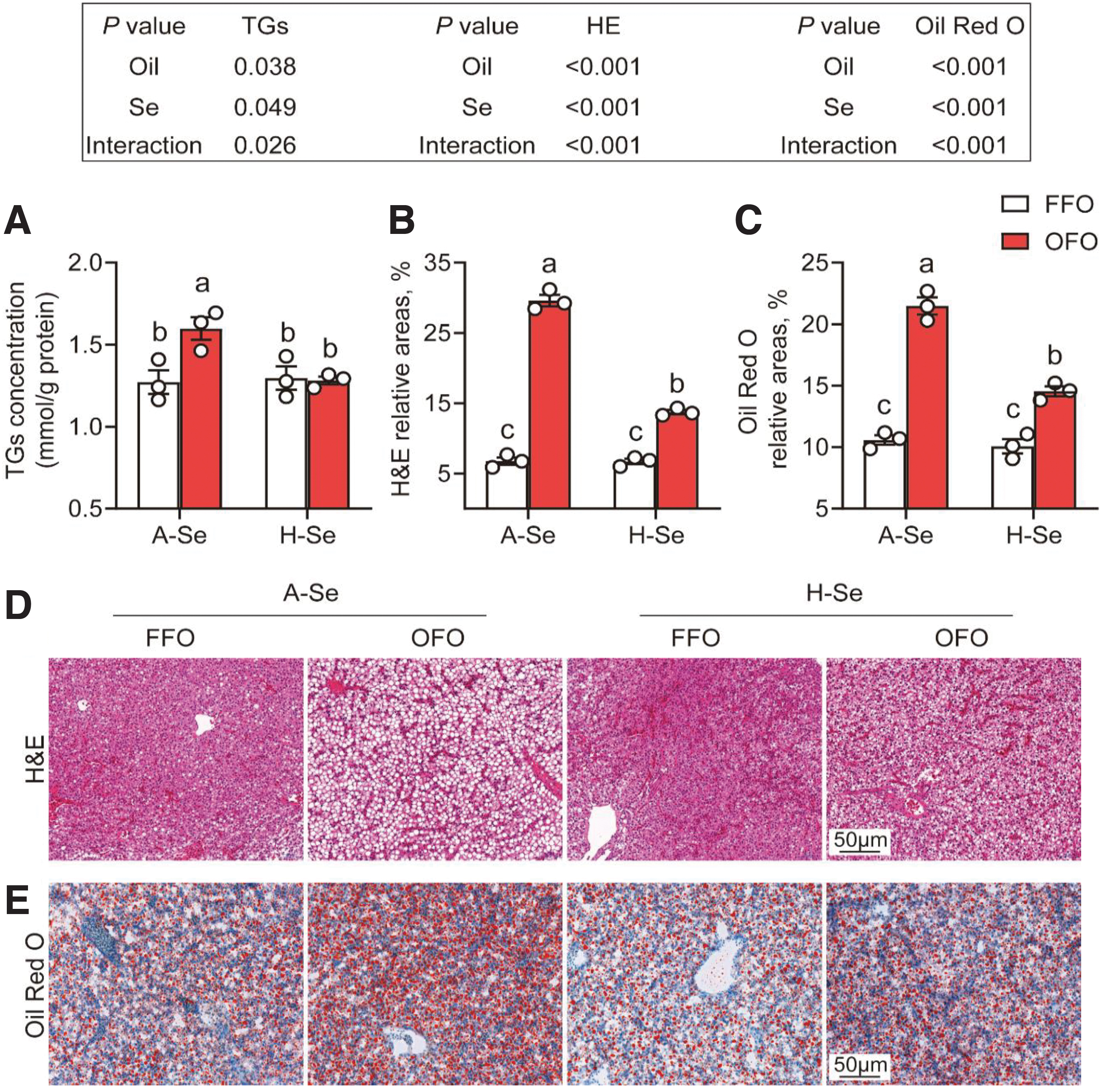
Compared with the A-Se+OFO group, H-Se+OFO diet significantly decreased hepatic lipid content (19.8%) (Fig. 2). Dietary Se and OFO did not interact to influence plasma glucose content, but they affected the contents of plasma cholesterol, TGs, and non-esterified fatty acids (NEFAs) (Supplementary Table S3). Compared with the control (A-Se+FFO group), A-Se+OFO diet increased plasma cholesterol (36.0%), TGs (77.0%), and NEFAs (92.3%).
Compared with the A-Se+OFO group, H-Se+OFO diet significantly decreased plasma cholesterol (12.8%), TGs (24.1%), and NEFAs (29.8%). Dietary Se and OFO also interacted to alter plasma alanine transaminase (ALT) and aspartate aminotransferase (AST) activities (Supplementary Table S3). Compared with the A-Se+FFO group, A-Se+OFO diet significantly increased plasma ALT (67.2%) and AST activities (45.7%).
Compared with the A-Se+OFO group, H-Se+OFO diet significantly decreased plasma ALT (30.1%) and AST activities (23.0%). Collectively, these findings suggested that dietary Se supplementation did alleviate OFO-induced lipid accumulation in the liver of yellow catfish.
Since studies suggested that OFO induced oxidative stress but A-Se addition reduced oxidative stress (Zhang et al., 2021a; Zhang et al., 2021b), we next determined the effects of Se addition on OFO-induced oxidative stress. As expected, dietary Se and OFO interacted to influence oxidative stress. Compared with the control (A-Se+FFO group), the A-Se+OFO diet significantly reduced the activities of catalase (Cat), glutathione peroxidase (Gpx), Txnrd2 (11.0%–50.1%) and total antioxidant capacity (T-AOC; 56.3%), and glutathione (GSH)/oxidized glutathione (GSSG) ratio (12.0%), and increased malondialdehyde (MDA; 53.1%), 4-hydroxy-2-nonenal (4-HNE; 22.2%), and protein carbonyl (41.7%) contents, but did not influence the total superoxide dismutase (T-Sod) activity (Supplementary Figs. S3A, S3B, S4A, S4B).
Compared with the A-Se+OFO group, the H-Se+OFO diet significantly increased the Cat, Gpx, Txnrd2 (12.6%–45.9%) and T-AOC (63.5%), and GSH/GSSG ratio (15.9%), and decreased MDA (22.2%), 4-HNE (14.1%), and protein carbonyl (17.8%) contents, and did not affect T-Sod activity (Supplementary Figs. S3A, S3B, S4A, S4B). Together, our data suggested that dietary Se supplementation attenuated OFO-induced lipotoxicity and oxidative stress.
Dietary Se supplementation attenuated OFO-induced mitochondrial damage and dysfunction in the liver of yellow catfish
Next, we explored the influences of Se and OFO on mitochondrial structure and function. Transmission electronic microscopy (TEM) observation indicated that, compared with the A-Se+FFO group, the mitochondria in the A-Se+OFO group were swollen and vacuolated, indicating mitochondrial damage (Fig. 3A). The mitochondrial numbers were significantly lower in the A-Se+OFO group than those in the A-Se+FFO group (38.7%).
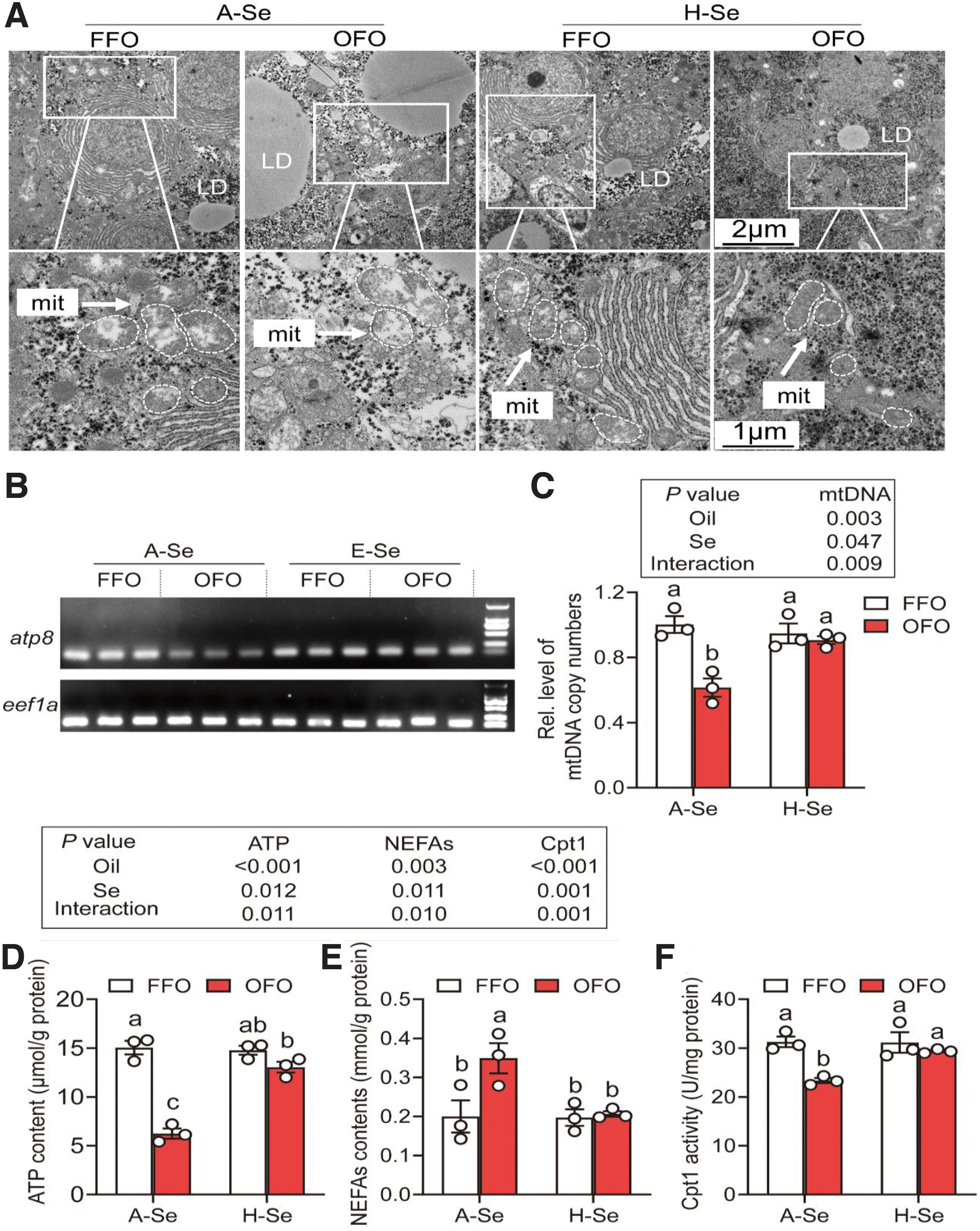
However, compared with the A-Se+OFO group, the mitochondrial damage and the decrease in mitochondria numbers were obviously attenuated in the H-Se+OFO group (47.5%). OFO significantly reduced the mitochondrial numbers, and Se supplementation reversed the OFO-induced decrease in mitochondrial numbers (Fig. 3B, C and Supplementary Fig. S5).
Further, we detected the effects of Se and OFO on mitochondrial function-related indices, such as messenger RNA (mRNA) expression levels of complexes subunits, adenosine-triphosphate (ATP) and NEFAs content, and carnitine palmitoyltransferases 1 (Cpt1) activity. Compared with the A-Se+FFO group, A-Se+OFO reduced the mRNA expression levels of complexes I/III/IV/V subunits (31.3%–51.7%) (Supplementary Fig. S6). Dietary Se supplementation attenuated the OFO-induced decrease of mRNA expression levels of complexes I/III/IV/V subunits (29.0%–67.7%) (Supplementary Fig. S6).
Compared with the A-Se+FFO group, A-Se+OFO reduced the ATP content (58.5%) and Cpt1 activity (25.5%), and it increased the NEFAs content (74.6%) (Fig. 3D–F), indicating that OFO led to mitochondrial dysfunction. Meanwhile, Se supplementation attenuated the OFO-induced mitochondrial dysfunction (Fig. 3D–F). Together, our results demonstrated that dietary Se supplementation attenuated OFO-induced mitochondrial damage and dysfunction.
Dietary Se supplementation attenuated OFO-induced suppression of Pparα signaling pathway in the liver of yellow catfish
We next investigated the mechanisms underlying Se- and OFO-induced variations of lipid metabolism. At first, we performed RNA sequencing (RNA-seq) to screen the differentially expressed genes (DEGs) and pathways, and the sequencing data were presented in Supplementary Table S4. The principal-component analysis showed that the samples clustered according to Se level in one dimension and OFO level in another dimension (Supplementary Fig. S7A), with all the triplicates clustering together.
Compared with the A-Se+FFO group, A-Se+OFO diet upregulated 311 genes and downregulated 421 genes (Supplementary Fig. S7B). Compared with the H-Se+FFO group, 329 genes were upregulated and 163 genes were downregulated in the H-Se+OFO group (Supplementary Fig. S7C). The 10 DEGs (5 up- and 5 downregulated DEGs) exhibited similar trends between the RNA-seq and the quantitative polymerase chain reaction (q-PCR) analysis (Supplementary Fig. S8), confirming the validity of the RNA-seq results.
Compared with the A-Se+FFO group, the A-Se+OFO group suppressed the PPAR signal and the ubiquitin mediated proteolysis pathway, and dietary Se supplementation rescued the inhibitory effects by A-Se+OFO (Supplementary Fig. S9).
Since the PPAR pathway is closely linked to lipid metabolism (Bougarne et al., 2018), we hypothesize that Se and OFO influence lipid metabolism through the PPAR pathway. We next sought to further determine the effects of Se and OFO on PPAR signals and lipid metabolism. Compared with the A-Se+FFO group, A-Se+OFO diet inhibited the PPAR pathway and decreased mRNA expression of the fatty acid β-oxidation-related genes (55.4%–77.6%), including acyl-CoA synthetase long chain family member 1 (acsl1), cpt1, cpt2, acyl coenzyme A dehydrogenase, long chain, and medium chain (acadl and acadm) (Fig. 4A, B).
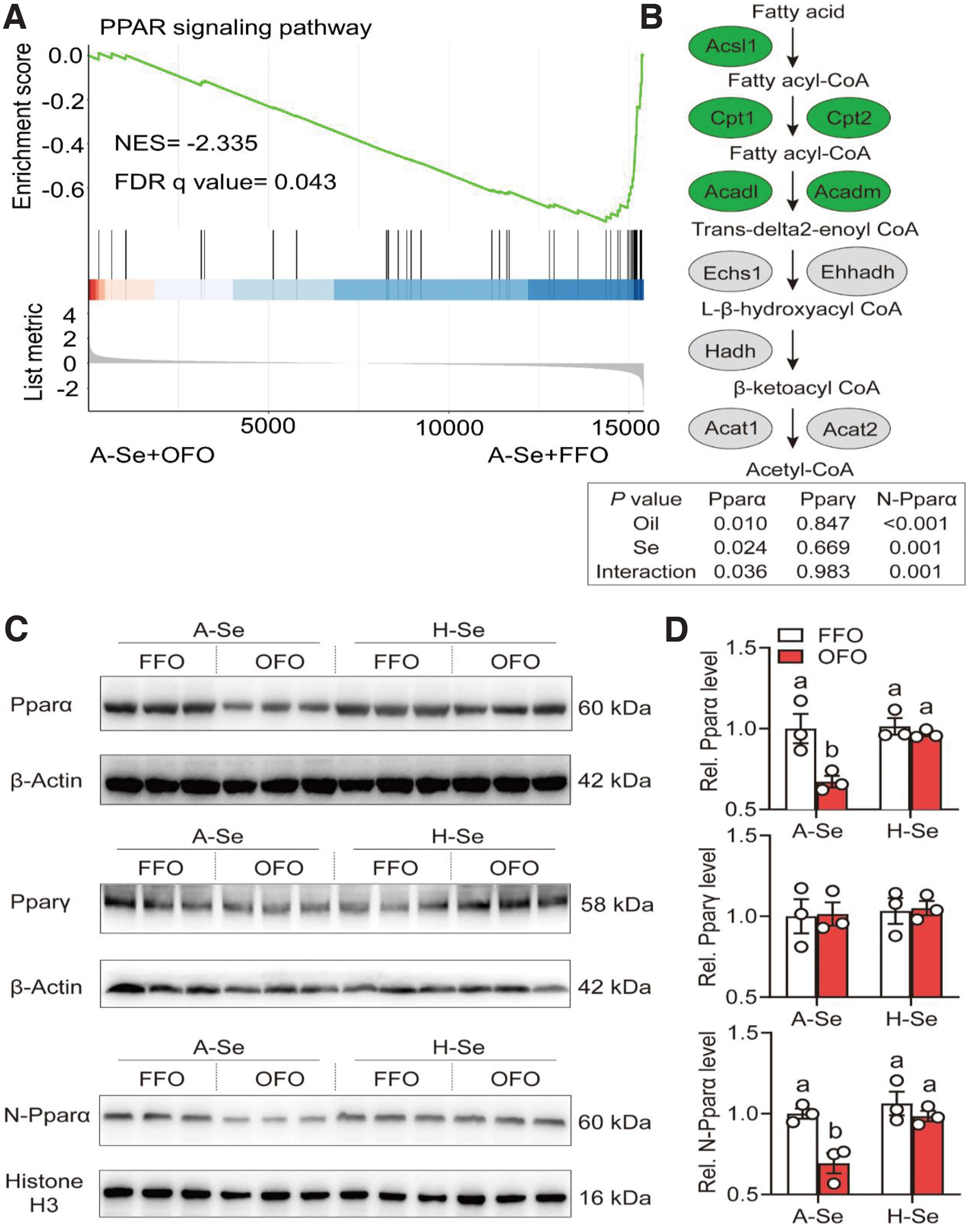
Se supplementation reversed the OFO-induced decline of PPAR pathway and mRNA expression of fatty acid β oxidation-related genes. Unexpectedly, the pparα and pparγ transcript levels exhibited no significant difference among the four groups. However, immunoblotting indicated that, compared with the A-Se+FFO group, A-Se+OFO diet reduced Pparα total protein expression (32.8%) and its nuclear location (30.6%), but it did not influence peroxisome-proliferator-activated receptor γ (Pparγ) protein expression (Fig. 4C, D).
Se supplementation reversed the OFO-induced reduction of Pparα total protein expression (43.7%) and its nuclear location (41.9%), and it did not influence Pparγ protein expression (Fig. 4C, D). Studies suggest that Pparα and Pparγ control lipid metabolism by forming Pparα/Pparγ-Rxrα hetero-dimers (Bougarne et al., 2018; Evans et al., 2004). Thus, we investigated whether dietary Se and OFO influenced Pparα/Pparγ-Rxrα complex. Compared with the A-Se+FFO group, A-Se+OFO diet reduced the formation of Pparα-Rxrα complex (47.3%) (Supplementary Fig. S10).
Compared with the A-Se+OFO group, H-Se+OFO diet attenuated the OFO-induced decrease of Pparα-Rxrα complex (46.2%) (Supplementary Fig. S10). Dietary Se and OFO did not interact to influence the formation of Pparγ-Rxrα complex (Supplementary Fig. S10). Collectively, our study indicated that Se attenuated OFO-induced suppression of the Pparα signaling pathway (via the inhibition of Pparα total protein expression and its nuclear location and the interaction between Pparα and Rxrα) and fatty acid β oxidation.
Dietary Se supplementation attenuated OFO-induced increase of protein K48-linked polyubiquitination and decrease of ubiquitin-specific protease 4 protein expression in the liver of yellow catfish
Since oxidative stress is closely related to ubiquitination and ubiquitination regulates lipid metabolism (Chen et al., 2021; Sarraf et al., 2013), we monitored the effects of dietary Se supplementation and OFO on the ubiquitination process. Compared with the A-Se+FFO group, A-Se+OFO diet inhibited the protein K48-linked deubiquitination pathway (Fig. 5A). Further analysis found that, compared with the A-Se+FFO group, A-Se+OFO diet reduced the mRNA expression of USP family members (15.3%–61.8%) (Fig. 5B).
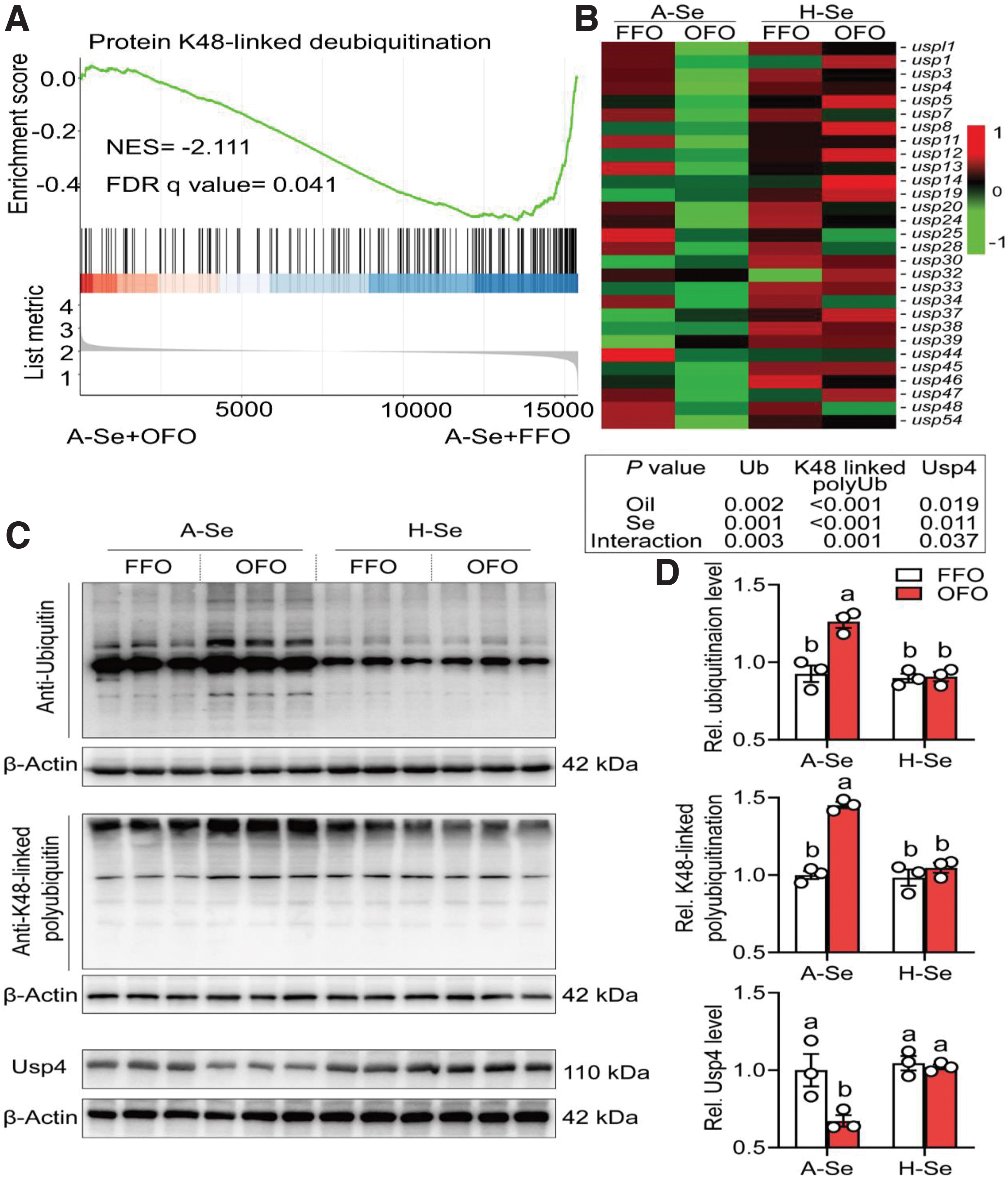
Dietary Se supplementation attenuated OFO-induced decrease of the protein K48-linked deubiquitination pathway and mRNA expression levels of USP family members. Moreover, compared with the A-Se+FFO group, A-Se+OFO diet significantly increased the ubiquitination (36.4%) and K48-linked polyubiquitination levels of total protein (45.1%) (Fig. 5C, D). Dietary Se supplementation attenuated OFO-induced increase of ubiquitination (28.2%) and K48-linked polyubiquitination (27.9%) levels of total protein (Fig. 5C, D). From these results, we speculated that dietary Se supplementation attenuated the OFO-induced increase of protein K48-linked polyubiquitination by increasing the related deubiquitination.
Ubiquitin-mediated proteolysis, regulated by E3 ubiquitin ligases and deubiquitinases, displays a crucial role in lipid metabolism (Chen et al., 2021; Sarraf et al., 2013). Therefore, we want to know which deubiquitinases mediate Se and OFO-induced variations of lipid metabolism. We found that Se and OFO interacted to influence the mRNA and protein expression of ubiquitin-specific protease 4 (Usp4), an important endogenous negative regulator of NAFLD (Zhao et al., 2018a). Compared with the A-Se+FFO group, A-Se+OFO diet decreased the mRNA (61.8%) and protein (32.8%) expression of Usp4.
Dietary Se supplementation attenuated OFO-induced decrease of mRNA (1.20-fold) and protein (51.8%) expression of Usp4. Zhao et al. (2018a) found that Usp4 expression was obviously downregulated in the livers from the NAFLD patients. Thus, we speculated that Usp4 mediated the Se- and OFO-induced variations of lipid metabolism. Strikingly, UbiBrowser 2.0 (
Dietary Se supplementation attenuated OFO-induced decrease of txnrd2 expression in the liver of yellow catfish
Next, our focus was to elucidate whether the effects of Se and OFO on lipid metabolism were regulated by specific selenoproteins. We used the RNA-seq to identify differentially expressed selenoproteins, and we found that Se and OFO did not interact to alter overall selenoprotein expression except for txnrd2 (Supplementary Fig. S12 and Supplementary Table S5). Compared with the A-Se+FFO group, A-Se+OFO diet decreased txnrd2 expression (58.0%) (Supplementary Fig. S12). Dietary Se supplementation attenuated OFO-induced decrease of txnrd2 expression (1.04-fold) (Supplementary Fig. S12).
Txnrd2, a mitochondrion-resident selenoprotein, exerts a critical role in mitochondrial redox homeostasis (Hellfritsch et al., 2015; Sibbing et al., 2011). Combined with the analysis of Txnrd2 enzyme activity mentioned earlier, we further proposed that txnrd2 might mediate Se and OFO-induced changes of oxidative stress.
Se attenuated oxidized eicosapentaenoic acid-induced lipotoxicity in primary hepatocytes of yellow catfish
To demonstrate the molecular mechanisms of Se and OFO-induced changes of lipid metabolism, we performed in vitro experiments using the yellow catfish hepatocytes. First, we determined an adequate concentration for EPA, oxidized eicosapentaenoic acid (oxEPA), and Se incubation. The MTT assay showed that 0–200 μM of EPA and oxEPA, and 0–5 μM of Se incubation did not markedly influence the cell viability (Supplementary Fig. S13A, C).
One hundred fifty micromolars oxEPA significantly increased TGs concentration (11.6%) compared with the 150 μM EPA group (Supplementary Fig. S13B), and 5 μM of Se reversed oxEPA-induced lipotoxicity (Supplementary Fig. S13D). Collectively, we used the 150 μM of EPA and oxEPA and 5 μM of Se as the subsequent concentration in our in vitro studies. Moreover, the Se (5 μM) and EPA concentrations (150 μM) used in cell culture experiments were of physiological relevance to those in the plasma (2.26–5.61 μM Se and 152.94–201.36 μM EPA) of yellow catfish (Supplementary Table S3).
Txnrd2 mediated Se and oxEPA-induced variations of lipid metabolism and mitochondrial ROS in primary hepatocytes of yellow catfish
Studies suggested that oxidative stress was associated with lipid metabolism (Chen et al., 2022; Zhang et al., 2021a), and that Txnrd2 was essential for maintaining the mitochondrial redox homeostasis (Hellfritsch et al., 2015; Sibbing et al., 2011). Therefore, we hypothesized that txnrd2 participated in Se- and oxEPA-induced changes of lipid metabolism and mitochondrial reactive oxygen species (mtROS).
To verify this hypothesis, we reduced txnrd2 expression via the small interfering RNA (siRNA), and we found that txnrd2 siRNA2 presented over 70.0% knockdown efficiency (Supplementary Fig. S14A). Thus, txnrd2 siRNA2 was used for txnrd2 knockdown experiments. Se treatment significantly alleviated oxEPA-induced decrease of txnrd2 gene expression (83.2%) and enzyme activity (35.4%) (Supplementary Fig. S14B, C). The txnrd2 knockdown significantly reduced gene expression of txnrd2 in EPA, oxEPA, EPA+Se, and oxEPA+Se groups (63.1%–73.8%) and exacerbated oxEPA-induced decrease of Txnrd2 activity (21.3%) (Supplementary Fig. S14B, C).
As expected, Se treatment alleviated but txnrd2 knockdown exacerbated oxEPA-induced lipotoxicity (8.50%) (Fig. 6A, D and Supplementary Fig. S15). From these findings, we could conclude that txnrd2 was involved in Se- and oxEPA-induced modulation of lipid metabolism.
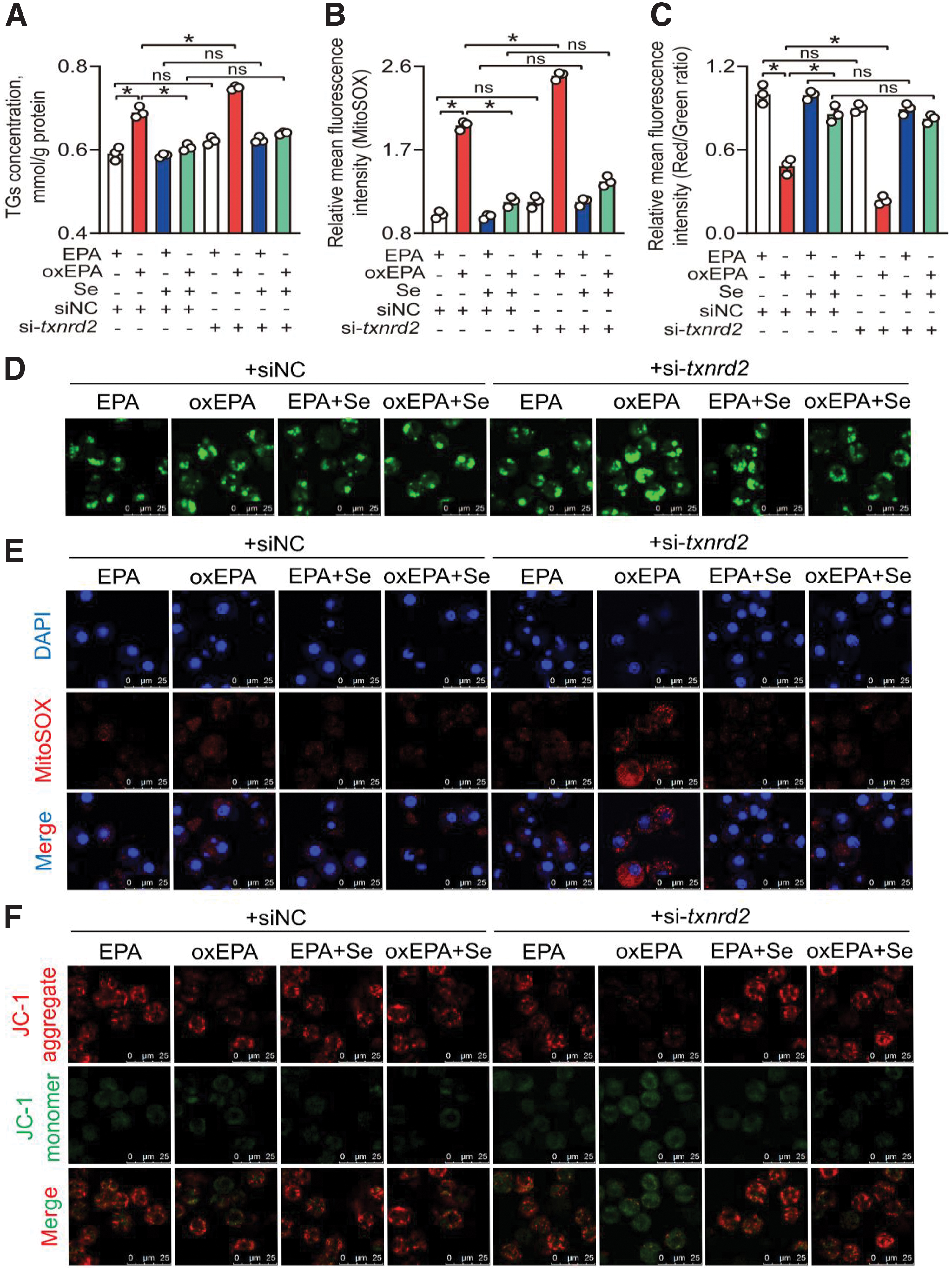
Next, we monitored the changes of mtROS production, mitochondrial function, and oxidative stress. Se treatment significantly alleviated oxEPA-induced increase of mtROS (41.9%), and MDA (8.53%), 4-HNE (7.10%), and protein carbonyl (13.6%) contents, whereas txnrd2 knockdown significantly exacerbated oxEPA-induced increase of mtROS (27.3%), and MDA (8.49%), 4-HNE (8.88%), and protein carbonyl (14.4%) contents (Fig. 6B, E and Supplementary Figs. S16A and S17).
Se treatment significantly alleviated oxEPA-induced decrease of mitochondrial membrane potential (MMP; 77.2%), ATP content (40.8%), mitochondrial DNA (mtDNA) copy numbers (87.4%), mRNA expression of complexes subunits (22.5%–73.9%), fatty acid β oxidation (33.4%), and Cpt1 activity (23.8%), and alleviated oxEPA-induced increase of NEFAs (33.6%), whereas txnrd2 knockdown significantly exacerbated oxEPA-induced decrease of MMP (51.0%), ATP content (18.9%), mtDNA copy numbers (17.5%), mRNA expression of complexes subunits (20.5%–35.0%), fatty acid β oxidation (26.0%), and Cpt1 activity (11.5%), but exacerbated oxEPA-induced increase of NEFAs (35.2%) (Fig. 6C, F and Supplementary Figs. S16B–F and S18).
Further, Se treatment significantly alleviated oxEPA-induced decrease of Usp4 (72.3%) and Pparα (1.25-fold) protein expression whereas txnrd2 knockdown significantly exacerbated oxEPA-induced decrease of Usp4 (28.9%) and Pparα (28.7%) protein expression (Supplementary Fig. S19). Collectively, our findings demonstrate that txnrd2 mediates Se- and oxEPA-induced variations of lipid metabolism and mtROS in yellow catfish hepatocytes.
mtROS mediated Se- and oxEPA-induced changes of lipid metabolism together with Usp4 protein expression in primary hepatocytes of yellow catfish
To substantiate the role of txnrd2-mediated mtROS in changes of lipid metabolism induced by Se and oxEPA, we incubated primary hepatocytes with Mito-TEMPO, a specific mtROS scavenger. Results indicated that Mito-TEMPO incubation reversed oxEPA-induced increase of lipotoxicity (10.1%), mtROS (50.7%), and MDA (13.9%), 4-HNE (7.47%), and protein carbonyl (13.3%) contents (Fig. 7A, B, D, E and Supplementary Figs. S20, S21A, and S22).
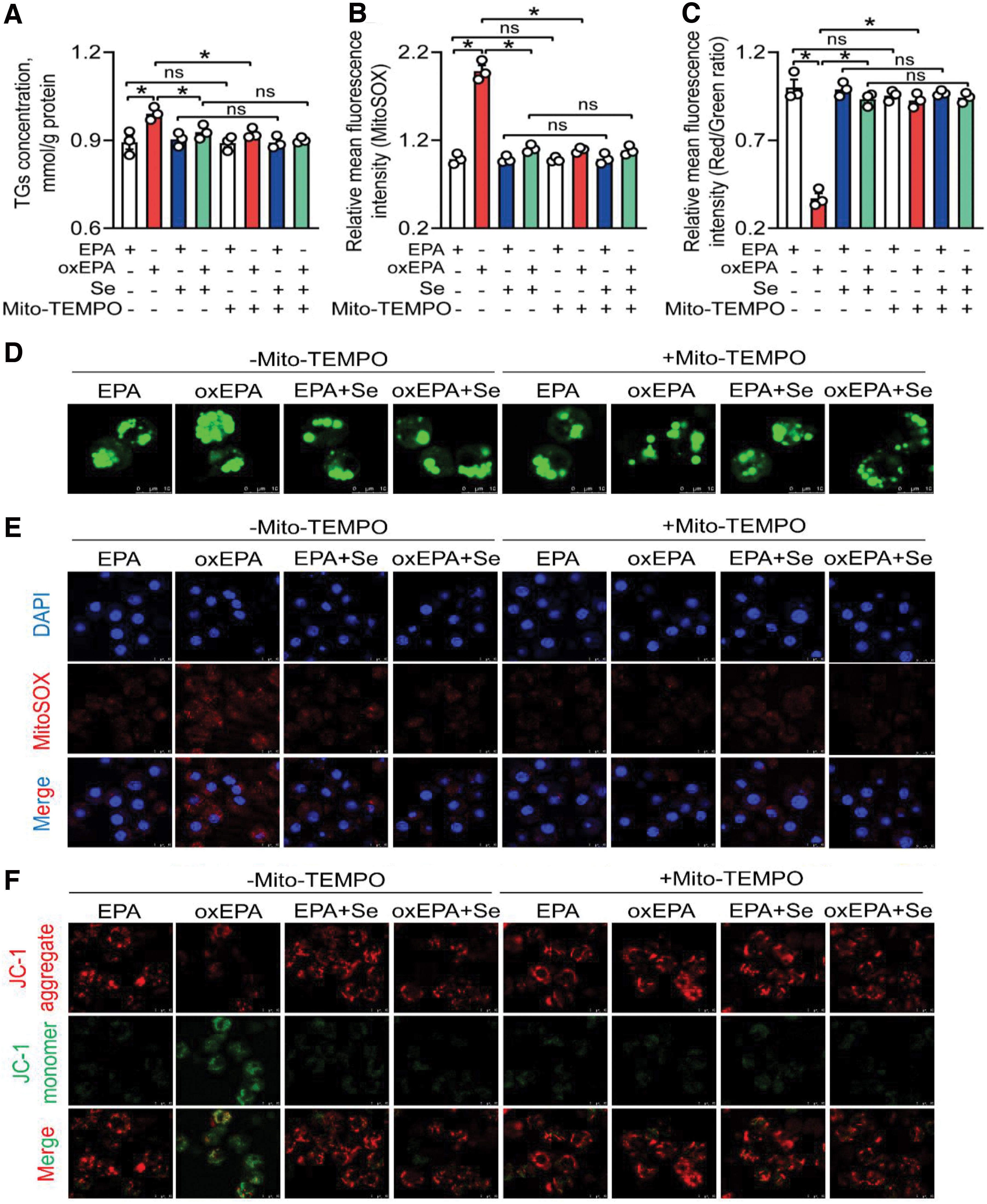
Meantime, Mito-TEMPO incubation reversed oxEPA-induced reduction of MMP (1.57-fold), ATP content (62.8%), and mtDNA copy numbers (77.2%) and alleviated oxEPA-induced increase of NEFAs (30.6%) (Fig. 7C, F and Supplementary Fig. S21B–E). Mito-TEMPO incubation reversed oxEPA-induced decrease of Usp4 (1.09-fold) and Pparα (2.22-fold) protein expression (Supplementary Fig. S21F, G). Together, mitochondrial oxidative stress mediated the Se- and oxEPA-induced variations of the lipid metabolism, Usp4 and Pparα protein expression in yellow catfish hepatocytes.
Usp4 mediated deubiquitination and stabilization of Pparα in HEK293T cells
Since our earlier studies clearly illustrated that Se incubation attenuated oxEPA-induced increase of mtROS through increasing the txnrd2 expression, and further reversed the oxEPA-induced decrease of lipotoxicity, we next explored the mechanism of mtROS-mediated inhibition of the fatty acid β-oxidation. Pparα is believed to be a critical transcription factor responsible for fatty acid β-oxidation and fine-tuned by ubiquitination (Blanquart et al., 2002; Zhao et al., 2018b).
Our bioinformatics analysis found that Usp4 was the potential deubiquitinase of Pparα. Therefore, we hypothesized that the mtROS-induced decrease of fatty acid β-oxidation was triggered by an increase in Pparα ubiquitination modification following a decrease in Usp4. Fluorescence confocal microscopy showed that Usp4 colocalized with Pparα in primary hepatocytes of yellow catfish (Fig. 8A). Co-immunoprecipitation assays indicated that the overexpressed Usp4 pulled down the over-expressed Pparα in the HEK293T cells (Fig. 8B, C), indicating that Usp4 is directly associated with Pparα.
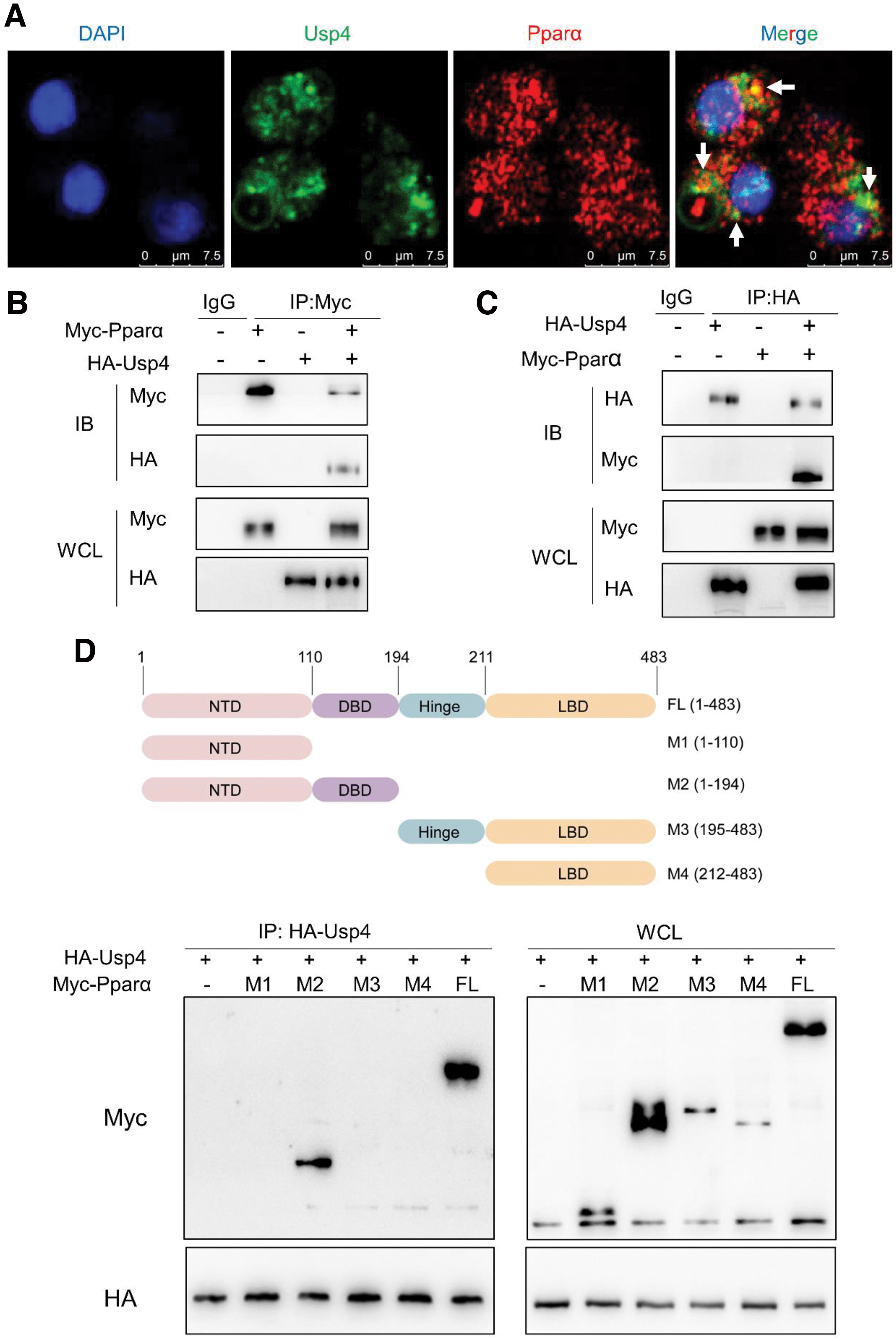
Domain mapping analyses suggested that the DNA-binding domain (aa111–194) of Pparα is required for Usp4 interaction (Fig. 8D). Subsequently, we determined whether Usp4 functioned to deubiquitinate Pparα, and we found that, in the absence of MG132, a proteasome inhibitor, Pparα polyubiquitination levels were decreased in Usp4-overexpressing HEK293T cells. In the presence of MG132, Pparα polyubiquitination levels were enhanced regardless of whether Usp4 was overexpressed in HEK293T cells (Supplementary Figs. S23A and S24A), indicating that Usp4 could decrease Pparα ubiquitination.
Studies suggested that the monoubiquitination, Lys48- and Lys63-linked polyubiquitination are the main forms of ubiquitination (Kim et al., 2011). Therefore, we used the ubiquitin mutant with all the lysine residues replaced by arginine except K48 (K48-only) or K63 (K63-only) to determine the binding specificity of Usp4. The results showed that, in the presence of Usp4 overexpression, Pparα polyubiquitination levels were decreased in wild-type ubiquitin and K48 ubiquitin mutant groups, but Pparα polyubiquitination levels showed a few changes in the K63 ubiquitin mutant group (Supplementary Figs. S23A and S24A), indicating that the K48-linked ubiquitin chains on Pparα were the major form of the ubiquitin linkages hydrolyzed by Usp4. Together, these findings suggested that Usp4 mediated the Pparα deubiquitination and stabilization.
Usp4-mediated Pparα deubiquitination is required for Se- and oxEPA-induced variations of lipid metabolism in primary hepatocytes of yellow catfish
Given the importance of Usp4 in stabilizing Pparα and Pparα role in controlling lipid metabolism (Bougarne et al., 2018), we investigated the role of Usp4-mediated Pparα deubiquitination in Se- and oxEPA-induced changes of lipid metabolism. To explore this, we detected the effects of usp4 knockdown on lipid metabolism by using the siRNA. We found that usp4 siRNA1 presented over 70.0% knockdown efficiency for usp4 (Supplementary Fig. S25A).
Thus, usp4 siRNA1 was used for usp4 knockdown experiments. The usp4 knockdown significantly reduced gene expression of usp4 in EPA, oxEPA, EPA+Se, and oxEPA+Se groups (65.9%–75.6%) and exacerbated oxEPA-induced decrease of Usp4 protein expression (42.2%) (Supplementary Fig. S25B–D). The TGs assay showed that usp4 knockdown exacerbated oxEPA-induced lipotoxicity (3.32%) (Fig. 9A–C), suggesting that Usp4 mediated the Se- and oxEPA-induced changes of lipid metabolism.
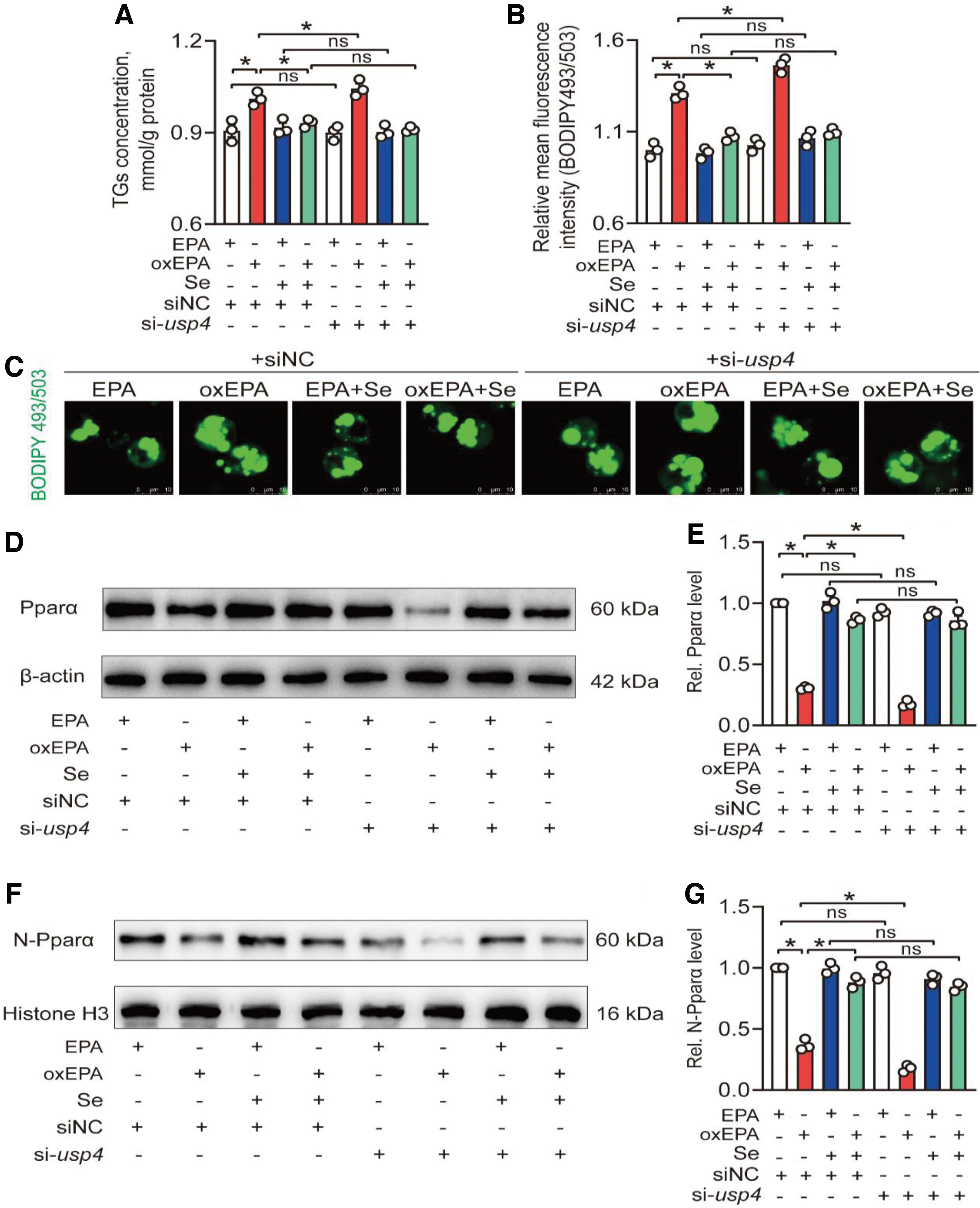
Moreover, usp4 knockdown exacerbated oxEPA-induced decrease of Pparα total protein expression (42.2%) and its nuclear location (51.8%). Se treatment significantly alleviated oxEPA-induced increase of Pparα K48-linked polyubiquitination (42.7%) but usp4 knockdown exacerbated oxEPA-induced increase of Pparα K48-linked polyubiquitination (28.6%) (Supplementary Fig. S26), suggesting that oxEPA reduced Pparα protein expression via Usp4-induced decrease of deubiquitination and consequent stabilization of Pparα.
Moreover, the pparα mRNA expression showed no significant differences among eight treatments (Supplementary Fig. S27A), but Se incubation significantly alleviated oxEPA-induced reduction of transcript levels of acsl1, cpt1, cpt2, acadl, and acadm (fatty acid β-oxidation related genes; 86.7% to 1.73-fold) (Supplementary Fig. S27B–F) and usp4 knockdown significantly exacerbated oxEPA-induced reduction of transcript levels of acsl1, cpt1, cpt2, acadl, and acadm (31.4%–40.6%) (Supplementary Fig. S27B–F), suggesting that oxEPA-induced decrease of Usp4 protein expression contributed to the decreased transcript levels of fatty acid β-oxidation related genes. Collectively, these findings indicated that Usp4-mediated Pparα deubiquitination was required for Se and oxEPA-induced changes of the lipid metabolism.
Discussion
In the present study, dietary Se supplementation ameliorated OFO-induced oxidative stress, mitochondrial dysfunction, reduction of fatty acid β-oxidation, and abnormal lipid deposition. Mechanistically, dietary Se supplementation restored Usp4 mediated-deubiquitination of Pparα and further inhibited ubiquitin-proteasome-dependent Pparα degradation, and thus enhanced fatty acid β-oxidation and alleviated OFO-induced lipotoxicity.
Our results revealed the underlying mechanism of Se supplementation in alleviating OFO-induced lipotoxicity and highlighted the crucial function of Se supplements in alleviating OFO-induced mitochondrial oxidative stress and suppression of fatty acid β-oxidation, which offer an alternative strategy against OFO-induced lipotoxicity in fish and human beings.
Our previous study reported that OFO intake resulted in liver lipotoxicity (Zhang et al., 2021a), but whether Se supplementation can improve OFO-induced liver lipotoxicity remains unknown. The current study showed that H-Se supplementation dramatically reversed OFO-induced liver lipotoxicity. To the best of our knowledge, the present study is the first report involved in Se supplementation alleviating OFO-induced hepatic lipotoxicity.
Similarly, Liu et al. (2021) reported that A-Se supplementation (0.3–1.2 mg/kg) significantly relieved high-fat diet-induced hepatopancreas injury and lipid deposition in the grass carp Ctenopharyngodon idella. From this perspective, it may suggest that an appropriate dietary Se supplementation will contribute toward maintaining lipid homeostasis.
Several studies have demonstrated that oxidative stress influenced lipid homeostasis and accelerated lipotoxicity (Chen et al., 2022; Zhang et al., 2021a). Our previous study clearly showed that OFO triggered oxidative stress in the liver and further led to lipotoxicity (Zhang et al., 2021a). In the present study, dietary OFO intake caused hepatic oxidative stress.
Compared with FFO, we found that OFO reduced the contents of n-3 PUFAs, including EPA and DHA, which was also reported in a previous study (Feng et al., 2020). n-3 PUFAs may act as antioxidants by regulating the antioxidant signaling pathway (Oppedisano et al., 2020). Thus, the decrease of n-3 PUFAs content in OFO might disturb the redox homeostasis of liver tissues. In addition, we detected the presence of some oxylipins in the OFO.
Since oxylipin contents are relatively low and its biological functions are still investigated (Nayeem, 2018), the role of oxylipins in OFO-induced oxidative stress remains to be further studied. Se, an essential trace element, has exhibited its powerful antioxidant properties among vertebrates (Kang et al., 2022; Li et al., 2021; Zhang et al., 2021b).
Hence, to explore the mechanism of Se supplementation in alleviating OFO-induced lipotoxicity, we questioned whether dietary Se supplementation reversed OFO-induced liver lipotoxicity through attenuating OFO-induced oxidative stress. Along these lines, our study showed that dietary Se reduced the OFO-induced increment in MDA, 4-HNE, and protein carbonyl contents. MDA, 4-HNE, and protein carbonyl are widely used as biomarkers for oxidative damage.
Therefore, the result suggested that dietary Se supplementation attenuated OFO-induced oxidative stress. An imbalance between the antioxidant defense system and ROS production induces oxidative stress (Chen et al., 2022). We found that dietary Se addition alleviated OFO-induced reduction of Cat, Gpx, Txnrd2 enzyme activities, and GSH/GSSG ratio. Therefore, Se supplementation-induced reduction of oxidative stress might be attributed to the increase of Cat, Gpx, Txnrd2 enzyme activities, and GSH/GSSG ratio.
Meanwhile, we found that dietary Se supplementation also attenuated the OFO-induced decline in txnrd2 mRNA expression. Previous studies have demonstrated that Txnrd2, a mitochondrion-resident selenoprotein, is crucial for maintaining mitochondrial redox homeostasis (Hellfritsch et al., 2015; Sibbing et al., 2011). Sibbing et al. (2011) pointed out that mutations in Txnrd2 decreased Txnrd2 activity, and they induced oxidative stress and cardiomyopathy.
In contrast, Txnrd2 overexpression reduced endoplasmic reticulum (ER) stress and oxidative stress in multiple myeloma cells (Fink et al., 2016). Thus, we hypothesized that Txnrd2-mediated mtROS production might account for the Se- and OFO-induced alterations of oxidative stress. In the in vitro study, we chose the 150 μM of EPA and oxEPA and 5 μM of Se as their incubation concentrations, which are of physiologic relevance to those in the plasma (2.26–5.61 μM Se and 152.94–201.36 μM EPA) of yellow catfish.
Therefore, these concentrations were appropriate to investigate the mechanism of Se and OFO affecting lipid metabolism in the liver of yellow catfish. We found that OFO groups (A-Se+OFO and H-Se+OFO groups) had lower plasma EPA concentration, and Se addition groups (H-Se+FFO and H-Se+OFO groups) had higher plasma Se concentration.
Combining the results mentioned earlier, the decreased plasma EPA concentration in OFO groups (A-Se+OFO and H-Se+OFO groups) and increased plasma Se concentration in Se addition groups (H-Se+FFO and H-Se+OFO groups) might be attributable to the oxidation of fish oil and dietary Se supplementation, respectively. Notably, Se metabolism and excretion constitute an important aspect that reveals how Se exerts its function.
However, to the best of our knowledge, at present, Se metabolism and secretion are not very clear. It is an important aspect of our future study. Our in vitro study found that Se incubation attenuated the oxEPA-induced decline in txnrd2 mRNA expression and enzyme activity, and increased mtROS, MDA, and TGs concentration. Likewise, Kiermayer et al. (2007) also found that A-Se supplementation significantly enhanced Txnrd2 activity in the liver of mice.
Gao et al. (2016) pointed out that Se supplementation alleviated the lead (Pb)-induced downregulation of txnrd2 mRNA expression levels in broiler chicken. Hormesis is a dose–response phenomenon characterized by a low-dose stimulation and a high-dose inhibition (Calabrese et al., 2010). Specifically, low doses of toxic stimuli activate the body's adaptive protective mechanisms against physical damage, but high doses of toxic stimuli inhibit the body's protective mechanisms.
Mitochondrial Txnrd2 is an important part of the vitagene network that mediates the protective mechanisms (Calabrese et al., 2010). Hence, we can infer that the high-dose inhibition may account for the OFO and oxEPA-induced decrease of txnrd2. Similarly, previous neuroprotection studies also suggested that physiological amounts of supplements are neuroprotective, whereas higher concentrations are clearly neurotoxic (Calabrese et al., 2007; Drake et al., 2003).
Moreover, txnrd2 knockdown further aggravated oxEPA-induced increment of the mtROS, MDA, and TGs concentration. Inhibition of mtROS generation by Mito-TEMPO incubation attenuated oxEPA-induced increase of MDA and TGs concentration. Similarly, Chen et al. (2022) demonstrated that Mito-TEMPO treatment dramatically relieved ZnO nanoparticle-induced increase of MDA and TGs concentration.
These findings suggested that Txnrd2 mediated Se- and oxEPA-induced changes of mtROS, oxidative stress, and lipid metabolism. Kiermayer et al. (2015) pointed out that heart-specific knockout of Txnrd2 increased mtROS production and decreased mitochondrial fatty acid catabolism. Collectively, our data indicated that Se supplementation attenuated OFO-induced mtROS, and it further attenuated OFO-induced oxidative stress and lipotoxicity through increasing txnrd2 expression.
Mitochondria are the major organelles for the ROS production, ATP synthesis, and fatty acid β-oxidation (Zhang et al., 2021a). Excessive ROS will damage mitochondrial structure, reduce MMP and the mRNA expression levels of complexes subunits, and decrease mtDNA copy numbers, which, in turn, decreases oxygen consumption and the fatty acid β-oxidation capacity, and ultimately leads to ATP depletion and mitochondrial dysfunction (Zhang et al., 2021a).
Accordingly, the reduction of fatty acid β-oxidation capacity and ATP content suggested mitochondrial dysfunction (Zhang et al., 2021a). In addition, it has been well established that mitochondrial dysfunction directly mediates oxidative stress-induced abnormal lipid deposition (Chen et al., 2022; Zhang et al., 2021a). Therefore, we explored the effects of Se and OFO on mitochondrial dysfunction and determined its role in Se and oxEPA-induced changes of lipid metabolism in our in vivo and in vitro studies, respectively.
Our in vivo study indicated that dietary OFO induced the mitochondrial injury, decreased mtDNA copy numbers and mRNA expression levels of complexes I/III/IV/V, fatty acid β-oxidation capacity, and ATP content, in agreement with other studies (Long et al., 2022; Zhang et al., 2021a). Dietary Se supplementation alleviated OFO-induced mitochondrial injury and dysfunction. Similarly, other studies suggested that H-Se diet protected against the 2,4,6-trinitrobenzene sulfonic acid-triggered dysfunction of the mitochondria (Tirosh et al., 2007).
In hippocampal HT22 neuronal cells, Se supplementation suppressed glutamate-induced cell death and prevented mitochondria structural damage (Ma et al., 2017). Further, our in vitro study showed that Se and Mito-TEMPO incubation attenuated oxEPA-induced mitochondrial injury, mitochondrial dysfunction, and lipotoxicity, similar to other reports (Chen et al., 2022).
However, txnrd2 knockdown exacerbated oxEPA-induced mitochondrial injury and dysfunction, and lipotoxicity, as observed in a previous study (Kiermayer et al., 2015). Together, these findings suggested that mtROS-mediated mitochondrial dysfunction was essential for Se- and OFO-induced changes of lipid metabolism.
Although studies have demonstrated that oxidative stress triggers lipid deposition by decreasing fatty acid β-oxidation capacity (Chen et al., 2022; Zhang et al., 2021a), the intrinsic molecular mechanism of oxidative stress-mediated reduction of fatty acid β-oxidation remains largely unknown. Our in vivo study indicated that OFO and Se interacted to affect the fatty acid β-oxidation and ubiquitin system, and they might modulate Pparα protein expression through ubiquitin-proteasome dependent degradation.
Recent studies report that the ubiquitin system is crucial for regulating protein turnover and is involved in obesity and lipid metabolism disorder (Chen et al., 2021; Sarraf et al., 2013). Pparα, a key regulator of fatty acid β-oxidation, is finely tuned by ubiquitination (Blanquart et al., 2002; Zhao et al., 2018b). In addition, several previous studies have shown that oxidative stress activates protein ubiquitination (Kästle et al., 2012; Sarraf et al., 2013).
Here, we identified Usp4 as the corresponding deubiquitinase, which mediated deubiquitination and stabilization of Pparα. Although studies have illustrated that PPARα can be modified by ubiquitination (Blanquart et al., 2002; Zhao et al., 2018b), our study is the first report to find that Usp4 is required for the deubiquitination of Pparα, which broadens our understanding of the Pparα regulatory mechanism.
Further, our study showed that Se alleviated OFO-induced reduction of protein levels of Usp4 and Pparα, and Se and Mito-TEMPO incubation alleviated oxEPA-induced reduction of Usp4 and Pparα protein levels. Therefore, we determined the importance of Usp4-mediated deubiquitination and stabilization of Pparα in oxidative stress triggered the reduction of fatty acid β-oxidation by blocking usp4 expression.
The results showed that usp4 knockdown aggravated oxEPA-induced lipotoxicity and oxEPA-induced decrease of Pparα protein expression and Pparα K48-linked polyubiquitination, indicating that Usp4-mediated Pparα deubiquitination was involved in Se- and oxEPA-induced variations of lipid metabolism. We also found that usp4 knockdown aggravated oxEPA-induced decrease of mRNA expression levels of acsl1, cpt1, cpt2, acadl, and acadm, fatty acid β oxidation-related genes.
Previous studies pointed out that Pparα modulated the transcript levels of these genes (acsl1, cpt1, cpt2, acadl, and acadm) (Aoyama et al., 1998; Bougarne et al., 2018). These findings suggested that Usp4-mediated Pparα deubiquitination was crucial in Se- and oxEPA-induced changes of fatty acid β oxidation.
Materials and Methods
Electronic laboratory notebook was not used.
Ethic statement
The present experimental protocols followed the Huazhong Agriculture University (HZAU) guideline for the use of experimental animals. The HZAU Ethics Committee approved our experimental protocols (identification code: Fish-2019-09-21).
Expt. 1: in vivo study
Feed formulation, animal culturing, and sampling
A 2 × 2 factorial experiment was conducted here. Four diets were produced with two Se levels, and each Se level had one FFO level and one OFO level, respectively. Se was added in the form of Na2SeO3 (214485, ≥99% in purity; Sigma). The FFO was oxidized by heating at 60°C and aerated for 4 days. The oxidation of fish oil was evaluated by the peroxide values (POV) (Zhang et al., 2021a).
Dietary Se contents were 0.24, 0.25, 0.49, and 0.48 mg/kg, and the POV were 3.85, 89.69, 3.57, and 86.58 meq/kg for the A-Se+FFO group (the control), A-Se+OFO group, H-Se+FFO group, and H-Se+OFO group, respectively (Supplementary Table S6). For yellow catfish culturing, 360 juvenile yellow catfish (mean initial body weight: 3.99 ± 0.01 g, 2-month-old) were cultured in the 12 tanks, with 30 yellow catfish per tank containing 300 L water. Each experimental diet was fed to triplicate tanks. Fish were fed to the satiation twice daily. The feeding experiment lasted for 10 weeks.
Sampling occurred at the end of the 10-week feeding experiment. After the euthanization of yellow catfish with the 100 mg/L tricaine methanesulfonate solution, the liver was sampled on ice, quickly frozen in liquid nitrogen, and kept in an −80°C freezer for the subsequent analysis. We analyzed TGs and Se contents, protein expression, enzymatic activities, histochemistry and ultrastructural observation, and transcriptomic sequencing.
Analysis of fatty acid composition and oxylipin contents in FFO, OFO, and oxEPA
The fatty acid composition of FFO and OFO was determined according to a previous study (Rincón-Cervera et al., 2019), with slight modifications. Briefly, 25 mg FFO or OFO and 1 mL n-hexane were added into a test tube and mixed. The mixture was methylated for 1 h at 100°C with 1 mL methylation reagent (acetyl chloride: methanol, 1:20, v/v) to produce fatty acid methylesters.
Then, they were cooled to room temperature, followed by the addition of 1 mL distilled water in each test tube. The mixture was centrifuged at 1200 g for 5 min. The following upper hexane layer containing the fatty acid methylesters was used for the analysis of the gas chromatography (GC-2010 plus; Shimadzu Emit Co., Ltd., Tokyo, Japan).
The oxylipins contents of FFO, OFO, and oxEPA were determined according to a previous study (Wu et al., 2018), with slight modifications. Briefly, 15 mg FFO or OFO or 20 μL oxEPA were added into a test tube, and then 190 μL methanol (containing 0.1% acetic acid, 0.1% butylated hydroxytoluene) and 200 μL 0.25 M sodium carbonate (dissolved with methanol/water at a ratio of 1:1, v/v) were added. Subsequently, they were vortexed homogeneously and heated at 60°C for 30 min. After these steps, 25 μL acetic acid was added into the tube and the pH was adjusted to the values between 4 and 6.
The resulting solution was used for the solid phase extraction, and the collected solution was subjected to the mass spectrometric analysis by high-performance liquid chromatography tandem triple quadrupole mass spectrometry using the QTRAP quadrupole/linear ion-trap tandem mass spectrometer (AB Sciex, Toronto, Canada).
Determination of TGs, NEFAs, plasma biochemical indicators, and Se contents
The TGs, NEFAs, glucose and cholesterol contents, and ALT and AST activities were determined with their corresponding kits (A110-1-1, A042-1-1, F006-1-1, A111-1-1, C009-2-1, C010-2-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The Se contents were determined as previously described (Zhang et al., 2021b).
Determination of enzymatic activities
For Cpt1 and Txnrd2 activity analysis, we first isolated the mitochondria from the liver and hepatocytes according to a previous study (Luongo et al., 2020). Cpt1 activity was measured, based on analyzing the initial CoA-SH formation by the 5,5′-dithio-bis-(2-nitrobenzoic acid) reaction at 412 nm, and expressed as the 1 μmol product formed per min per mg mitochondrial protein at 28°C.
The Txnrd2 activity was measured at 412 nm using the Txnrd Assay Kit (A119-1-1; Nanjing Jiancheng Bioengineering Institute). The Bradford Protein Assay Kit (A045-2-1; Nanjing JianCheng Bioengineering Institute) was used for the analysis of the soluble protein concentration.
Analysis of oxidative stress and ATP content
For the analysis of oxidative stress parameters, 0.1 g liver samples were homogenized in 0.9 mL ice-cold buffer (2 mM EDTA, 0.25 M sucrose, 0.02 M Tris-HCl, 0.5 mM phenyl methyl sulfonyl fluoride, 0.1 M sodium fluoride, and 0.01 M β-mercapto-ethanol, pH 7.4). They were then centrifuged at 2500 g at 4°C for 10 min. The supernatant was used for the following analysis.
The Cat and T-Sod activities were measured at 405 and 550 nm, respectively, using Cat and T-Sod assay kits (A007-1-1, A001-1-1; Nanjing Jiancheng Bioengineering Institute). The Gpx and T-AOC activities were determined at 340 and 405 nm with Gpx and T-AOC assay kits (S0058 and S0121; Beyotime Biotechnology, Shanghai, China), respectively.
GSH and GSSG contents were measured by the commercial kit (S0053; Beyotime Institute of Biotechnology; Shanghai, China). The MDA and ATP contents were determined using the Lipid Peroxidation MDA Assay Kit and the ATP Assay Kit (S0131S and S0026; Beyotime Institute of Biotechnology). The 4-HNE and protein carbonyl contents were determined using the 4-HNE enzyme-linked immunosorbent assay (ELISA) Kit (ml038440-1; Shanghai Enzyme-Linked Biotechnology, Shanghai, China) and the Protein Carbonyl assay Kit (A087-1-2; Nanjing Jiancheng Bioengineering Institute).
Transcriptomic sequencing and analysis
We undertook the transcriptomic sequencing and analyses in the BIOMARKER TECHNOLOGIES (Beijing, China), and the details were shown in our publication (Zhang et al., 2022). All these reads were deposited in the Sequence Read Archive of the NCBI database (SRA Accession: PRJNA850468). We determined the DEGs based on the absolute value of log2 (fold change) ≥1 and p value <0.05.
We performed the analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Set Enrichment Analysis to identify the enrichment pathways relevant with the signal transduction and metabolism. We selected the 10 candidate genes for the q-PCR validation. We also selected eight housekeeping genes (gapdh, elfa, b2m, tuba, 18S rRNA, rpl7, hprt, and ubce) to analyze their expression stability at the transcriptional level. After analysis via the online tool geNorm, we found that the b2m and elfa mRNA levels were the most stable, and accordingly used their geometric mean to normalize the gene expression levels.
The 2−ΔΔCt method was utilized to calculate the fold-change of their mRNA expression to the control (A-Se+FFO group). Their primer sequences were presented in Supplementary Table S7.
Histological and TEM observation
The liver tissues and hepatocytes were stained with the H&E and ORO for the histological observation, and TEM was used to observe the ultrastructure of tissues and hepatocytes, based on the protocols described in our previous study (Zhang et al., 2022). We examined ten fields randomly from each sample and then used the software Image J to quantify the relative areas for lipid droplets (LDs) from the ORO staining and the vacuoles from H&E staining.
Immunoblotting
The immunoblotting analysis was undertaken according to our previous study (Zhang et al., 2022). The primary antibodies included: rabbit anti-Pparα (1:1000, 15540-1-AP; Protein Technology, Wuhan, China); rabbit anti-Pparγ (1:1000, 16643-1-AP; Protein Technology); mouse anti-Usp4 (1:2500, 66822-1-Ig; Protein Technology); rabbit anti-Ubiquitin (1:1500, A18185; ABclonal, Wuhan, China); rabbit anti-Ubiquitin (linkage-specific K48; 1:5000, ab140601; Abcam, London, United Kingdom); rabbit anti-Histone H3 (1:6000, 17168-1-AP; Protein Technology); rabbit anti-Rxrα (1:600, 21218-1-AP; Protein Technology); rabbit anti-β-Actin (1:10,000, AC026; ABclonal); mouse anti-6 × His Tag (1:10,000, 66005-1-Ig; Protein Technology); rabbit anti-Myc Tag (1:10,000, AE070; ABclonal); and mouse anti-HA Tag (1:10,000, 66006-2-Ig; Protein Technology).
Secondary antibodies were: Goat anti-mouse IgG (H+L; 1:10,000, SA00001-1; Protein Technology), the HRP-conjugated anti-rabbit IgG antibody (Light chain specific; 1:10,000, 93702; Cell Signaling Technology, Danvers, MA), or Goat anti-rabbit IgG (H+L; 1:10,000, SA00001-2; Protein Technology). The immunoreactive bands were visualized via the enhanced chemiluminescence instrument (Cell Signaling Technology), and the densitometry was analyzed via the Image J software (National Institutes of Health).
mtDNA copy number assay
The relative levels of mtDNA copy number were determined by q-PCR according to a previous study (Eaton et al., 2007). The mitochondrial atp synthase f0 subunit 8 (atp8) gene was used to measure mtDNA copy number, which was normalized to the eukaryotic translation elongation factor 1 alpha 1 (eef1α1). The primer sequence information for atp8 and eef1α1 was presented in Supplementary Table S8.
Expt. 2: in vitro study
Preparation of oxEPA
The oxEPA was obtained from the purified and fresh EPA, based on a previous study (Sethi et al., 2002). By measuring thiobarbituric acid-reactive substances, we found that the MDA contents were 0.12 and 1.62 μM for 100 μM fresh EPA and oxEPA, respectively. Before the fresh EPA or oxEPA treatment, the fresh EPA or oxEPA was diluted with 10% fetal bovine serum to their corresponding concentration in the media.
Cell culture and MTT assay
The human embryonic kidney 293T cell line (HEK293T cells) and yellow catfish primary hepatocytes were cultured, and their cell viabilities were determined via the MTT methods, as previously described (Zhang et al., 2022).
The siRNA transfection and plasmid construction
The txnrd2 and usp4 siRNAs were synthesized in the GenePharma Biotech Company (Shanghai, China), and their sequences were listed in Supplementary Table S9. We constructed the expression plasmid for pcDNA3.1-Pparα-Myc and pcDNA3.1-Usp4-HA. The brief procedure was followed next: At first, we subcloned the open reading frame sequences encoding yellow catfish Pparα and Usp4 into the pcDNA3.1 (+) vector with the Myc-tag and HA-tag sequences inserted at the N-terminus of Pparα and the C-terminus of Usp4, respectively.
The Pparα mutants were produced in the pcDNA3.1-Pparα-Myc plasmid via the Mut Express II Fast Mutagenesis Kit (Vazyme). We used the Lipofectamine 2000 reagent (11668019; Thermo Fisher Scientific, Waltham, MA) for the transient transfections. After the transfection with 4 μg plasmids or 100 nM siRNA oligonucleotides for 48 h, we collected the cells, and we analyzed the interference efficiency using the q-PCR and/or Western blotting assays. The primer sequences for these plasmids were shown in Supplementary Table S10.
Detection of LDs, mtROS, and MMP
We used 5 mg/mL BODIPY 493/503 (D3922; Thermo Fisher Scientific), 5 μM MitoSOX (M36008; Thermo Fisher Scientific), and 5 μg/mL JC-1 (C2006; Beyotime) to detect the intracellular LDs, mtROS, and MMP, respectively, and the protocols were based on our previous publication (Chen et al., 2022). The maximum excitation/emission wavelengths of MitoSOX are 396 and 610 nm, respectively.
For JC-1, the maximum excitation/emission wavelengths of JC-1 aggregate are 585 and 590 nm, respectively. The maximum excitation/emission wavelengths of JC-1 monomer are 514 and 529 nm, respectively. The fluorescent densities of the stained cells were visualized via the laser scanning confocal microscope (Leica), and quantified via the software Image J.
Immunoprecipitations
We performed the immunoprecipitation analysis, based on the protocols in our publication (Zhang et al., 2021a). The cells were lysed in the RIPA buffer solution with the addition of the protease inhibitor cocktail on the ice for 30 min. Then, the cellular lysates were centrifuged at 12,000 g at 4°C for 10 min. The lysates were incubated with the anti-Rxrα, anti-Pparα, anti-HA Tag, or anti-Myc Tag antibody at 4°C overnight, followed by the incubation together with the protein A/G agarose beads for 4 h.
The beads were washed five times with the lysis buffer solution. The immunoprecipitated proteins were incubated with an anti-Pparα, anti-Pparγ, anti-Rxrα, anti-HA Tag, anti-Myc Tag, anti-6 × His Tag, or anti-Ubiquitin (linkage-specific K48) antibody, and they were used to measure the corresponding protein expression via the Western blot analysis.
Statistical analysis
The SPSS 19.0 software (IBM, Armonk, NY) was used to perform the statistical analysis. Results were shown as the mean ± standard error of the mean. Before the statistical analysis, the Kolmogorov-Smirnov test was utilized to detect the normality of the data, and Bartlett's test was performed to measure the homogeneity of the variances among the treatments. Student's t tests were used to compare the difference in fatty acids and oxylipins contents in the FFO, OFO, and oxEPA (unpaired, two-tailed).
Two-factor analysis of variance (ANOVA) was undertaken to analyze the data in our in vivo study (Expt. 1). When the two-factor ANOVA revealed the significant p-interaction (p < 0.05), we used the Duncan's multiple-range test to analyze the discrepancies among the groups (Expt. 1). We performed the data analysis in the in vitro study (Expt. 2) via the one-way ANOVA. The significance level was set at p < 0.05.
Availability of Data and Materials
The data that support the findings of this study are available from the corresponding author on request.
Footnotes
Authors' Contributions
D.-G.Z.: conceptualization, investigation, methodology, writing—original draft, and writing—review and editing. W.S.K.: methodology, writing—review and editing. X.-J.L.: methodology, software, and investigation. E.Z.: methodology, writing—review and editing. T.Z.: methodology, software, investigation, and visualization. Y.-C.X.: methodology, software, and investigation. X.-L.W.: methodology, software, and investigation. W.-H.L.: methodology, software, and investigation. Z.L.: funding acquisition, supervision, project design and administration, and writing—review and editing.
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
The study is funded by Shenzhen Institute of Nutrition and Health, Huazhong Agricultural University (SZYJY2023016) and the National Key R&D Program of China (2018YFD0900400).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Supplementary Figure S14
Supplementary Figure S15
Supplementary Figure S16
Supplementary Figure S17
Supplementary Figure S18
Supplementary Figure S19
Supplementary Figure S20
Supplementary Figure S21
Supplementary Figure S22
Supplementary Figure S23
Supplementary Figure S24
Supplementary Figure S25
Supplementary Figure S26
Supplementary Figure S27
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Table S10
OriginalImagesforBlots
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.