
Abstract
Significance:
Central nervous system (CNS) diseases are disorders of the brain and/or spinal cord and include neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and multiple sclerosis. Nuclear factor erythroid 2–related factor 2 (NRF2) is a transcription factor belonging to the cap-n-collar family that harbors a unique basic leucine zipper motif and plays as a master regulator of homeostatic responses.
Recent Advances:
Kelch-like ECH-associated protein 1 (KEAP1) is an adaptor of the Cullin3 (CUL3)-based ubiquitin E3 ligase that enhances the ubiquitylation of NRF2, which promotes the degradation of NRF2 to suppress its transcriptional activity in the absence of stress. Cysteine residues of KEAP1 are modified under stress conditions, and NRF2 degradation is attenuated, allowing it to accumulate and induce the expression of target genes. This regulatory system is referred to as the KEAP1-NRF2 system and plays a central role in protecting cells against various stresses. NRF2 also negatively regulates the expression of inflammatory cytokine and chemokine genes and suppresses pathological inflammation. As oxidative stress, inflammation, and proteostasis are known to contribute to neurodegenerative diseases, the KEAP1-NRF2 system is an attractive target for the treatment of these diseases.
Critical Issues:
In mouse models of neurodegenerative diseases, Nrf2 depletion exacerbates symptoms and enhances oxidative damage and inflammation in the CNS. In contrast, chemical or genetic NRF2 activation improves these symptoms. Indeed, the NRF2-activating chemical dimethyl fumarate is now widely used for the clinical treatment of MS.
Future Directions:
The KEAP1-NRF2 system is a promising therapeutic target for neurodegenerative diseases.
Introduction
It has been difficult to develop therapeutic drugs for disorders of the central nervous system (CNS), in part owing to the presence of the blood-brain barrier. The disorders that affect the brain or spinal cord are collectively referred to as CNS diseases, including neurodegenerative diseases, Alzheimer's disease (AD), Parkinson's disease (PD), and multiple sclerosis (MS). Novel therapeutic targets in the brain and spinal cord for alleviating neurodegeneration and neuroinflammation have been explored for years.
Nuclear factor erythroid 2 (NF-E2)–related factor 2 (NRF2) is a cap-n-collar (CNC) family transcription factor (Sykiotis and Bohmann, 2010; Yamamoto et al., 2018) and regulates the expression of a set of antioxidant and detoxification enzyme (Ishii et al., 2000; Itoh et al., 1997) and proteostasis-related factors (Pajares et al., 2017). NRF2 plays critical roles in protecting cells and tissues against environmental stresses, including electrophiles and reactive oxygen species (ROS).
NRF2 is regulated mainly by Kelch-like ECH-associated protein 1 (KEAP1) (Itoh et al., 1999). KEAP1 is a component of the Cullin3 (CUL3)-based ubiquitin E3 ligase and acts as an adaptor component of ubiquitin E3 ligase, which regulates NRF2 ubiquitination and its degradation. Therefore, KEAP1 suppresses the transcriptional activity of NRF2 in the absence of stress (Itoh et al., 1999; Kobayashi et al., 2004). When cells are exposed to oxidative and/or electrophilic stress, the cysteine residues of KEAP1 are modified, and the KEAP1-mediated ubiquitination of NRF2 is suppressed. Thereafter, NRF2 is stabilized, translocates into the nucleus, and induces the expression of a set of target genes. This regulatory system is referred to as the KEAP1-NRF2 system and plays a central role in protecting cells against environmental stresses (Uruno and Motohashi, 2011; Yamamoto et al., 2018).
In addition, NRF2 negatively regulates the expression of inflammatory cytokine and chemokine genes (Kobayashi et al., 2016) and represses pathological inflammation (Itoh et al., 2004). NRF2 also suppresses inflammation in mouse models of autoimmune diseases, such as Scurfy and NOD mice (Suzuki et al., 2017; Yagishita et al., 2019).
Accumulating lines of evidence clearly support the notion that the activation of NRF2 signaling suppresses oxidative stress and inflammation or protects metabolic tissues from oxidative stresses (Liu et al., 2022; Uruno et al., 2015; Yagishita et al., 2014). It is known that oxidative stress, inflammation, and proteostasis are largely involved in the pathogenesis of neurodegenerative diseases, such as AD, PD, and MS, and the KEAP1-NRF2 system appears to be a promising therapeutic target for neurodegenerative diseases. In fact, dimethyl fumarate (DMF), the NRF2-inducing compound, has been used for the clinical therapy of MS. In this review article, we outline the roles of NRF2 in neurodegenerative diseases.
Discovery of the CNC Family Transcription Factors p45 NF-E2 and NRF2
The erythroid-oriented transcription factor p45 or NF-E2 heterodimerizes with MAFF, MAFG, and MAFK, which are small MAF (sMAF) transcription factors (Igarashi et al., 1994; Itoh et al., 1995). The p45-sMAF heterodimer binding sequence and NF-E2 binding motif have been identified to be TGCTGA(G/C)TCAT/C (Fig. 1A), in the middle of which is a 12-O-tetradecanoylphorbol-13-acetate response element (TRE; Fig. 1B) (Mignotte et al., 1989a; Mignotte et al., 1989b). An important observation is that the NF-E2 binding motif shows high similarity with cis-regulatory elements identified in the regulatory regions of detoxification enzymes, named antioxidant response elements (AREs) (Rushmore et al., 1991) or electrophile response elements (EpREs) (Friling et al., 1990). This observation led to the identification of CNC-sMAF transcription factors as critical regulators of phase II detoxification enzyme expression. It has been proposed that the NF-E2 binding motif and AREs/EpREs should be collectively referred to as CNC-sMAF binding elements (CsMBEs; Fig. 1A) (Katsuoka et al., 2022; Otsuki et al., 2016).

Among CNC family members, NRF2 is assumed to be the factor that regulates detoxification enzyme gene expression in metabolic organs and tissues because NRF2 is most abundantly expressed in these tissues upon exposure to stresses (Itoh et al., 1995; Moi et al., 1994; Yamamoto et al., 2018). This hypothesis was established through analyses of Nrf2 gene knockout mice (Itoh et al., 1997). The indispensable roles of sMAF factors have been demonstrated by the analysis of triple sMAF gene knockout mice and cells (Yamazaki et al., 2012). These elaborate genetic mouse studies unequivocally demonstrated the functional contributions of the NRF2-sMAF heterodimer to the expression of detoxification and antioxidant enzyme genes (Fig. 1C). The expression of these genes is markedly abolished in Nrf2 knockout mice (Itoh et al., 1997). It is now widely accepted that NRF2 plays critical roles in protecting against xenobiotic, electrophilic, and oxidative stresses.
Multiple Functions of NRF2
When cells are exposed to xenobiotics, the phase I detoxifying reaction mediated by cytochrome P450 enzymes metabolically activates the xenobiotics. The metabolites of the phase I reaction acquire an electrophilic character and become good substrates of phase II enzymes, which are mostly transferases. These phase I products are subsequently metabolized by phase II conjugation reactions, such as glutathione (GSH) conjugation, sulfation, or glucuronidation. NRF2 positively regulates the expression of phase II detoxification enzyme genes, such as NQO1, which encodes NAD(P)H:quinone oxidoreductase 1; GSTs, which encode glutathione S-transferases; and UGTs, which encode UDP glucuronosyltransferases (Itoh et al., 1997; Kwak et al., 2001).
In addition to the expression of phase II detoxification enzymes, NRF2 also induces the expression of GSH synthesis–related genes, including Slc7a11, which encodes cystine/glutamate exchange transporter, and the γ-glutamylcysteine synthetase subunits GCLC and GCLM (Hayashi et al., 2003; Ishii et al., 2000; Sasaki et al., 2002). NRF2 also induces the expression of antioxidant genes, such as HMOX1, which encodes heme oxygenase-1; TXNRD1, which encodes thioredoxin reductase 1; GPX2, which encodes glutathione peroxidase 2; and PRDX1, which encodes peroxiredoxin 1 (Alam et al., 1999; Cho et al., 2002; Ishii et al., 2000; Sakurai et al., 2005; Soriano et al., 2008; Thimmulappa et al., 2002). As Nrf2 knockout mice show high sensitivity to toxic chemicals and oxidative stress (Cho et al., 2004; Enomoto et al., 2001; Wakamori et al., 2022), these observations strongly suggest that NRF2 protects cells against various environmental stresses through the induction of cytoprotective enzyme expression (Fig. 2).

Recently, chromatin immunoprecipitation/deep-sequencing analysis revealed the existence of more NRF2 target genes (Suzuki and Yamamoto, 2015). Relatedly, in addition to detoxification and antioxidant enzyme genes, CsMBEs act on the regulatory regions of inflammatory cytokine genes, including IL6 and IL1B, which encode interleukin (IL) 6 and interleukin 1β, respectively, and chemokine genes, such as CCL2, which encodes monocyte chemoattractant protein 1 (Kobayashi et al., 2016). Of importance, NRF2 negatively regulates the expression of these proinflammatory cytokine/chemokine genes.
The induction of NRF2 suppresses the lipopolysaccharide-induced expression of IL6, IL1B, and CCL2, demonstrating that NRF2 contributes to the suppression of pathological inflammation. NRF2 contributes to pentose phosphate pathway, glycogen, and gluconeogenesis-related enzyme gene expressions (Furusawa et al., 2014; Mitsuishi et al., 2012; Uruno et al., 2016; Uruno et al., 2015; Uruno et al., 2013). In addition, NRF2 also regulates expressions of proteasome (PSMs) (Kwak et al., 2003), sequestosome 1 (SQSTM1), also named as p62 (Ishii et al., 2000), and the autophagy adaptor protein NDP52 (CALCOCO2) (Jo et al., 2014). NRF2 thus plays critical roles in regulations of metabolism and proteostasis (Bathish et al., 2022; Cuadrado, 2022).
Neh Domains of NRF2 and Identification of KEAP1
To clarify the mechanism by which various stresses activate NRF2, a structure-function analysis of NRF2 was performed. Domain structure-function analyses exploiting both human/mouse NRF2 (Moi et al., 1994) and the chicken NRF2 homologue (ECH; erythroid-derived CNC homology protein) (Itoh et al., 1995) have revealed the presence of six functional domains in NRF2, which are referred to as Neh (NRF2-ECH homology) domains (Fig. 3A) (Itoh et al., 1999). The Neh2 domain acts as a degron of NRF2 and is important for the regulation of NRF2. Neh1 is a basic region and leucine zipper domain that facilitates the binding of NRF2 and DNA. The Neh6 domain plays an important role in the interaction with β-transducin repeat-containing protein (β-TrCP) and acts as a KEAP1-independent degron (Chowdhry et al., 2013; Kuga et al., 2022; Rada et al., 2011). Neh4 and Neh5 are trans-activation domains that interact with transcriptional coactivators, including CREB-binding protein (CBP) (Katoh et al., 2001).

As the Neh2 domain acts as a degron and is important for the regulation of NRF2 activity, we conducted a yeast two-hybrid screening to identify the proteins that interact with this domain. Sequencing of positive colonies identified in the screening revealed that a single protein, KEAP1, predominantly interacts with the Neh2 domain (Itoh et al., 1999).
The KEAP1 molecule contains domains such as a Broad complex, Tramtrack and Bric-a-brac (BTB) domain, intervening region (IVR), double glycine repeat (DGR) domain, and carboxyl-terminal region (CTR; Fig. 3B) (Itoh et al., 1999). KEAP1 interacts with CUL3 and acts as a substrate adaptor for a CUL3-based ubiquitin E3 ligase (Fig. 3C). KEAP1-based E3 ubiquitin ligase specifically ubiquitinates NRF2 and allows NRF2 to undergo rapid proteasomal degradation (Iso et al., 2016). KEAP1 binds NRF2 via its C-terminal DGR and CTR domains (collectively named the DC domain), forming a homodimer with its N-terminal BTB domain (Fig. 3B, C). KEAP1 also acts as a sensor for various stresses, including ROS and toxic electrophiles (Saito et al., 2015; Suzuki et al., 2019; Takaya et al., 2012). Thus, KEAP1 is a ubiquitin ligase adaptor and acts as a sensor for various environmental stresses.
In normal unstressed cells, NRF2 protein levels are maintained at low levels owing to the KEAP1-mediated negative regulation of NRF2 through ubiquitination and proteasomal degradation. Two motifs in the Neh2 domain directly interact with two KEAP1 DC domains (Padmanabhan et al., 2006; Tong et al., 2006). In the absence of stress, the KEAP1-based E3 ligase ubiquitinates lysine residues in the Neh2 domain, and NRF2 is subsequently degraded through the proteasome pathway. Oxidative and electrophilic stresses disrupt two-site binding of NRF2 with KEAP1. As will be described later, autophagy chaperone p62 or small chemical inhibitors of KEAP1-NRF2 inhibit the protein–protein interaction (PPI) between NRF2 and KEAP1, inducing NRF2 stabilization and activation. This mechanism has been referred to as the Hinge and Latch mechanism (Horie et al., 2021).
KEAP1 Harbors Multiple Residues That Act as Stress Sensors
KEAP1 contains multiple cysteine residues that serve as unique sensors for various environmental stresses, including electrophiles, ROS, and reactive nitrogen species (RNS) (Uruno and Motohashi, 2011). The Cys151 residue in the BTB domain and the Cys273 and Cys288 residues in the IVR have been identified as important cysteine residues for sensing electrophiles. Cys151 contributes to NRF2 activation by electrophiles, including diethyl maleate (DEM), tert-butylhydroquinone (tBHQ), sulforaphane, DMF, and oleanolic triterpenoid 1-[2-cyano-3,12-dioxooleane-1,9(11)-dien-28-oyl] imidazole (CDDO-Im) (Kobayashi et al., 2009; Takaya et al., 2012). Cys288 has been shown to contribute to NRF2 activation by 15-deoxy-Δ 12,14 -prostaglandin J2 (15d-PGJ2) (Suzuki and Yamamoto, 2017).
Cys151, Cys273, and Cys288 are involved in the nitro fatty acids to mediate NRF2 induction (Suzuki and Yamamoto, 2017). In addition to these electrophiles, KEAP1 cysteine residues also play critical roles as sensors for hydrogen peroxide (H2O2), a representative ROS. Of interest, H2O2 modifies four cysteine residues of KEAP1, Cys226, Cys613, or Cys622/624, two of which sense H2O2 and activate the NRF2 signaling pathway (Fig. 3C) (Suzuki et al., 2019).
Some tricarboxylic acid cycle metabolites are known to bind KEAP1 cysteine residues and activate NRF2 signaling. Fumarate accumulates at high levels in renal carcinomas associated with mutations in the FH1 gene, which encodes fumarate hydratase, and in the kidneys of Fh1 knockout mice. Under these conditions, KEAP1 cysteine residues are succinylated as a result of fumarate binding, leading to inhibition of the ubiquitin ligase activity of KEAP1, which leads to the accumulation of NRF2 (Adam et al., 2011; Ooi and Furge, 2012; Ooi et al., 2011). It has also been reported that itaconate levels are elevated in inflammatory cells owing to upregulation of aconitate decarboxylase 1 (Irg1/Acod1), which metabolizes aconitate to itaconate, and accumulating itaconate alkylates the KEAP1 Cys151, Cys257, Cys273, Cys288, and Cys297 residues and induces NRF2 expression (Mills et al., 2018). It was reported that cell-permeable dimethyl itaconate activates NRF2 signaling in primary astrocytes to exert neuroprotective effects (Darvish Khadem et al., 2022).
KEAP1 Is an Important Target for Drug Development
Based on the molecular mechanism summarized previously, the KEAP1-NRF2 system has emerged as an important target for drug discovery. The elaborate molecular mechanism underlying KEAP1-mediated NRF2 activation reveals that NRF2 activators are potential drugs for the treatment of oxidative stress-related and inflammation-related diseases. In fact, compounds that are known to modify KEAP1 cysteine residues have been applied clinically (Majkutewicz, 2022). In addition, bardoxolone methyl and omaveloxolone (RTA-408), derivatives of synthetic triterpenoid CDDO, are in clinical trials for chronic kidney disease associated with type 2 diabetes mellitus (Nangaku et al., 2020) and clinical trials for Friedreich's ataxia (Lynch et al., 2022), respectively.
In addition to modifications of these, cysteine residue, p62, a molecular chaperone of autophagosomes, has been shown to be involved in the regulation of cellular NRF2 levels. When p62 aggregates, serine residues in the STGE motif in the KEAP1-interacting region of p62 are phosphorylated. p62 phosphorylated at these residues binds tightly to the NRF2-binding pocket of KEAP1 (Fig. 3C) and stops the degradation of NRF2 (Ichimura et al., 2013; Komatsu et al., 2010). The binding affinity of p-p62 for KEAP1 is increased, resulting in suppression of the PPI between KEAP1 and NRF2. Recently, nuclear magnetic resonance spectroscopy titration experiments revealed that low molecular weight or chemical inhibitors of the KEAP1-NRF2 PPI effectively disrupt the interaction between the NRF2 DLGex (Fukutomi et al., 2014) and KEAP1 DC motifs (Horie et al., 2021). Chemical inhibitors of the KEAP1-NRF2 PPI seem to be more specific but less toxic than inhibitors that bind to KEAP1 cysteine residues, indicating that this class of chemicals is attractive candidate NRF2 activators (Schmoll et al., 2017).
Perturbation of NRF2 Regulation Leads to Neurodegenerative Diseases
The stress response mediated by the KEAP1-NRF2 system plays important roles in various organs and tissues facing the outside of the body (Yamamoto et al., 2018). In addition, this system has been found to play a role in the CNS (Taguchi et al., 2010; Uruno et al., 2021; Uruno et al., 2020). RNA sequencing (RNA-seq) transcriptome analysis revealed that cortical neurons less abundantly express Nrf2 mRNA than astrocytes and microglia (Cuadrado et al., 2019; Zhang et al., 2014), suggesting that stress response is weaker in neurons than in astrocytes and microglia (Fig. 4). This may be owing to epigenetic inactivation of the NRF2 promoter early in cortical neuronal development (Bell et al., 2015).
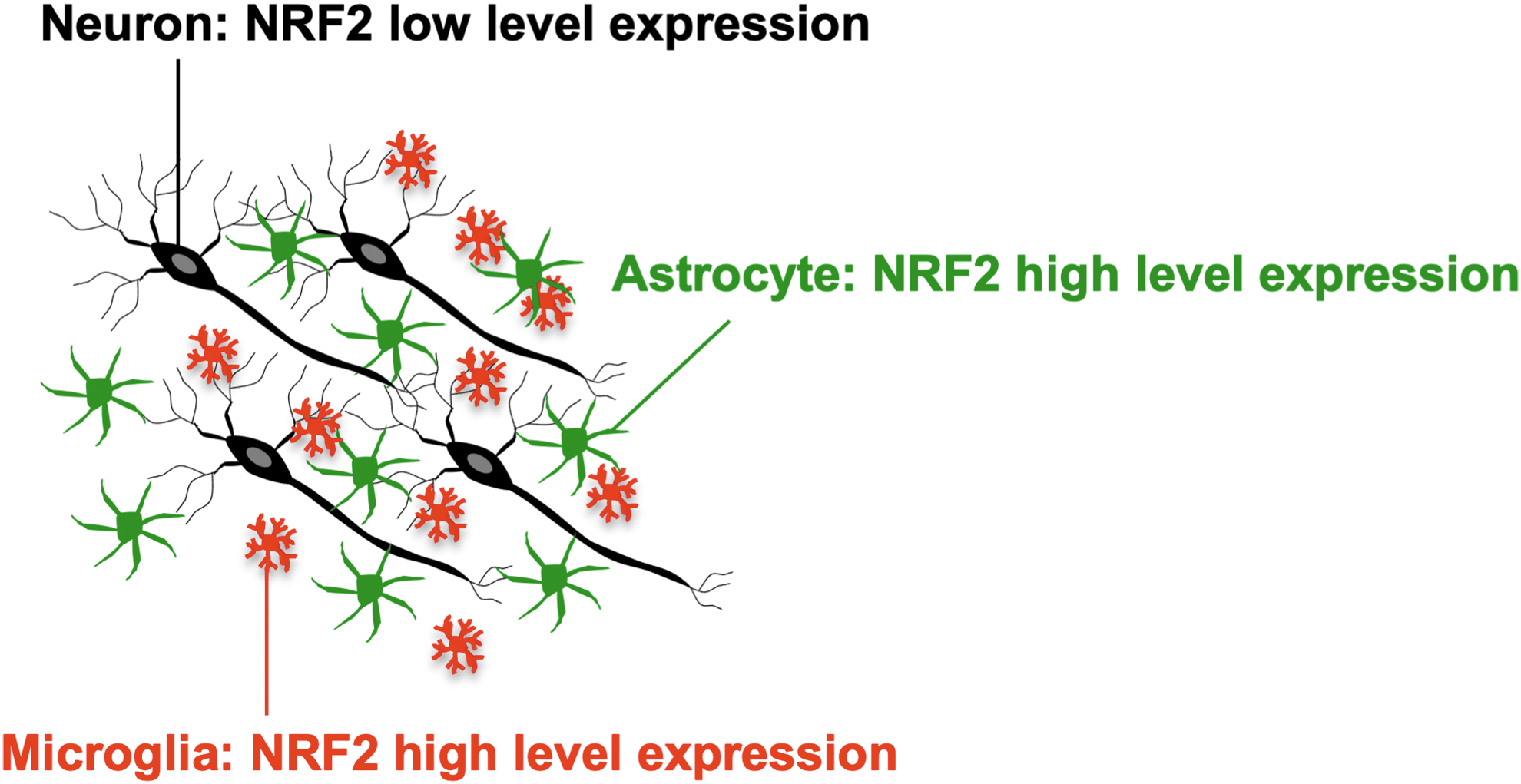
Indeed, although Keap1 depletion potently enhances the expression of NQO1, an NRF2 target gene, in astrocytes, it does not increase NQO1 expression in neurons (Yagishita et al., 2017). Keap1 depletion also induces NQO1 expression in microglia (Uruno et al., 2020). These observations suggest that the KEAP1-NRF2 system contributes to the expression of cytoprotective enzyme genes and maintenance of CNS homeostasis. Therefore, dysfunction or perturbation of this system is somehow related to neurodegenerative diseases.
DMF Is a Therapeutic Drug for MS
MS is a chronic inflammatory and degenerative disease of the CNS characterized by multiple spatial and temporal lesions (Chen et al., 2016) caused by demyelination owing to autoimmune-mediated inflammation and infiltration of lymphocytes and macrophages (McPherson and Anderton, 2013). In addition to inflammation, oxidative stress has been shown to contribute to the pathogenesis of MS (Arnold et al., 2014). The pathogenesis of MS is complex, and MS can be characterized by both relapses and progressive accumulation of disability independent of relapses. Therefore, multiple targets for suppressing both inflammatory and degenerative mechanisms need to be considered (Oh and Bar-Or, 2022).
Fumaric acid esters have been used for the treatment of psoriasis, a T cell–mediated skin disease (Balak, 2015). Fumaric acid esters have also been suggested to be efficacious in the treatment of MS (Schilling et al., 2006). Fumaric acid derivates, such as DMF, monoethylfumaric acid, and methyl hydrogen fumarate, also exist. It has been shown that oral administration of DMF can effectively treat psoriasis (Nieboer et al., 1989) and prevent CNS inflammation in experimental autoimmune encephalomyelitis (EAE) model mice (Schilling et al., 2006). DMF has been successfully used to suppress the clinical symptoms of MS (Fox et al., 2012; Gold et al., 2012; Hutchinson et al., 2014).
MS and the KEAP1-NRF2 System
Of importance, DMF induces NRF2 expression and activates NRF2 signaling. As given in Figure 5, elaborate biochemical analyses have revealed that DMF modifies the Cys151 residue of KEAP1 and inactivates the ubiquitin ligase activity of KEAP1, which leads to the accumulation and activation of NRF2 (Saito et al., 2015; Suzuki et al., 2019; Takaya et al., 2012). Indeed, DMF upregulates the expression of NRF2 target genes in the peripheral blood of humans (Hammer et al., 2018). DMF alleviates inflammation and symptoms in EAE model mice (Schilling et al., 2006; Vainio et al., 2022). Another NRF2 activator, TFM-735, has also been shown to ameliorate MS phenotypes in EAE model mice (Higashi et al., 2017).
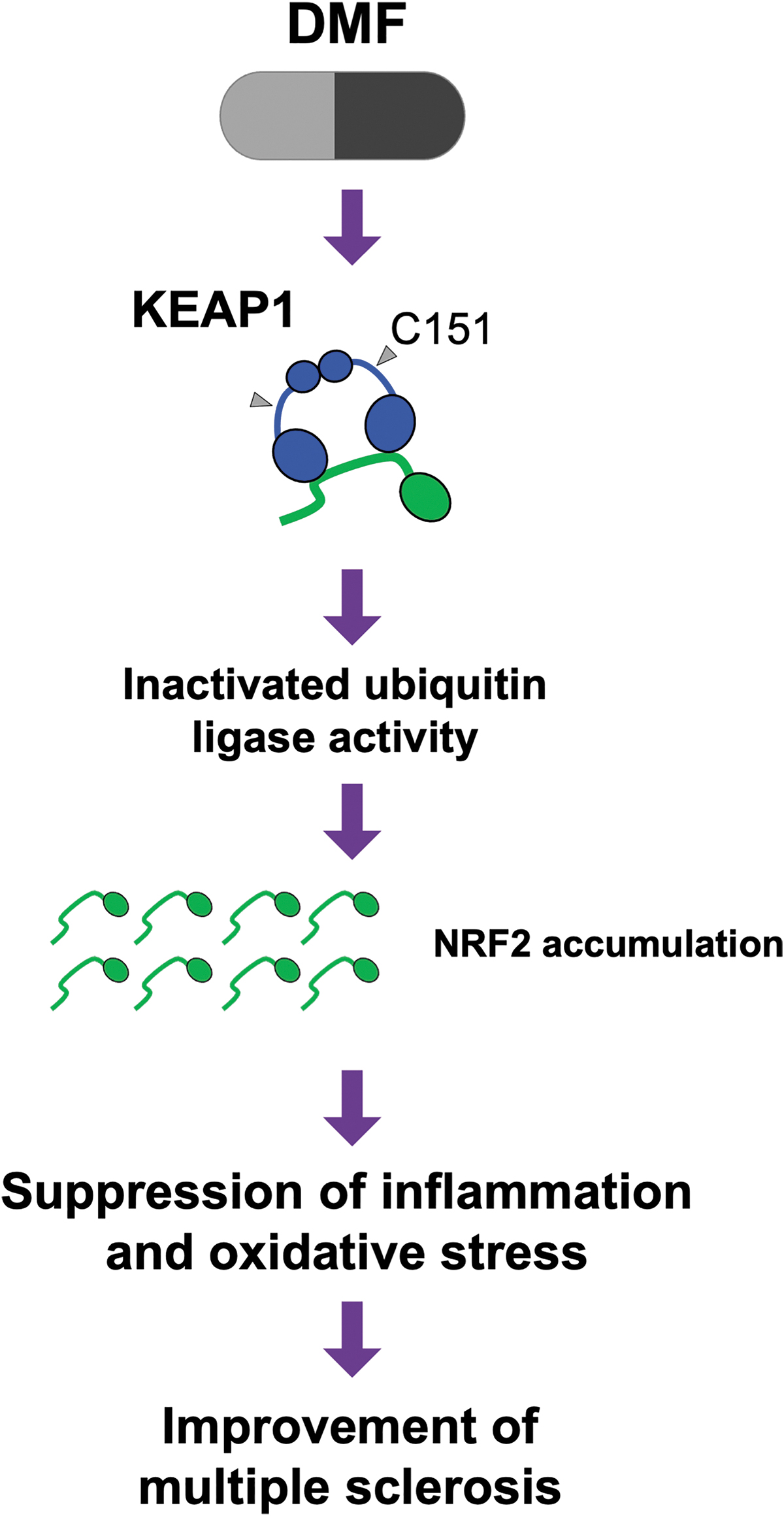
In renal tumors harboring mutations in the FH1 gene, which encodes fumarate hydroxylase 1, and in the kidneys of Fh1-depleted mice, fumarate accumulates at very high levels, and cysteine residues of KEAP1 are efficiently and heavily succinylated (Adam et al., 2011; Ooi and Furge, 2012; Ooi et al., 2011). Under these conditions, NRF2 is robustly activated. These findings suggest that the fumarate derivatives considered or used for clinical application may exert their efficacy through activation of NRF2 signaling by binding to cysteine residues of KEAP1 and inactivating KEAP1. It was reported that 6″-O-succcinylapigenin, which is a succcinylated derivative of a natural compound apigenin, induces translocation of NRF2 to the nucleus.
Administration of 6″-O-succcinylapigenin decreases KEAP1 expression and increases heme oxygenase-1 (HO-1) expression in brain and improves neurological scores in middle cerebral artery occlusion model rats (Zhang et al., 2019). Although the mechanism how 6″-O-succcinylapigenin decreases expression of KEAP1 remains unclear, these results suggest that succcinylation is a good target for neurodegenerative diseases. It seems quite plausible that the KEAP1-NRF2 system mediates the therapeutic effects of DMF. In contrast, it has been reported that the therapeutic effect of DMF in the EAE model is not inhibited by the depletion of Nrf2 (Schulze-Topphoff et al., 2016). Targeted knockout of Hca2, which encodes hydroxycarboxylic acid receptor 2, was shown to attenuate the therapeutic effect of DME in EAE model mice (Chen et al., 2014).
Although chemical/pharmacological models of NRF2 induction are useful for studying the therapeutic effects of drugs, pharmacological induction methods inherently have off-target effects. For instance, DMF has off-target effects on hydroxycarboxylic acid receptor 2 (Chen et al., 2014), NRF2 activators have off-target effects on peroxisome proliferator–activated receptor-γ (Wang et al., 2000). Thus, genetic NRF2 induction models utilizing KEAP1 hypomorphic or Keap1 knockdown mouse lines are useful for avoiding the off-target effects of chemicals (Taguchi et al., 2010; Uruno et al., 2016). Therefore, model mice in which NRF2 is genetically activated have been examined.
In the Keap1FA/FA mouse line displays, NRF2 is activated in whole tissues; thus, this mouse line is useful for evaluating the effects of NRF2 activation (Taguchi et al., 2010). Moreover, EAE was modeled in Keap1FA/FA mice. Although wild-type mice displayed elevation of EAE clinical scores and inflammation, the elevation of EAE scores and inflammation were effectively suppressed in Keap1FA/FA mice (Kobayashi et al., 2016), strongly supporting the notion that NRF2 activation prevents the development of MS symptoms in mice. Thus, the KEAP1-NRF2 system has emerged as an important therapeutic target for MS.
PD and the KEAP1-NRF2 System
PD is a progressive neurodegenerative disorder that is characterized by a triad of motor symptoms, specifically rigidity, bradykinesia, and tremor. PD is caused by abnormalities in the α-synuclein protein and the loss of dopaminergic neurons (Lotharius and Brundin, 2002). α-Synuclein, which is highly expressed in the brain, is a 140-amino acid protein lacking cysteine and tryptophan (Breydo et al., 2012). As given in Figure 6, misfolded α-synuclein proteins oligomerize, and these oligomerized proteins form fibrils (Lee and Trojanowski, 2006). α-Synuclein is a major component of Lewy bodies, which are found in the neurons of patients with PD (Lee and Trojanowski, 2006). Clinical trials of antibodies against α-synuclein for PD treatment are ongoing (Brys et al., 2019; Jankovic et al., 2018; Simuni et al., 2021), but therapeutic intervention appears to be difficult owing to the discrepancy in timing between the accumulation of α-synuclein and the onset of symptoms. Multiple therapeutic targets seem to be necessary for the treatment of PD.
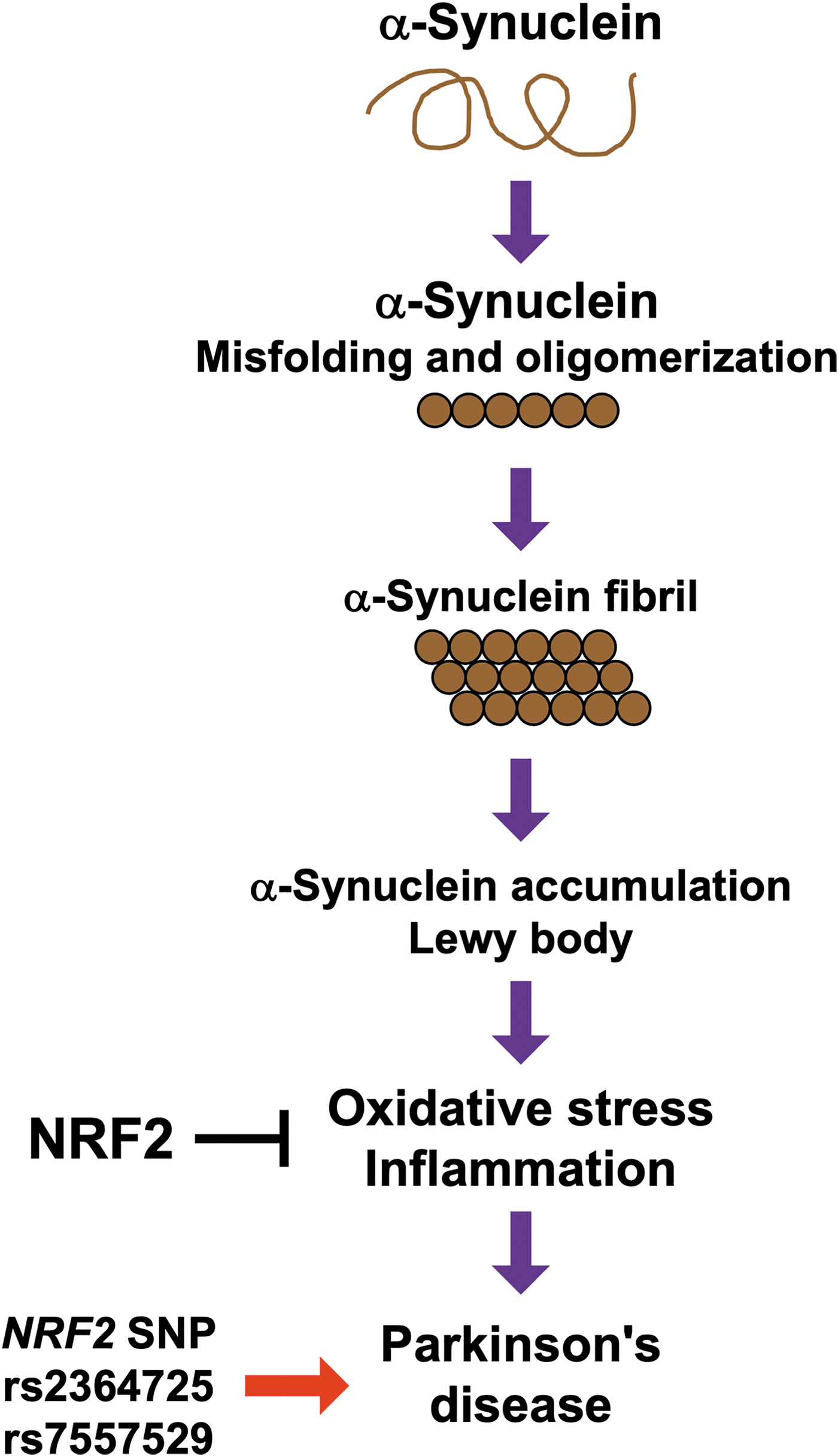
Both oxidative stress and inflammation have been shown to contribute to the pathogenesis of PD, and mitochondrial dysfunction in neurons may also be involved in the disease (Bento-Pereira and Dinkova-Kostova, 2021; Lotharius and Brundin, 2002; Tufekci et al., 2012). One recent study indicated that NRF2 plays important roles in mitochondrial function (Petrillo et al., 2020). Clinical studies have also revealed the importance of NRF2 in PD. The expression levels of NRF2, NQO1, glutamate-cysteine ligase, and glutathione reductase are elevated in the circulating leukocytes of patients with PD (Petrillo et al., 2020), suggesting that oxidative stress is involved in the pathogenesis of PD and that NRF2 signaling is activated in response to oxidative stress, although not sufficiently to fully overcome the stress. Furthermore, NRF2 single nucleotide polymorphisms (SNPs), including rs2364725 and rs7557529, are associated with the incidence of PD (Todorovic et al., 2015; von Otter et al., 2010a).
Nrf2 knockout mouse study in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)–induced PD model has been reported (Rojo et al., 2010). This study displayed that Nrf2 knockout aggravates neuroinflammation and neuron damage in brains of the MPTP model mice. It was also reported that administration of sulforaphane to mice increases NRF2, NQO1, and HO-1 expressions in the basal ganglia, indicating sulforaphane activates NRF2 signaling in brains. Sulforaphane exerts protective effects against MPTP-induced death of nigral dopaminergic neurons in wild-type mice. However, the effects of sulforaphane are not observed in Nrf2 knockout mice. Sulforaphane also suppresses astrogliosis and microgliosis and decreases proinflammatory cytokine level (Jazwa et al., 2011).
An animal PD model study with stereotaxic delivery of an adeno-associated viral (AAV) vector for expression of human α-synuclein in the ventral midbrain was executed (Lastres-Becker et al., 2012). The AAV human α-synuclein model displays that Nrf2 knockout exacerbates degeneration of nigral dopaminergic neurons and dopaminergic neuron. In response to exogenous expression of α-synuclein, Nrf2 knockout microglia fails to activate the expression of HO-1 and NQO1. Postmortem brain tissue samples from PD patients showed increased HO-1 expression in astrocytes and microglia.
The effects of DMF administration in the AAV vector for expression of human α-synuclein in the ventral midbrain of mice was also studied (Lastres-Becker et al., 2016). An administration of DMF exerts protective effects in nigral dopaminergic neurons against toxicity of α-synuclein and suppresses neuroinflammation after delivery of the AAV vector expressing human α-synuclein to ventral midbrain in mice, and Nrf2 knockout cancels these protective effects.
The effects of the synthetic triterpenoid CDDO methyl amide (CDDO-MA), a potent activator of NRF2 and its signaling cascade, on the MPTP-induced PD model have been studied. Oral administration of CDDO-MA protects against MPTP-induced nigrostriatal dopaminergic neurodegeneration and pathological α-synuclein accumulation and rescues striatal damage caused by oxidative stress and impairment of GSH metabolism (Yang et al., 2009).
Mechanistic studies on the role of NRF2 in the progression of PD have given rise to an intriguing possibility. The synthetic triterpenoid CDDO trifluoroethyl amide (CDDO-TFEA) and the electrophile tBHQ promote endogenous NRF2-dependent α-synuclein clearance in astrocytes but not in neurons (Baxter et al., 2021). The stress response of the KEAP1-NRF2 system appears to be weaker in neurons than in astrocytes, which may be owing to the high level of histone acetylation of the NRF2 locus in neurons. Therefore, treatment of neurons with histone deacetylase (HDAC) inhibitors may induce derepression of NRF2 gene expression. Of importance, epigenetic NRF2 derepression restores the ability of neurons to be affected by NRF2 activators and enables NRF2 activators to induce α-synuclein clearance. Therefore, HDAC inhibition is an alternative therapeutic strategy for PD, as it enhances the effect of the KEAP1-NRF2 system. As given in Figure 6, these findings indicate that NRF2 affects neurons to protect against PD development; thus, the KEAP1-NRF2 pathway is as a new therapeutic target for PD.
Noncanonical NRF2 Regulation in the Brain and PD
In addition to the KEAP1-NRF2 system, the canonical pathway responsible for activating NRF2, noncanonical mechanisms responsible for regulating NRF2 signaling have been reported in the brain. Two noncanonical regulators of NRF2 signaling in the brain may play important roles in preventing PD development. One of these regulatory mechanisms is mediated by the BACH1-sMAF heterodimer, which lacks transactivation activity. BACH1 is a CNC family transcription factor, and the BACH1-sMAF heterodimer binds to the CsMBE. BACH1-sMAF acts as an NRF2 repressor within cells. Bach1 gene knockout results in activation of the NRF2 pathway and prevents MPTP-induced PD in mice (Ahuja et al., 2021). A substituted benzimidazole was found to suppress BACH1, and oral administration of this BACH1 inhibitor was found to activate NRF2 signaling and prevent MPTP-induced PD in mice.
The other regulator of NRF2 signaling is the potent inhibitor of dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A), altered generation of neurons 2 (ALGERNON2), which has been identified as a novel drug candidate for PD. ALGERNON2 activates NRF2 in the brain, enhances neuronal survival, suppresses the production of proinflammatory cytokines, and rescues neurodegeneration in an MPTP-induced PD model. As ALGERNON2 stabilizes the cyclin D1/p21 complex, it may activate NRF2 through p21 (Nakano-Kobayashi et al., 2020). Other noncanonical mechanisms of NRF2 activation have also been reported (Al-Mubarak et al., 2021; Habas et al., 2013), but they need to be further verified.
AD and the KEAP1-NRF2 System
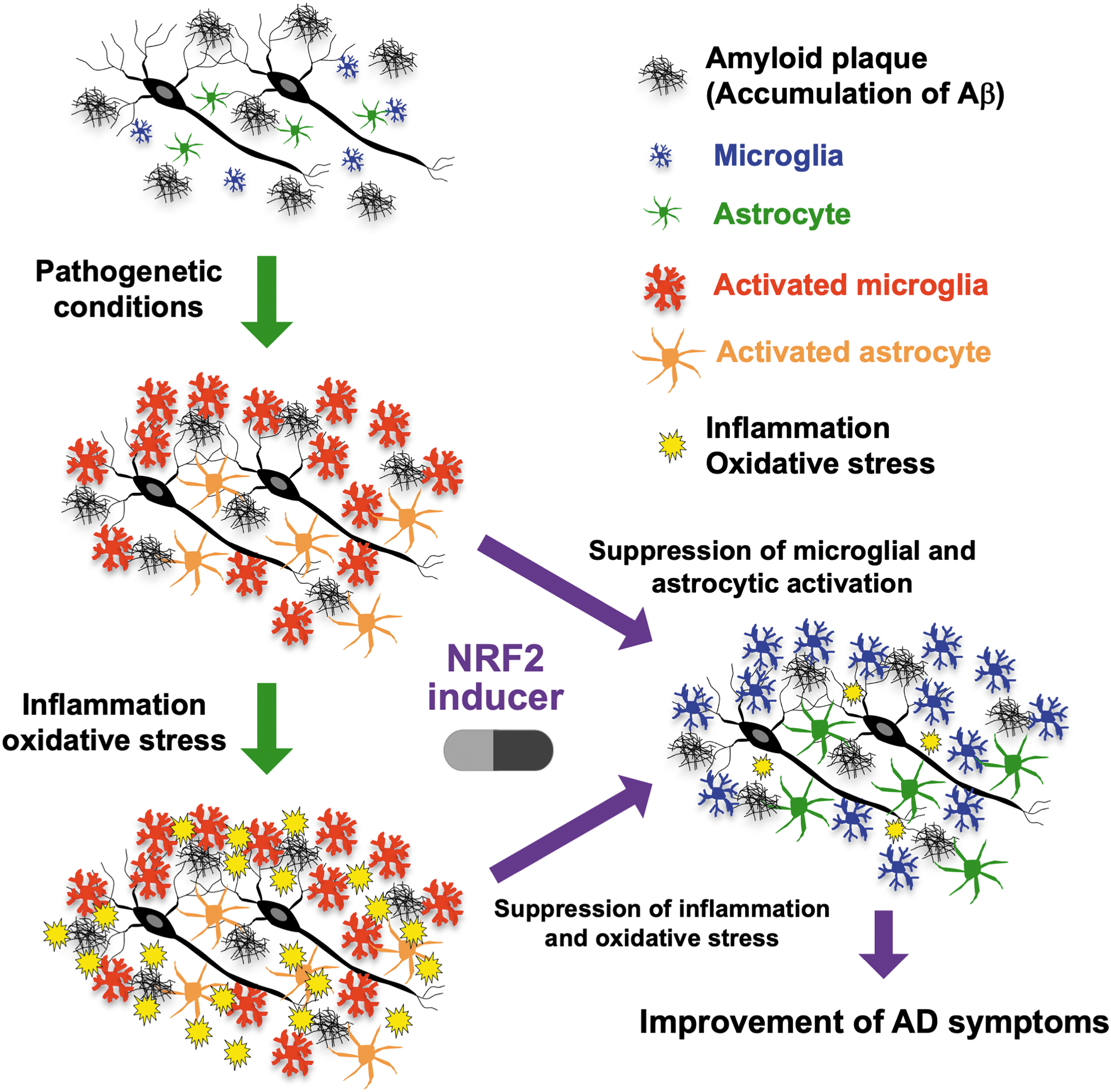
AD is the most common disease associated with cognitive impairment and dementia. The incidences of mild cognitive impairment and dementia are increasing worldwide because of the aging of the population. As given in Figure 7, AD has been recognized as one a socially important disease.
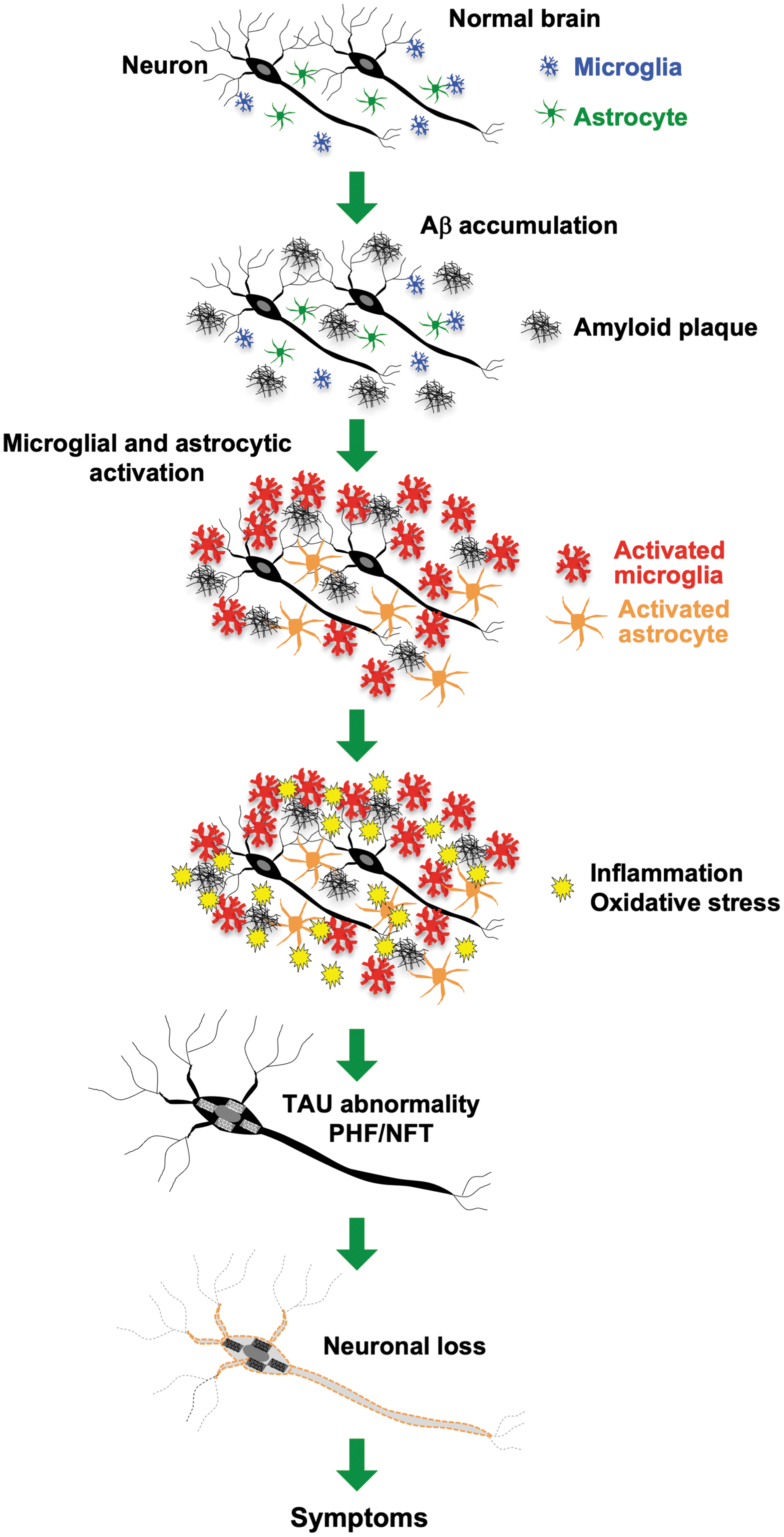
It is known that abnormalities in amyloid β (Aβ) and tau protein contribute to the pathogenesis of AD (Bloom, 2014; Selkoe, 1991). In AD, accumulation of Aβ induces microglial and astrocyte activation, which is followed by the development of inflammation and oxidative stress in the brain (Markesbery, 1999; Meraz-Ríos et al., 2013). These alterations induce secondary degenerative changes, including abnormalities in tau protein, for example, the formation of paired helical filaments (PHFs) and neurofibrillary tangles (NFTs). PHF and NFT formation induce structural and functional changes and subsequent neuronal loss, leading to the onset of symptoms and signs of dementia. Because NRF2 is known to exert antioxidant and anti-inflammatory effects, it seems reasonable to hypothesize that activation of NRF2 signaling may alleviate AD pathology.
In 2007, through immunobiological analyses, NRF2 expression in the nucleus and cytoplasm of neurons was examined. In the normal hippocampus, NRF2 is predominantly expressed in the nucleus; however, NRF2 is predominantly localized in the cytoplasm of hippocampal neurons in the AD brain, suggesting that NRF2 accumulation in the nucleus is attenuated in the AD brain (Ramsey et al., 2007). The expression of NRF2 and the activities of the antioxidant enzymes glutathione peroxidase and catalase were found to be suppressed in the superior temporal gyrus in patients with AD (Youssef et al., 2018). Overexpression of mutant P301S tau increases KEAP1 acetylation at K312 and increases KEAP1 levels while decreasing NRF2 levels in the hippocampus and primary neurons (Xie et al., 2022). These wide-ranging observations suggest that the KEAP1-NRF2 system is activated in AD, but that its activity may not be sufficient to protect neurons from degeneration.
It has been reported that NRF2 mRNA expression is increased in the temporal cortices of patients with AD (Castillo et al., 2017). Nrf2 mRNA levels are also elevated in the cortices of AppNL-G-F/NL-G-F knock-in AD model mice (Castillo et al., 2017). Although the cellular activity of NRF2 is regulated mainly through its proteasomal degradation (Kobayashi et al., 2004), enhanced NRF2 signaling may also increase Nrf2 mRNA expression levels through two CsMBE-like sequences in the regulatory region of Nrf2 (Kwak et al., 2002). Therefore, an increase in Nrf2 mRNA expression levels may be caused by the KEAP1-NRF2 system–mediated stress response in the cerebral cortices of patients with AD and model animals.
Of interest, it has been found that the SNP rs242561 results in the generation of a CsMBE, a binding motif for the NRF2-sMAF heterodimer, in a regulatory region of the MAPT gene, which encodes the microtubule-associated protein tau (Wang et al., 2016). This SNP, which results in a polymorphic CsMBE allele, is associated with high mRNA expression of MAPT and is in complete linkage disequilibrium with a highly protective allele identified in multiple GWASs of neurodegenerative diseases.
To monitor diagnosis and prognosis of AD, blood biomarker analysis was studied in early AD patients. The study was performed by a targeted transcriptome analysis on mild AD patients and matched controls (Milanesi et al., 2021). Many inflammation and redox genes were differentially expressed in blood of patients with AD, and based on the gene expression profile, NRF2 and NF-κB, the master regulators of redox and inflammation homeostasis, are disturbed in the blood samples of AD. These redox and inflammation genes related to NRF2 and NF-κB could be useful biomarkers for monitoring diagnosis and prognosis of mild AD patients.
A case–control study with 725 AD patients and 845 controls for genetic variation for NRF2 and KEAP1 genes was performed, and SNPs and haplotypes were analyzed for associations with disease risk and AD cerebrospinal fluid biomarkers (von Otter et al., 2010b). NRF2 and KEAP1 are not associated with risk of AD. However, one haplotype allele of NRF2 is associated with earlier age of AD onset, supporting that common variants of the NRF2 gene might affect disease progression.
Similar to these changes in human AD patients, Nrf2 depletion increases phosphorylated tau protein levels in the mouse brain (Jo et al., 2014). The NRF2 activator sulforaphane upregulates the expression of the autophagy adaptor protein NDP52 (CALCOCO2), which contains three CsMBE-like sequences in its regulatory region, in neurons. Overexpression of NDP52 facilitates the clearance of phosphorylated tau in the presence of an autophagy stimulator. These findings from mouse model studies in combination with those from human studies support the notion that activation of the NRF2 signaling pathway may ameliorate AD pathology.
NRF2 Loss-of-Function Mouse Model and AD
Studies using Nrf2 gene knockout mice have revealed that NRF2 protects mice from neuronal damage. APPV717I and TAUP301L double transgenic AD model mice were crossed with Nrf2 knockout mice to generate compound Nrf2 knockout (Nrf2 KO) AD model mice, and it was found that the Nrf2 KO::AD mice display increased Aβ and phosphorylated tau levels. Similarly, oxidative stress and inflammation are enhanced in the brains of these compound mutant mice. Of note, young adult Nrf2 KO::AD mice show impairment of spatial learning and memory (Rojo et al., 2017). Nrf2 KO::AD mice died 2 months earlier than APPV717I and TAUP301L mice and exhibited motor deficits and terminal spinal deformities. Astrogliosis and microgliosis are exacerbated in the brains of Nrf2 KO::AD mice (Rojo et al., 2018).
Compound Nrf2 KO APP/PS1 and 5xFAD transgenic mice have also been generated. Nrf2 deficiency significantly exacerbates cognitive impairment and spatial learning and memory deficits in APP/PS1 transgenic mice without disrupting motor function. Aβ levels and microglial marker expression are increased in the brains of Nrf2 KO::APP/PS1 transgenic mice, and these changes are linked to cognitive impairment (Branca et al., 2017).
It was reported that NRF2 negatively regulates the expression of the Bace1 gene, which encodes β-secretase. β-Secretase is an enzyme that cleaves amyloid precursor protein, and its expression is increased in the brains of patients with AD (Bahn et al., 2019). Nrf2 deficiency increases Aβ levels and the number of amyloid plaques in the brains of 5xFAD transgenic mice and exacerbates cognitive impairment. These wide-ranging observations further support the hypothesis that NRF2 activation inhibits the progression of AD pathology.
NRF2 Gain-of-Function Mouse Model and AD
To develop drugs for the treatment of AD, the effect of NRF2 gain-of-function mutations has been studied utilizing various AD model animals. It has been reported that daily oral gavage of the NRF2-inducing compound DMF alleviates cognitive impairment in APPV717I and TAUP301L double transgenic mice (Rojo et al., 2018). DMF also decreases the expression of inflammation and glial cell markers in APPV717I and TAUP301L transgenic mice, indicating that DMF suppresses inflammation and pathological glial activation in the brains of AD model mice. The electrophile 6-methylsulfinylhexyl isothiocyanate (6-MSITC), which is found in horseradish, has been found to induce NRF2 signaling (Hou et al., 2011). We found that when 6-MSITC was orally administered to AppNL-G-F/NL-G-F mice, cognitive impairment was alleviated, and the number of activated microglia in the brains decreased (Uruno et al., 2020).
Sulforaphane is a natural electrophilic compound contained in broccoli sprouts. Sulforaphane interacts with the Cys151 residue of KEAP1, resulting in NRF2 signaling activation (Takaya et al., 2012). Sulforaphane ameliorates cognitive impairment in 5xFAD and 3xTg-AD mice and reduces Aβ accumulation in the brains of these AD model mice (Bahn et al., 2019).
The synthetic triterpenoid CDDO and its derivatives also activate the KEAP1-NRF2 system (Honda et al., 1998; Liby et al., 2005). For instance, CDDO-MA activates the NRF2 signaling pathway (Yang et al., 2009), and when administered to Tg19959 AD model mice, CDDO-MA was found to decrease the number of amyloid plaques and Aβ42 levels (Dumont et al., 2009). CDDO-MA relieves microgliosis and oxidative stress and improves spatial memory retention in Tg19959 mice.
The currently available evidence suggests that the KEAP1-NRF2 system exerts beneficial therapeutic in AD (Fig. 8). For instance, studies on Keap1FA/FA mice, in which NRF2 is genetically activated by Keap1 knockdown, have provided important insight. Keap1FA/FA mice were crossed with AppNL-G-F/NL-G-F knock-in AD model mice (Saito et al., 2014) to generate AppNL-G-F/NL-G-F::Keap1FA/FA mice. Although control AppNL-G-F/NL-G-F mice show cognitive impairment in the passive avoidance task, AppNL-G-F/NL-G-F::Keap1FA/FA mice, in which NRF2 is activated, do not display such deficits. In the brains of these AD model mice with NRF2 activation, GSH levels are elevated, but oxidative stress marker levels are decreased (Uruno et al., 2020). Whereas the expression of the inflammatory cytokine gene Il6 is increased in the brains of control AD mice, Il6 expression is suppressed in the brains of AD mice with NRF2 activation.
Effects of Space Travel and Aging on the NRF2-Mediated Stress Response
NRF2 plays important roles in the aging process and is an important target for elucidating the mechanisms of aging-related neurodegenerative disorders. It has been clearly shown that the physical changes associated with aging are substantially accelerated in space. Aging occurs faster and its effects are more marked during space travel than on Earth. Therefore, space is a good model for studying accelerated aging. In space, our body is exposed to various stresses, such as microgravity and cosmic radiation. Microgravity rapidly induces osteoporosis and muscle atrophy, whereas cosmic radiation induces oxidative stress and DNA damage.
We hypothesized that NRF2 may protect our body from the stresses associated with space travel. To understand the effects of NRF2 against stresses associated with space travel, six wild-type and six Nrf2 KO mice were launched from the Kennedy Space Center and kept in the International Space Station (ISS) “Kibo” Experiment Module for 31 days. All mice were returned safely to Earth (Suzuki et al., 2020). We collected blood samples from the tail before launch, during the stay in the ISS, and after the return to Earth, and plasma metabolome analysis was performed. In addition, after the return to Earth, inferior vena cava plasma and tissue samples were taken from mice, and metabolome and transcriptome analyses were conducted (Hayashi et al., 2021; Suzuki et al., 2022; Suzuki et al., 2020, Uruno et al., 2021).
Lipid metabolome analyses of plasma and brain samples from wild-type mice that experienced spaceflight showed that plasma phospholipid levels were elevated during spaceflight, whereas brain phospholipid levels were decreased after spaceflight; however, these changes were attenuated in Nrf2 KO mice (Uruno et al., 2021). The relationship between accelerated aging in space and neurodegenerative diseases remains unclear; nonetheless, these changes in lipid levels observed after spaceflight are similar to those observed in the brains of AD model mice (Uruno et al., 2020).
Transcriptome analyses showed that the expression of NRF2 target detoxification, metabolism, and antioxidant enzyme genes was induced in wild-type mice after spaceflight but that the induction of the expression of these genes was severely attenuated in Nrf2 KO mice (Suzuki et al., 2020). In addition, metabolome analysis revealed that plasma concentrations of glycine and succinate were decreased after spaceflight but that these changes were abolished in Nrf2 KO mice. Intriguingly, metabolome analyses of human plasma samples collected in a cohort study, the Tohoku Medical Megabank Project, showed that glycine and succinate levels were decreased in the plasma of individuals aged 60–80 years compared with the plasma of those aged 20–40 years, suggesting that the metabolic changes observed in the plasma of mice after spaceflight are comparable with age-related metabolic changes in humans (Suzuki et al., 2020).
Conclusions
The role of the KEAP1-NRF2 system in neurodegenerative diseases is an attractive research topic. In animal models of neurodegenerative diseases, NRF2 activation exerts therapeutic effects. NRF2 is also important factor for monitoring diagnosis and prognosis of neurodegenerative diseases. DMF is clinically used for treatment of MS, and DMF and other type of NRF2 inducers seem to be applicable for treatment of PD and AD. CDDO derivative omaveloxolone may exert therapeutic efficacy for treatment of neurodegenerative diseases. Based on these observations, the KEAP1-NRF2 system has become an attractive therapeutic target.
Footnotes
Authors' Contribution
A.U. and M.Y. conceptualized the content and wrote the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Platform Project for Supporting Drug Discovery and Life Science Research [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from the Japan Agency for Medical Research and Development (AMED), Grant No. JP22ama121038; the Tohoku Medical Megabank Project from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the AMED, Grant Nos. JP20km0105001 and JP21tm0124005; Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS), Grant Nos. 19H05649 and 20K07352; the Takeda Foundation and the Naito Foundation.