
Abstract
Significance:
Several therapeutic strategies for cancer treatments have been developed with time, and significant milestones have been achieved recently. However, with these novel therapies, not all cancer types respond and in the responding cancer types only a subset is affected. The failure to respond is principally the result that these cancers develop several mechanisms of resistance. Thus, a focus of current research investigations is to unravel the various mechanisms that regulate resistance and identify suitable targets for new therapeutics.
Recent Advances:
Hence, many human cancer types have been reported to overexpress the inducible nitric oxide synthase (iNOS) and it has been suggested that iNOS/nitric oxide (NO) plays a pivotal role in the regulation of resistance. We have postulated that iNOS overexpression or NO regulates the overexpression of pivotal anti-apoptotic gene products such as B-cell lymphoma 2 (Bcl-2), B-cell lymphoma extra large (Bcl-xL), myeloid cell leukemia-1 (Mcl-1), and survivin. In this report, we describe the various mechanisms, transcriptional, post-transcriptional, and post-translational, by which iNOS/NO regulates the expression of the above anti-apoptotic gene products.
Critical Issues:
The iNOS/NO-mediated regulation of the four gene products is not the same with both specific and overlapping pathways. Our findings are, in large part, validated by bioinformatic analyses demonstrating, in several cancers, several direct correlations between the expression of iNOS and each of the four examined anti-apoptotic gene products.
Future Directions:
We have proposed that targeting iNOS may be highly efficient since it will result in the underexpression of multiple anti-apoptotic proteins and shifting the balance toward the proapoptotic gene products and reversal of resistance. Antioxid. Redox Signal. 39, 853–889.

I. Introduction
Drug resistance in cancer has become the driving force for developing new therapeutics, as well as a basis in the understanding of cancer pathophysiology. The effectiveness of many conventional therapies can be hindered by both acquired and innate drug resistance (Glickman and Sawyers, 2012; Ward et al., 2021). The long list of the reported underlying mechanisms of resistance include, among others, increased drug efflux (Gottesman and Pastan, 2015), drug inactivation (Shelton et al., 2016), mutation of target sites (Wood, 2015), and resistance to cytotoxic death (Debatin, 2004; Sui et al., 2013).
Conventional therapies, such as chemotherapy, radiotherapy, and cell-mediated immunotherapy, all induce anti-cancer cytotoxic effects, including primarily the induction and promotion of programmed cell death, a process known as apoptosis (Binju et al., 2019; Kerr et al., 1994). Apoptosis itself is a highly regulated process involving several signaling pathways that are mobilized by pro- and anti-apoptotic proteins. The expression and the cellular localization of these proteins determine whether a cell survives or dies by apoptosis.
Due to the nature of cancer and its association with uncontrolled proliferation and disrupted cell cycle machinery function, apoptosis is also commonly dysregulated in cancerous cells and especially in chemoresistant cells (Green and Evan, 2002; Jan and Chaudhry, 2019). Moreover, the abnormal upregulation of anti-apoptotic proteins, otherwise known as pro-survival proteins, has been shown to induce cytoprotective effects against chemotherapeutic agents.
Therefore, their expression levels have been considered as prime determinants of the development of drug resistance in tumor cells (Fisher et al., 1993; García-Aranda et al., 2018; Li et al., 2020; Lotem and Sachs, 1992).
Nitric oxide (NO) is an endogenous gaseous free radical, synthesized intracellularly by the three NO synthase (NOS) isoforms, namely, endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS), and neuronal nitric oxide synthase (nNOS). NO exhibits well defined roles in physiological processes, whereas its contribution in the pathophysiology of several diseases, including cancer, is still very complex and quite perplexing. Among the three NOS isoforms, iNOS expression levels have been mostly implicated in the regulation of multiple cancer functions, with both promoting and inhibiting actions having been described (Hickok and Thomas, 2010).
Recent advances over the regulatory effects of NO in cancer have unrevealed linkages between the overexpression/induction of iNOS/NO signaling and the expression profiles of several anti-apoptotic proteins that may interfere with the drug-resistant phenotype of several cancer types (Bonavida, 2020; Fantappiè et al., 2015; Iyer et al., 2008; Singh and Gupta, 2011).
The current review aims at highlighting the interplay between iNOS/NO expression and apoptosis induction, emphasizing on the iNOS/NO-mediated regulation of four major anti-apoptotic proteins, namely B-cell lymphoma 2 (Bcl-2), B-cell lymphoma extra large (Bcl-xL), myeloid cell leukemia-1 (Mcl-1), and survivin. The underlying mechanisms of the NO-mediated regulation of the aforementioned genes are further discussed at both the transcriptional, post-transcriptional, and post-translational levels, whereas bioinformatic analysis was performed to delineate putative correlations among the expressions of several involved genes in various cancers.
Moreover, in the context of the therapeutic efforts for reversal of drug resistance in cancer, various means of targeting either iNOS or the targeted anti-apoptotic proteins are also presented.
II. Apoptosis
A. General features of apoptosis and differentiation from necrosis
Apoptosis largely plays a homeostatic role, as well as a role in development, disease defense, and cell health regulation. There are numerous triggers of apoptosis, as well as several activating pathways. Failure of proper apoptosis activation can lead to the development of several disease states, including the growth of multiple types of cancer (Fabregat et al., 2007; Picard and Shirihai, 2022; Reed, 1999; Yang et al., 2010). Deoxyribonucleic acid (DNA) damage, unregulated cell proliferation, exposure to toxins, and various diseases are all known as apoptosis inducers (Elmore, 2007; Xiao et al., 2019; Zheng et al., 2018).
Apoptosis progression is characterized by several morphological changes in cellular structures, identified mainly in early light and electron microscopy studies (Häcker, 2000; Kerr et al., 1972; Voss and Strasser, 2020). Cell shrinkage followed by pyknosis are major morphological hallmarks associated with apoptosis induction. Pyknosis occurs as the cell's nuclei and chromatin condense and can be observed in necrotic cells, as well.
Organelles and other cell structures are condensed and packaged within the cytoplasm and remain membrane bound, unlike cells undergoing necrosis (D'Arcy, 2019; Obeng, 2020). After pyknosis, cell chromatin material undergoes karyorrhexis, in which the nucleus begins to disintegrate and crumble. Subsequently, cells show blebbing along the plasma membrane and begin to break into fragments called apoptotic bodies.
The process is called budding. Apoptotic bodies contain cytoplasm, embedded organelles, and also nucleic fragments. These fragments then undergo phagocytosis by either macrophages or surrounding healthy cells, allowing for the reuptake and reutilization of cellular matter characterizing a non-inflammatory result (Hengartner, 2001; Savitskaya and Onishchenko, 2015).
B. Apoptotic signaling
The extrinsic pathway
The extrinsic apoptotic pathway is initiated by specific binding of death ligands to corresponding death receptors on the surface of the targeted cell, leading to cell death. The main death ligands belong to the tumor necrosis factor (TNF) family and include the Fas ligand (FasL), the TNF related apoptosis inducing ligand (TRAIL), and TNF.
The major specific death receptors responsible for TNF family ligand binding include Fas, TRAIL-R, and TNFR1, respectively (Goldar et al., 2015; Liu et al., 2017; Shlyakhtina et al., 2017). FasL binding causes the recruitment of the adapter protein Fas associated death domain (FADD), whereas TNF ligand binding leads to the recruitment of the TNF receptor associated death domain (TRADD). FADD associates with procaspase-8 through interaction and dimerization of the death effector domains (DED).
These interactions form an activated complex, the death inducing signaling complex (DISC), and pro-caspase-8 and pro-caspase 10 cleavages yielding activated caspase-8 and caspase-10, respectively (Kruidering and Evan, 2000; Loftus et al., 2022; Wajant, 2002; Walczak, 2013). Caspase-8 and caspase-10 are then able to activate several downstream executioner caspases, for example, caspases 3, caspase 7, and poly ADP-ribose polymerase (PARP) cleavage (Kaufmann et al., 1993; Loftus et al., 2022) leading to DNA fragmentation and apoptosis.
TNFR1 activation and TRADD recruitment induces the formation of a nuclear factor-kappa B (NF-κB) signaling complex called complex I in the plasma membrane. TRADD, Receptor interacting protein kinase 1 (RIP1), and TRAF2 are components of complex I (Bertheloot et al., 2021; Pobezinskaya and Liu, 2012). The TNFR1 pathway can mediate signals for both cell survival and cell death. Generally, complex I mediates signals for cell survival, whereas Complex II mediates signals for cell death.
In cells fated for apoptosis, complex I is internalized or endocytosed contributing to complex II formation (Schneider-Brachert et al., 2004; Siegmund et al., 2022). Proapoptotic complex II forms in the cytoplasm and FADD is recruited (Micheau and Tschopp, 2003; Wajant and Siegmund, 2019). Caspase-8 or caspase-10 is then cleaved within complex II, and downstream executioner caspases are eventually cleaved and activated.
The intrinsic pathway
The main characteristics of the intrinsic pathway are mitochondrial outer membrane permeabilization (MOMP), cytochrome-c release, formation of apoptosome complex, and activation of caspase-9 (Zaman et al., 2014). Intrinsic apoptosis senses a wide range of internal stress signals that are usually produced by cellular stresses, such as DNA damage, high levels of reactive oxygen species (ROS), endoplasmic reticulum (ER) stress, or nutrient starvation.
All types of intracellular stress signals eventually converge at the mitochondria where the fate of the cell is decided. Members of the Bcl-2 family can be both pro- and anti-apoptotic, and a balance between their levels determines whether apoptosis occurs or not.
The Bcl-2 family of proteins are highly associated with the apoptosis regulation in the mitochondria. Bcl-2 proteins can generally be categorized into pro-apoptotic, anti-apoptotic, and Bcl-2 homology 3 (BH3) only proteins. The common pro-apoptotic Bcl-2 proteins include the Bcl-2 homologous antagonist killer (Bak), Bcl-2 related protein X (BAX), Bcl-2 antagonist of cell death (BAD), BH3 interacting-domain death agonist (Bid), BCL-2-interacting mediator of cell death (Bim), and Bcl-2-interacting killer (Bik) members, whereas Bcl-2, Bcl-x, Bcl-w, Bcl-xL, and Bcl-x short (Bcl-xS) are known as Bcl-2 proteins with anti-apoptotic function (Aouacheria et al., 2005; Blaineau and Aouacheria, 2009; Hinds et al., 2007; Strasser and Vaux, 2018).
The levels of several Bcl-2 proteins are essential in regulating the MOMP and the release of cytochrome c and other apoptotic factors (Cory and Adams, 2002; Morris et al., 2021; Schuler and Green, 2001). Pro-apoptotic proteins BAX and Bak are necessary for the MOMP (Jourdain and Martinou, 2009; Landes and Martinou, 2011; Morris et al., 2021). The MOMP mediates movement of mitochondrial pro-apoptotic factors into the cytosol.
The ratios of anti-apoptotic to pro-apoptotic proteins can be important measures for the strength of the apoptosis signals. Members of the Bcl-2 family are also regulated by the p53 tumor suppressor protein (Czabotar et al., 2014; Schuler and Green, 2001). Puma and Noxa, BH3 only proteins, are strongly associated with p53-mediated apoptosis through increased BAX expression (Capuozzo et al., 2022; Li, 2021; Liu et al., 2003; Oda et al., 2000).
Other groups of proteins like Smac/DIABLO and HtrA2/Omi have been shown to inhibit IAPs (inhibitors of apoptosis proteins) resulting in the promotion of the apoptotic signal (Dohi et al., 2004; Jaiswal et al., 2015; Schimmer, 2004; Srinivasula et al., 2001; van Loo et al., 2002) Smac/DIABLO controls the proliferation of cancer cells by regulating the phosphatidylethanolamine synthesis (Pandey et al., 2021).
III. Expression of the Anti-Apoptotic Proteins Bcl-2, Bcl xL, Mcl-1, and Survivin in Cancer
A. B-cell lymphoma 2
Bcl-2 was identified as a proto-oncogene, as its increased expression confers cytoprotective effects and poor outcomes in patients with various cancer types (Reed et al., 1988; Roh et al., 2021). Bcl-2 functions as an anti-apoptotic protein that promotes survival signals, and blocking apoptosis (Hockenbery et al., 1990; Singh et al., 2019). Bcl-2 expression has been positively associated with cancer progression in multiple cancer types including among others, prostate (Campbell and Leung, 2021; McDonnell et al., 1992; Rosser et al., 2003), breast (Joensuu et al., 1994; Sun et al., 2019), melanomas (Hartman and Czyz, 2013), and small cell lung carcinomas (Jiang et al., 1995; Lochmann et al., 2018; Rudin et al., 2021), and several other cancer types (Gazzaniga et al., 1996; Gupta et al., 2021; Reed et al., 1991; Sinicrope et al., 1995; Zhang et al., 2021).
Frequent chromosomal translocations (14:18) involve the Bcl-2 gene locus, as observed in follicular B-cell lymphomas (Correia et al., 2015; Tsujimoto et al., 1985), serving as a potential source of Bcl-2 overexpression in B-lymphocytes.
In addition to the role of Bcl-2 in regulating apoptosis, studies have shown that its interaction with other molecules can contribute to its tumorigenic activity. Bcl-2 has been observed to cooperate with c-myc to promote cell survival and proliferation in pre-B cells, oral squamous cell carcinoma (OSCC) and hematopoietic cells (Chen et al., 2017b; Vaux et al., 1988; Zhang et al., 2019b).
More recently, Choi and Yoon (2011) linked FK506-binding protein 38 (FKBP38) with Bcl-2 stabilization and resistance to kinetin riboside and etoposide (Choi et al., 2010; Shirane-Kitsuji and Nakayama, 2014). In addition, multiple studies have determined that Bcl-2 overexpression is linked to chemo-immuno-resistance in several cancer types and lines. Earlier studies on Bcl-2 have shown overexpression of endogenous Bcl-2 in human malignant glioma cells was linked to resistance for: Fas/APO-1 antibody-mediated apoptosis, chemotherapeutics, and irradiation treatments (Campos et al., 1993; Miyashita and Reed, 1993; Weller et al., 1995).
Further, Bcl-2 expression and Bcl-2 related pathways have been implicated in chemoresistance in several cancers (Qian et al., 2022; Zhang et al., 2021) including hepatocellular carcinomas (Ma et al., 2008; Sun et al., 2011b), cisplatin resistance in ovarian cancer (Yang et al., 2019; Yang et al., 2002), and paclitaxel resistance in breast cancer (Gu et al., 2020; Tabuchi et al., 2009).
B. B-cell lymphoma extra large
Likewise, Bcl-xL functions as a strong negative regulator of apoptotic signals (Yu et al., 2022). Though similar to Bcl-2 in structure and in function, Bcl-xL is also able to regulate apoptosis independently of Bcl-2 (Boise et al., 1993; Kale et al., 2018). Bcl-xL has been determined to induce apoptosis resistance in multiple cancer types. Both Bcl-2 and Bcl-xL expression was shown to be linked to melanoma progression from primary melanoma to metastatic melanomas (Lucianò et al., 2021; Zhang and Rosdahl, 2006).
Further, Bcl-xL is also implicated in tumor angiogenesis through the regulation of chemokine interleukin 8 (CXCL8) (Gabellini et al. 2018; Gabellini et al., 2008; Giorgini et al., 2007), as well as affecting cell migration in both breast cancer cells (Bessou et al., 2020) and pancreatic cancer cells (Choi et al., 2016). In addition, Bcl-xL has been shown to promote tumor metastasis in these cancer types as well. The mechanism of metastatic promotion was determined to be outside of Bcl-xL's standard role and anti-apoptotic activity (Choi et al., 2016), highlighting the regulatory effects of Bcl-2 family proteins outside of their traditional apoptotic mechanisms.
Generally, Bcl-xL has been shown to function as a pro-tumoral agent promoting metastasis and tumor invasion in breast cancer cells (Bessou et al., 2020; Fernández et al., 2000), as well as increased cancer progression in melanoma (Leiter et al., 2000; Lucianò et al., 2021), colorectal cancers (Ramesh et al., 2021; Zhang et al., 2008), and prostate cancer (Campbell and Leung, 2021; Castilla et al., 2006).
C. Myeloid cell leukemia-1
In this context, Mcl-1 expression in cancer cells is also shown to have pro-survival and pro-tumoral effects (D'Aguanno and Del Bufalo, 2020; Thomas et al., 2013). Although the general function of Mcl-1 is similar to Bcl-2, Bcl-xL, and survivin, Mcl-1 has been identified to have roles independent of Bcl-2. Studies have shown that Mcl-1 and Bcl-2 are often differentially expressed, indicating an in vivo difference in the roles of Mcl-1 and Bcl-2 as apoptosis regulators (Krajewski et al., 1995; Wang et al., 2021).
Consistent with its role in promoting cell survival, Mcl-1 overexpression is regularly observed in several cancers (Beroukhim et al., 2010; Wang et al., 2021). Recent studies have shown that high Mcl-1 expression is associated with poor prognosis in acute myeloid leukemia (AML) patients (Li et al., 2019b). Accordingly, high Mcl-1 levels are observed in several other cancers, and its overexpression is associated with poor prognosis of patients' gastric (Lee et al., 2015; Likui et al., 2009; Maeta et al., 2004), breast (Campbell et al., 2018; Winder and Campbell, 2022), and non-small cell lung cancers (NSCLCs) (Wen et al., 2019).
Further, Mcl-1 expression has also been linked to therapeutic chemoresistance of various tumors (Fu et al., 2022; Kaufmann et al., 1998; Ma et al., 2016; Michels et al., 2014; Wang et al., 2021; Yecies et al., 2010).
D. Survivin
Contrary to the aforementioned proteins, which are all classified as Bcl-2 family proteins, survivin is defined as an IAP (Erlandsson et al., 2022). Survivin suppresses apoptosis through its interaction with caspases, preventing their cleavage and activation (Jaiswal et al., 2015; Tamm et al., 1998). Consequently, survivin is able to inhibit both extrinsic and intrinsic pathways of apoptosis. Survivin is also involved in the process of cell division and cell proliferation, as well as recently being identified as a regulator of several immune cells (Garg et al., 2016; Gil-Kulik et al., 2019; Gurbuxani et al., 2005; Leung et al., 2007; Miletic et al., 2016; Singh et al., 2022; Song et al., 2005).
Survivin overexpression is also associated with promotion of cancer metastasis, angiogenesis, and general progression in several cancer types (Chu et al., 2012; Fernández et al., 2014; Mehrotra et al., 2010; Wang et al., 2012; Yu et al., 2021). Survivin expression is widespread throughout numerous cancer types (Khan et al., 2021; Warrier et al., 2020; Wheatley and Altieri, 2019), including, among others, lung (Fung et al., 2021; Monzó et al., 1999), breast (Martínez-Sifuentes et al., 2022; Nasu et al., 2002; Oparina et al., 2021; Tanaka et al., 2000), ovarian (Cohen et al., 2003; Gąsowska-Bajger et al., 2021), and colorectal cancers (Antonacopoulou et al., 2011; Sarela et al., 2000; Zhou and Lin, 2015).
The dysregulation of Bcl-2, Bcl-xL, Mcl-1 and survivin is observed over multiple malignancies; however, their expression profiles may vary among cancer types (de Necochea-Campion et al., 2013; Frenzel et al., 2009). Accordingly, the overexpression of the earlier gene products has been tied with poor prognosis, cancer progression, and resistance to chemotherapeutic drugs, thus paving the way for their potential use as cancer biomarkers (Campbell et al., 2010; Dunne et al., 2018; Maji et al., 2018; Zarogoulidis et al., 2015).
To highlight another protein that may be relevant under iNOS/NO signaling; Fas-associated death domain-like interleukin-1β-converting enzyme (FLICE) like inhibitory protein (FLIP) is associated with regulation of the extrinsic pathway of apoptosis. FLIP is shown to prevent apoptosis induction through inhibition of procaspase-8 recruitment (French and Tschopp, 1999; Safa, 2013). FLIP is implicated in the development of chemoresistance in several cancers (Abedini et al., 2004; Safa, 2020; Safa, 2013).
Similar to Bcl-2 and the other antiapoptotic proteins examined, FLIP overexpression is also observed in multiple cancer types (Elnemr et al., 2001; Shirley and Micheau, 2013; Zhou et al., 2004; Zong et al., 2009).
IV. iNOS and NO
A. Inducible nitric oxide synthase
Each NOS isoform is associated with different primary NO functions in various physiological systems (Förstermann and Sessa, 2012; Król and Kepinska, 2021). Both nNOS and eNOS isoforms, known as “constitutive NOS,” are calcium/calmodulin dependent and synthesize NO in quick bursts (Abu-Soud et al., 1997; Cinelli et al., 2020; Kone et al., 2003). iNOS is unique among the NOS isoforms, as it is calcium independent and releases NO in much higher concentrations than eNOS and nNOS. iNOS has been also shown to be a functional part of both the inflammatory system and immune response (Cinelli et al., 2020; MacMicking et al., 1997; Xue et al., 2018). iNOS expression has been identified in a variety of cell types (Cinelli et al., 2020), including hepatocytes (Geller et al., 1993; Nussler et al., 1992), smooth muscle cells (Koide et al., 1993; Macnaul and Hutchinson, 1993), chondrocytes (Charles et al., 1993), neurons (Koprowski et al., 1993; Minc-Golomb et al., 1994), cardiac myocytes (Balligand et al., 1994), and immune cells (Xiong et al., 2004; Xue et al., 2018).
iNOS dysregulation has been further associated with several cancers and their functions related to tumor progression and metastasis, with contrasting pro- and anti-tumor activities have also been described (Juang et al., 1997; Klotz et al., 1998; Kojima et al., 1999; Le et al., 2005; Li and Xu, 2005; Liao et al., 2019; Minhas et al., 2020; Swana et al., 1999; Thomsen et al., 1994; Vanini et al., 2015; Zhang et al., 2014). Although iNOS activity can be generally regulated by substrate and cofactor availability, iNOS expression and transcriptional regulation are typically dependent on cell type and cell species (Kleinert et al., 2004; Minhas et al., 2020; Pautz et al., 2010).
The iNOS-mediated NO synthesis is tightly regulated by the availability of the substrates and the essential cofactors, as well as by the utilizability of the enzyme itself and their inducers, such as inflammatory cytokines (Cinelli et al., 2020; Schmidlin and Wiesinger, 1994), as discussed later. Given that iNOS levels are correlated with tumorigenesis (Crowell et al., 2003; Lechner et al., 2005; Minhas et al., 2020; Vanini et al., 2015), cell proliferation, and tumor progression (Cobbs et al., 1995; Kielbik et al., 2019; Klotz et al., 1998; Thomas and Wink, 2017; Thomsen et al., 1995) the deep understanding of the regulation of iNOS expression and iNOS activity are critical, as it may pave the way for the development of novel cancer therapeutics based on iNOS targeting (Ding et al., 2021; Wang et al., 2020).
The availability of the primary substrate L-Arginine, as well as the L-arginine transporters cationic amino acid transporter (CAT) 1 and CAT2 and CAT3 are associated with the iNOS activity. Tetrahydrobiopterin (BH4) has been shown to act as an essential cofactor for NO generation by iNOS. Further, in most cells, iNOS activity is dependent on BH4 availability, as BH4 is required for the iNOS dimerization and enzyme activation (Cho et al., 1995; Correa-Aragunde et al., 2013; Tzeng et al., 1995; p. 1).
The inhibition of iNOS activity by NO has been reported, thus suggesting a negative feedback loop between NO and iNOS (Assreuy et al., 1993; Cinelli et al., 2020). The negative feedback interactions can be attributed to a number of mechanisms originating from NO-induced pathways that mediate, among others, upregulation of cyclic guanosine monophosphate (cGMP) and increased p53 activity, which is known to act as an iNOS transcriptional repressor (Forrester et al., 1996; Li et al., 2015).
The proteasome pathway and the 26S proteasome subunit play a primary role in iNOS degradation (Ciechanover and Schwartz, 1998; Soundararajan et al., 2023). Early research done by Musial and Eissa (2001) discovered that proteasomes play a regulatory role on iNOS expression both transcriptionally and post-translationally. On the transcriptional level, proteasome interactions can contribute to iNOS expression via the activation of the NF-κB pathway and through the degradation of inhibitor of kappa B (IκBα).
In addition, the proteasome regulates iNOS post-translationally through the degradation of the iNOS protein itself (Li et al., 2021; Neish et al., 2000; Xie et al., 1994). More recently, iNOS degradation in macrophages was observed to be regulated through autophagy (Wang et al., 2019).
Studies on iNOS expression profiles have illuminated the role of non-coding RNAs (ncRNAs) on iNOS regulation. Although expression is cell-type and species type dependent, numerous microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have been discovered to play a large role in the regulation of iNOS expression as well as a connection between these ncRNAs and the development of a number of diseases, and tumor and cancer progression (Jiang et al., 2019; Khan et al., 2019).
lncRNAs are transcripts ≥200 nucleotides long (Sana et al., 2012). Like other ncRNAs, lncRNAs participate in the regulation of gene expression at an epigenetic, transcriptional, and post-transcriptional levels. lncRNAs often exert their effect upstream of various miRNA. As a result, lncRNAs are linked with the progression of several diseases, cancer progression, and the expression of a number of genes and gene products (Jiang et al., 2019). Several lncRNAs have been discovered to activate or inhibit iNOS/NOS2 expression.
lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has been shown to inhibit the iNOS/NF-κB signaling pathway in human endometrial cells (Yu et al., 2019), whereas lncRNA urothelial cancer associated 1 (UCA1) was able to reverse the inhibitory effects of miR-204 on iNOS, by acting as a competitive endogenous RNA in human AML cells (Liang et al., 2020).
B. Nitric oxide
NO has been shown to have a number of various regulatory effects, in part due to the formation of reactive nitrogen species (RNS). Some relevant reactive downstream derivatives of NO are peroxynitrite and dinitrogen trioxide. Some relevant reactions include: NO reaction with superoxide to form peroxynitrite and also reaction with nitrogen dioxide (NO2) to form dinitrogen trioxide (Habib and Ali, 2011; Somasundaram et al., 2019; Squadrito and Pryor, 1998; Subapriya et al., 2002).
The reaction of RNS downstream of NO can lead to nitration and oxidation of DNA (Mintz et al., 2021; Weiming et al., 2002). In particular, DNA reaction with peroxynitrite causes general DNA damage and even potential single-strand DNA breakages (Ahmad et al., 2019; Cantoni and Guidarelli, 2008; Chen et al., 2011b; Ullrich and Kissner, 2006).
NO signaling can be mediated by two primary pathways: cGMP dependent and cGMP independent (Obermajer et al., 2013; Stamler, 1994; Thomas et al., 2008; Vanini et al., 2015). NO in the cGMP-independent signaling primarily regulates through protein modification by NO and RNS derived from NO (Khan et al., 2020). One important modification is S-nitrosylation formed by the reaction of NO with the thiol of a cysteine residue (Fernando et al., 2019; Stamler et al., 2001).
Nitrosylation and de-nitrosylation of proteins are involved in the regulation of a range of different physiological functions from angiogenesis to immune response (Hernansanz-Agustín et al., 2013). In addition, S-nitrosylation dysregulation is implicated in cancer progression and metastasis (Fernando et al., 2019; Wang, 2012).
Further, more recent evidence has shown that NO is also involved in epigenetic regulation (Socco et al., 2017). Vasudevan et al. (2015) showed that NO can regulate genes in cancer through direct induction of histone post-translational modifications (Palczewski et al., 2019). Moreover, NO can further impact epigenetic regulation through involvement with DNA methylation, and modifications of miRNA expression, or other ncRNAs and/or indirectly by regulating chromatin-modifying enzymes (Vasudevan et al., 2016).
V. iNOS Expression in Human Cancers
The role of NO in cancer is complex. It has been shown that NO can have varying effects on tumorigenesis and cancer progression. These effects are heavily mediated by several factors, including the concentration of NO, location of NO exposure, as well as cell type and species (Cinelli et al., 2020). Because NO is a prevalent molecule in cancer biology, its generation by iNOS has been heavily studied in multiple cancer types.
iNOS has been shown to be overexpressed in a number of human cancers. iNOS overexpression may promote tumor growth and development through varying pathways depending on cancer type and the state of the tumor microenvironment (TME). Increased iNOS activity is observed in malignant/invasive human breast lesions (Loibl et al., 2002), and is also associated with generally poor prognosis in triple negative breast cancer (TNBC) patients (Garrido et al., 2017).
It is also suggested that iNOS activity and NO production are upregulated in advanced breast cancer patients (Alagöl et al., 1999; Lin et al., 2022), as well as high iNOS expression being associated with increased metastasis, disease recurrence, and decreased overall patient survivability (Garrido et al., 2017; Switzer et al., 2012).
Researchers also determined that smoking may increase iNOS levels through observation of lung cancer tissues comparing tissue from smokers and nonsmokers (Chen et al., 2008). Puhakka et al. (2006) suggested that increased iNOS expression as well as nNOS were linked to lung carcinogenesis. More recently, smoking was linked to increased iNOS polymorphisms in the development of bladder cancer (Huang et al., 2014b).
In addition, iNOS overexpression was observed in human oral premalignant epithelial lesions, suggesting that iNOS overexpression could be linked to human oral carcinogenesis (Chen et al., 2002; Kawanishi et al., 2017). Interestingly, iNOS overexpression has also been tied to several anti-tumor effects as well. In breast cancer models, progesterone activation of iNOS was shown to increase apoptosis (Mishra et al., 2020; Pance, 2006).
Additional studies also showed that overexpressed iNOS in breast carcinomas promotes apoptosis (Vakkala et al., 2000; Zhang et al., 2019a). In vitro studies also detected iNOS protein in OSCC cells, as well as determining that NO exposure induced p53 accumulation and thereby activated apoptosis in these cancer cells (Yang et al., 2015; Zhao et al., 2005). Overexpression of iNOS is also heavily linked to induced cytotoxicity in cells, and in vitro studies also show that iNOS expression can enhance chemotherapeutic cytotoxicities, such as cisplatin (Adams et al., 2009; Kielbik et al., 2019).
VI. NO and Apoptosis Signaling
A. NO regulation of apoptosis
NO impacts the apoptotic pathways via binding, S-nitrosylation of involved proteins, and by formation of the highly reactive species peroxynitrite. Cell exposure to both endogenous and exogenous NO has direct influence on the mitochondria, leading in some cases to cytochrome c release and the induction of apoptosis. One major target of NO is cytochrome oxidase (COX), and through its binding to it, NO may regulate cellular respiration (Brown and Cooper, 1994; Cleeter et al., 1994; Salvemini et al., 2013).
NO has also been reported to inhibit both Complex IV and Complex I of the electron transport chain (ETC) (Clementi et al., 1998; Tengan and Moraes, 2017). NO-mediated inhibition of Complex IV is reversible, but long-term exposure to NO leads to S-nitrosylation of several thiols, and to long-term Complex I inhibition. The inhibition of the ETC and NO binding to COX is tied to increased super-anion and radical production (Holotiuk et al., 2019; Moncada and Erusalimsky, 2002; Poderoso et al., 1996).
The production of these oxidants, in addition to elevated NO levels, promotes peroxynitrite formation (Ahmad et al., 2019; Patel et al., 1999). Peroxynitrite was demonstrated to activate the P38 and c-Jun-N-terminal kinase (JNK)/mitogen activated protein kinases (MAPKs) and subsequently involved in the initiation of the intrinsic apoptotic pathway (Go et al., 1999; Janakiram and Rao, 2012; Shacka et al., 2006; Xia et al., 1995). So, in many cases, NO has been shown to act as an inducer of apoptosis.
Along with the pro-apoptotic role of NO in various cell models, NO has also been demonstrated to have anti-apoptotic effects as well. It has also been suggested that NO can inhibit apoptosis through the activation of the cGMP dependent pathway, leading to downregulation of several caspases as well as through post-translational modifications on a number of pro-apoptotic proteins (Azad et al., 2006; Dash et al., 2003; Khan et al., 2020).
In addition, NO has been further reported to exert dual and opposite roles in p53 accumulation in several cells (Khan et al., 2020; Messmer et al., 1996), with its inhibitory effects attributed to the induction of NO-mediated GC to AT mutations in the p53 gene (Choudhari et al., 2013). NO's pro or anti-apoptotic effects seem to be dependent on a number of conditions such as cell type, cell environment, concentration of NO, etc.
Kim et al. (2001) found that NO can prevent cytochrome c release through inhibition of caspase-8 and thereby obstructing the downstream pro-apoptotic mechanism of Bid-cleavage. Kim et al. (1998) have also demonstrated NO upregulation of the anti-apoptotic protein Bcl-2 levels in Michigan Cancer Foundation-7 (MCF-7) cells and hepatocytes (Khan et al., 2020). NO's effect on apoptosis does seem to vary depending on a number of conditions; however, several studies do show that there is a notable connection between apoptotic and NO signalings. The cross-talk of NO and several anti-apoptotic molecules is discussed later.
S-nitrosylation of caspase-3 and caspase-9
S-nitrosylation has been shown to play a role in the endogenous regulation of mitochondrial caspases (Mannick et al., 2001; Plenchette et al., 2015). Caspase-3 is one of the primary executioner caspases and is a downstream effector of both the intrinsic and extrinsic pathways of apoptosis (Brentnall et al., 2013). Several studies have demonstrated that NO is able to inhibit Caspase-3 through S-nitrosylation (Mitchell and Marletta, 2005; Saligrama et al., 2014; Zech et al., 1999). Caspase-3 S-nitrosylation inhibits its apoptotic effects and promotes cell survival. In vivo experiments on glioma cells showed that S-nitrosylation of caspase-3, induced by increased iNOS activity, contributes to tumor supportive modulation of microglia (Shen et al., 2016).
In addition, caspase-3 S-nitrosylation was demonstrated to be associated with myeloperoxidase (MPO) and iNOS expression in breast cancers (Saed et al., 2010). Inhibition of both iNOS and MPO decreased caspase-3 S-nitrosylation and increased apoptosis in epithelial ovarian cancer cells.
Similarly, endogenous caspase-9 activity is also inhibited through S-nitrosylation (Fernando et al., 2019; Kim and Tannenbaum, 2004; Török et al., 2002). Another study by Kim and Tannenbaum (2004) showed that S-nitrosylation of procaspase-9 in HT-29 human colon adenocarcinoma (COAD) cells inhibits its cleavage into active caspase-9 and thereby downregulates its apoptotic effect. Török et al. (2002) showed that S-nitrosylation of procaspase-9 was able to inhibit apoptosis and prevent cytochrome c release in cholangiocarcinoma (CHOL).
More recently, S-nitrosylation and caspase-9 were found to be involved in regulating apoptotic activity through transnitrosylation from procaspase-9 to X-linked inhibitor of apoptosis protein (XIAP) in a study of cerebral ischemia-reperfusion (Zhang et al., 2016b). Authors found that the transfer of the S-nitrosyl group resulted in procaspase-9 cleavage and activation and subsequent XIAP inhibition.
S-nitrosylation of XIAP and cellular inhibitor of apoptosis protein
XIAP, like survivin, is another member of the IAP family. XIAP functions to inhibit apoptosis through downregulation of caspase activity, such as inhibition of caspase-3, caspase-7, and caspase-9 (Holcik et al., 2001; Ren et al., 2014; Riedl et al., 2001; Salvesen and Duckett, 2002). NO has been shown to regulate XIAP activity through S-nitrosylation. S-nitrosylation of XIAP has been implicated as a contributor to several neurodegenerative diseases.
In neuronal cells, S-nitrosylation of XIAP has been shown to inhibit its anti-apoptotic activity and prevent caspase inhibition by XIAP (Nakamura et al., 2021; Nakamura et al., 2010; Tsang et al., 2009). Further, Nakamura et al. (2021) found that transnitrosylation between caspase-3 and XIAP may play a role in regulation of apoptosis. The inhibitory effect of S-nitrosylation was shown to be transferred from caspase-3 to XIAP, thereby promoting caspase-3 activity and shifting toward a more pro-apoptotic phenotype.
In addition, a recent study using the human embryonic kidney cell line HEK293t has shown that S-nitrosylation of XIAP at Cys 213 was found to directly disrupt XIAP mediated inhibition of caspase-3 (Wu et al., 2015). S-nitrosylation of XIAP has been mainly studied with regards to neuronal death and neurodegenerative disease.
Another IAP family member cellular inhibitor of apoptosis protein (cIAP) is also regulated by NO via S-nitrosylation. In a recent study, treatment with NO donor glyceryl trinitrate led to S-nitrosylation of cIAP1 and the promotion of apoptosis (Romagny et al., 2018). S-nitrosylation of cIAP1 inhibited RIP1 Lys63-linked ubiquitination, leading to the formation of TNFα-mediated death domain. Additionally, S-nitrosylation of cIAP1 was able to occur through iNOS stimulation in EMT6H mammary cancer cells. Interestingly, S-nitrosylation was observed to inhibit the E3 ubiquitin ligase activity in both XIAP and cIAP, highlighting one conserved mechanism of S-nitrosylation on these two IAP members.
VII. Regulation of Anti-Apoptotic Gene Products by NO
A. Regulation of Bcl-2 by NO
Bcl-2 plays a role in inhibiting NO-induced apoptosis and NO-mediated initiation of the intrinsic pathway of apoptosis is dependent on the downregulation of Bcl-2 and other Bcl-2 family anti-apoptotic proteins, as well as the activation of BAX and Bak, and pro-caspase-9 cleavage and activation (Fig. 1) (Messmer et al., 1996; Snyder et al., 2009). Further, NO has been shown to be able to regulate Bcl-2 expression through multiple mechanisms and has been reported to exert both positive and negative regulatory effects.

Early studies showed that NO exposure led to several changes in the protein expression of Bcl-2 family proteins, including Bcl-2. Previous research done by Tamatani et al. was among the first to show that treatment with an NO donor was linked to Bcl-2 protein family regulation. They determined that the increased BAX to Bcl-2 ratios in neuronal cells was caused by signaling cascades activated by NO, during NO-induced apoptosis (Tamatani et al., 1998).
Another early study (Genaro et al., 1995) found a link between exogenous NO donors incubated in splenic B cells with sustained Bcl-2 messenger RNA (mRNA) levels, though the exact mechanisms of Bcl-2 regulation by NO in these studies were not determined. However, since then, several pathways have been identified and suspected to be relevant to NO regulation of Bcl-2 transcription, and S-nitrosylation has been identified as an important regulatory mechanism for Bcl-2 activity (Azad et al., 2006). Though a conclusive map of NO's complete effects on Bcl-2 has not been outlined as of yet.
The ability for NO to regulate Bcl-2 on the level of transcription is not well understood. Upregulation of Bcl-2 transcription in cancers has been associated with activation of the phosphoinositide 3-kinase (PI3K)/Akt (protein kinase B) pathway (Bratton et al., 2010; Huang et al., 2020; Zhou and Zhao, 2019). The PI3K/Akt pathway is also involved in Bcl-2 transcriptional regulation in the pathology of other diseases including cardiac hypertrophy (Meng et al., 2021a) and also regulating neuronal survival (Ciani et al., 2002).
In a recent study on cardiac hypertrophy, Meng et al. (2021a) indicated that Bcl-2 was a downstream effector of signaling mediated by the PI3K/Akt pathway. Using analysis of differentially expressed genes of the PI3K/Akt pathway, Meng et al. (2021a) identified several transduction signals that initiated Bcl-2- mediated cell survival in the development of cardiac hypertrophy. Some of these signals include Akt inhibition of BAD and retinoid X receptor alpha (RXR-alpha) (Wang et al., 2017), and also promotion of the cAMP response element binding protein (CREB) activity (Meng et al., 2021b; Wilson et al., 1996).
Further studies have shown that NO is also able to regulate CREB activity in cerebellar neurons, as well as additionally regulating Bcl-2 activity (Ciani et al., 2002). Ciani et al. demonstrated that NO induces pro-survival effects through activation of CREB via phosphorylation. Further, they identified Bcl-2 as an important gene under transcriptional control by CREB and have shown to be additionally regulated by the NO-cGMP signaling cascade. Previous studies also support the importance of the PI3K/Akt pathway in the regulation of Bcl-2, cell survival, and apoptosis (Dudek et al., 1997; Kulik et al., 1997).
Moreover, in a study on human breast cancer, Bratton et al. (2010) clearly showed that PI3K/Akt activation led to upregulation of Bcl-2 at the level of transcription.
The PI3K/Akt pathway has also been linked to cell survival and also shown to be activated by iNOS/NO (Prueitt et al., 2007) in pancreatic cancers (Wang et al., 2016), colon cancers (Ying et al., 2007), ovarian cancers (Engels et al., 2008), and in neuroblastomas (Yoo et al., 2018). More recent studies have identified the role of iNOS in melanoma survival and have also highlighted the PI3K/Akt pathway as a primary effector pathway for iNOS regulation. Ding et al., (2021) support that NO production (by iNOS) activates Akt-kinase through stimulation of the PI3K/Akt pathway.
The activation of the PI3K/Akt pathway was also discovered to be accompanied by S-nitrosylation of the tumor suppressor gene PTEN; both of these regulatory signals of NO can contribute to the poor survival rates in melanoma patients expressing iNOS. The exact mechanism of PI3K/Akt activation by iNOS is currently unknown, though several studies suggest a number of possibilities. An earlier study that linked iNOS to PI3K/Akt suggested a cGMP-dependent mechanism of PI3K activation (Kawasaki et al., 2003), though other cGMP-independent mechanisms have been theorized as well (Deora et al., 1998).
Interestingly under NO regulation, the PI3K/Akt pathway has also been reported to regulate Bcl-2 in a different manner. In a study on novel NO donors, furoxan-based NO-donating β-elemene hybrids, in vitro treatment with the donor resulted in significant obstruction of p-Akt activation (Chen et al., 2017a). Authors also report that treatment resulted in decreased Bcl-2 expression, while BAX was upregulated.
The mechanism behind Bcl-2 downregulation by the Novel NO donor was the inhibition of Akt phosphorylation of BAD. Prevention of BAD phosphorylation increases its interactions with Bcl-2 leading to its downregulation. Though the PI3K/Akt pathway is intimately tied to Bcl-2 transcription, this study highlights the complex nature of NO regulation of Bcl-2, as well as the utility of NO based therapies. The NO-donating β-elemene derivatives displayed significant anti-tumoral effects, in part through PI3K/Akt inhibition and downstream Bcl-2 inhibition.
In addition, iNOS regulates signal transducer and activator of transcription 3 (STAT3) activity through S-nitrosylation (Kim et al., 2014). STAT3 has been directly linked to Bcl-2 and Mcl-1 transcription (Bhattacharya et al., 2005). S-nitrosylation of STAT3 prevents its phosphorylation and activation, which also inhibits downstream Bcl-2 and Mcl-1 expressions. In vitro studies have shown that treatment with the NO donor S-nitrosoglutathione led to STAT3 S-nitrosylation and was also correlated with reduced cell proliferation as well as increased apoptosis in head and neck squamous carcinoma cells (HNSC) (Singh et al., 2015).
In another recent study in prostate cancer, a low concentration NO was observed to increase Bcl-2 expression through upregulated Runt-related transcription factor 2 (RUNX2) (Nesbitt et al., 2016). RUNX2 was shown to be able to associate with and transactivate the Bcl-2 promoter region, upregulating transcription (Browne et al., 2012). The mechanism of a low concentration NO-mediated upregulation of Bcl-2 through RUNX2 stimulation has also been confirmed by previous studies during osteoblast apoptosis (Ho et al., 2009).
Post-transcriptional regulation by miRNAs
Several miRNAs have been associated with Bcl-2 regulation; however, very few of these miRNAs have been studied in connection with iNOS and NO signaling. miRNA-mediated Bcl-2 regulation has been shown to take place both upstream and downstream of iNOS. Studies on selenium deficiency in porcine cerebellums highlighted a key relationship between miR-294, iNOS, and Bcl-2. MiR-294 expression was observed to target and downregulate iNOS and also downregulate apoptosis. Zichan et al. (2021) observed that selenium deficiency decreased miR-294 expression, which, in turn, led to increased iNOS expression and downstream suppression of Bcl-2, thus suggesting miR-294 as an upstream regulator of Bcl-2 and apoptosis.
Further, Li et al. investigated the role of iNOS/NO signaling on several downstream miRNAs miR-34, miR-203, and miR-1301 in colon cancer. They showed that NO/stress-induced apoptosis was mediated by expression of these three miRNAs, in a p53-dependent manner. In addition, loss of p53 was observed to inhibit NO expression of these miRNAs, as well as preventing NO-induced apoptosis. miR-1301 was found to promote apoptosis through downregulation of Bcl-2 and Bcl-xL (Fang et al., 2012). NO-mediated induction of miR-34 and miR-203 were also determined to downregulate Bcl-2 expression (Ji et al., 2009; Li et al., 2015; Tazawa et al., 2007).
Post-translational regulation
NO may further regulate Bcl-2 expression at a post-translational level. In an early study, Cahuana et al. demonstrated that treatment with both endogenous and exogenous NO led to carbonylation of Bcl-2 in RINm5F cells. Bcl-2 carbonylation or oxidation by NO has been suggested as a mark/tag for subsequent proteasomal degradation. High concentrations of endogenous NO, produced from IL-1B exposure, showed evidence of Bcl-2 carbonylation occurring before downregulation of the Bcl-2 protein (Cahuana et al., 2004).
Conversely, NO was found to upregulate Bcl-2 activity and stability in both chromium (vi)- induced and cisplatin-induced apoptosis, in human epithelial lung cancer cells H460. Both Azad et al. and Chanvorachote et al. determined that Bcl-2 upregulation is caused by S-nitrosylation of Bcl-2 by NO. Potential residues for S-nitrosylation include: cysteine 229 and cysteine 158. NO exposure and S-nitrosylation of Bcl-2 are accompanied by inhibited degradation and inhibited ubiquitination of Bcl-2, thereby increasing cell resistance to apoptosis.
Alternatively, NO was observed to have no effect on Bcl-2 phosphorylation in both cases (Azad et al., 2006; Chanvorachote et al., 2006). Further research has confirmed these findings, Iyer et al. (2008) demonstrated that S-nitrosylation by NO is a key mechanism in regulation of Bcl-2 ubiquitination and degradation, and a key mechanism in the overall inhibition of apoptosis by NO.
In addition, a study done by Tian et al. (2012) highlighted further the role of iNOS/NO in Bcl-2 post-translational regulation. Tian et al. determined a strong connection between melanoma differentiation associated gene-7 (MDA-7)/IL24, a cytokine associated with anti-cancer effects, and downregulation of iNOS and downstream Bcl-2 denitrosylation. Using ZD55-IL-24, an adenovirus containing the IL-24 gene (Zhong et al., 2010), they found that treatment led to decreased iNOS expression, increased Bcl-2 denitrosylation, and increased Bcl-2 ubiquitination in malignant melanoma and renal carcinoma cells.
Moreover, Bcl-2 denitrosylation was also shown to be mediated by increased TrxR1 levels, a denitrosylating protein. In the same study, Tian et al. (2012) confirmed the importance of iNOS/NO signaling via the use of iNOS-siRNA (small interfering RNA) knockout experiments. iNOS knockout showed increased Bcl-2 denitrosylation and a subsequent decrease in Bcl-2 protein levels through proteasomal degradation.
Lastly, NO exposure has also been shown to regulate Bcl-2 degradation through downregulation of the MAP Kinase Phosphatase-3 (MKP-3) and MKP-3 mRNAs in human endothelial cells. MKP-3 promotes Bcl-2 degradation through extracellular signal regulated kinase (ERK) 1/2 dephosphorylation. ERK1/2 increases Bcl-2 stability through phosphorylation of Bcl-2, which prevents ubiquitination and degradation of the protein.
In this study, Rossig et al. tested and found that exogenous NO donor sodium nitroprusside (SNP) prevented ERK1/2 dephosphorylation in human umbilical vein endothelial cells treated with TNFα. Therefore, exogenous NO is able to prevent Bcl-2 degradation through downregulation of MKP-3 and stabilization of ERK1/2 (Rossig et al., 2000). A following study also showed a relation between NO and ERK1/2 in Bcl-2 regulation. In a study on bronchial asthma, Maa et al. (2003) determined that inhibition of endogenous NO led to increased Bcl-2 expression in eosinophils of asthma patients. Maa et al. highlighted the p38 MAPK and ERK pathways as mechanisms of apoptosis regulation and Bcl-2 regulation in eosinophils.
They also highlighted ERK1/2, in particular, as a regulator of Bcl-2 protein synthesis. ERK1/2 has also been shown to decrease levels of Bcl-2, Mcl-1, and Bcl-xL in pancreatic cancer cells (Boucher et al., 2000).
B. Regulation of Bcl-xL by NO
Bcl-xL is another anti-apoptotic member of the Bcl-2 family of proteins with a major regulatory role in the intrinsic pathway of apoptosis (Fig. 2). Bcl-xL and Bcl-2 share a similar structural domain. Despite this similarity, Bcl-xL can act independently of Bcl-2 and plays several other unique regulatory roles including regulation of autophagy, calcium signaling, regulation of cell death in neuronal cells, and several other functions (Boise et al., 1993; Lee and Fairlie, 2019; Michels et al., 2013).
Bcl-xL is one isoform of the Bcl-X gene; one other isoform being Bcl-xS (Warren et al., 2019). The variance in isoforms occurs due to alternative splicing of Bcl-X, where Bcl-xL acts as an anti-apoptotic Bcl-2 family protein, while Bcl-xS is primarily pro-apoptotic (Chang et al., 1999; Stevens and Oltean, 2019). Bcl-xL acts to inhibit apoptosis primarily through inhibition of the MOMP. Bcl-xS, on the other hand, is able to bind to and inhibit Bcl-xL and other anti-apoptotic Bcl-2 family members (Lindenboim et al., 2001; Plötz et al., 2012).
It seems that Bcl-xS and Bcl-xL expressions may be regulated in opposing fashions by NO. In human neuroblastoma cells, NO donor SNAP treatment led to an increased level of Bcl-xL and conversely the level of Bcl-xS was observed to decrease (Rodríguez-Martín et al., 2000). Another study also found that DEA-NO exposure led to a decreased Bcl-xL/Bcl-xS ratio (Canals et al., 2001).
The regulatory effects of NO on Bcl-xL is not well understood, though there is a definite connection between NO signaling and Bcl-xL in cancer. One in vitro experiment showed that levels of Bcl-xL were observed to be significantly decreased during treatment with NO donor in colonic cancer cells (Wang and MacNaughton, 2005). This downregulation of Bcl-xL by NO along with p53 accumulation were determined to be important contributors to the apoptosis inducing effects of NO.
Bcl-xL overexpression is common in several cancer types, including chondrosarcoma (de Jong et al., 2018), glioblastoma and melanoma (Trisciuoglio et al., 2017), Non-Hodgkin's lymphoma (Hernandez-Luna et al., 2013), liver (Shimizu et al., 2010), colon (Zhang et al., 2008), and colorectal cancers (Scherr et al., 2016). Bcl-xL overexpression in cancer has been tied to increased tumor metastasis, cancer progression, poor survival rates, and resistance to several cytotoxic therapies. Recent studies have determined that Bcl-xL is able to affect cancer and promote tumor malignancies, independent of its typical anti-apoptotic effects (Choi et al., 2016).
The ability of Bcl-xL to promote cancer progression in a separate manner than anti-apoptotic regulation provides an additional hurdle for the potential therapeutic targeting of Bcl-xL.
Similar to the other anti-apoptotic proteins, Bcl-xL confers chemotherapeutic resistance by inhibition of cytochrome c release and inhibition of the mitochondrial pathway of apoptosis. In comparison with other anti-apoptotic Bcl-2 proteins such as Bcl-2 and Mcl-1, Bcl-xL is a particularly strong anti-apoptotic regulator and possesses stronger chemoresistance (Boise et al., 1993). In murine B-cells, immunosuppressant-induced apoptosis was prevented by Bcl-xL expression, though it was not prevented by Bcl-2 expression (Gottschalk et al., 1994). Scherr et al. (2016) also determined that Bcl-xL was the only overexpressed Bcl-2 anti-apoptotic protein in colorectal cancer cell lines, indicating Bcl-xL ability to affect cancer types distinct from Bcl-2.
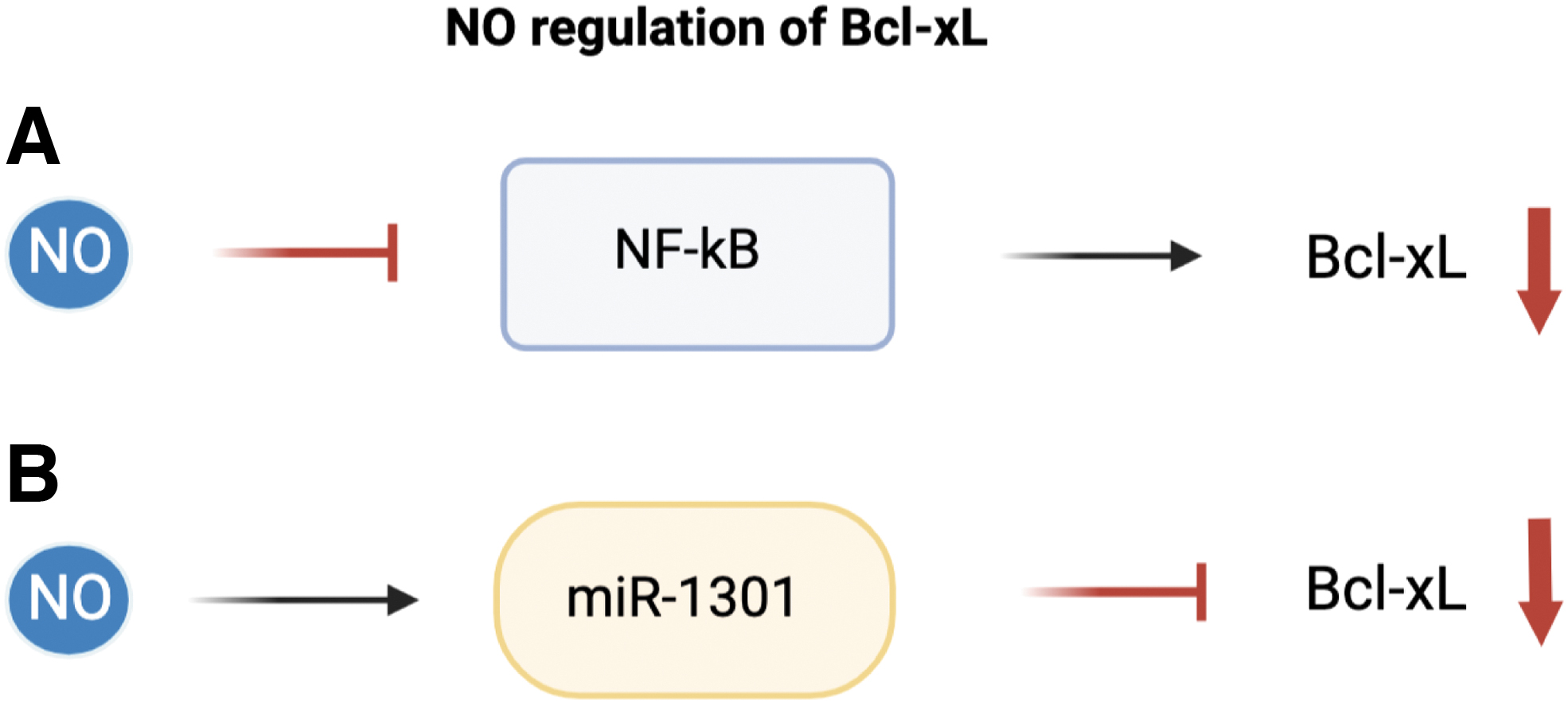
NO has also been shown to regulate apoptosis through the regulation of NF-κB. NF-κB has anti-apoptotic activity associated with induction of anti-apoptotic proteins including IAPs and Bcl-xL (Karin and Lin, 2002; Olson and Garbán, 2008). Several other studies have also supported that NF-κB inhibition also led to downregulation of various anti-apoptotic genes such as XIAP, and Bcl-xL and may also include Bcl-2 and survivin (Mackenzie et al., 2008; Sethi et al., 2008; Tsubaki et al., 2014).
Bcl-xL has been shown to be regulated through the NF-κB pathway at the level of transcription NF-κB binding sites on the Bcl-xL promoter were shown to be significant in Tax induced Bcl-xL expression in human T-cells (Jurkat cell line), and more recently NF-κB signaling has strongly been tied to Bcl-xL regulation through both canonical and non-canonical NF-κB signaling in chronic lymphocytic leukemia (Haselager et al., 2021; Mori et al., 2001). The connection between both NO and Bcl-xL with NF-κB signaling may highlight one mechanism of regulation.
Further, Huerta-Yepez et al., (2004) reported that NO inhibition of NF-κB was able to suppress Bcl-xL expression. Huerta-Yepez et al. studied the sensitizing effects of NO on prostate carcinoma cell lines. They highlighted the inhibition of the NF-κB-Bcl-xL pathway by NO donor as a major contributor to sensitization of CaP prostate cancer cells to TRAIL-mediated apoptosis. Further, authors reported that Bcl-xL expression was downregulated via NO inhibition of NF-κB, and downstream inhibition of Bcl-xL transcription.
More recently, treatment with NO donor was observed to sensitize prostate cancer cells to cisplatin induced apoptosis (Huerta-Yepez et al., 2013). Again, NO donor was demonstrated to inhibit both Ying Yang 1 and Bcl-xL expression through upstream inhibition of NF-κB. These reports also highlight the potential of NO donors in reversing chemoresistance.
Post-transcriptional regulation by miRNAs
Bcl-xL has been shown to be regulated by a number of miRNAs, such as the Let-7 family of miRNAs (Shimizu et al., 2010). Though several areas of miRNA regulation have been shown, a strong relationship between NO epigenetic regulation and Bcl-xL remains to be studied. Similar to Bcl-2 regulation, Li et al. identified miRNA suppression of Bcl-xL. Several miRNAs were identified to be regulated by NO in a p53 dependent pathway.
Li et al. (2015) determined miR-1301 as one of these regulators of Bcl-xL under NO induced stress. Additional studies also determined miR-1301 as an inhibitor of Bcl-xL expression in HepG2 cells (Fang et al., 2012). Though there is evidence for NO-related miRNA mediated regulation of the Bcl-2 family of proteins, there are still significant gaps in this area of study.
Post-translational regulation
S-nitrosylation has been observed to regulate Bcl-2 protein degradation (Snyder et al., 2009). Even though Bcl-2 and Bcl-xL are somewhat similar, the effects of S-nitrosylation on Bcl-xL is unclear. In general, the function of NO in regulating Bcl-xL post-translationally remains obscure. One potential mechanism of post-translational regulation is shown in neuronal death. Bcl-xL may be regulated by NO indirectly through ROS stimulation, leading to cleavage of the Bcl-xL protein (Cleeter et al., 1994; Park et al., 2017). A comparison of the similar structures of Bcl-2 and Bcl-xL may hint at similar mechanisms or effects of S-nitrosylation, though currently there is a lack of in-depth studies showing the effects of NO on the post-translational regulation of Bcl-xL.
C. Regulation of Mcl-1 by NO
Mcl-1 is an anti-apoptotic member of the Bcl-2 family of proteins (Fig. 3). Although Mcl-1 and Bcl-2 genes share conserved coding regions, there are several differences located mainly in the promoter region. In addition, Mcl-1 has been shown to have a shorter-half life than Bcl-2 or Bcl-xL (Bolomsky et al., 2020; Kozopas et al., 1993).
Mcl-1 exhibits its anti-apoptotic nature through inhibition of Bak and BAX (Germain et al., 2008). Mcl-1 has been shown to directly interact and inhibit Bak through the formation of a Bak-Mcl-1 complex in mammalian cells (Cuconati et al., 2003). Germaine et al. (2008) also determined that Mcl-1 was able to inhibit cytochrome c release by inhibiting BAX, though the exact mechanism is uncertain. In hematopoietic cells, Mcl-1 has also been shown to be able to associate with BAX (Bolomsky et al., 2020; Zhou et al., 1997).
Overall, Mcl-1 is a pro-survival protein and its overexpression is linked to inhibition of apoptosis in various cancers. Research done by Pervin et al. (2011) has shown that Mcl-1 stabilization in human breast cancer cells inhibits the apoptotic signal.
Three isoforms of Mcl-1 have been identified, namely Mcl-1L, Mcl-1S, and Mcl-1ES (Kim et al., 2009). Generally, Mcl-1 is categorized as anti-apoptotic, though alternative splicing of the Mcl-1 gene has led to the discovery of two pro-apoptotic isoforms: Mcl-1S and Mcl-1ES while Mcl-1L has shown anti-apoptotic effects. Kim et al. (2009) demonstrated that Mcl-1ES coexpression with Mcl-1L increased Mcl-1ES ability to induce apoptosis, with Mcl-1L also failing to prevent apoptosis.
The ability for NO to regulate the different isoforms of Mcl-1, or the splicing of other related anti-apoptotic proteins has not been thoroughly studied, and it may be an interesting target for future study.
The regulatory role of NO in Mcl-1 transcription is not clear, though several potential regulatory pathways are discussed next. In vitro, NO exposure through the NO donor SNAP was observed to downregulate both Mcl-1 and Bcl-2 in human chondrocytes (Cillero-Pastor et al., 2011). The exact mechanism of downregulation was not determined, though Cillero-Pastor et al. suspect production of ROS and RNS to be implicated in Bcl-2 and Mcl-1 downregulation. In addition, NO-induced signaling pathways, including cGMP, P38/MAPK, and PI3K/Akt, may be implicated in Mcl-1 regulation.
Other pathways known to be activated by NO such as MAPK (Doronzo et al., 2011) and PI3K/Akt (Ding et al., 2021) have also been shown to play a role in Mcl-1 regulation. However, a direct connection between NO and Mcl-1 through these signaling pathways has not been established. Han et al. (2020) showed that inhibition of MAPK signaling pathways led to inhibition of Mcl-1 expression in Mycobacterium tuberculosis infected RAW264.7 macrophages.
Mcl-1 expression has also been shown to be regulated through the PI3K/Akt signaling pathway as well. Moreover, Mcl-1 was observed to be downregulated in 3-Bromopyruvate induction of apoptosis in breast cancer cells, through the downregulation of p-Akt and through the PI3K/Akt signaling pathway (Liu et al., 2014).
Post-transcriptional regulation: epigenetics; miRNAs
Current research on the direct post-transcriptional regulation of Mcl-1 by iNOS is unclear. Several miRNAs have been identified to regulate Mcl-1 expression post-transcriptionally: miR-29a/b, miR-125b (Gong et al., 2013), miR-193b (Chen et al., 2011a), miR-26a (Gao et al., 2013), miR-133B (Crawford et al., 2009), and others (Mittal et al., 2021). Of these miRNAs, a direct relationship with either iNOS and NO signaling has not yet been established.
Though several miRNAs potentially play roles in both NO and Mcl-1 regulation. Endogenous miR-29 has been shown to regulate Mcl-1 expression in both malignant and non-malignant cell lines (Mott et al., 2007). More recently, miR-29 has been identified to target iNOS regulation, where downregulation of miR-29 is correlated with increased iNOS expression (Duan et al., 2017). Further research on miRNA and additional epigenetic mechanisms would be beneficial to the future study of NO and Mcl-1.
Post-translational regulation
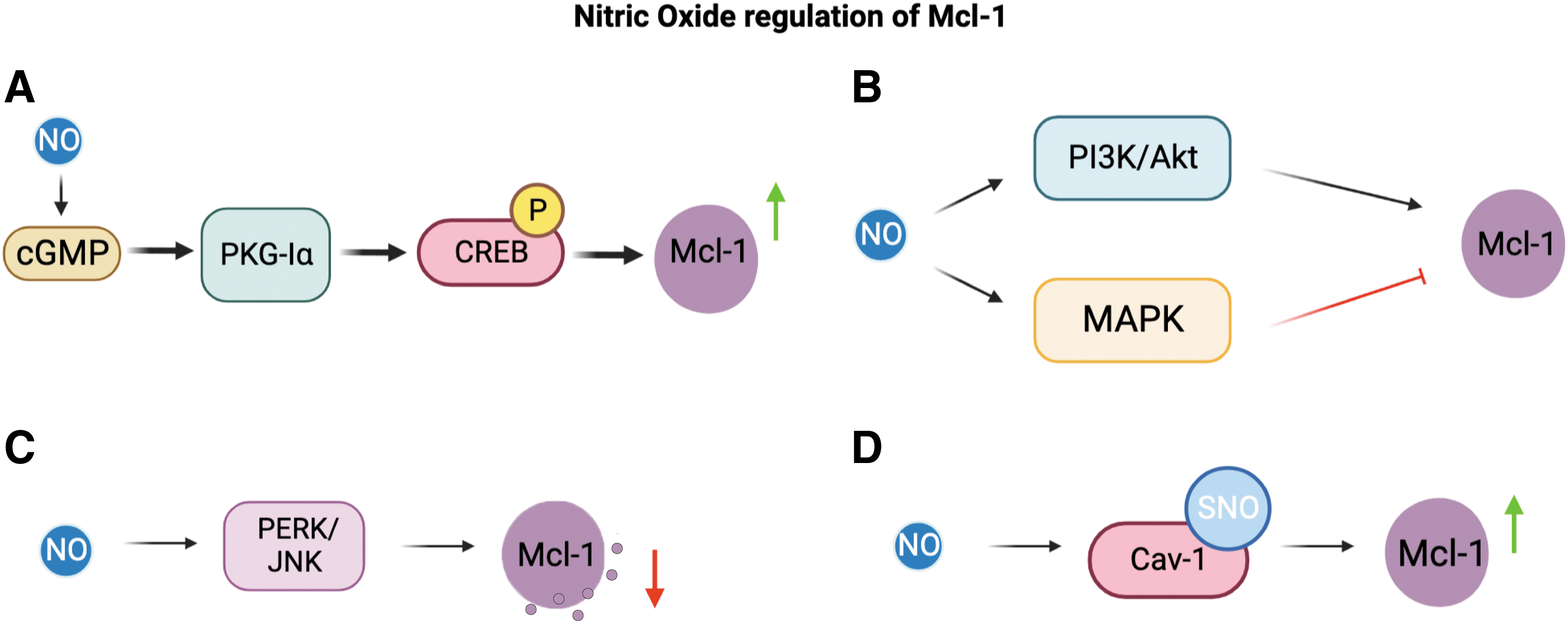
Su et al. (2011) determined a link between increased endogenous iNOS after exercise, and increased activity of the anti-apoptotic protein Mcl-1 (additionally Grp78, A1, and IL-8). Su et al. suggested that Mcl-1 upregulation after exercise induction of iNOS/NO in neutrophils is primarily caused through the NO/cGMP-mediated signaling. Further, another study also examined the NO/cGMP pathway in the regulation of apoptosis.
Wong et al. (2012) examined the NO/cGMP/PKG-Iα signaling pathway in regulation of apoptosis in NSCLC cells. They found that inhibition of the cGMP/PKG-Iα pathway led to decreased Mcl-1 expression in vitro. As previously described in Bcl-2 regulation, CREB phosphorylation was determined to regulate Mcl-1 protein expression. CREB phosphorylation was believed to be in reliance on PKG-Iα (Wong et al., 2012).
NO is able to regulate Mcl-1 expression through the cGMP/PKG-Iα signaling and CREB phosphorylation. Survivin expression, and several other IAPs, were also examined and shown to be regulated in a similar manner. Both survivin and Mcl-1 expressions are upregulated by the NO/cGMP/PKG-Iα pathway, and they play an important role in the prevention of apoptosis in NSCLC cells (Wong et al., 2012).
Interestingly, Wong et al. (2012) also examined the cGMP/PKG-Iα pathway on regulating Bcl-2 and Bcl-xL. They determined that siRNA knockout of PKG-Iα led to some decrease in Bcl-2 expression; however, DT-2 exposure, an inhibitor of PKG-Iα activity, showed no change in expression levels of Bcl-2 and Bcl-xL. The difference in regulation of these anti-apoptotic members was thought to be due to differences in half life; Mcl-1 having a shorter half-life than Bcl-2 or Bcl-xL (Bolomsky et al., 2020).
Snyder et al. (2009) showed that NO exposure leads to increased Mcl-1 degradation. Snyder et al. determined that NO induction of Mcl-1 degradation occurs in an alternative pathway than the previously determined NOXA pathway. Through experimentation using the NO Donor diethylenetriamine (DETA)-NO, Snyder et al. also determined that Mcl-1 degradation by NO takes place in the absence of BAX/Bak and thereby points to degradation occurring upstream of BAX/Bak activation.
This coincides with Mcl-1 inhibitory interactions with BAX and Bak; it makes sense that Mcl-1 degradation aids in BAX/Bak activation. NO did not induce degradation of Mcl-1 in the absence of JNK1, suggesting that NO is able to negate the anti-apoptotic Mcl-1 protein, in part, through the ASK1/JNK pathway.
Mcl-1 has been previously shown to be regulated by the JNK pathway and the protein kinase R-like endoplasmic reticulum kinase (PERK)/eIF2a phosphorylation in INS-1E cells. Prior findings indicate that the JNK pathway is responsible for Mcl-1 phosphorylation and subsequent inactivation during oxidative stress (Inoshita et al., 2002). Allagnat et al. (2011) further observed that cytokine-induced inhibition of Mcl-1 expression could be prevented with the presence of JNK inhibitor peptide.
NO production after exposure of pancreatic beta cells to cytokine stimuli leads to activation of the PERK and JNK signaling pathways, thus causing downstream Mcl-1 inhibition. This NO-induced apoptosis in pancreatic beta cells was independent of c-GMP or p53 signaling, whereas the cytokine-induced apoptosis in the same cells was strongly affected by the activation of the ER stress pathways, as NO depletes ER Ca2+ stores, thereby causing ER stress in the cells (Oyadomari et al., 2001).
The earlier findings suggest that NO-mediated Mcl-1 regulation may play a role in the ER stress-induced apoptosis. Further, exposure of INS-1E cells to pro-inflammatory cytokines led to inhibition of Mcl-1, attributed in part to NO activation of the JNK pathway, and NO interaction with the ER stress pathway, leading to eIF2a activation (Allagnat et al., 2011).
NO is also able to regulate Mcl-1 protein degradation via Caveolin-1 (Cav-1). NO upregulation of Cav-1 and Mcl-1 also plays a large role in reducing anoikis in human NSCLCs (Powan and Chanvorachote, 2014). NO was observed to attenuate Cav-1 degradation through S-nitrosylation of the protein. S-nitrosylation was shown to prevent ubiquitination and subsequent degradation (Chanvorachote et al., 2009). Cav-1 is then able to inhibit Mcl-1 degradation. The mechanism by which Cav-1 stabilizes Mcl-1 is through inhibition of Mcl-1 ubiquitination and also potentially phosphorylation (Chunhacha et al., 2012).
D. Regulation of survivin by NO
Survivin, also referred to as BIRC5, is involved in cell division and survival maintenance, caspase downregulation, and apoptosis inhibition (Fig. 4). Survivin is a member of the IAP protein family. Near its N-terminus, survivin contains a single baculovirus inhibitor of apoptosis (BIR) repeat domain. These BIR domains are characteristic of all IAPs and are necessary sites for protein interactions, and protein binding that lead to apoptosis inhibition (Cossu et al., 2019; Hinds et al., 1999; Li et al., 1999; Schimmer, 2004).
Under physiological states, survivin inhibits caspases' apoptotic activities and expression indirectly, through cooperation with other proteins (Wheatley and Altieri, 2019). Marusawa et al. (2003) have proposed the hepatitis B X-interacting protein (HBXIP) as one essential cooperating protein for survivin's anti-apoptotic effects. HBXIP association with survivin prevents Apaf-1 activation and suppression of Caspase-9.
In addition, researchers determined that survivin also interacts and forms a complex with the XIAP. The formation of this complex allows survivin to increase XIAP stability as well as promote XIAP inhibition of caspases (Dohi et al., 2004; Martínez-García et al., 2019).
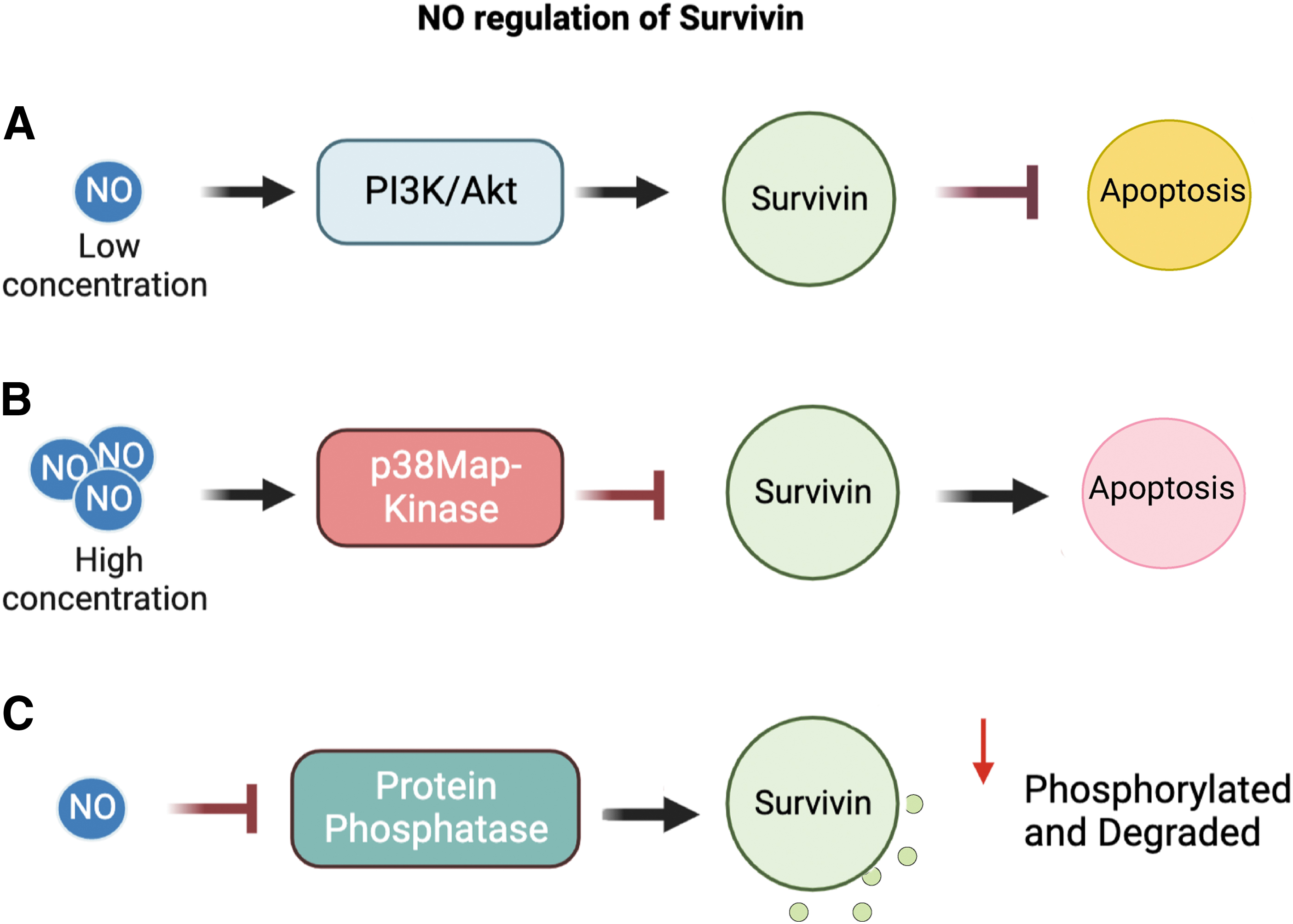
Research done by Wang et al. (2011a) on rat hepatocytes suggests a direct relationship between both iNOS and NO concentration with survivin expression. Low concentrations of NO/iNOS had an anti-apoptotic effect. iNOS/NO regulation of survivin expression at low concentrations is mediated through the PI3K/Akt/NF-κB pathways (Wang et al., 2011a). Through iNOS plasmid transfection and NO donor experiments on rat hepatocytes, Wang et al. found that under low concentrations of iNOS/NO survivin expression was increased.
Further, cells treated with higher NO donor dosage and larger iNOS plasmid transfection led to decreased survivin expression. Therefore, iNOS may regulate survivin via a concentration dependent mechanism.
Conducting studies on taxol- and cisplatin-induced apoptosis in HNSC, Fetz et al. (2009) determined that low concentrations of iNOS/NO generated cytoprotective effects through regulation of survivin expression. Low expression of iNOS was determined to create chemoresistance to cisplatin and taxol-induced apoptosis directly through regulation of survivin expression. RNA interference (RNAi)-mediated reduction of survivin levels reversed NO cytoprotective effects in HNSC cells. Depletion of survivin also led to increased cell sensitivity to drug-induced apoptosis, indicating that survivin may act as a downstream anti-apoptotic effector of NO/iNOS.
Fetz et al. (2009) also confirmed that NO upregulation of survivin expression is mediated through the PI3K/Akt pathway (Brady et al., 2008; Wang et al., 2011a). NO phosphorylation of PI3K/Akt mediates downstream upregulation of survivin expression. PI3K/Akt inhibition reversed survivin expression and increased sensitivity to apoptosis. PI3K alone was observed to increase survivin mRNA levels (Zhao et al., 2010).
Moreover, under high NO concentrations, the suppression of survivin was linked to activation of the p38MAP-kinase pathway. Inhibition of p38MAP-Kinase with SB203580 prevented (high concentration) NO suppression of survivin expression (Chao et al., 2004; Han et al., 2018).
A common characteristic of NO-mediated regulation of survivin in multiple cell types is NO's concentration-dependent effects. Low NO concentrations (generated both endogenous or exogenous) were observed to increase survivin expression, thereby leading to increased cytoprotective effects as well as tumor progression and cell survival. Generally, high concentrations of NO suppressed survivin expression leading to apoptosis; though the exact mechanism of survivin control by NO is not currently known. NO activation of the PI3K/Akt1/NF-κB pathway and p38MAP-Kinase pathway has been shown to regulate downstream survivin expression (Engels et al., 2008; Liu et al., 2021).
In addition, in the human ovarian cancer cell lines OVCAR3 and SKOV3, NO has been shown to have varying effects that are heavily concentration dependent. High concentrations of NO promote phosphorylation of the p38 MAPK pathway, leading to suppression of survivin. Low concentrations of NO had an opposite effect, promoting cytoprotectivity. Low concentrations of NO, from low iNOS expression, promote expression of survivin through the increased phosphorylation of the PI3K/Akt pathway (Engels et al., 2008; Han et al., 2018).
Further research has also highlighted the role of the transcription factor STAT3 in survivin regulation. In research done on Arctigenin, a lignan with potential anticancer effects, researchers determined a link between iNOS downregulation, STAT3 activation and survivin expression. In human ovarian cancer cells OVAR3 and SKOV3, Huang et al. (2014a) found that Arctigenic treatments promoted apoptosis through downregulation of iNOS and survivin, as well as prevention of STAT3 activation/phosphorylation.
This process of Arctigenin that caused apoptosis was reversed when treated with exogenous NO. This effect hints at a direct relationship between survivin expression and iNOS/NO expression. The experiment also highlights STAT3 as an important transcription factor for iNOS signaling and regulation of the anti-apoptotic protein survivin.
Post-transcriptional regulation by miRNAs
As previously mentioned, iNOS is involved in the regulation of several miRNAs responsible for downregulation of anti-apoptotic proteins, including survivin (Li et al., 2015). Specifically, NO was observed to induce miR-203 overexpression. miR-203 expression was shown to promote apoptosis through direct targeting of survivin. In a previous experiment on human pancreatic cancer cells, Xu et al. (2013) determined that miR-203 was able to downregulate survivin through targeting of a binding site located in the 3′UTR (untranslated region).
Post-translational regulation
In an in vitro study on dendritic cell-mediated apoptosis, exogenous NO donor was observed to increase proteasomal degradation of survivin. AC20 cells were treated with an exogenous NO donor and exhibited decreased levels of survivin. It was determined that NO promotes survivin degradation through enhanced proteasome-dependent pathways (Huang et al., 2005). Huang et al. proposed that NO enhances survivin degradation through inhibition of protein phosphatases.
Following phosphatase activity inhibition, protein-kinase activity may be upregulated leading to increased phosphorylation of survivin by these kinases: cdc2 and cyclin B (Cheung et al., 2013; O'Connor et al., 2000; Pannone et al., 2007; Wang et al., 2011b). Increased phosphorylation promotes ubiquitination and subsequent degradation of survival. Interestingly, survivin phosphorylation on Thr34 has been associated with increased survival and cytoprotective effects (Barrett et al., 2009). Other studies have further shown cyclin-dependent kinase (CDK) p34 cdc2 to be downstream of the PI3K/Akt pathway (Zhang et al., 2016a) and Cdc2 along with cyclin B were able to phosphorylate and upregulate survivin expression (O'Connor et al., 2000).
VIII. Targeting Anti-Apoptotic Proteins and iNOS/NO
iNOS and NO signaling play a large role in cancer biology and pathophysiology, and in the regulation of apoptosis. Signaling effects of downstream iNOS are complex, and NO's bilateral effects prove to be a large question in completely resolving the NO signaling pathways. Cell type, species, TME, and tumor type are necessary in examining the outcomes of NO in cancer progression as well as cancer treatment (Khan et al., 2020; Vanini et al., 2015).
The dysregulation of apoptosis in cancers has been highly associated with tumor progression, tumor metastasis, and also the development of chemoresistance. Regulation of apoptosis through anti-apoptotic proteins has emerged as an important mechanism for understanding cancer resistance to therapeutics (D'Aguanno and Del Bufalo, 2020; Shahar and Larisch, 2020).
In addition, the study of the anti-apoptotic machinery and the relationship between iNOS/NO signaling and the regulation of several anti-apoptotic proteins Bcl-2, Bcl-xL, Mcl-1, and survivin provide opportunities for reversing resistance and sensitizing cancer cells to current therapeutics.
A. General mechanisms of anti-apoptotic proteins mediating cancer resistance to cytotoxic therapies
Under normal physiological conditions, Bcl-2, Bcl-xL, Survivin, and Mcl-1 play essential roles in apoptosis, cell development, and differentiation. It is only the dysregulation and often overexpression of these anti-apoptotic proteins that are associated with pro-cancer effects and protection from apoptosis. Due to the nature of cancer cells and the development of tumors, it makes sense that pro-survival signals would be favored and the expression of anti-apoptotic proteins would be increased.
In general, the upregulation of anti-apoptotic gene products induces cytoprotective effects by resisting apoptotic triggers. Therapeutics treatments that rely on stimulating apoptosis, then, are severely hindered by anti-apoptotic gene product upregulation. The dysregulation and overexpression of these anti-apoptotic proteins can be initiated through several mechanisms.
Early findings identified a Bcl-2 translocation t(14:18) and determined its association with Non-Hodgkin's lymphomas (Weiss et al., 1987). However, further research identified the same t(14;18) translocation to be common in normal healthy humans also (Liu et al., 1994). Other subsequent research also determined that this translocation and related Bcl-2 expression to be a weak marker for lymphomas (Pezzella et al., 1990), and overall translocation associated with other anti-apoptotic protein overexpression is inconsistent over multiple cancer types (Campbell and Tait, 2018).
Another, more common, mechanism resulting in anti-apoptotic protein overexpression is gene amplification or gene copy number increase. In a study done on somatic-copy number alterations in cancer cells, Beroukhim et al.(2010) found that regions containing genes for Mcl-1 and Bcl-xL were found to be amplified across a number of tumor types (Munkhbaatar et al., 2020). Interestingly, Bcl-2 amplifications were found to be more rare.
Single nucleotide polymorphisms may also play a role in overexpression of anti-apoptotic proteins. Several polymorphisms in the survivin promoter have been identified and are associated with survivin upregulation in multiple cancer types (Altieri and Marchisio, 1999; Jaiswal et al., 2015). Xu et al. (2004) determined a strong link between the -625G/C polymorphism in the survivin promoter to be associated with high survivin expression in esophageal cancers. Other survivin related polymorphisms are associated with age of onset of breast cancer (Sušac et al., 2019).
Other mechanisms of overexpression are primarily transcriptionally related. One example is the anti-apoptotic protein Mcl-1 that has been shown to be upregulated in leukemia cells through upstream activation of the STAT3/NF-κB pathways (Allen et al., 2011). Upregulation of these anti-apoptotic proteins induced by the dysregulation of various signaling pathways provides opportunities for targeting using iNOS/NO based therapeutics. Due to many overlaps in signaling pathways between NO and the transcription of anti-apoptotic proteins, such as the pathways JNK/STAT3, NF-κB/P53, PI3K/Akt, there are opportunities for further examination.
B. Targeting Bcl-2, Mcl-1, Bcl-xL, and survivin
Various studies have been and are currently being done on Bcl-2 family proteins and some anti-apoptotic Bcl-2 family inhibitors are in development and are undergoing both preclinical as well as clinical trials (Knight et al., 2019; Opferman, 2016). One major approach to Bcl-2 family inhibition is the development of BH3 mimetics. BH3 mimetics aim at disrupting anti-apoptotic Bcl-2 family proteins by replicating the natural inhibitory effects of BH3 only proteins (Patel et al., 2021).
Many of these treatments have found varying success, however, several roadblocks have also been identified. In studies on the anti-apoptotic Bcl-2 inhibitor, ABT-263, effects were shown to be limited by induced thrombocytopenia following dosing (Petros et al., 2010; Suvarna et al., 2019; Zhang et al., 2007). Venetoclax, another novel Bcl-2 inhibitor, has found success, being approved by the FDA for both the treatment of patients with chronic lymphocytic leukemia and AML (Roberts et al., 2016; Souers et al., 2013; Ward et al., 2021).
Success with Venetoclax is mediated by the development of several other resistance mechanisms. Data suggest that a G101V mutation in Bcl-2 confers resistance to Venetoclax by reducing affinity in chronic lymphocytic leukemia patients (Blombery et al., 2019). In addition, the TME conditions have also been shown to mediate Venetoclax sensitivity and resistance (Leverson and Cojocari, 2018).
Moreover, several drugs and treatments for the specific targeting of Mcl-1 are currently being studied. Mcl-1 treatments include: CDK inhibitors, deubiquitinase inhibitors, antisense oligonucleotide treatment, and BH3 mimetics including several other Bcl-2 family inhibitors (Bolomsky et al., 2020; Quinn et al., 2011). CDK inhibitors indirectly target Mcl-1, decreasing Mcl-1 transcription. One CDK inhibitor of Mcl-1, Flavopiridol, is currently undergoing clinical trials (Bisol et al., 2020; Kitada et al., 2000).
In addition, deubiquitinase inhibitors are able to increase Mcl-1 degradation thereby downregulating the protein. WP1130 is a deubiquitinase inhibitor that has been shown to inhibit deubiquitinase USP9X, a protein that has been observed to prevent Mcl-1 degradation by removal of ubiquitin (Hu et al., 2021; Sun et al., 2011a).
The advantage of some Bcl-2 family inhibitors is the ability to target multiple anti-apoptotic proteins. ABT-737, another BH3 mimetic drug, exhibits strong binding affinity to both Bcl-2, Bcl-xL, and Bcl-w (Adams et al., 2019; Oltersdorf et al., 2005). ABT-737 is currently undergoing clinical trials. One resistance mechanism has developed as Mcl-1 upregulation has been exhibited following ABT-737 exposure (Adams et al., 2019; Yecies et al., 2010).
The ability for novel treatment options to target a wide variety of anti-apoptotic proteins may be essential for preventing acquired resistances. Further, there are potentially synergies to be studied by combining various drug and treatment strategies. Knockdown of USP9X, a Mcl-1 deubiquitinase protein, has been shown to sensitize colon carcinoma and leukemia cell lines to ABX-737 treatments (Schwickart et al., 2010; Wei et al., 2020). This evidence may indicate that the deubiquitinase inhibitor W1130 and ABX-737 could be a successful co-treatment option, though this would need to be further studied.
BH3 mimetics have been demonstrated to inhibit the three key anti-apoptotic Bcl-2 family proteins discussed earlier. Gossypol and its newer derivatives Appogossypol and Sabutoclax all bind to Bcl-2, Bcl-xL, and Mcl-1 (Balakrishnan et al., 2008; Liu et al., 2022; Wei et al., 2010). The success of Gossypol derivatives lies in its ability to inhibit multiple anti-apoptotic members of the Bcl-2 family. Previous BH3 mimetics, such as ABX-737, were challenged by subsequent upregulation of Mcl-1 following treatment.
Further, the use of these treatments in combinational therapies has shown significant success. A study done by Dash et al., (2011) determined that combinational treatment with Sabutoclax sensitized prostate cancer to MDA-7/IL-24, an anti-cancer cytokine. Gossypol and other Gossypol derivatives are currently undergoing both Preclinical and Clinical trials (Martínez-Escardó et al., 2021; Renner et al., 2022).
C. Targeting iNOS/NO
Upregulation and overexpression of anti-apoptotic proteins in cancer presents a challenge to chemo and immunotherapies. The overexpression of survivin, Mcl-1, Bcl-2, and Bcl-xL has been shown to promote cancer progression as well as promote cytotoxic resistance. Downregulation of these anti-apoptotic gene products would be beneficial in reversing resistance and sensitizing cells to chemo-immuno-therapeutics. Targeting iNOS and NO to restore regulation of these anti-apoptotic gene products can provide an opportune method for reversing resistance.
Overexpression of iNOS/NO is associated with cytotoxicity and promotion of apoptosis, and increased expression of iNOS in certain cancer cell lines has been shown to downregulate the expression of survivin, Bcl-2, Bcl-xL, and Mcl-1. Increasing intratumoral levels of NO through iNOS overexpression is a useful strategy for therapy (Szabo, 2016).
For example, iNOS and NO levels were shown to be increased and contribute to the antitumor effects of Bacillus Calmette Guerin immunotherapy treatment of bladder cancer and urothelial carcinoma (Hosseini et al., 2006; Shah et al., 2014). In addition, gene insertion of iNOS expression at intratumoral sites is hypothesized to be a potential technique for enhancing radiation-induced apoptosis in colorectal cancer cells (Chung et al., 2003), and inhibition of iNOS is also implicated in improved response to radiotherapy (Pereira et al., 2020).
iNOS gene transfection in tumor cells has also previously been shown to induce cell cytotoxicity as well as induce death in neighboring bystander cells (Bazak et al., 2017; McCarthy et al., 2011; Xie et al., 1997). Though this effect may be cell dependent, the highly diffusible nature of NO may allow for diverse anti-metastatic treatments through this induction of the iNOS gene. With the goal of increasing NO levels, treatment with exogenous NO donors may be beneficial in promoting cytotoxicity similar to that of iNOS induction.
Several studies have shown that treatment with NO donor can lead to NO-induced apoptosis (Beppu et al., 2022; Li et al., 2019a; Nishio and Watanabe, 1997; Zhang et al., 2019a).
In addition to targeting iNOS as a means for therapeutics, there has been strong research focusing on the use of exogenous NO donors as potential therapeutics (Wink et al., 1997). Early studies showed that treatment with NO donors was able induce cytotoxicity and sensitivity to cytotoxic agents. Further research determined that treatment with NO-donating nonsteroidal anti-inflammatory drugs, also known as NSAIDS, and NO-donating aspirin (NO-ASA) caused inhibition of iNOS itself (Spiegel et al., 2005; Vanini et al., 2015). The self-inhibitory role of NO donors can be one potential way of targeting endogenous iNOS activity.
In addition, NO-ASA treatment in HT-29 human colon cancer cells led to chemoprevention through inhibition of iNOS expression and iNOS activity. Other early studies identified that NONOate NO donors and S-nitrosothiol NO donors were able to enhance cisplatin toxicity (Wink et al., 1997). Though there has been success with in vitro models, there are still concerns over application of NO donors in vivo. Lee and Pfeifer have identified limitations of the NO donor SNP.
SNP has been determined to be linked to increased genotoxicity, as well as having mutagenic properties through formation of RNS and ROS (Huang et al., 2017; Lee and Pfeifer, 2007; Miller and Megson, 2007). The use of NO donors as treatment is a growing field, and using NO donors to target downstream anti-apoptotic Bcl-2, Bcl-xL, Mcl-1, and Survivin is of interest.
iNOS regulation and NO signaling have been identified as major players in the development of novel cancer therapeutics. Treatment with statins has also been identified as a source for iNOS induction. These drugs have been shown to increase tumoricidal and apoptotic activities through iNOS induction and increased iNOS stimulation of statins in MCF-7 cells (Gorabi et al., 2019; Kotamraju et al., 2007). In several TNBC cell lines, fluvastatin and simvastatin treatments showed increased cytotoxicity due to iNOS expression (Kanugula et al., 2014).
Another treatment, N-(4-hydroxyphenyl)retinamide (4-HPR), has been observed to inhibit breast cancer cell growth through iNOS and eNOS inductions. NO production also plays a large role in 4-HPR's apoptotic effects in breast cancer cells (Cao et al., 2021; Simeone et al., 2002). The chemotherapeutic agent 5-fluorouracil (5-FU) induces iNOS and apoptosis in liver cancer cells. Co-treatment with L-Arg has been shown to sensitize liver carcinoma cells to apoptosis by 5-FU through increased iNOS activity (Chang et al., 2012; Jiang et al., 2002; Yin et al., 2007).
Conversely, overexpression of iNOS and NO has been linked to sustained levels of Bcl-2, Bcl-xL, survivin, and Mcl-1. Inhibition of iNOS, under these conditions, may then be beneficial in restoring sensitivity to apoptosis. Multiple iNOS inhibitors have been developed, many of which are currently being studied (Cinelli et al., 2020). These inhibitors target iNOS through two primary methods: competitive inhibition, and dimerization inhibition. In vitro testing has shown that inhibition of endogenous iNOS, using the inhibitor L-NMMA and L-NAME, in TNBC tissue was associated with decreased tumor growth and metastasis (Granados-Principal et al., 2015).
IX. Bioinformatic Analysis
Examining the apoptosis-related gene set (The Cancer Genome Atlas [TGCA],
Specifically, we found some positive correlations between iNOS and survivin (BIRC5) in lung squamous cell carcinoma (LUSC), rectum adenocarcinoma (READ), and thyroid carcinoma (THCA), and a negative correlation in thymoma (THYM); but also, positive correlations in the normal tissue of COAD and esophageal carcinoma (ESCA), and a negative correlation in the normal tissue of kidney renal papillary cell carcinoma (KIRP) (Tables 1 and 2).
BIRC5 Versus Inducible Nitric Oxide Synthase Cancer
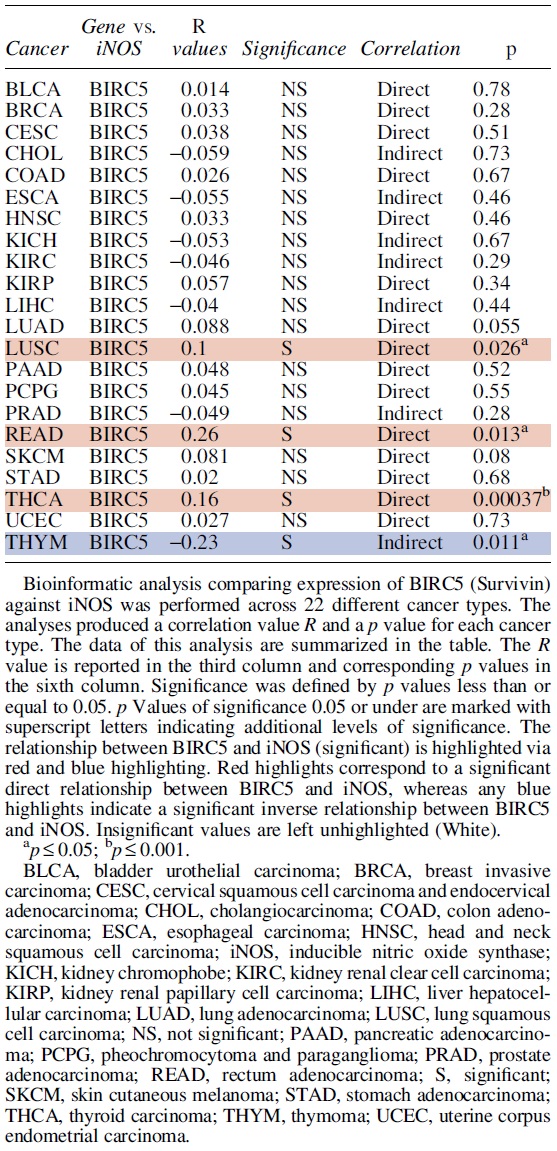
BIRC5 Versus Inducible Nitric Oxide Synthase Normal
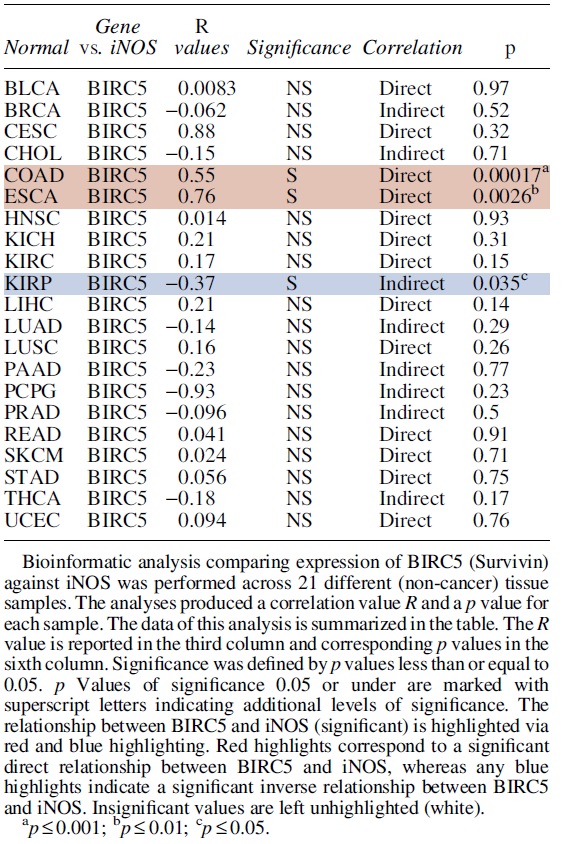
MCL1 was also negatively correlated with iNOS in COAD, kidney renal clear cell carcinoma (KIRC), KIRP, Prostate adenocarcinoma (PRAD), READ, THCA, and THYM; but also, in the normal tissue of PRAD and uterine corpus endometrial carcinoma (UCEC) (Tables 3 and 4).
MCL1 Versus Inducible Nitric Oxide Synthase Cancer
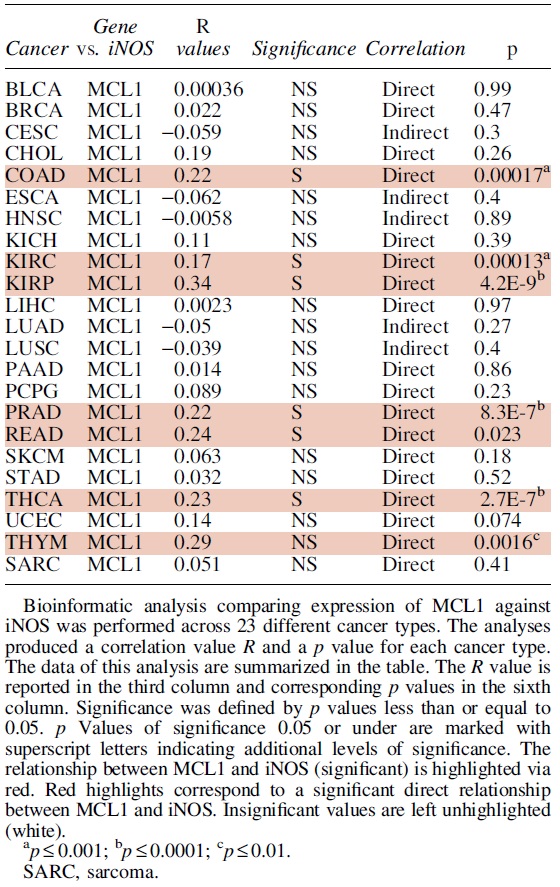
MCL1 Versus Inducible Nitric Oxide Synthase Normal
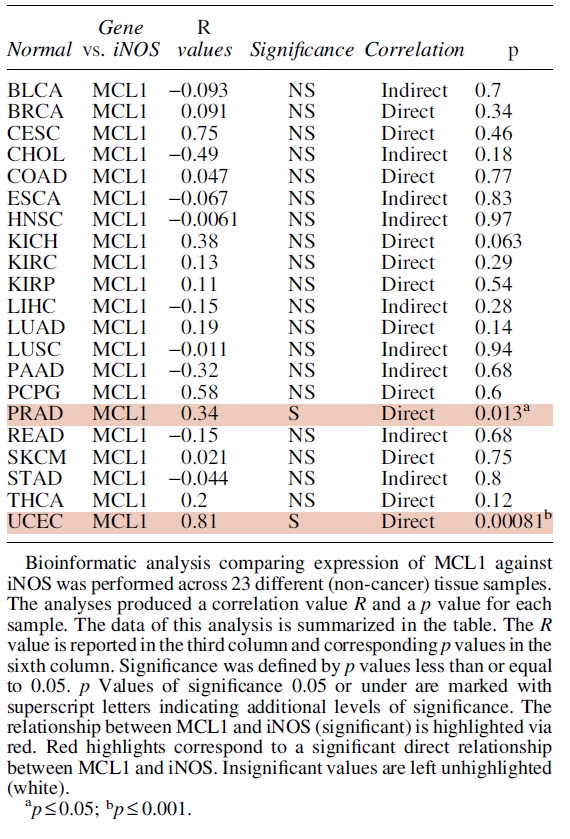
BCL2L1 was correlated with iNOS in ESCA, KIRC, THCA, and UCEC, and anti-correlated in CHOL and THYM. These two genes were also correlated in the normal tissue of Bladder Urothelial Carcinoma (BLCA), Kidney Chromophobe (KICH), KIRC, LUSC, LUSC, Skin Cutaneous Melanoma (SKCM), SKCM, Stomach adenocarcinoma (STAD), and THCA (Tables 5 and 6).
BCL2L1 Versus Inducible Nitric Oxide Synthase Cancer
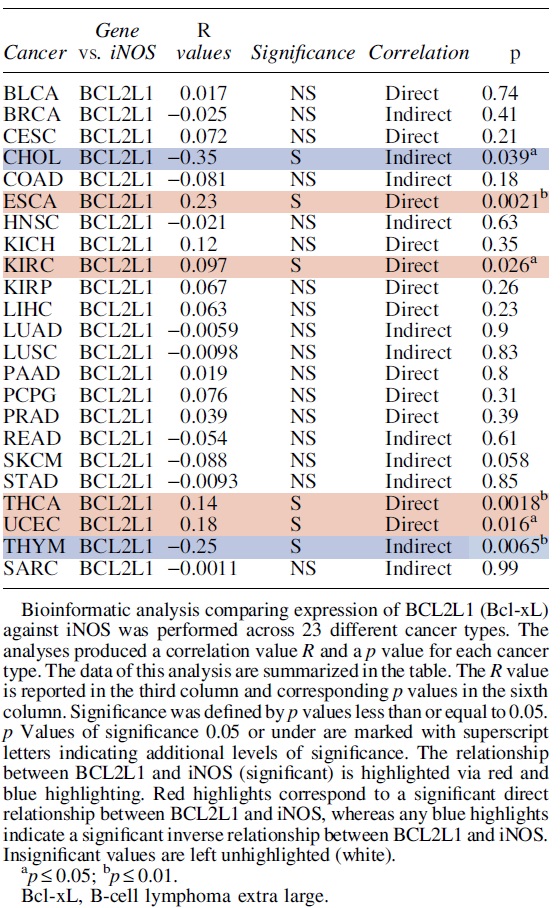
BCL2L1 Versus Inducible Nitric Oxide Synthase Normal
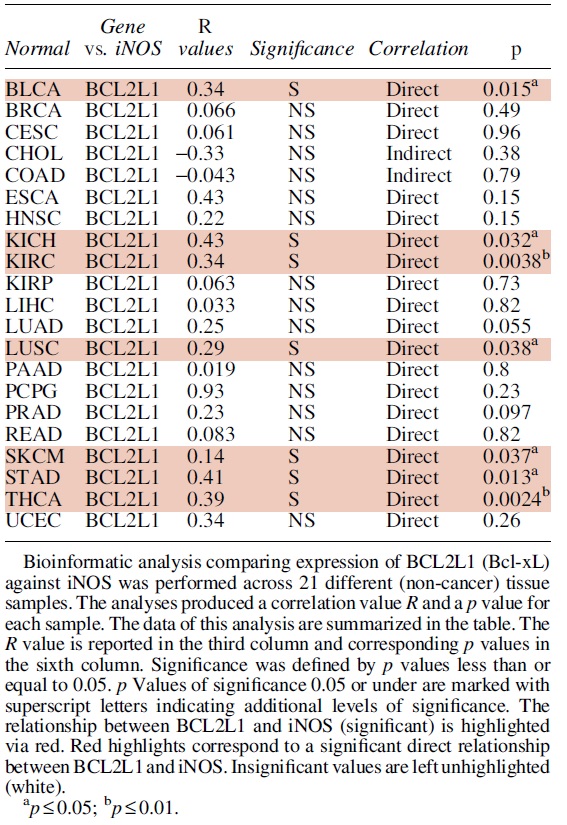
On the other hand, Bcl-2 was significantly correlated with iNOS in cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), head and neck squamous cell carcinoma (HNSC), KIRC, KIRP, PRAD, and THYM, but also in the normal tissue of KICH, liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), LUSC, PRAD, SKCM, THCA, and UCEC. These two genes were anti-correlated in the normal tissue of CHOL (Tables 7 and 8).
BCL2 Versus Inducible Nitric Oxide Synthase Cancer
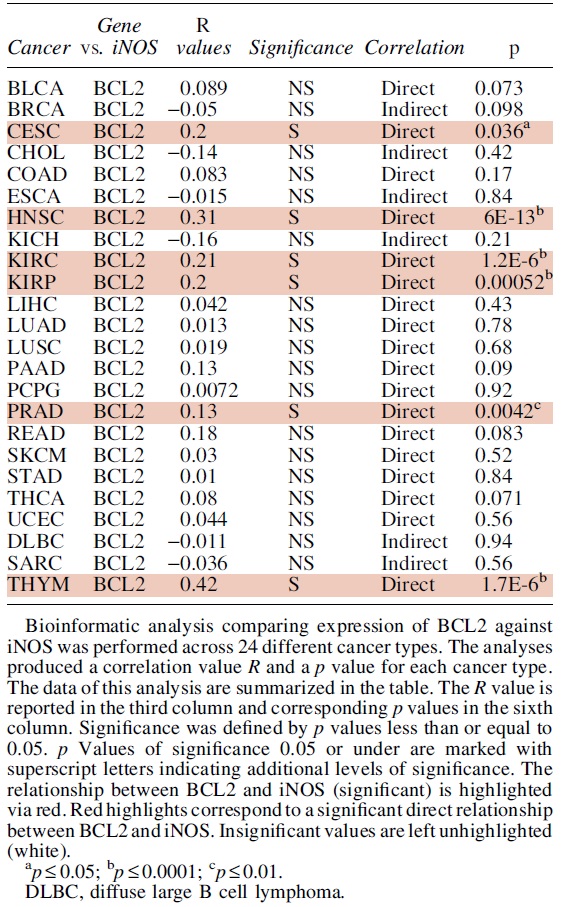
BCL2 Versus Inducible Nitric Oxide Synthase Normal
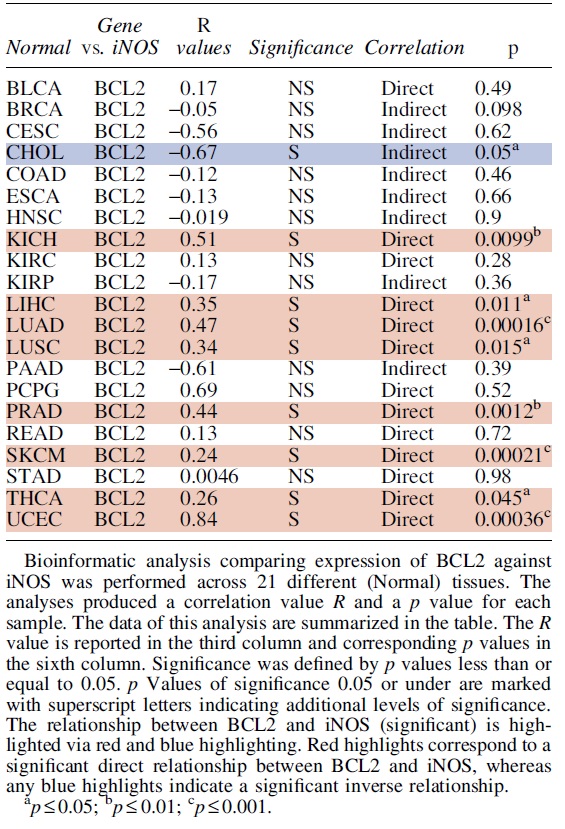
In addition, as shown in Figure 5 (Supplementary Table S1), we explored the differential expression of iNOS (+NOS2), the anti-apoptotic genes BCL2, BCL2L1 (Bcl-xL), MCL1, and BIRC5 (Survivin), and also several other relevant apoptotic genes BAD, BAX, IAP, FADD, FAS, TRAIL, and MEF2 across 14 cancer types (BLCA, breast invasive carcinoma [BRCA], COAD, ESCA, HNSC, KICH, KIRC, KIRP, LIHC, LUAD, LUSC, PRAD, STAD, and THCA) in the TCGA having >10 pairs of tumor-normal samples. NOS2 (iNOS) was significantly upregulated in ESCA (fold change [FC] = 84.28), KIRC (FC = 2.25), and LIHC (FC = 4.53). BIRC5 (Survivin) was significantly upregulated in lung cancer (LUAD; FC = 17.21 and LUSC; FC = 28.22; p < 0.05), as well as in breast cancer (BRCA; FC = 9.06), LIHC (FC = 17.97) and kidney cancers (KICH, FC = 4.52; KIRC, FC = 5.74; KIRP, FC = 13.95). It also exhibited high levels of expression (2 < FC <4) in BLCA, COAD, ESCA, HNSC, PRAD, and STAD.
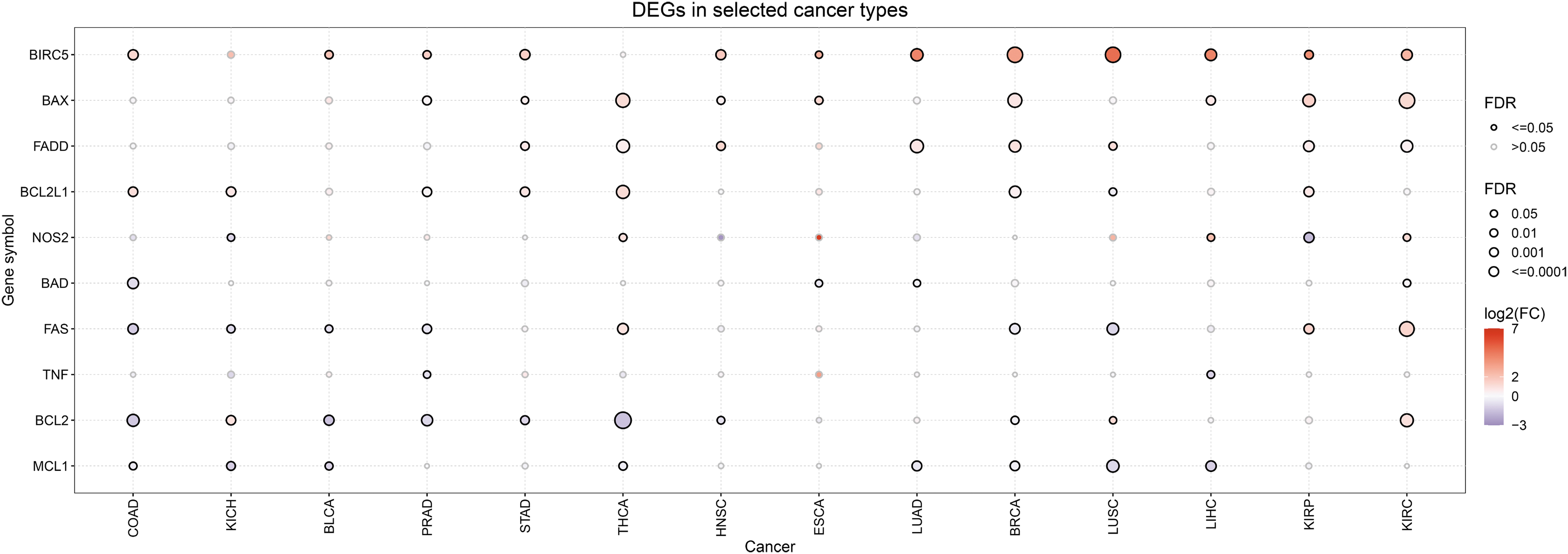
In addition, BAX was significantly upregulated in ESCA (FC = 2.40), KIRC (FC = 2.28), KIRP (FC = 2.70), and THCA (FC = 2.21). Also, BCL2 was significantly upregulated in LUSC (FC = 2.31); whereas BCL2L1 (Bcl-xL) was significantly upregulated in COAD (FC = 2.08) and THCA (FC = 2.20). We also found that FADD was significantly upregulated in HNSC (FC = 2.46) and LUSC (FC = 2.13); whereas FAS was significantly upregulated in KIRC (FC = 2.55) and KIRP (FC = 2.57). Finally, TNF was significantly upregulated in ESCA (FC = 9.58).
X. Conclusions and Future Perspectives
Stronger relationships between iNOS and NO in their roles in the regulation of the anti-apoptotic proteins discussed herein (Bcl-2, Bclxl, Mcl-1, and survivin) are complex and still many pathways remain to be uncovered. Based on the current findings discussed via targeting iNOS and/or NO to reverse cancer resistance, they have a good potential to be a critical mechanism in challenging current issues with resistance to therapeutic treatments.
Although we have demonstrated that iNOS/NO is intimately involved in the regulation and expression of anti-apoptotic proteins, however, the role of iNOS/NO signaling on anti-apoptotic proteins should not be regarded in isolation. Clearly and challenging, many relevant signaling pathways and alternative effects of NO signaling play both parallel and opposing roles in apoptotic regulation.
Therefore, for any clinical applications of either iNOS inhibitors or NO donors to target the overexpressed anti-apoptotic gene products to reverse resistance clinically, it is important to continue investigations on the conditions and schedules to be used, the type and stage of the cancer, and the various deleterious side effects emanating from these interventions. Nonetheless, although several specific inhibitors against various anti-apoptotic gene products have been developed and tested preclinically and clinically, these monospecific agents may not be effective as other anti-apoptotic gene products will not be inhibited and the cancer cells will remain resistant.
Thus, it may be more useful to target a single moiety, like iNOS or NO, that will inhibit various anti-apoptotic gene products simultaneously and should result in the reversal of resistance.
The overexpression of anti-apoptotic gene products in various cancers has been well documented and reviewed (Kapoor et al., 2020; Shahar and Larisch, 2020; Wei et al., 2010). For instance, Bcl-2 overexpression is associated with both chemo-immune resistance and poor prognosis in some cancers (Choi and Yoon, 2011; Choi et al., 2010; Karakas et al., 1998; Ma et al., 2008; Sup et al., 2005; Tabuchi et al., 2009; Yang et al., 2002).
Likewise, Bcl-xL is also involved in inhibiting apoptosis and also involved in the pathogenesis of some cancers such as melanoma (Lee et al., 2019; Leiter et al., 2000; Lucianò et al., 2021). Mcl-1 plays similar but distinct roles in apoptosis (Krajewski et al., 1995; Widden and Placzek, 2021) though it functions like Bcl-2 and Bcl-xL in various cancers and is associated with poor prognosis (Placzek et al., 2010). Survivin, which does not belong to the Bcl-2 family (Bcl-2, Bcl-xL and Mcl-1), mediates its anti-apoptotic activity via the prevention of caspase cleavages and activities, unlike the earlier Bcl-2 family members that act on the mitochondria (Jaiswal et al., 2015; Shin et al., 2001; Tamm et al., 1998).
Clearly, the earlier anti-apoptotic gene products mediate their activities via overlapping and uniquely different mechanisms and also via their interactions with other molecules (Aranovich et al., 2012; Jaiswal et al., 2015; O'Brien and Kirby, 2008; Zhang et al., 2019b).
The role of iNOS/NO in the regulation of Bcl-2, Mcl-1, Bcl-xL, and survivin was examined for each at the transcriptional, post-transcriptional, and post-translational levels. For Bcl-2, high levels of NO have been reported to induce apoptosis via inhibition of the PI3K/Akt pathway and downstream downregulation of Bcl-2 expression (Chen et al., 2017a).
However, in prostate cancer, low concentration NO is associated with tumor survival via the activation of RUNX2 and Bcl-2 upregulation (Nesbitt et al., 2016). Interestingly, iNOS-mediated S-nitrosylation of STAT3 activity resulted in the inhibition downstream of Bcl-2 expression (Kim et al., 2014). Bcl-2 expression is also mediated via iNOS regulation of several p53 dependent miRNAs (Li et al., 2015). In addition, NO mediates Bcl-2 proteasomal degradation at the post translational level through several mechanisms, including carbonylation and activation of MKP-3.
Conversely, NO has also been reported to stabilize Bcl-2 in lung cancer cells during promoting chemoresistance to cisplatin through S-nitrosylation of Bcl-2 (Azad et al., 2006; Boucher et al., 2000; Cahuana et al., 2004; Chanvorachote et al., 2006; Maa et al., 2003; Rossig et al., 2000; Tian et al., 2012; Zhong et al., 2010).
For Bcl-xL NO also mediates inhibition of Bcl-xL expression (Huerta-Yepez et al., 2004; Karin and Lin, 2002; Olson and Garbán, 2008). The primary mechanism is via the inhibition of NF-κB by NO and downstream inhibition of its target Bcl-xL (Huerta-Yepez et al., 2013; Huerta-Yepez et al., 2004). Several miRNAs have been reported to regulate Bcl-xL expression in connection with NO, though the complete relationship between NO related miRNAs and the regulation of anti-apoptotic proteins is not well understood as a whole (Li et al., 2015).
Little is known about the regulatory effects of NO on the Bcl-xL protein post-translationally, especially in relation to cancer, though it has also been suggested that Bcl-xL is degraded by the NO-induced ROS in neural cells (Park et al., 2017).
For Mcl-1, NO inhibits Mcl-1 expression in some cell systems via complex mechanisms, including cGMP signaling and the interaction with several survival pathways (Cillero-Pastor et al., 2011). Also similar to Bcl-2 and Bcl-xL, NO regulated miRNAs have been implicated in the regulation of Mcl-1 (Li et al., 2015). Further, NO primarily regulates Mcl-1 through mediated Mcl-1 degradation in the cytoplasm (Chanvorachote et al., 2009; Powan and Chanvorachote, 2014; Snyder et al., 2009).
For survivin, at low levels of NO, survivin is induced via the PI3K/Akt/NF-κB pathways (Brady et al., 2008; Chao et al., 2004; Fetz et al., 2009; Huang et al., 2014a; Wang et al., 2011a). Contrastly, at high concentrations of NO, survivin expression is inhibited via the phosphorylation of the p38 MAPK pathway (Bhowmick and Girotti, 2013; Chao et al., 2004). Like stated earlier, survivin expression is also regulated by NO-mediated expressions of various miRNAs (Li et al., 2015; Xu et al., 2013) and NO-mediated degradation of survivin (Huang et al., 2005).
Altogether, it is clear that iNOS/NO is involved in the regulation (up or down) of the expression of anti-apoptotic gene products examined in this report. It is also clear that cross-talks exist among the various regulatory mechanisms mediated by iNOS/NO.
NO's ability to epigenetically regulate anti-apoptotic proteins is also unclear. The function of noncoding RNAs, as well as DNA methylation or histone modification is obscure in the relationship between NO and anti-apoptotic protein regulation. Though the potential for iNOS targeting to regulate anti-apoptotic protein activity and restore apoptotic effects is promising, there continue to be significant gaps in establishing a complete picture of NO interaction with anti-apoptotic signaling.
Our various analyses in the regulation of the four anti-apoptotic gene products by iNOS/NO were explored through bioinformatic analyses in subsets of human cancers. The direct correlations were not evident in the majority of the different types of cancers examined, though many correlations were consistent with our analyses in distinct cancer types. We also explored the differential expression of several other relevant apoptotic associated genes to illustrate further connections between these genes that may be points of interest for future study.
In conclusion, we have demonstrated that the level of iNOS/NO expression in various cancers may dictate the regulation of various anti-apoptotic gene products and their roles in the pathogenesis of cancer and resistance to apoptotic stimuli by therapeutics.
These findings emphasize the potential utility of targeting iNOS/NO in the development of novel therapeutics, though there remain many unanswered questions regarding the agents used, their selectivities, the choice of NO, iNOS inducers or inhibitors, and the development of biomarkers to determine the use of NO donors or NO inhibitors. In addition, there is also the need to develop novel targeting agents that are not only selective but also may be complexed with other cytotoxic agents.
Footnotes
Acknowledgment
The authors acknowledge the Department of Microbiology Immunology and Molecular Genetics, David Geffen School of Medicine for their assistance in the preparation of this article.
Authors' Contributions
The original design of this review was done by B.B. and I.B. The draft writing of the article was done by I.B. and B.B. The editing and review of the article was performed by I.B., S.B., M.L., A.Z., and B.B. The bioinformatic analysis was performed by A.Z. All the authors contributed to the finalization and revision of the article.
Author Disclosure Statement
There are no conflicts of interest by all authors.
Funding Information
There was no funding by any agencies.
Supplementary Material
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.