
Abstract
Oxidative stress is involved in the development of several pathologies. The different reactive oxygen species (ROS) produced during oxidative stress are at the origin of redox post-translational modifications (PTMs) on proteins and impact nucleic acids and lipids. This review provides an overview of recent data on cysteine and methionine oxidation and protein carbonylation following oxidative stress in a pathological context. Oxidation, like nitration, is a selective process and not all proteins are impacted. It depends on multiple factors, including amino acid environment, accessibility, and physical and chemical properties, as well as protein structures. Thiols can undergo reversible oxidations and others that are irreversible. On the contrary, carbonylation represents irreversible PTM. To date, hundreds of proteins were shown to be modified by ROS and reactive nitrogen species (RNS). We reviewed recent advances in the impact of redox-induced PTMs on protein functions and activity, as well as its involvement in disease development or treatment. These data show a complex situation of the involvement of redox PTM on the function of targeted proteins. Many proteins can have their activity decreased by the oxidation of cysteine thiols or methionine S-methyl thioethers, while for other proteins, this oxidation will be activating. This complexity of redox PTM regulation suggests that a global antioxidant therapeutic approach, as often proposed, is unlikely to be effective. However, the specificity of the effect obtained by targeting a cysteine or methionine residue to be able to inactivate or activate a particular protein represents a major interest if it is possible to consider this targeting from a therapeutic point of view with our current pharmacological tools. Antioxid. Redox Signal. 41, 152–180.
Introduction
The balance between oxidizing and reducing reactions, so called redox homeostasis, is essential for the regulation of several cell functions. Even though reactive oxygen species (ROS) and reactive nitrogen species (RNS) are regulating many physiological conditions (such as cell cycle, glycolysis, Calvin cycle, and protein turnover), their excessive production can lead to various pathologies, including, diabetes, cancers, and cardiovascular and neurodegenerative diseases (Kumar et al., 2022; Ochoa et al., 2018). ROS, including hydroxyl radical (HO•−), superoxide anion (O2 •−), and hydrogen peroxide (H2O2), are produced by several enzymatic systems: NAPDH oxidases (NOX1 to 5 and DUOX1 and 2), xanthine oxidase, superoxide dismutases (SOD1 to 3), cytochrome P450. RNS are nitric oxide-derived (•NO) molecules. The main RNS is peroxynitrite (ONOO–), obtained by the reaction of nitric oxide (generated by nitric oxide synthases) with superoxide anion.
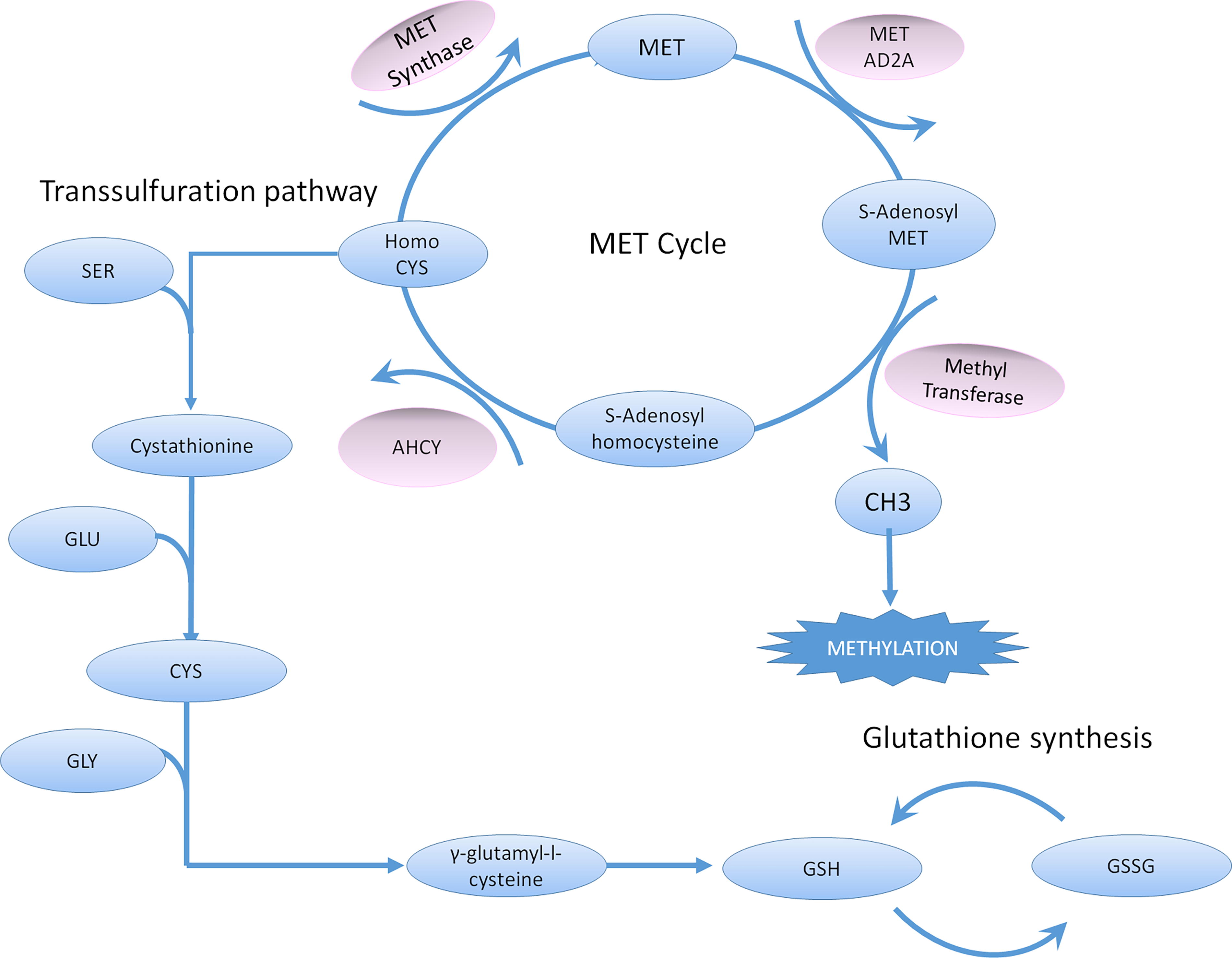
Involvement of ROS and RNS in physiological and pathological conditions is notably mediated by the direct oxidation of DNA, lipids, and proteins by these molecules (Juan et al., 2021). In this review, we will only focus on the post-translational modifications of proteins targeted by ROS and RNS. These redox-induced post-translational modifications (PTM) can be reversible or irreversible. They can lead to the activation, the inhibition, or the degradation of oxidized proteins. The redox-induced PTM are mainly targeting the thiol groups of two amino acids: cysteine and methionine. However, lysine, arginine, threonine, proline, tyrosine, phenylalanine, and tryptophan residues can also be modified (through carbonylation and nitration). We will first describe the most observed modifications before studying their most recent reported pathological implications. A synthetic knowledge of cysteine and methionine metabolism has been described (Lauinger and Kaiser, 2021) and is represented in Figure 1.
Redox-Induced Amino Acid Modifications
Cysteine post-translational modifications
ROS and RNS can induce the oxidation of cysteine thiol. However, only 10%–20% of the thiol groups are oxidized in cellular conditions, the remaining cysteines being inert and/or inaccessible (Jones, 2008). ROS can induce four major reversible and irreversible modifications of cysteine residues (see Fig. 2). The first one is S-sulfenylation. This oxidative reaction mediated by H2O2 is reversible (notably by antioxidants such as ascorbate) (Anschau et al., 2020) and leads to the formation of a sulfenic acid (Cys—SH ↔ Cys—SOH). S-sulfenylation can induce conformational modifications in the protein structure.
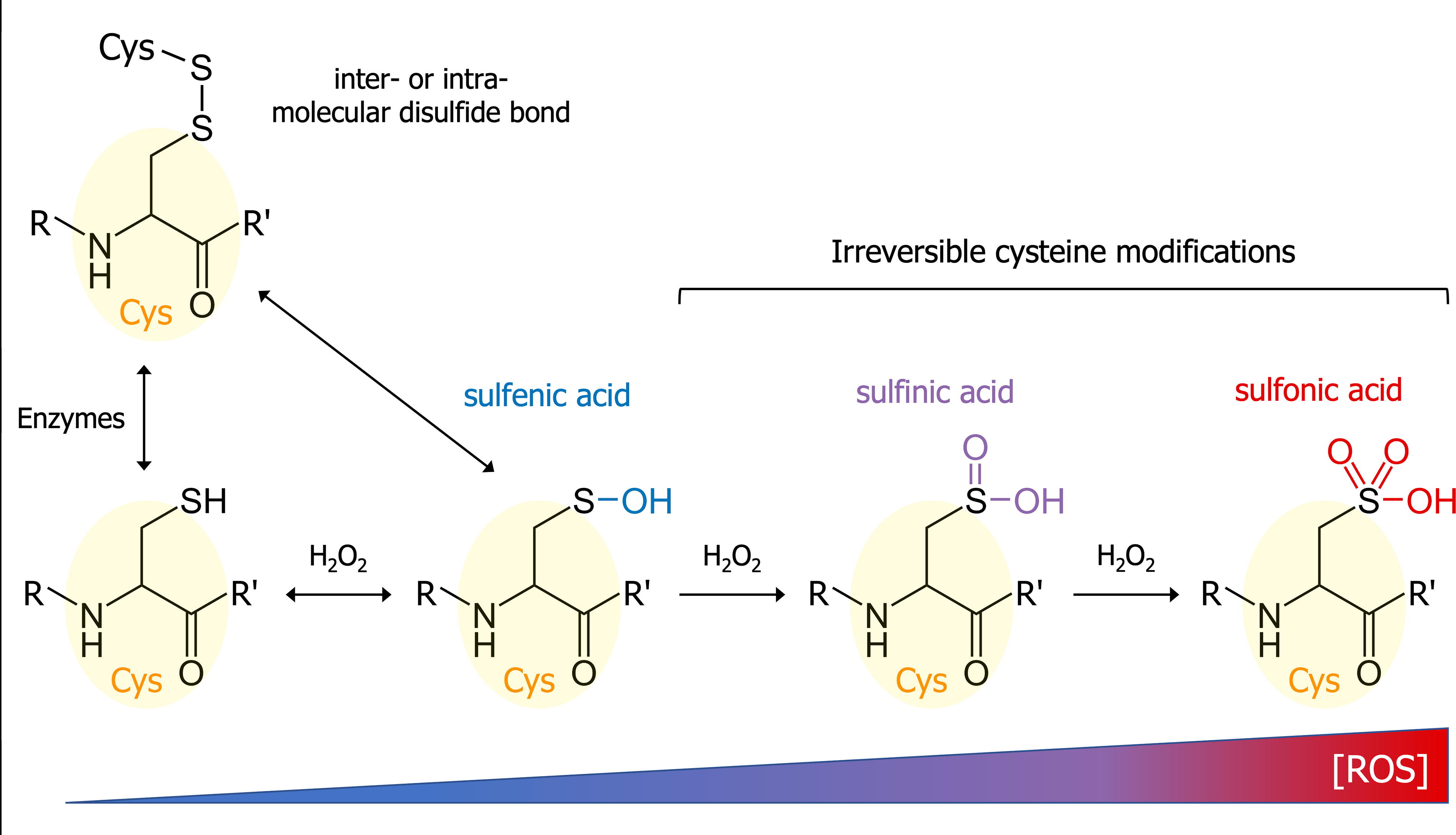
As cysteines are found in the active sites of many enzymes, this modification can lead to major changes in protein activities (Lo Conte and Carroll, 2013). Recent studies have highlighted the involvement of cysteine S-sulfenylations in the activation of Src kinase (Heppner et al., 2018; Yang et al., 2020) and PDI (Yang et al., 2022), and in the binding of CTR1 to VEGFR2 (Das et al., 2022).
Sulfenic acids are highly unstable. Cysteine sulfenic acids may therefore rapidly interact with other cysteine thiol residues to form inter- or intramolecular disulfide bonds (Cremers and Jakob, 2013). However, several factors such as cysteine pKa and accessibility and steric hindrance will influence the cystine formation. This oxidant-mediated disulfide bond formation, occurring in the cell cytoplasm, is distinct from the enzyme-driven cystine formation observed in the endoplasmic reticulum during protein folding.
Cysteine sulfenic acid may be irreversibly overoxidized by hydrogen peroxide to form a sulfinic acid (S-sulfinylation; Cys—SOH ↔ Cys—SOOH). Protein S-sulfinylation was at first considered a protein purification artifact or a transient state between sulfenylation and sulfonylation. However, around 5% of cysteine residues were shown to be sulfinylated in rat liver proteins (Hamann et al., 2002). Despite a higher stability than RSOH, RSOOH is more complicated to characterize, and only a few proteins were shown to be sulfinylated: the peroxidase family of peroxiredoxin (Prx) (Kriznik et al., 2020), the Parkinson’s Disease-involved protein DJ-1 (Barbieri and Luchinat, 2019), and the matrix metalloprotease MMP-7 (Fu et al., 2001).
In the presence of high H2O2 concentrations, the sulfinic acid will be oxidized to sulfonic acid (Cys—SOOH ↔ Cys—SOOOH). This S-sulfonylation reaction is the last step of the sequential oxidation of cysteine thiol, leading to an irreversibly damaged protein that will be promised to a proteasomal degradation (Xiong et al., 2011).
The RNS are also able to induce protein PTM by targeting cysteine residues. The main modification is cysteine S-nitrosylation. This reversible addition of nitric oxide on the thiol of cysteines lead to the formation of nitrosothiol groups (Cys—SH ↔ Cys—SNO). S-nitrosylation is involved in signal transduction and in the regulation of considerable physiological and pathological conditions. To date, over 3,000 peptides and proteins from different species were shown to be S-nitrosylation targets (647 in different tissues from mouse), mainly by using mass spectrometry-based methodologies (Devarie-Baez et al., 2013; Doulias et al., 2013).
In a recent study, 535 unique S-nitrosylation sites among 434 proteins were identified and associated with human pancreatic adenocarcinoma pathogenesis (Tan et al., 2019). Cysteine sensitivity to nitrosylation varies greatly depending on the proteins. Structural analyses of nitrosylated peptides have highlighted the influence of protein tertiary structures in this phenomenon. An hydrophobic environment and the presence of a nearby positive charge could facilitate NO concentration and thiol deprotonation, respectively (Marino and Gladyshev, 2010). Cysteines with low pKa could also be more sensitive to nitrosylation.
Finally, the very reactive peroxynitrite was shown to induce cysteine S-sulfenylation through the following reaction: Cys—SH + ONOO− → Cys—SOH + NO2 − (Zeida et al., 2013).
Under highly oxidative conditions, reactive cysteines can also be S-glutathionylated. The tripeptide glutathione (GSH) or its oxidized form (glutathione disulfide, GSSG) can react with cysteine thiols or with cysteine sulfenic acid, leading to protein S-glutathionylation (Musaogullari and Chai, 2020).
Finally, it was shown that protein cysteine residues can undergo persulfidation (or S-sulfhydration). This reversible modification, characterized by the addition of a sulfur atom to the thiol group of the cysteine (Cys—SH → Cys—SSH), was proposed to be the main mechanism for H2S signaling. Several proteins were shown to be persulfidated such as parkin (on Cys-59, Cys-95, and Cys-182) in sample from patients with Parkinson’s disease and GSK3ß (on Cys-218) in sample from patients with Alzheimer’s disease (Chung et al., 2004; He et al., 2023; Petrovic et al., 2021; Vandiver et al., 2013).
Methionine post-translational modifications
Less known than cysteine modifications, methionine oxidations were often linked to protein damage. However, recent studies have shown the involvement of methionine oxidations in the regulation of protein functions. Indeed, the oxidation of this hydrophobic amino acid induces alterations in its chemical and physical properties, leading to modifications in protein structure and stability (Chao et al., 1997; Samson et al., 2014).
There are two oxidized forms of methionine: methionine sulfoxide and methionine sulfone. Methionine oxidation mechanisms are summarized in Figure 3. Methionine can be readily oxidized by hydrogen peroxide to form methionine sulfoxide (also called MetO) through the following reaction: Met—S — CH3 + H2O2 ↔ Met—SO — CH3 + H2O. Methionine S-sulfoxidation is reversed by repair enzymes called methionine sulfoxide reductases (MSR) (Griendling et al., 2016). The concentration of MetO depends on the balance between ROS amounts and reductase activity.

The levels of methionine sulfoxide were shown to be more elevated during aging and inflammation where the oxidative stress conditions are known to be high (Moskovitz and Smith, 2021). Several proteins and peptides were shown to have their functions altered by methionine oxidation: α2-macroglobulin, calmodulin, cytochrome c, GAPDH, glucagon, and hemoglobin (Levine et al., 2000).
If not reduced by methionine sulfoxide reductases, MetO can be further and irreversibly oxidized by H2O2 to methionine sulfone (Met—SO — CH3 → Met—SOO — CH3), leading to protein degradation.
The levels of methionine oxidation are complicated to evaluate and often overestimated due to in vitro oxidation occurring during protein extraction. A quantitative analysis of methionine oxidation was recently conducted on >3,800 methionine-containing peptides of the human proteome. The authors have shown that the level of in vivo methionine oxidation was low in unstressed human cells, between 3.5% and 5% (Bettinger et al., 2020).
A recent study showed that sulfoxidized proteins are more likely to also have another PTM. Indeed, 98%, 88%, and 74% of MetO proteins are also phosphorylated, ubiquitinated, and acetylated, respectively. This cross talk between methionine oxidation and other modifications could be a cellular defense mechanism against oxidative stress (Veredas et al., 2017).
Protein carbonylation
Oxidative stress is also responsible for the modification of amino acids other than cysteine and methionine. ROS, and more particularly hydroxyl radicals, can notably induce the irreversible addition of a carbonyl group (C = O, carbonylation) on the lateral chains of lysine, arginine, threonine, proline, and tryptophan. The levels of carbonylated proteins can be used as a marker of oxidative stress. The most common protein carbonylation observed affects the basic amino acids lysine and arginine. The reaction of these two amino acids with hydroxyl radical leads to the replacement of their amino groups by aldehyde functions and to the formation of α-aminoadipic semialdehyde (AAS; LysCHO) from lysine and γ-glutamic semialdehyde (GGS) from arginine (Fig. 4) (Akagawa, 2021). AAS and GGS formation represent about 60% of total protein carbonylation in the liver (Kehm et al., 2021).
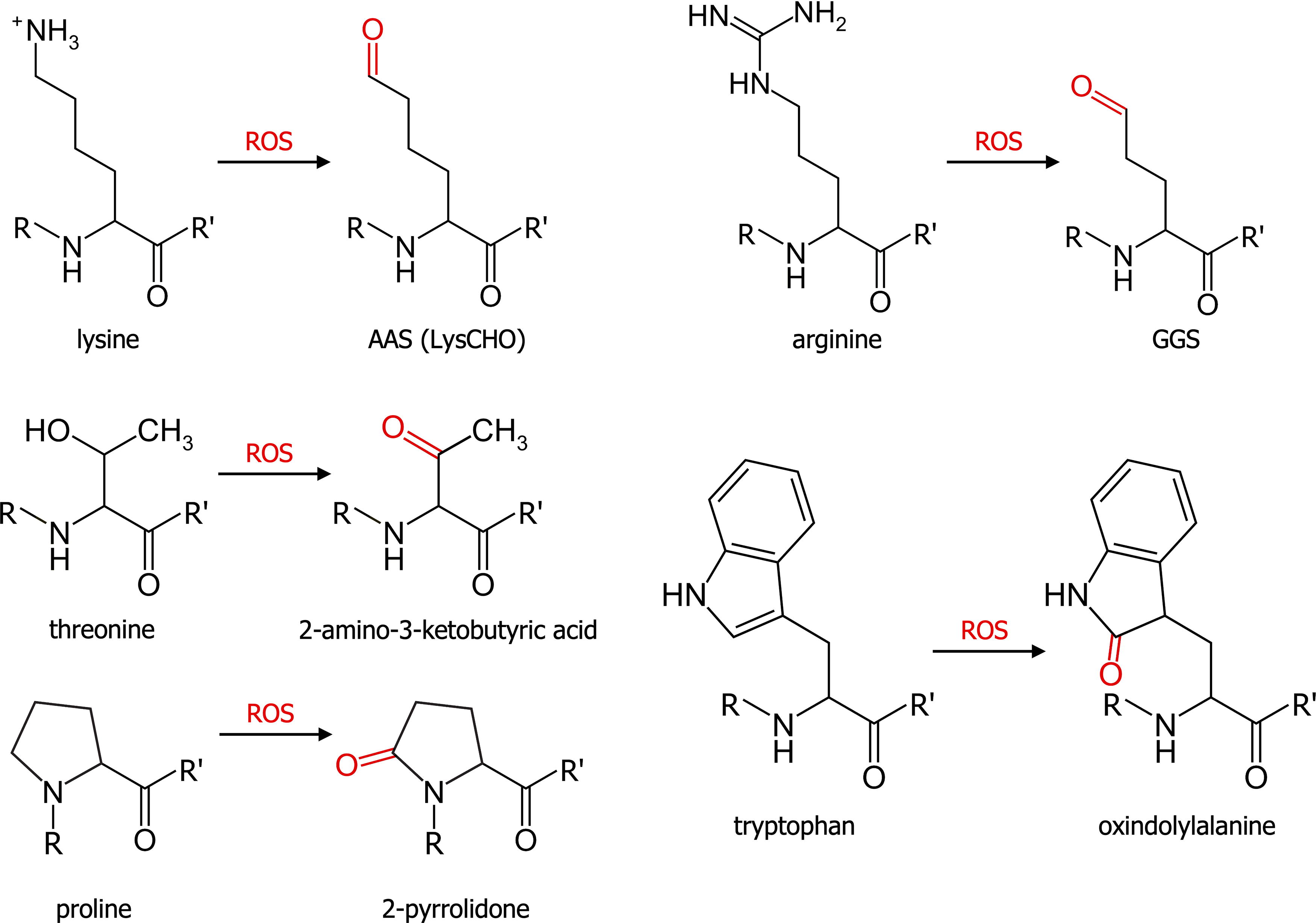
Free radicals are also able to induce the addition of ketone groups on proline and tryptophan (leading to the formation of 2-pyrrolidone and oxindolylalanine, respectively) and the oxidation of threonine hydroxyl into a ketone group (forming 2-amino-3-ketobutyric acid, Fig. 4) (Akagawa, 2021). A recent study showed that carbonylation does not occur randomly; it depends on amino acid composition, environment, and accessibility. The authors showed notably that carbonylation sites are mainly located in region rich in positively charged amino acids (lysine and arginine) (Weng et al., 2017).
Carbonylation may cause protein partial unfolding, leading to the reduction or the loss of its function. These useless proteins can be repaired or degraded, notably by proteasome. Studies have shown that, while mildly carbonylated proteins are proteolyzed by proteasome, excessively oxidized proteins may aggregate and accumulate in the cell (Dalle-Donne et al., 2006). Protein carbonylation was shown to be linked to aging, inflammation, diabetes, and smoking (Cabiscol et al., 2014; Dalle-Donne et al., 2017; Hecker and Wagner, 2018; Thanan et al., 2012).
Nitration of aromatic amino acids
As seen previously, RNS-induced protein modifications include cysteine and methionine oxidations. However, RNS are also able to induce the nitration of four aromatic amino acids directly or indirectly: tyrosine, tryptophan, phenylalanine, and histidine. The most studied RNS-induced nitration is the reaction of tyrosine with peroxynitrite forming 3-nitrotyrosine (3-NT; Fig. 5). The nitration of this amino acid reduces the -OH group pKa, impedes tyrosine phosphorylation, and triggers conformational changes (Radi, 2013). Tyrosine nitration is selective and depends on protein structure and environment.
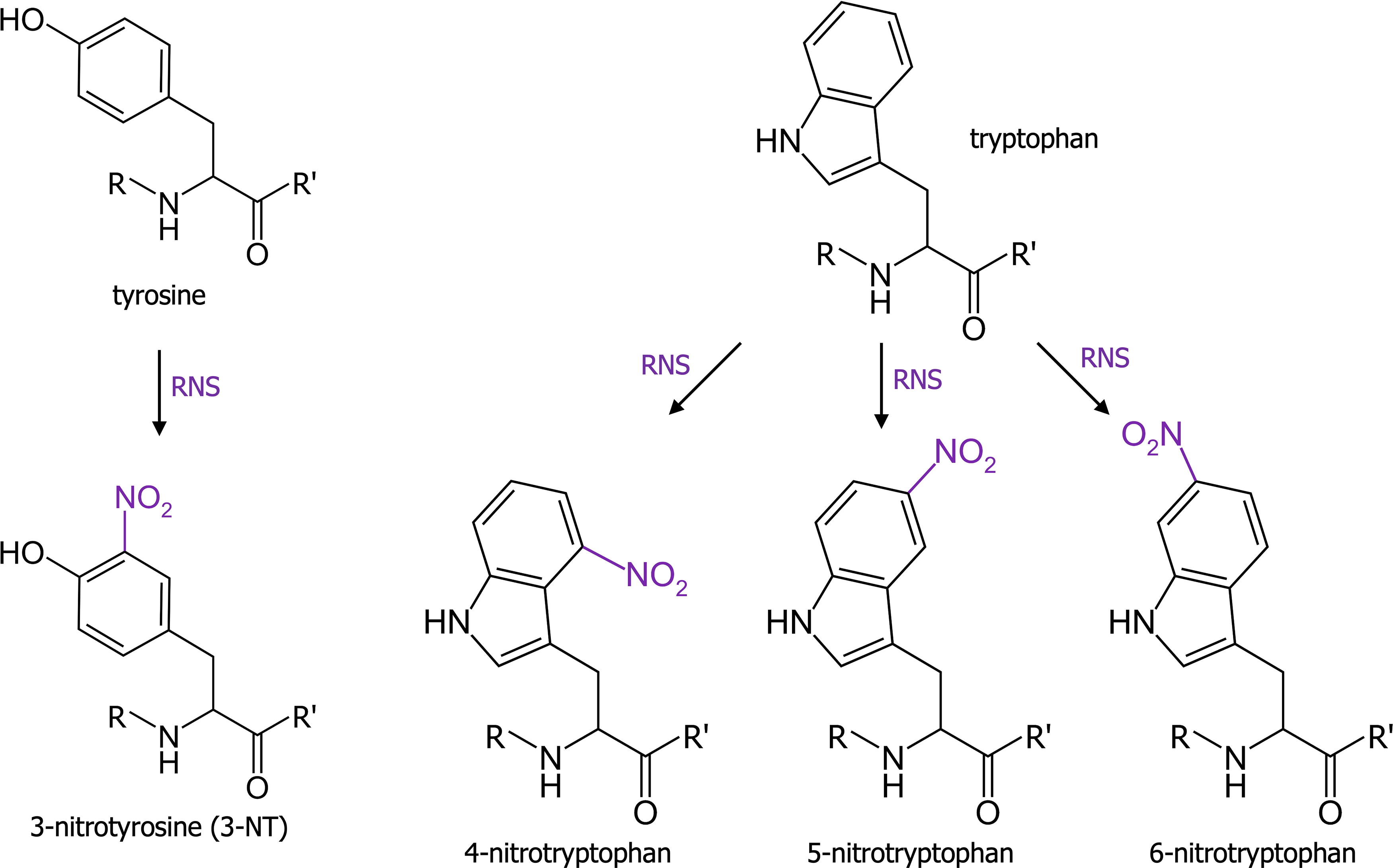
It was notably observed during aging and in inflammatory conditions. Studies have shown that nitration of superoxide dismutase 2 (SOD2) was linked to the inactivation of this enzyme, while cytochrome c nitration induces a lack of its normal electron transport capacities and a gain of a peroxidatic activity (Radi, 2013). Contrary to tyrosine, which can be nitrated only on one specific carbon (carbon 3), tryptophan has several potential reactive sites. Indeed, the reaction of peroxynitrite with free tryptophan can produce 1-, 2-, 4-, 5-, 6-, and 7-nitrotryptophan. However, 4-, 5-, and 6-nitrotryptophan (represented in Fig. 5) are the most common nitrated tryptophan observed in proteins (BSA, hemoglobin, myoglobin, FGF-1, vinculin) (Nuriel et al., 2011).
Histidine and phenylalanine react indirectly with peroxynitrite to form oxo- and nitro-histidine, and p-, m-, and o-tyrosine, as well as nitrophenylalanine (Alvarez and Radi, 2003; van der Vliet et al., 1994).
Other redox-induced modifications
Oxidative stress can also induce less frequent protein modifications, such as the formation of di-tyrosine (di-Tyr; observed in calmodulin), di-tryptophan (di-Trp; multiple isomers), and di-histidine (di-His) (Kehm et al., 2021; Malencik and Anderson, 2003). Cross links can also be observed between histidine and cysteine, arginine, or lysine.
It should be noted that other PTMs indirectly induced by oxidative stress, such as SUMOylation, palmitoylation and farnesylation, were shown to be linked to several pathologies (Dietrich and Ungermann, 2004; Feligioni and Nisticò, 2013; Jeong et al., 2021). However, these modifications will not be discussed in this review as we are focusing on modifications directly induced by ROS and RNS.
Impacted Proteins
As stated previously, oxidation and nitration are selective processes and not all proteins are modified. It depends on several factors, including amino acid environment, accessibility, and physical and chemical properties, as well as protein structures. To date, hundreds of proteins have been shown to be modified by ROS and RNS. In this study, we present only the main ones (classified according to their types) and the impact of redox-induced PTMs on protein functions and activity (see Tables 1–3). In this review, we will focus especially on the implication of these modifications in cancers and neurodegenerative diseases.
Main Redox Post-Translational Modifications Inducing Inactivation or Inhibition on Protein Activity and Functions
The impacted amino acid positions are indicated for human proteins. *: Amino acid position for JAK2. TBD, to be determined; S-S bond, disulfide bond.
Main Redox Post-Translational Modifications Modulating Protein Aggregation
The impacted amino acid positions are indicated for human proteins. S-S bond, disulfide bond.
Main Redox Post-Translational Modifications Inducing an Increase in Protein Activity and Functions
The impacted amino acid positions are indicated for human proteins. TBD, to be determined; S-S bond, disulfide bond.
Peroxidases and dismutases
Peroxiredoxins (Prx) are cysteine-based peroxidases known to reduce the concentration of H2O2 in the cells by catalyzing its reduction into H2O. Several studies have shown that Prx activity can be regulated by H2O2 itself. Indeed, hydrogen peroxide induces the oxidation of Prx1 Cys-51 into sulfenic acid (Yang et al., 2002). This residue can also be overoxidized into sulfinic acid, leading to its inhibition and aggregation. The peroxidatic cysteines of Prx2 and 3 were also shown to be oxidized into sulfenic acids, reducing their reactivity approximately 1,000 times (Peskin et al., 2013).
Interestingly, two SODs were shown to be regulated by ROS and RNS. These enzymes are also involved in the regulation of H2O2 concentration, but unlike Prx family, they catalyze its production from superoxide anions and water. In the presence of abnormally high amount of H2O2 (up to 1 mM), SOD1 cysteine 111 residue is irreversibly oxidized, leading to enzyme inhibition and disulfide bond-independent aggregation (Chen et al., 2012). Similarly, SOD 2 is also regulated by PMT. However, this enzyme is inactivated is the presence of high concentrations of RNS. Indeed, 1 mM of peroxynitrite induces the loss of 90% of SOD2 activity. This inactivation is due to the nitration of Tyr-74 to 3-NT (Yamakura et al., 1998).
Isomerases
Protein disulfide isomerase (PDI) is a crucial enzyme for the correct folding of proteins, catalyzing the formation and the breakage of disulfide bonds as the newly synthesized proteins fold. This enzyme, linked to several pathologies like thrombosis and cardiac diseases, was recently shown to be regulated by oxidation. Indeed, the transient sulfenylation of PDI Cys-56 was detected following hydrogen peroxide exposure, leading to the inhibition of PDI reductase activity and to the promotion of its oxidase activity (Yang et al., 2022). The proline isomerase Pin1 was also shown to be regulated by oxidation, notably by the sulfenylation of its catalytic cysteine (Cys-113). In the presence of high concentration of H2O2, the sulfinylation of this residue was observed. The oxidation of Cys-113 inactivates Pin1, notably its ability to isomerize Tau (Chen et al., 2015; Innes et al., 2015).
Metabolism enzymes
Several metabolic enzymes were shown to be regulated by redox-induced PTM over the past 20 years. The enzyme GAPDH (glyceraldehyde-3-phosphate dehydrogenase), crucial for glycolysis and gluconeogenesis, was shown to be oxidized on its catalytic cysteine (Cys-152). Under oxidative conditions, this cysteine residue can be reversibly oxidized to sulfenic acid and then overoxidized to sulfinic and sulfonic acid, leading to the irreversible inactivation of the enzyme. Interestingly, S-sulfonated GAPDH was shown to translocate to subcellular domains where it could provoke apoptosis (Lia et al., 2020; Tristan et al., 2011).
On the opposite, aldose reductase (AR) and cytochrome c were shown to be activated by oxidation. The sulfenylation of cardiac AR cysteine residues (mainly Cys-298) activates the enzyme notably during myocardial ischemia (Wetzelberger et al., 2010). The regulation of cytochrome c is more complicated. This enzyme can be oxidized by sulfoxidation of its Met-80 residue, increasing the peroxidase activity. However, in the absence of substrate, the methionine can be further oxidized to methionine sulfone, leading to enzyme inactivation (Parakra et al., 2018). Peroxynitrite, known to inhibit mitochondrial respiration and induce apoptosis, can also target cytochrome c by nitrating Tyr-67 into 3-NT. This nitration induces a conformation change, displacing methionine 80 residue and increasing the enzyme peroxidatic activity (Cassina et al., 2000).
Two recent publications have shown that pyruvate kinase M2 (PKM2) was also strongly regulated by oxidative modifications. This glycolysis enzyme is required for the optimization of pyruvate kinase (PK) activity and is crucial for the metabolic shift between glycolysis and pentose phosphate pathway observed under oxidative conditions. The authors of the first study have shown that the oxidation of two cysteine residues (Cys-358 and Cys-424) inhibits PKM2, thus leading to the reduction of PK activity (Irokawa et al., 2021). On the contrary, a second publication has shown that the selective oxidation of Met-239 residue sustains PKM2 in an active tetramer (He et al., 2022).
Proteases
Despite being found active at inflammatory sites, the cysteine-protease cathepsin K was shown to be sensitive to oxidative stress. Indeed, the sulfenylation of Cys-25 by H2O2 strongly inactivates cathepsin K. This inhibition was shown to be reversible. However, in the presence of high concentration of hydrogen peroxide, two-thirds of the enzyme was irreversibly inactivated by sulfinylation or sulfonylation of the same cysteine residue. Moreover, H2O2 was also shown to impair the processing and the maturation of the zymogen (Godat et al., 2008).
Similarly, the ubiquitin-specific proteases 1 and 7 (USP1 and USP7) were shown to be inactivated by sulfenylation of their catalytic cysteines (Cys-90 and Cys-223, respectively) (Cotto-Rios et al., 2012). Recent studies have highlighted the role played by these proteases in the development of cancers (Lu et al., 2021; Meng and Li, 2022). The calcium-dependent protease calpain-1 was also shown to be inactivated by oxidation. In the presence of H2O2, the formation of a disulfide bond between the Cys-108 and the active site Cys-115 reduces calpain-1 activity and autolysis (Lametsch et al., 2008). Finally, oxidation can lead to the activation of the matrix metalloprotease MMP-7 by converting the thiol residue of pro-MMP7 cysteine switch to sulfinic acid (Fu et al., 2001).
Phosphatases
Oxidative stress has a strong impact on cell kinome, notably by regulating (mainly inhibiting) the activity of several phosphatases. PTP1B, one of the most central protein tyrosine phosphatases, was shown to be inhibited by the conversion of its catalytic cysteine (Cys-215) into sulfenic acid. This inactivation is reversible; however, in the presence of high concentration of ROS, the Cyst-215 can be irreversibly converted into sulfinic or sulfonic acid (Liu et al., 2022).
The two phosphatases Cdc25B and Cdc25C were also shown to be inactivated by ROS. In oxidative conditions, the sulfenylation of their catalytic cysteines followed by the formation of a disulfide bond with a second cysteine leads to the closure of the active site of the two phosphatases and their reversible inactivation (Buhrman et al., 2005; Savitsky and Finkel, 2002). All the phosphatases are subjected to inactivation upon oxidation, as reported in Table 1, except PP2A, which can be activated by nitration of Tyr-265 (see Table 3).
Several MAPK phosphatases (MKP) were also shown to be inactivated by ROS. Indeed, the sulfenylation of catalytic cysteines of MKP-1 (also called DUSP1 for dual-specificity phosphatase 1), MKP-3 (DUSP6), MKP-5 (DUSP10), and MKP-7 (DUSP16) causes their reversible inhibition (Kamata et al., 2005; Seth and Rudolph, 2006). These inhibitions lead to the sustained activation of JNK and p38 MAPK (Dolado et al., 2007). Redox-induced inhibitions by sulfenylation of catalytic cysteines were also reported for the phosphatases SHP-1 and SHP-2 (linked to B cell proliferation), and the tumor suppressor PTPRJ (DEP-1) in acute myeloid leukemia (Crump et al., 2012; Godfrey et al., 2012).
The protein phosphatase 2A (PP2A) is differently regulated by ROS and RNS. While ROS inhibits PP2A activity by inducing the formation of an intramolecular disulfide bond or disulfide bonds to other brain proteins, RNS activate this enzyme by provoking the nitration of Tyr-265 residue (Deng et al., 2019; Foley et al., 2007). This activation is notably associated with nitrosative stress-induced endothelial dysfunction.
Cell signaling pathways
PI3K/Akt signaling pathways are strongly involved in the regulation of cell cycle and cell proliferation, and are therefore linked to the development of numerous cancers. Several actors of this signaling pathway were impaired by oxidative stress. This is notably the case for Akt1 and Akt2. Both kinases were inactivated by oxidation. However, their inhibition mechanisms by ROS and RNS differ. The inhibition of Akt1 (PKBα) is due to a succession of reactions starting with the S-nitrosylation of Cys-296 by nitric oxide.
Once nitrosylated, Cys-296 reacts with Cys-310 to form a disulfide bond, leading to the dephosphorylation of Thr-308 and the inactivation of the enzyme (LU et al., 2013). While Akt1 is inhibited by nitric oxide, the inhibition of Akt2 (PKBβ) is due to the ROS produced notably by NADPH oxidases. These ROS oxidize the Cys-124, therefore inducing the formation of a disulfide bond between this cysteine and the cysteine Cys-297 or Cys-311. This inactivated Akt2 could then be recycled or degraded (Wani et al., 2011). PI3K/Akt signaling pathway is also impacted by oxidative stress through the regulation of the tumor suppressor PTEN. Similar to the regulation of several other phosphatases described previously, PTEN is reversibly inhibited by the H2O2-induced oxidation of its active Cys-124 and the subsequent formation of an intramolecular disulfide bond with Cys-71 (Zhang et al., 2020). This redox-induced PTEN inhibition leads to the activation of Akt.
ERK/MAP kinase signaling pathway was also shown to be regulated by ROS and RNS. A recent study shows that both ERK1 (p44 MAPK, MAPK3) and ERK2 (p42 MAPK, MAPK1) undergo active reaction with H2O2 to form sulfenic acids (notably after PDGF treatment), leading to the reversible kinase inhibition (Keyes et al., 2017). This study also shows that ERK1/2 phosphorylation is not linked to oxidization. The targeted cysteines are not yet identified; however, Cys-159 (murine ERK2 numbering) could be involved. Another study proved that under nitrosative stress, ERK1 and 2 are S-nitrosylated by nitric oxide on the Cys-183 (for ERK1). This S-nitrosylation sabotages ERK phosphorylation, leading to kinase inhibition and apoptosis induction (Feng et al., 2013). The nuclear translocation of ERK could also be impacted by this modification. As nitric oxide levels are low in tumor microenvironment, it could be interesting to trigger NO production to reduce tumoral development.
EGF receptor was also shown to be strongly regulated by ROS, and a recent study has deciphered the mechanisms of this regulation. This study has shown that exogenous or endogenous H2O2 induces the reversible sulfenylation of a unique cysteine residue, Cys-797, leading to an EGFR autophosphorylation and its kinase activation (Truong et al., 2016). Concentrations from 1 to 10 μM of H2O2 stimulate EGFR activity in a dose-dependent manner, reaching a maximal activation of threefold. However, at concentrations exceeding 25 μM, H2O2 reduces EGFR kinase activity, probably through further oxidation of the Cys-797. Several cancer cell types expressing mutated forms of EGFR presented stronger sulfenylation in response to EGF. Cys-797 sulfenylation induced by H2O2 was also shown to reduce the effects of EGFR inhibitors such as afatinib.
The proto-oncogene Src is also regulated by redox-induced PTM. Indeed, a recent work has shown that Src kinase is activated by ROS produced by NADPH oxidases through the sulfenylation of two cysteines, Cys-185 and Cys-277 (chicken numbering, corresponding to Cys-188 and Cys-280 in human Src), allowing the phosphorylation of Tyr-416 (Tyr-419 in human Src) through conformational changes (Heppner et al., 2018). Several other cysteines were also identified as potential oxidation sites (Heppner, 2021). Similar to Src, several Src family kinases (SFKs) were shown to be oxidized on highly conserved cysteine residues: YES1 (Cys-255 and Cys-287), FYN (Cys-246 and Cys-404), LYN (Cys-419 and Cys-468) and BLK (Cys-482) (Heppner, 2021).
Likewise, NF-κB pathway is regulated by two different cysteine oxidations, leading to opposite effects. The DNA-binding ability of the transcription factor NF-κB was shown to be directly inhibited through the S-glutathionylation of Cys-62 of its p50 subunit (Pineda-Molina et al., 2001), while the same pathway can be activated by the inactivation of its inhibitor IKKβ. This inhibition is due to the S-glutathionylation of Cys-197 (Reynaert et al., 2006). Pro-oxidative conditions can therefore regulate the expression patterns of different genes.
Janus kinases (JAKs) are crucial for cytokine-induced proliferation, and they are known to be negatively regulated by oxidative stress, notably by nitric oxide (Duhé et al., 1998). It was shown that JAK2 was inhibited through the sulfenylation of two cysteines located in the catalytic domain of this enzyme (Cys-866 and Cys-917). A disulfide bond can also be formed between the two residues of this redox switch. Similar regulations by oxidation were observed for JAK1 and JAK3. However the involved residues have to be determined, notably for JAK3, as a sequence alignment showed it lacks the second cysteine of the redox switch (Duhé, 2013).
Calmodulin (CaM) is a part of calcium signal transduction pathway, mediating several processes, including inflammation and apoptosis. This protein was shown to be regulated by oxidation of three of its nine methionine residues. The reversible oxidation of Met-77 in methionine sulfoxide reduces the ability of calmodulin to activate calcium/calmodulin kinase IIα (CaMKIIα) (Marimoutou et al., 2018). Similarly, the oxidation of Met-144 and Met-145 to methionine sulfoxides reduces the ability of calmodulin to activate PM-Ca-ATPase. Most of this effect seems to be due to the oxidation of Met-144 (Bartlett et al., 2003).
Apoptosis regulators
Apoptosis regulation is capital to avoid excessive cell death or uncontrolled cell proliferation. Apoptosis was shown to be regulated at diverse levels by oxidation. The tumor suppressor p53 was notably shown to be inactivated by ROS-induced oxidation. P53 DNA binding core domain contains three zinc binding cysteines (Cys-176, Cys-238, Cys-242 + His-179). Upon oxidation by H2O2, two intramolecular disulfide bonds are formed between Cys-176 and Cys-242, and Cys-182 and Cys-238, leading to the loss of zinc and the reduction of p53 DNA binding ability (Scotcher et al., 2013).
Such an inhibition of p53 transcription activity was also observed in glioma malignant cells in the presence of peroxynitrite (Cobbs et al., 2003). The Cys-277 was also shown to be oxidized, through S-glutathionylation or formation of intermolecular bonds with other proteins such as 14-3-3 (Shi et al., 2021). The pro- and antiapoptotic proteins Bax and Bcl-2 are also regulated by redox-mediated PTM. Indeed, while Bax is activated by the H2O2-induced oxidation of its cysteine residue 62 leading to its translocation and apoptosis induction (Nie et al., 2008), the antiapoptotic Bcl-2 is inhibited by hydrogen peroxide through the sulfenylation of its Cys-158 and Cys-229 (Luanpitpong et al., 2013).
Structural proteins
Several redox-induced regulations of the major cytoskeleton component actin were identified. Actin Cys-374 is the most reactive, undergoing different oxidation reactions according to the oxidant molecule: sulfenylation, sulfinylation, sulfonylation, glutathionylation, nitrosylation, and carbonylation. Cys-374 can also form an intramolecular bond with Cys-285 or intermolecular bonds with other actin molecules. The oxidation of Cys-374 leads to a reduction of actin polymerization and its nitrosylation inhibits actin binding to profilin-1 (García-Ortiz et al., 2017; Rouyère et al., 2022). Actin can also be oxidized on its methionine residues 44 (Met-44) and 47 (Met-47), increasing its depolymerization (Hung et al., 2013).
Intermediate filaments are also targeted by oxidant molecules, and vimentin was shown to be sensitive to oxidation, particularly on its reactive Cys-328. In the presence of hydrogen peroxide, this particular cysteine residue can form disulfide bonds, impairing vimentin polymerization into filaments (Viedma-Poyatos et al., 2020).
Chaperones
The heat shock proteins (Hsp) are chaperone proteins, protecting the cells during stressful conditions. Several of them are sensitive to oxidation, notably Hsp60, Hsp70, and Hsp90. In the presence of nitric oxide, Hsp60 is S-nitrosylated on Cys-237, improving its mitochondrial targeting and its binding to Tfam (Suliman et al., 2010). Similarly, Hsp90 can be S-nitrosylated by nitric oxide leading to its inhibition. This nitrosylation was putatively assigned to Cys-597. However, the authors of the study were unable to totally rule out the involvement of Cys-596 (Martínez-Ruiz et al., 2005). Hsp72, which participates in the degradation of misfolding or damaged proteins following a stress, was shown to be activated by sulfenylation of its Cys-306. This oxidation induces rearrangements, allowing the subsequent oxidation of Cys-276. These cysteine oxidations of Hsp72 (that can be induced by methylene blue) favor the degradation of its bound substrates such as tau (Miyata et al., 2012).
DJ-1, also called PARK7 due to its involvement in Parkinson’s disease, was recently shown to be sensitive to oxidation. Under oxidative conditions, the Cys-106 of DJ-1 is oxidized into sulfinic acid and can be overoxidized to form a sulfonic acid. The overoxidation of DJ-1 destabilizes its active dimeric structure, leading to a function loss (Kiss et al., 2017).
Sirtuins
The sirtuin family is constituted by seven highly conservated deacylase enzymes called SIRT1 to SIRT7. Several studies have correlated a decreased sirtuin deacylase activity to several aging-related diseases, including Parkinson’s disease. Recent publications have shown that ROS and RNS are involved in the regulation of these enzymes. SIRT1 was shown to be carbonylated on a cysteine residue (probably Cys-482) by hydrogen peroxide and smoke cigarette extract, leading to the enzyme inhibition and degradation by the proteasome (Caito et al., 2010).
Similarly, SIRT2, 6, and 7 were recently shown to be carbonylated under oxidative conditions, leading to their degradations (Baeken et al., 2021). SIRT1, 3, 5, and 6 were also shown to be inhibited by NO-induced cysteine nitrosylation (Kalous et al., 2020). Tyrosine nitration of these enzymes were also observed (Kalous et al., 2021). Finally, SIRT6 was shown to be sulfenylated in the presence of 500 μM of H2O2, on cysteine residues 18 (Cys-18) or Cys-144, according to the cell types (Kalous et al., 2021; Long et al., 2017).
One of the goals of PTM is to increase the functionality of the proteome to run different biological functions of the normal cell. These PTM consist in the covalent attachment of functional groups that will undergo phosphorylation, acetylation, glycosylation, acylation, ubiquitination, SUMOylation, and oxidation of protein targets. These PTM can take place at any moment of the protein life cycle. As a result, the physiological PMT can, for example, regulate the subcellular localization, the protein folded conformation, the protein–protein interaction, and protein activities. Pathological PTM of one or more target proteins can lead to diseases, including cancers and neurological disorders such as Alzheimer’s, Parkinson’s, and prion diseases.
Pathological Implications of Redox Post-Translational Modifications
PTMs are important regulators of signal transduction pathways, gene expression, and cell cycle progression. Through these modifications, proteins can be activated or inhibited, and protein–protein interactions can be facilitated or prevented. PTMs can include phosphorylation, oxidation, acetylation, glycosylation, ubiquitination, methylation, and SUMOylation (Meng et al., 2022). In this third part of the review, we focused on recent findings regarding cysteine oxidation, methionine oxidation, and carbonylation’s involvement in pathologies. At the clinical level, there is little experimental evidence of a direct role for redox PTMs in pathology. However, due to the large number of proteins affected by the redox PTMs (see Tables 1–3), these modifications can have a significant impact on the progression of many diseases, including cancer, Alzheimer’s and other neurodegenerative diseases, diabetes, and liver or heart diseases.
Cancers: Recent advance in redox PTM involvement
The involvement of oxidative stress in tumor progression and its impact on the efficacy of anticancer therapies have been known for a long time. Oxidative stress impacts numerous signaling pathways and proteins, leading to increased DNA damage and impaired repair mechanisms. Indeed, cancer progression involves impact on proliferation, apoptosis, immune escape, metabolic dysregulation, genome instability, angiogenesis, migration and metastasis, inflammation, epigenetic regulation, senescence, and interaction with the microbiome, as recently reviewed (Hanahan, 2022).
All these mechanisms have been shown to be impacted by ROS production and oxidative stress (Bekhet and Eid, 2021; Wang et al., 2021). Excessive ROS production in healthy cells can either lead to cell death or to the modification of proteins and nucleic acids that are subsequently involved in the development of cancer cells. In fact, it is classically accepted that cancer cells show a redox balance more in favor of oxidation and a higher ROS level than healthy cells. Tumor cells seem to be more resistant than normal cells to oxidative stress.
In cancer progression, redox PTMs can influence a wide range of molecular processes, such as cell growth and survival, DNA damage, and inflammation, leading to tumorigenesis. For example, redox PTMs have been linked to the activation of the PI3K/AKT pathway, which is important for cell survival and proliferation. In addition, redox PTMs can modulate DNA damage and repair, angiogenesis, and metastasis. However, an excessive increase in ROS production remains toxic for these cells and many anticancer molecules induce an increase in ROS, which contributes to their therapeutic effects. Despite this, beyond a certain ROS threshold, tumor cells can resist treatment by modulating the expression and activity of repair and antioxidant enzymes. This antagonistic effect of ROS in cancer development, therapy, and chemoresistance is synthetized in Figure 6.

Cysteine redox PTM in cancer
Cysteine oxidation has been involved in the modification of many signal transduction effector proteins (kinases or receptors), thus explaining an involvement in tumor progression. However, ROS production is also sometimes involved in antitumoral regulation. Direct modification of epidermal growth factor receptor by H2O2 at a critical active site cysteine (Cys-797) has been shown to enhance its tyrosine kinase activity in human A431 ovarian cancerous cell lines (Paulsen et al., 2011). These molecular studies indicate that sulfenylation of Cys-797 (i.e., conversion to R-SOH) can enhance kinase function, and that subsequent conjugation with GSH (to form S-glutathionylated EGFR; R-SSG) is likely responsible for restoring inactive EGFR and preventing further irreversible oxidation (Truong et al., 2016).
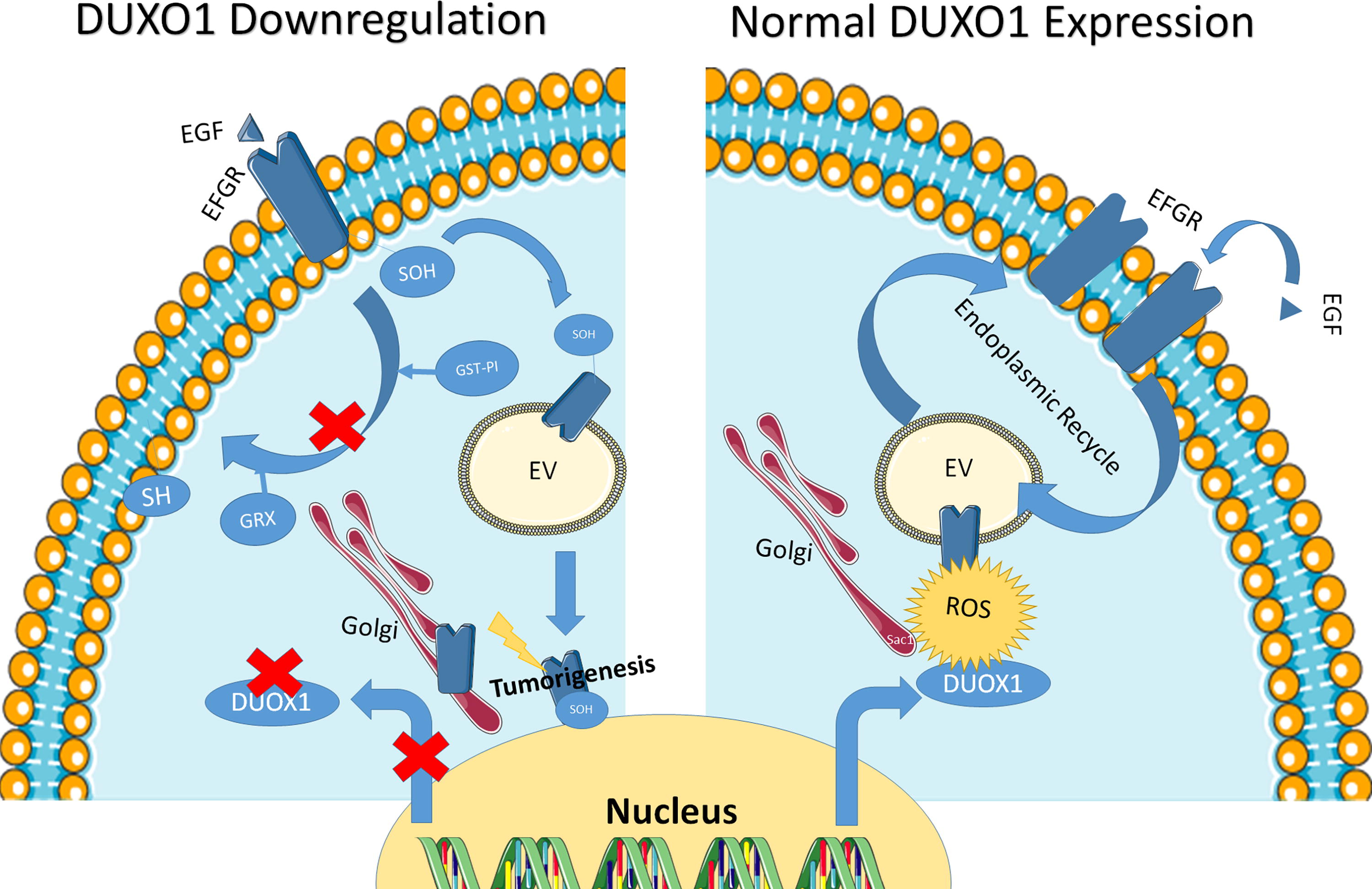
More recently, Little et al. showed Duox1 downregulation accelerates the turnover of cysteine oxidation in lung cancer cell lines, leads to an inappropriate EGFR receptor nuclear localization, and decreases EGFR receptor trafficking, recycling, and signaling (Little et al., 2019). Altered EGFR internalization in DUOX1-deficient lung cancer cells is associated with enhanced GSTP1-mediated S-glutathionylation of EGFR-SOH and subsequent reduction to EGFR-SH, whereas such turnover of EGFR-SOH is delayed in DUOX1-expressing cells and is independent of GSTP1 (Little et al., 2019).
The use of Cys-797S EGFR mutants did not suppress all detectable sulfenylation of the receptor, suggesting that other cysteines might be involved. In addition, an indirect effect is possible since the phosphatidylinositol 4-phosphate [PtdIns(4)P] phosphatase Sac1 is subjected to cysteine oxidation in a signaling hug involving EGFR and Duox1 (Park et al., 2019). Thus, antitumoral ROS production by Duox1 is needed to maintain an efficient EGFR recycling through cysteine oxidation residue on EGFR and Sac1. Those results were summarized in Figure 7.
Pyruvate kinase M2 (PKM2) is the last rate-limiting enzyme of the aerobic glycolysis converting ADP and phosphoenolpyruvate in ATP and pyruvate. PKM2 exists in tetramer or in dimer, the tetramer being the active form in glycolysis, while the dimer can enter the nucleus and act as a transcription factor (Zhang et al., 2019). Thus, PKM2 tetramer, through its glycolytic activity, will favor tumor growth by promoting the Warburg effect (a shift from oxidative mitochondrial metabolism to aerobic glycolysis more in favor of tumor proliferation due to a better energetic supply for the tumor) (Fukushi et al., 2022).
It has also been reported that PKM2 dimer has low enzymatic activity, but it can shift in the nucleus under EGFR stimulation to activate B-catenin pathway. Recent report showed that oxidation of two cysteine residues (Cys-358 and Cys-424) inhibits PKM2 enzymatic activity and sensitizes the tumor cells to Redox-dependent chemotherapies such as cisplatin (Irokawa et al., 2021). Thus, PKM2 under EGFR stimulation represents another example involving cysteine oxidation in an antitumoral effect.
Another recent example of antitumoral impact of ROS production and cysteine oxidation relies on the direct cysteine sulfynilation of free cysteine induced by the cysteine dioxygenase 1 (CDO1). CDO1 limits tumor progression downstream the NRF2/Keap1 pathway in a model of lung cancer in mice. Lung tumors developed in Keap1R554Q/LSL-KrasG12D/Trp53flox mice showed that CDO1 antagonizes the growth and survival of tumor-bearing Keap1Mut by producing toxic sulfinic acid and depleting the cell from NADPH (Kang et al., 2019).
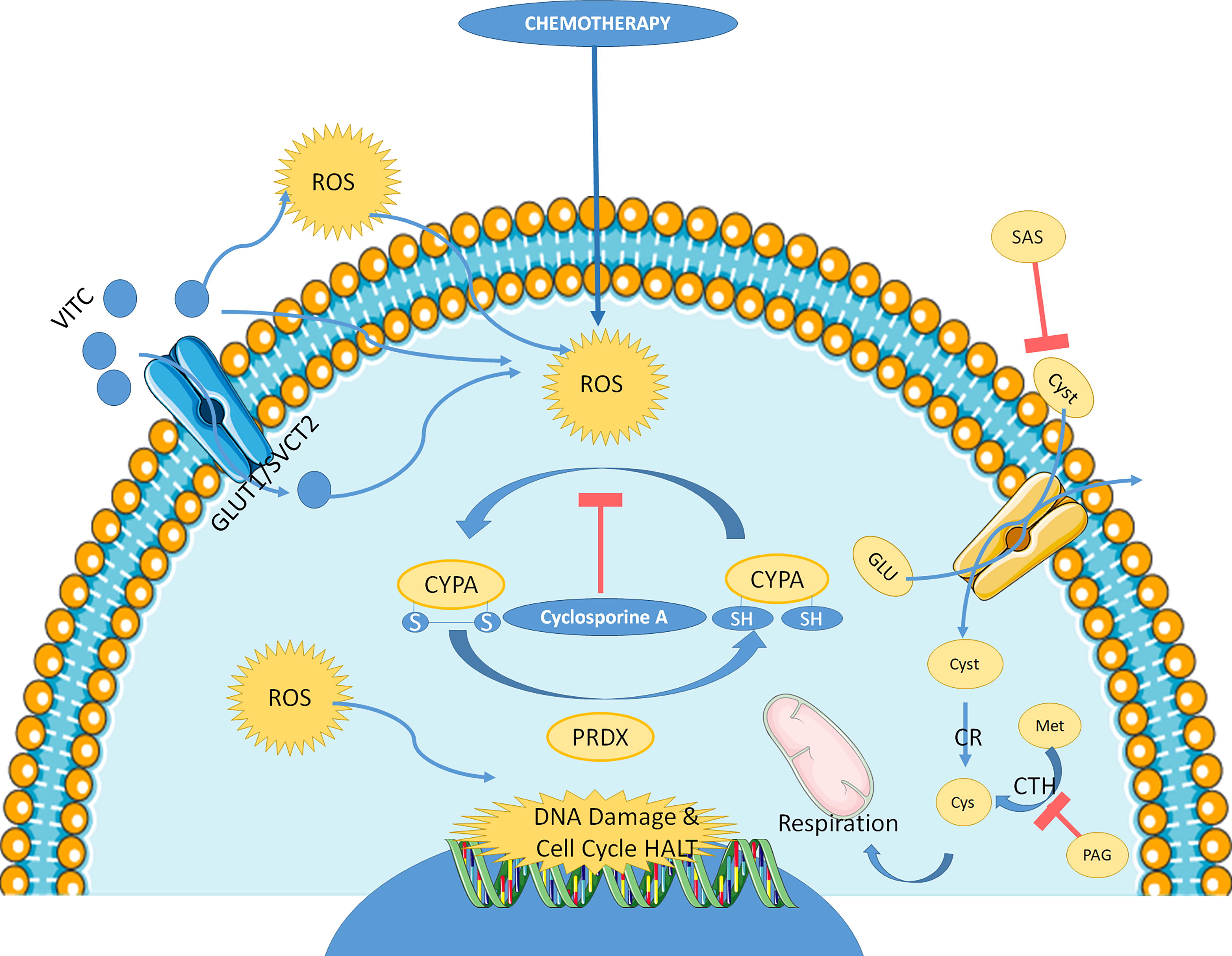
Cysteine deprivation represents an effective way to limit antioxidant defense of cells and increase a harmful oxidative stress in different types of cancers. Thus, cysteine oxidation might represent an important way to decrease the level of active cysteine, while increasing the presence of harmful compounds toxic to the tumoral cells. Recent data showed that cyclophilin A (CypA) cysteine oxidation through disulfide bond formation reduces ROS levels and increases colorectal cancer cell survival to oxidant insult and chemotherapies. This effect is amplified by peroxiredoxin-2, which recycle reduced CypA. Overall survival in patients suffering from colorectal cancer is correlated with the level CypA/peroxiredoxin-2 (Peng et al., 2021).
Cyclosporin A inhibits CypA oxidation and maintains chemotherapy sensitivity and presents a synergistic effect with 5-Fluorouracil and oxaliplatin in a BALB/c nude mice ubcutaneously injected with LoVo oxaliplatin-resistant colorectal human cells (Peng et al., 2021). The reduced form of vitamin C, ascorbic acid, alters the redox state of cysteine-containing proteins in triple-negative breast cancer cell lines and induces cell toxicity. This effect is inversely correlated with peroxiredoxin-1 expression (El Banna et al., 2019). The impact of CYPA cysteine oxidation in the context of chemotherapy is summarized in Figure 8.
In general, cysteine represents in cancerous cells a major actor controlling cellular antioxidant defenses. As such, redox PTM of cysteine presents some ambivalent effect. Historically, the impact of ROS on signaling, particularly in cancer, has identified the cysteines at the active center of phosphatases as major targets of ROS, responsible at the cellular level for phosphatase inhibition, and hence signaling in favor of kinases and phosphorylation of numerous cellular proteins (Olsen et al., 2013).
In cancer, these phosphorylations are considered to be protumoral (Dustin et al., 2020). The more recent work we report here on EGFR, PKM2, CypA, or CDO1 suggests that cysteine oxidation may also play an antitumoral role. The ratio of oxidative insult level compared with the level of available cysteine might represent an important point impacting cell fate and limiting therapeutic intervention at the level of cysteine oxidation due to the complexity of this regulation. Different studies showed that cysteine depletion might be a good alternative to that complex situation. Indeed, besides being a major actor for antioxidant defenses (cysteine being the rate-limiting amino acid for glutathione synthesis), cysteine is also a major regulator of cell metabolism through hydrogen sulfide production, and thus, its tumoral level might represent a good biomarker for cancer progression and treatment (An et al., 2022).
Cysteine depletion has shown promising results for ovarian cancer by impacting the glycolytic metabolism and the oxidative phosphorylation at the same time. Indeed, inhibiting cystine cellular uptake and endogenous production through the trans-sulfuration pathway in ovarian cancer cells led to cell death by apoptosis in cells using oxidative phosphorylation and by ferroptosis and necrosis in glycolytic cells (Novera et al., 2020). Liu et al. recently developed a new highly specific “Lock and Key” fluorescent probe for cysteine (Liu et al., 2019). Using that probe, they showed that inhibiting cystine transporter to deplete intracellular cysteine is more efficient than depleting intracellular GSH for sensitizing cancer cells to chemotherapy (Liu et al., 2019). The development of probe to follow cysteine within the tumor would represent an interesting approach to understand the complex role of cysteine in cancer development and treatment.
Methionine redox modification in cancer
Methionine sulfoxide reductase (MSR) A and B limit methionine sulfoxide formation. Loss of MSR function is associated with a variety of pathological conditions such as heart disease, liver injury, and cancer (De Luca et al., 2010; Erickson et al., 2008; Lei et al., 2007; Singh et al., 2017). Loss of MSRA drives the metastatic spread of pancreatic ductal adenocarcinoma organoids established from various stages of the disease. Loss of MSRA remodels glucose metabolism and results in the oxidation of a methionine residue (M-239) that allosterically activates pyruvate kinase M2 (PKM2) to promote pancreatic ductal adenocarcinoma metastasis in mouse models (He et al., 2022).
Conversely, oxidation of a PKM2 cysteine leads to inhibition of its activity (Anastasiou et al., 2011). In a cohort of patients with triple-negative breast cancer, immunoglobulin-like transcript 4 (ILT4) has been shown to increase tumor progression and PKM2 expression in vivo (Zhang et al., 2023).
As for cysteine, methionine depletion has also been evaluated in the context of triple-negative breast cancer (Malin et al., 2021). Methionine deprivation increases ROS production and thioredoxin reductase expression through NRF2/ATF4 pathway. This approach sensitizes cancer cells to auranofin treatment (a thioredoxin reductase inhibitor). The same complex situation was observed in studies targeting PKM2 in lung cancer. PKM2 overexpression led to a poor prognosis in patients with non–small cell lung cancer (NSCLC) following curative resection (Ito et al., 2023). In addition, inhibition or downregulation of PKM2 in in vivo studies has shown interesting results in inhibiting tumor progression (Dai et al., 2023; Wang et al., 2023). However, another study also showed that PKM2 activators can have antitumor activity on this type of tumor (Li et al., 2018). This last study could support the idea that PKM2 methionine oxidation might have some antitumoral activity by favoring the tetrameric form.
The complexity of studying PKM2 relies on the fact that the tetrameric form (with glycolytic activity) and the dimeric form (with transcription factor activity) might have a different impact on cancer cell fate. In the previous studies, the increase or decrease of tetrameric form might in fact be relative to the modification of the dimeric form and its impact in the nucleus. This point might also be relevant to methionine oxidation of PKM2, which might decrease the dimeric form in the nucleus. These results suggest that the oxidation of methionine and its metabolism present a complex regulation, as in the case of cysteine, making difficult the use of these redox PTM to date as a therapeutic target in cancerology.
Carbonylation involvement in cancer
In agreement with elevated levels of oxidative stress in cancerous cells, higher total protein carbonylation has been reported in several types of cancer (Nathan et al., 2011). Some specifically carbonylated proteins have been identified in cholangiocarcinoma (Thanan et al., 2012). More recently, Aryal et al. showed that selective carbonylation of three specific proteins (HSP90β, filamin A, and bifunctional glutamate/proline-tRNA ligase) was observed in breast tumor tissue and cell lines compared with adjacent healthy tissue. However, the mechanism controlling the selectivity to protein oxidation and carbonylation is not well understood. Since plasmatic level in filamin A is a specific and sensitive marker for metastatic breast and prostate cancer, the study of the level of carbonylation of filamin A in this context represents an important evolution (Aryal and Rao, 2018).
Neurodegenerative diseases: Recent advances in redox PTM involvement
Redox PTM can lead to the development of neurodegenerative diseases, such as Alzheimer’s and Parkinson’s, by causing a decrease in the activity of proteins associated with neuronal function. Redox modifications can also lead to the overactivation of proteins that cause inflammation and cell death, leading to neuronal death. In addition, redox modifications can lead to the accumulation of misfolded proteins, which can further cause neuronal dysfunction and damage (Deshmukh et al., 2017; Simpson and Oliver, 2020; Zhou et al., 2022).
Finally, many neurodegenerative diseases involved the accumulation of protein aggregates as a marker of evolution of the pathology leading to neuronal defect. Different prone-to-aggregate protein such as tau, TDP43, or beta-amyloid peptides are subjected to PTM leading filament aggregation involved in neuronal cell death and neurodegenerative diseases (Fig. 9). In neurology, tau function is linked to the formation of abnormal protein deposits in the brain, called neurofibrillary tangles. These tangles are characteristic of several neurodegenerative diseases, such as Alzheimer’s disease, Pick’s disease, and progressive supranuclear palsy. The tau protein is normally present in neurons and plays an important role in the stabilization of microtubules, which are essential structures for the transport of molecules and organelles in neurons.

However, in neurodegenerative diseases, tau protein detaches from microtubules and aggregates into tangles, disrupting the normal functioning of neurons, and leads to their death. Research on tau function in neurology aims to better understand the mechanisms underlying the formation of neurofibrillary tangles and to develop treatments to prevent or slow the progression of these diseases. Therapeutic approaches targeting the tau protein are currently in development, including antibodies that can reduce the accumulation of tau in the brain (Goedert and Spillantini, 2019, 2000; Mena and Strafella, 2022; Morris et al., 2011; Wang and Mandelkow, 2016).
TDP43 (TAR DNA-binding protein 43) is a protein involved in the regulation of gene expression and the alternative splicing of RNA. It is mainly located in the cell nucleus and is expressed in many tissues, including the brain. However, studies have shown that TDP43 is implicated in several neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia, and Alzheimer’s disease. In these diseases, TDP43 is found as abnormal deposits in neurons and glial cells.
Studies have also shown that the dysregulation of TDP43 can lead to impaired synaptic function and neurotoxicity, thereby contributing to the pathogenesis of these neurodegenerative diseases (Arai et al., 2006; Igaz et al., 2011; Liao et al., 2022; Meneses et al., 2021; Neumann et al., 2006; Prasad et al., 2019; Wang et al., 2008). Increasing evidence indicate that there may be a link between ROS-induced oxidative stress and the accumulation of misfolded proteins in the form of aggregates (Abramov et al., 2020). Thus, the next sections describe the recent findings on cysteine and methionine oxidation in neurogenerative diseases. The results of those studies have been synthetized for Parkinson’s and Alzheimer’s diseases in Figures 10 and 11, respectively.


Cysteine redox PTM in neurodegenerative diseases
At the molecular level, different studies showed the impact of cysteine oxidation on protein structure, folding and aggregation of prone-to-aggregate proteins. Marinelli et al. took advantage of the use of a yeast protein FF domain (50–70 residues), whose folding and aggregation properties are well known and which possess four alpha helix and only one cysteine to study the impact of cysteine oxidation on the structure of that domain (Marinelli et al., 2018). Sulfonic acid is the most highly oxidized species of thiols, and its formation is irreversible.
We have still little information on how site-specific cysteine sulfonic acid (CySO3H) formation may impact protein conformation. Irreversible oxidation of a single free Cys as cysteine-sulfone can dramatically impact the folding and stability of the FF globular domain, leading to its aggregation into amyloid-like structures under physiological conditions. The computational analysis performed in this study suggests that many unrelated proteins containing exposed free Cys residues are susceptible to suffer from this deleterious conversion. These results suggest that when ROS levels increase, because of aging or stress, cells might live on the edge of a proteostasis catastrophe, even if proteins are not extensively oxidized.
DJ-1 cysteine oxidation
DJ-1 is a multifunctional protein that plays an important role in the regulation of cell survival, chaperone function, protease activities, mitochondrial functions, antioxidative properties, iron homeostasis, and regulation of transcription to the protein. In the nervous system, DJ-1 is expressed in neurons and glial cells, and its dysfunction is associated with several neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease.
In Parkinson’s disease, DJ-1 is involved in the regulation of the PTEN-induced putative kinase 1 (PINK1)/Parkin protein kinase signaling pathway, which is essential for the degradation of mitochondria damaged by autophagy. Mutations in DJ-1 are associated with an autosomal recessive form of Parkinson’s disease, and loss of DJ-1 function may lead to accumulation of dysfunctional mitochondria and increase in oxidative stress in neurons. Overexpression of DJ-1 may reduce Aβ production and improve neuronal survival in 5XFAD, Alzheimer’s disease model mice. Apart from Parkinson’s disease and Alzheimer’s disease, DJ-1 is also implicated in other neurological diseases, such as ALS and muscular dystrophy of Becker where oxidative stress is implicated (Antipova and Bandopadhyay, 2017; Baulac et al., 2009; Bonifati et al., 2003; Cheng and Zhang, 2021; Huang and Chen, 2021; Sandrelli and Bisaglia, 2023).
Numerous studies have ascribed various roles, including antioxidative properties, chaperone function, protease activities, mitochondrial functions, and regulation of transcription to the protein. The DJ-1 protein undergoes oxidation and post-translational modifications that are important for its function. Not only is DJ-1 linked to familial Parkinson’s disease, but it is also associated with the pathogenic mechanisms of sporadic Parkinson’s disease and other neurodegenerative disorders where oxidative stress is implicated. DJ-1 has been reported to have protein deglycase and DNA deglycase activity (De Lazzari and Bisaglia, 2017; Richarme et al., 2018). In humans, DJ-1 has been implicated in several pathologies such as cancer, Parkinson’s disease, and ALS. DJ-1 mutants have been identified in Parkinson’s disease.
The DJ-1 protein undergoes oxidation and post-translational modifications that are important for its function (Antipova and Bandopadhyay, 2017). The cysteine 106 of DJ-1 located in the active site is critical for protein function. This cysteine has been reported in three oxidation states (Cys-SH, Cys-sulfine, and Cys-sulfone). However, the crystal structure of DJ-1 in the oxidized state is essentially identical to that of the protein in the reduced state (Lin et al., 2012; Premkumar et al., 2011). Recently, using nuclear magnetic resonance spectroscopy with DJ-1 in solution at physiological temperature, Barbieri and Luchinat showed that the comparison of the 1H,15N chemical shifts between the two states reveals non-negligible changes in distinct parts of the protein backbone upon Cys106 oxidation.
Tau cysteine oxidation
The tau protein, a well-known effector of Alzheimer’s disease, forms toxic protein aggregates in neurons. While hyperphosphorylation of tau is well identified in the process of aggregation, little is known about the role oxidation may play in aggregation. Formation of intramolecular disulfide bond in four-repeat tau monomers (htau40) leads to fibril formation. The fibrils are less stable than fibrils formed under reducing conditions, but are highly effective in seeding oxidized tau monomers. The oxidized fibrils are different from fibrils formed under reducing conditions, in that they are more fragile and less stable toward proteolysis and denaturation (Weismiller et al., 2021).
Thus, different molecular studies highlight that cysteine oxidation represents a loss of structuration and further aggregation of proteins involved in neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases.
This point has been confirmed at cellular and patient level in different studies. Cryo-EM studies of tau filaments from human patients revealed that the two tau cysteine residues are not structurally equivalent since Cys-322 is incorporated into the core of the fibril, whereas Cys-291 is disordered and located in the fuzzy coat (Fitzpatrick et al., 2017). The two tau cysteine residues are important for its intrinsic acetyltransferase activity and for the proper localization of tau on microtubules (Cohen et al., 2013; Martinho et al., 2018).
The recently identified cysteine-modifying MAPT mutation C291R in a patient diagnosed with corticobasal degeneration (Marshall et al., 2015) further highlights the importance of Cys residues in tau physiology and pathology. In vitro studies revealed that a C291R mutant of four repeat tau aggregated into different kinds of protofibrils than wild-type tau construct (Karikari et al., 2020). Blocking Cys-322 mutation significantly abolishes tau toxicity, while with Cys-291 substitution, tau retains its toxicity (Prifti et al., 2021). Recent finding showed that the Mical drosophila protein comprises a conserved multidomain flavoprotein monooxygenase, which oxidizes tau Cys-322, leading to a highly aggregated form of the protein (Prifti et al., 2022).
Besides the prone-to-aggregate proteins, cysteine oxidation also affects cell signalization protein, structural proteins, proteases, and sirtuins (see Tables 1–3).
CaMKII cysteine oxidation
Ca2+/calmodulin-dependent protein kinase II (CaMKII) function is altered in Parkinson’s disease. A recent study by Di Maio et al. showed that the formation of a disulfide bridge (Cys-30 to Cys-289) across two adjacent CaMKII subunits occludes the binding site for calmodulin. The authors further showed that the interaction of calmodulin with CaMKII is lost in postmortem Parkinson’s disease brain specimen (Di Maio et al., 2020).
Parkin cysteine oxidation
Parkin is a protein ubiquitin ligase that plays an important role in regulating protein breakdown in cells. In the nervous system, Parkin is particularly important for the survival of dopaminergic neurons in the substantia nigra of the brain, which are implicated in Parkinson’s disease. Several studies have shown that mutations in the Parkin gene are associated with an inherited form of Parkinson’s disease, characterized by an early loss of neurons dopamine. Studies have also shown that loss of Parkin function may be implicated in other neurological disorders, such as Alzheimer’s disease and ALS (Chen and Dorn, 2013; Dawson and Dawson, 2014; Kitada et al., 1998; Zhang et al., 2015). In Parkinson’s disease, the protein Parkin, an ubiquitin protein ligase, protects the cells from oxidative stress (Schapira and Gegg, 2011).
Five cysteine residues of Parkin are subjected to irreversible oxidation as sulfinic and sulfonic acid and protect the cells. Parkin contributes to redox homeostasis downstream of oxidative stress production, and induces lower carbonyl contents in mouse and human brain tissues, and this effect is independent of its ubiquitin ligase activity (Kodsi et al., 2020).
Parkin is S-nitrosylated in vitro, as well as in vivo in a mouse model of Parkinson’s disease and in brains of patients with Parkinson’s disease and diffuse Lewy body disease. Moreover, S-nitrosylation occurring in the IBR and/or Ring2 domain inhibits parkin’s ubiquitin E3 ligase activity and its protective function (Chung et al., 2004). On the contrary, sulfhydration of parkin involving Cys-95, Cys182, and Cys-59 enhances its catalytic activity. Parkin sulfhydration is markedly depleted in the brains of patients with Parkinson’s disease, suggesting that this loss may be pathologic (Vandiver et al., 2013).
Cofilin cysteine oxidation
Cofilin1 was identified as a key player in different neurological diseases, e.g., Alzheimer’s or Parkinson’s disease (Maloney and Bamburg, 2007; Minamide et al., 2000; Schönhofen et al., 2014). Cofilin1, a structural protein associated to actin cytoskeleton organization, impacts mitochondrial function and induces oxidative neuronal cell death. Cofilin1 acts as a redox sensor in oxidative cell death pathways of ferroptosis and promotes glutamate excitotoxicity. Oxidation of Cys-139 and Cys-147 of cofilin1 are crucial in mediating the direct damage of mitochondria (Hoffmann et al., 2021). Other recent findings showed that oxidation at the Cys-80 position oxidation induces the loss of secondary structures in cofilin-1 and promotes cofilin-1 aggregation to forms amyloid-like structures in vitro (Kaushik et al., 2021).
Sirtuin cysteine oxidation
Sirtuin represents a family of protein implicated in gene and metabolic regulation through their capacity to remove acyl groups from lysine residues in proteins in an NAD+-dependent manner. Such activity may alter individual protein properties as well as the histone–DNA interaction.
Baeken et al. showed that neuronal SIRT degradation in Parkinson’s disease models is executed by autophagy rather than the proteasome. This effect relies on oxidative stress linked to cysteine oxidation and further carbonylation of carbonylation-prone amino acid (Baeken et al., 2021; Kalous et al., 2021).
Other cysteine oxidation
Finally, redox regulation besides neurodegeneration is also involved in peripheral nerve repair. Neuronal outgrowth is an integrin-dependent process. It depends on the ECM composition and on adhesome formation along the protrusions and within the growth cones. The study of the redox properties of adhesome in PC12 cells upon NGF stimulation showed that among the 1700 proteins analyzed in the adhesome, around 300 of them were sulfenylated on cysteine residues (Meißner et al., 2021).
Methionine redox PTM in neurodegenerative diseases
Synuclein methionine oxidation
Synuclein is a protein present in neurons, which plays an important role in the regulation of neurotransmission (Bae et al., 2012; Burré, 2015). It is also involved in the pathogenesis of several neurodegenerative diseases, including Parkinson’s disease and dementia with Lewy bodies (Spillantini et al., 1997). Synuclein is an intrinsically disordered protein, which means that it does not have a fixed three-dimensional structure and that it can adopt different conformations depending on its environment. This property allows it to interact with many other proteins and regulate their functioning (Stefanis, 2012).
In neurons, synuclein is mainly located in the nerve endings, where it participates in the regulation of the release of neurotransmitters. She acts in binding to synaptic vesicles, which contain the neurotransmitters, and modulating their fusion with the cell membrane to release the neurotransmitters into the synaptic space. However, when synuclein accumulates abnormally in neurons, it can form toxic aggregates called Lewy bodies. These aggregates disrupt the functioning of normal neurons and lead to their degeneration and death. This is what happens in Parkinson’s disease and Lewy body dementia, where Lewy bodies are present in neurons of brain regions involved in the control of movement and cognition.
Several studies have sought to understand the mechanisms underlying the formation of Lewy bodies and the toxicity of synuclein (Lashuel et al., 2013). Some have identified mutations in the synuclein gene, which are associated with an inherited form of Parkinson’s disease, while others have shown that post-translational modifications of synuclein, such as phosphorylation and nitration, can promote the formation of Lewy bodies (Henderson et al., 2019; Olanow and Brundin, 2013; Pajarillo et al., 2019; Polymeropoulos et al., 1997; Schmid et al., 2013).
A central hallmark of Parkinson’s disease is the intraneuronal aggregation within the substantia nigra of the protein α-synuclein into amyloid-rich Lewy bodies (Spillantini et al., 1998). Oxidized forms of methionine (Met) and tyrosine (Tyr) have been shown to be abundant in Lewy body deposits (Al-Hilaly et al., 2016), consistent with a role for oxidative stress in Parkinson’s disease (Barnham et al., 2004). At the molecular level, in vitro study, Met and Tyr oxidation are early and rapid events in Cu2+/H2O2-mediated damage to α-synuclein, while carbonyl formation is limited (Tiwari et al., 2018). α-synuclein (SN) contains four methionine (Met-1, Met-5, Met-116, and Met-127) and a high-affinity Cu2+ binding site located in the N-terminal (Nter) region of α-synuclein where Met-1 and Met-5 are located (Mason et al., 2016; Rasia et al., 2005).
The formation of oxidants from H2O2 by Cu2+ bound at this site could result in rapid met-sulfoxidation (within a minute) of these two Nter Met residues followed by slower oxidation of the Cter Met-116 and Met-127 completed after 6 h (Binolfi et al., 2016). Sulfoxides with metal ions have been suggested to alter fibrillation process of α-synuclein (Breydo et al., 2012). Tiwari et al. also showed that intra- and intermolecular dityrosine formation is observed upon Cu2+/H2O2/ascorbate oxidation, but oligomerization of α-synuclein is prevented in the presence of ascorbate compared to oxidation without ascorbate. Thus, mild oxidation of methionine and tyrosine residues of α-synuclein favors oligomeric α-synuclein formation, which can further impact mitochondrial functions through ATP synthase oxidation.
It has been shown that this impact on mitochondria of inducible pluripotent stem cell (iPSC)-derived neurons will further increase oxidative stress and cause α-synuclein to switch from its physiological role to a pathological toxic gain of function (Ludtmann et al., 2018). One important point in the occurrence of methionine oxidation seems to rely on the species of the oxidant. The use of tert-Butyl hydroperoxide at the molecular level, for example, is a selective methionine-specific oxidant, capable of oxidizing surface exposed and partially buried methionine residues to sulfoxide form stoichiometrically (Keck, 1996). Using bovine beta-lactoglobulin as a model of amyloid fiber formation, it was shown that oxidation of methionine residues modifies the fibril formation propensity, and modulates the route of amyloid fibrillation and morphologies of final aggregates (Maity et al., 2021).
Hungtintin methionine oxidation
Huntingtin is a protein that is implicated in several neurodegenerative diseases, including Huntington’s disease, Alzheimer’s disease, and Parkinson’s disease. The normal function of huntingtin is not yet fully understood, but it is known to play a role in intracellular signaling, intracellular transport, and the regulation of gene expression. In Huntington’s disease, a mutation in the huntingtin gene leads to the production of an abnormal form of the protein that accumulates in brain cells, causing damage and cell death.
Several studies have shown that mutated huntingtin interacts with other proteins and disrupts intracellular signaling pathways, contributing to Huntington’s disease pathology (Li and Li, 2004; Ross and Tabrizi, 2011). In Alzheimer’s disease, huntingtin is involved in the regulation of gene expression and intracellular signaling. Studies have shown that huntingtin is involved in regulating the expression of genes involved in the formation of amyloid plaques, which are a hallmark of Alzheimer’s disease (Cattaneo et al., 2005; Zuccato and Cattaneo, 2009). In Parkinson’s disease, huntingtin is involved in the intracellular transport of proteins and organelles. Studies have shown that huntingtin is involved in the transport of mitochondria, which are essential for energy production in cells. Mutations in the huntingtin gene may disrupt mitochondrial transport and contribute to Parkinson’s disease pathology (Li and Li, 2011; Trushina and McMurray, 2007).
In a recent study, Adegbuyiro et al. analyzed how methionine oxidation of huntingtin is affected by the presence of lipid membranes (Adegbuyiro et al., 2022). They showed using in situ atomic force microscopy that Met-8 oxidation reduced fibril formation at the expanse of shorter fibrils and oligomer formation. Without oxidation, granular huntingtin aggregates developed on the bilayer surface, while in the presence of H2O2, distinct plateau-like regions initially developed on the bilayer surface, which gave way to rougher patches containing granular aggregates (Adegbuyiro et al., 2022).
Therapeutic intervention in neurodegeneration through molecules impacting redox regulation is often proposed as a future therapeutic goal. In Alzheimer’s disease, the role or amyloid beta aggregation is well known and the possible involvement of oxidative stress has been recently reviewed (Mandal et al., 2022). They propose that to protect Met35 of amyloid beta peptide from oxidation, an increase in brain GSH would be protective against Alzheimer’s disease. However, due to the conflicting behavior observed following amino acid oxidation, this therapeutic approach seems to be rather limited.
Another recent study proposes that S-methyl-
Other methionine oxidation
Finally, an interesting proteome-wide measurement of methionine oxidation has been performed in aging mouse brains. Oxidation stoichiometries for specific methionine residues are highly consistent between individual animals and methionine sulfoxides are enriched in clusters of functionally related gene products, including membrane and extracellular proteins. There were no significant changes in methionine oxidation in brains of old mice. These results suggest that under normal conditions, methionine oxidation may be a biologically regulated process rather than a result of stochastic chemical damage (Bettinger et al., 2022).
Carbonylation involvement in neurodegenerative diseases
Owing to their irreversible formation, protein carbonyls have a unique stability and a wide range of downstream functional consequences. Primary (direct metal-catalyzed oxidation of amino acid side chains) and secondary (the addition of reactive aldehydes to amino acid side chains) protein carbonylation are recognized as markers of pathological ROS production in a variety of cell types and tissues (Dalle-Donne et al., 2006; Wong et al., 2012). During the past five years, carbonyl measurements have been used to delineate upstream oxidative stress mechanism in Parkinson’s and Alzheimer’s diseases, spinal muscular atrophy, and retinal degenerative disease.
A comparative study in plasma from patients with Parkinson’s and Alzheimer’s diseases revealed a sex-specific difference of protein carbonyl levels. Lower values were measured exclusively in females with Alzheimer’s disease, whereas males with Alzheimer’s disease displayed significantly higher values compared to healthy controls and those with Parkinson’s disease (Sharma et al., 2020). The cysteine oxidation of DJ-1 protein in Parkinson's disease was shown to impact the carbonylation of SERCA, an endoplasmic reticulum Ca2+ channel that plays an important role in Ca2+ homeostasis. SERCA enzymatic activity is significantly reduced in DJ-1β mutant flies, as well as in a human cell Parkinson’s disease model based on DJ-1-deficiency.
Higher carbonylation levels of SERCA were also observed in DJ-1-deficient cells compared with controls. Thus, impaired SERCA activity due to oxidative modification may play a role in Parkinson’s disease (Solana-Manrique et al., 2021). In motor neurons, it has been shown that spinal muscular atrophy induces a mitochondrial dysfunction of complex I, an increase in ROS production and carbonyl protein levels, and a decrease in protein synthesis. Modulation of ROS production in these neurons allows to restore protein synthesis levels in an mTOR-dependent manner (Thelen et al., 2020).
Finally, an increase in carbonyl amino acid level has been associated with photoreceptor cell death induced by N-methyl-N-nitrosourea in male C57BL/6 mice. HSP70 presents a protective effect on neuronal cells and is cleaved by calpain. Protein carbonylation of HSP70 leads to a more efficient degradation of HSP90 by calpain than noncarbonylated HSP70, thus increasing neuronal cell death (Furukawa et al., 2018). HSP70 (heat shock protein 70) is a chaperone protein that plays an important role in protecting cells against cellular stress. She is involved in many cellular functions, including regulation of the inflammatory response, protection against oxidative damage, and repair of damaged DNA (Belenichev et al., 2023; Niu et al., 2006). In neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and ALS, HSP70 is involved in the regulation of proteostasis, i.e., the regulation of protein quality in cells.
Several studies have shown that the expression of HSP70 is increased in the brain cells of patients with neurodegenerative diseases, suggesting a protective role of this protein in these diseases (Gupta et al., 2020; Lindberg et al., 2015; Turturici et al., 2011). Studies have also shown that increased HSP70 expression can reduce the aggregate protein richness induced in these diseases, such as beta-amyloid protein in Alzheimer’s disease and alpha-synuclein protein in Parkinson’s disease (Gao et al., 2015). However, other studies have proposed that the expression of HSP70 may also be involved in the progression of these diseases, by promoting the formation of amyloid plaques and protein aggregation (Turturici et al., 2011; Wilhelmus et al., 2007). Therefore, the exact role of HSP70 in neurodegenerative diseases remains to be determined.
Other pathologies: Recent advances in redox PTM involvement
Recent studies on redox PTM in pathologies other than cancer and neurodegeneration mostly involve the increase in protein carbonylation (see Fig. 12). Although ROS are supposed to be involved in the mechanism and pathogenesis of many diseases, the nature of the relationship is not always well known. In patients with diabetic nephropathy, Almogbel et al. measured the levels of protein carbonylation in patients and compared them with disease markers HbA1C, postprandial blood glucose (PPBG), and disease duration.
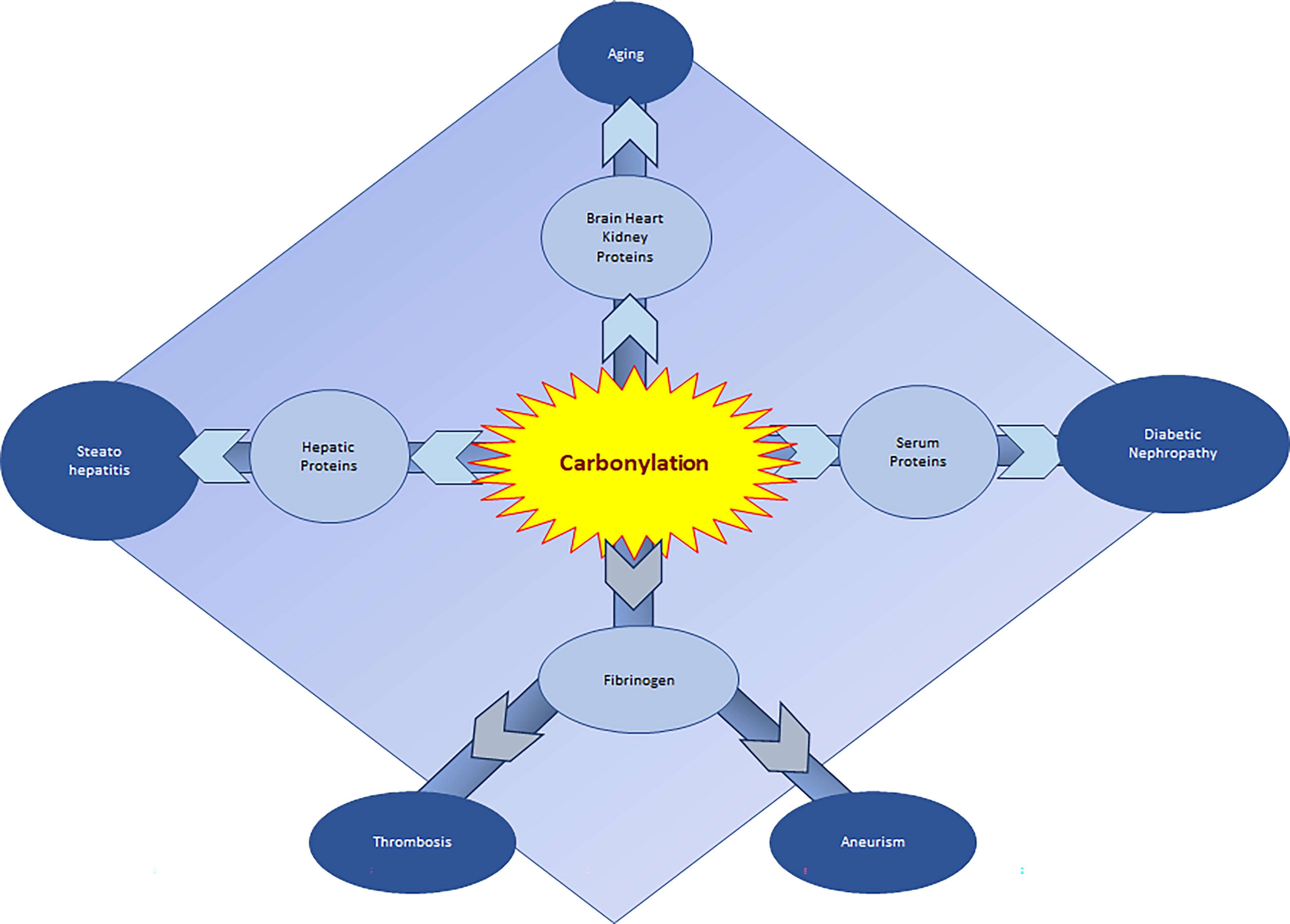
This study showed a positive correlation between protein carbonylation and diabetic nephropathy. The higher carbonylation in patients with higher HbA1C, blood glucose, diabetes duration, or serum creatinine indicates that oxidative modifications in proteins play a key role in the progression of diabetic nephropathy (Almogbel and Rasheed, 2019). Specific carbonylation of fibrinogen has been involved in longer persistence of thrombi (Suzuki et al., 2020). In vitro, lysine fibrinogen carbonylation by acrolein resulted in the generation of fibrinolysis-resistant fibrin by attenuating the activation of Glu1-plasminogen. This effect was confirmed in aorta of patients suffering from abdominal aortic aneurisms (Suzuki et al., 2020). Carbonylation of mitochondrial proteins are often associated with mitochondrial dysfunction and pathologies. Hepatic oxidative stress is believed to be one of the factors driving steatosis to nonalcoholic steatohepatitis.
A proteomic approach on hepatocyte cell line showed that most of the carbonylated proteins involved in nonalcoholic steatohepatitis pathogenesis target pathways controlling energy metabolisms. Activity of the ATP synthase subunit α (ATP5A), a key protein in cellular respiration, particularly affected fatty acid exposure by increased carbonylation of residues located in the nucleotide-binding region of the protein. The reduction of carbonylation of this protein increased its activity (Chienwichai et al., 2019). Finally, the level of carbonylation was assessed in brain, heart, and kidney tissue of 40 Wistar rats of varying ages (Kolawole et al., 2019). This study revealed a direct proportional relationship between age and protein carbonylation in all tissues studied, suggesting that carbonyl level might be a marker in the process of aging, contrary to methionine oxidation, which is more related to lifelong physiological regulation (Bettinger et al., 2022).
Conclusion
Because of the large number of proteins impaired by pathological redox PTMs, it is obvious to imagine the radical impact of these modifications on the initiation and progression of many diseases, including cancer, diabetes, liver and heart diseases, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s. Indeed, in disease, redox PTMs are impaired in response to upstream pathological events, altering, for example, the function of opposing classes of enzymes. So the identification of molecular bases, which lead to aberrant regulation of redox PTMs in the diseases, and revealing of the code of this redox PTMs will be fundamental to understand the etiology of different disorders (Prabakaran et al., 2012). In fact, the identification of the cellular and molecular bases that are impaired by the diseases could help find new drug targets that can provide a therapeutic benefit aimed at either blocking or reversing the pathological redox PTM code in diseases.
Footnotes
Authors’ Contribution
The article was written by C.C., H.K., L.F., and L.L. The figures were designed by C.C. and L.L. The article was reviewed by H.K.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This research received no external funding.