
Abstract
Significance:
Sirtuins are NAD+-dependent histone deacetylases regulating important processes in cellular biology such as inflammation, metabolism, oxidative stress, and apoptosis.
Recent Advances:
Despite initially being discovered to regulate transcription and life span via histone deacetylase activities, emerging data continually uncover new targets and propose additional roles. Due to the outstanding importance of the sirtuins in the control of the inflammatory response, their roles in the pathogenesis of several inflammatory-based diseases have become an area of intense research. Although sirtuins have been traditionally regarded as anti-inflammatory players, several recent findings suggest that their role in inflammation is complex and that in some cases sirtuins can indeed promote inflammation.
Critical Issues:
In this article, we provide an update on the latest findings concerning the new mechanisms of action and concepts about the role of sirtuins during inflammation. We focus on the impact that inflammatory-based processes exert on the liver, adipose tissue, and nervous system as well as on macrophage function and activation. Also, we discuss available data pointing to the dual role that, in particular contexts, sirtuins may have on inflammation control.
Future Directions:
Since the knowledge of sirtuin impact on metabolism is continually expanding, new venues of research arise. Besides become being regarded as candidates of therapeutic targets, posttranscriptional control of sirtuin expression by means of microRNAs challenges our traditional concepts of sirtuin regulation; importantly, the emerging role of NAD+ metabolism in aging and longevity has added a new dimension to the interest in sirtuin function. Antioxid. Redox Signal. 39, 1185–1208.
Introduction
Sirtuins (SIRTs), the NAD+-dependent histone deacylases, comprise a family of proteins involved in several critical signaling pathways both in prokaryotes and eukaryotes. In mammals, seven homologs of yeast Sir2 named SIRT1 to SIRT7 have been identified. Notably, this protein family plays a wide variety of important roles in cellular biology that largely exceed the canonical pathway of chromatin regulation through histone deacetylation. Increasing evidence suggests that sirtuins are pivotal in regulating the cellular response to stress, through epigenetic control of gene expression and transcriptional silencing of a broad array of core metabolic functions such as energetic metabolism, oxidative stress, aging, apoptosis, and inflammation. For this reason, sirtuins are increasingly regarded as potential therapeutic targets for different conditions, including cancer, cardiovascular disease, and respiratory disease, among others (Bonkowski and Sinclair, 2016).
Due to the outstanding role of the sirtuins in the control of the inflammatory response, their roles in the pathogenesis of several inflammatory-based diseases have become an area of intense research. Such interest is exemplified by the current increasing number of studies reporting uncovered mechanisms of action of already known drugs that involve sirtuin function, additional new pathways modulated by sirtuins, or the discovery of new small molecules able to modify sirtuin activity. Since the role of sirtuins in the inflammatory response is extensively reviewed, we focus to provide a brief update of the latest findings concerning new mechanisms of action or concepts about sirtuin roles during inflammation. We focus mainly on inflammatory-based conditions on adipose tissue, nonalcoholic fatty liver disease (NAFLD), and macrophage function and activation.
Sirtuins Are NAD+-Dependent Deacylases with Critical Roles in Metabolism and Inflammation
The seven members of the sirtuin family of mammals, that is, SIRT1 to SIRT7, are homologous to the yeast Sir2, the founding member of the family. The mammalian sirtuins are ubiquitous enzymes without a defined cell-type specificity (Uhlén et al., 2015). All of them share a highly conserved NAD+ binding site and catalytic core domain (Wang et al., 2019). Despite the characteristic enzymatic activity of sirtuins are the lysine deacetylation and the use of the universal cosubstrate NAD+, they can exhibit different preference for the acyl group in the Nɛ-acyl-lysine substrate. For example, among the mammalian sirtuins, SIRT5 was found to prefer the Nɛ-malonyl-lysine, Nɛ-succinyl-lysine, and Nɛ-glutaryl-lysine substrates to the Nɛ-acetyl-lysine substrate, and SIRT6 was found to prefer the Nɛ-myristoyl-lysine substrate to the Nɛ-acetyl-lysine substrate.
Of note, ADP-ribosylation could also be catalyzed by sirtuins as described for SIRT4 (Haigis et al., 2006), SIRT6 (Liszt et al., 2005; Mao et al., 2011) and SIRT7 (Simonet et al., 2020), in which the ADPR group of NAD+ gets transferred onto nucleophilic amino acid side chains on themselves or onto their protein substrates (Fig. 1). An extensive review of the chemical biology of sirtuins can be read in Chen et al. (2015). Also, the subcellular location and enzymatic functions vary among sirtuins. SIRT1, SIRT6, and SIRT7 are mainly nuclear; SIRT2 is predominantly cytosolic, whereas SIRT3, SIRT4, and SIRT5 are localized in mitochondria (Fig. 2). Although sirtuin compartmentalization is relevant because it works as an additional regulatory step of their biological functions, such localization is not strict and may vary depending on the physiological context of the cell.


Importantly, NAD+ levels are regulated independently in different cell compartments (Venter et al., 2014), however, the different NAD+ pools are still interconnected (Cambronne and Kraus, 2020; Gomes et al., 2013).
Since NAD+ is the common substrate of all sirtuins, changes in NAD+ concentrations, or NAD+/NADH ratios in cells, will affect several critical functions of sirtuins. NAD+ levels fluctuate during many physiological processes. Intracellular NAD+ levels are significantly affected by nutritional and environmental stimuli and these changes are reflected into NAD+-dependent enzymatic activities, which in turn lead to changes in cellular metabolism. Therefore, proper tuning of intracellular NAD+ concentrations appears critical to maintain cell homeostasis (Imai and Guarente, 2014; Nacarelli et al., 2019). Reported NAD+ concentrations in cells vary between studies and methodologies, but are generally in the range of 50–120 μM, making this a very abundant molecule in cells (Cambronne and Kraus, 2020).
There are two main pathways for the synthesis of NAD+, the so-called de novo pathway that utilizes the essential amino acid L-tryptophan to generate quinolinic acid that is further metabolized into NAD+, and the salvage pathway considered the main source of NAD+ in mammals, which utilizes nicotinamide, nicotinic acid, and nicotinamide riboside (Nikiforov et al., 2015).
NAD+ levels remain constant when used as a coenzyme, but in nonredox reactions, its levels are depleted from the cellular pool, requiring continuous synthesis of the dinucleotide. The reactions that consume NAD+ in cells and tissues are mediated by several enzymes that are involved in the nonoxidative roles of NAD+ (Chini et al., 2017). Among them, CD38 and PARPs are those that appear to contribute most significantly to NAD+ degradation. In contrast, sirtuins appear to have a minor role in NAD+ degradation, since they are low-turnover enzymes. Another emerging factor involved in NAD+ metabolism is the sterile alpha and TIR motif-containing 1 (SARM1) protein. It has been reported that SARM1 is involved in the mechanisms that link NAD+ depletion and Wallerian degeneration of axons.
Interestingly, the induction of SARM1-NAD+ase activity is triggered by an innate immune response driven by proinflammatory ligands and activation of toll-like receptors (TLRs) present in axons (Sambashivan and Freeman, 2021).
Recently, it has been described that cellular NAD+ levels decline during chronological aging and in progeroid states (Camacho-Pereira et al., 2016; Chini et al., 2017). NAD+ decline was also observed in animals submitted to a high-fat diet that is known to accelerate aging. Senescent cells are characterized by an upregulation of proinflammatory cytokines, which is termed the senescence-associated secretory phenotype (SASP). Therefore, NAD+ also performs as a metabolic knob fine-tuning of inflammation during senescence and in aging-associated inflammatory conditions, sirtuins being one of their downstream regulators.
Inflammation as a Homeostatic Process
Inflammation is an essential and innate immune response that operates at the cell, organ, and organism level and implies a defensive response during infection or trauma, while tending to return to homeostasis. It comes at the cost of a transient decline in function, so when the injury is held in time and the decline in functions persists, it can contribute to the pathogenesis of diseases (Medzhitov et al., 2012). The inflammatory response is composed of several inseparable pathways involving inflammatory cells, inflammatory mediators induced by sensor cells, and target tissues affected by such mediators.
A successful inflammatory response requires the sequential stages of “initiation,” “adaptation,” and “resolution,” which are coordinated programs that initiate shortly after inflammatory responses begin (Sugimoto et al., 2016). Besides, as the inflammatory response deviates from homeostasis, the extent and duration of this departure lead to the concept of acute and chronic inflammation. For example, the chronic inflammatory responses observed during obesity, diabetes, or aging are persistent low-grade prolonged conditions (Johnson et al., 2012; Nacarelli et al., 2019). On the contrary, the acute inflammatory response is of a shorter duration, greater severity, and farther departure from homeostasis (Joffre and Hellman, 2021). Sirtuins participate in all the stages of the inflammatory process, however, their roles may differ depending on the context the inflammation takes part in, for example, the nature of the insult, the tissues affected, or the cells involved.
Sirtuins and the Molecular Basis of Inflammation
After environmental sensing of danger and signaling from external membrane or internal protein receptors, cytosol and nuclear signaling pathways are upregulated, while many other genes are repressed aiming to reprogram proinflammatory resistance responses. In monocytes or macrophages or any organ-specific cells expressing TLRs, sensing structural molecules present in pathogens (typically lipopolysaccharide [LPS]) or endogenous molecules resulting from cell damage (nucleic acids, proteins) leads to reprogram many genes associated with anabolism and catabolism, including sirtuins (Teena et al., 2020). Adaptive immune cells also increase the expression of many overlapping and cell-specific cytokines and metabolic pathways. Importantly, successful resolution of inflammation returns to homeostasis, but epigenetically restored homeostasis may differ from that from which inflammation departed (Carson et al., 2011; Li et al., 2022).
In response to the inflammatory cues, inflammatory cells release specialized substances, including vasoactive amines and peptides, eicosanoids, proinflammatory cytokines, and acute-phase proteins, which mediate the inflammatory process. Sirtuins are usually regarded as anti-inflammatory regulators because they prevent inflammation or come into scenery to reprogram transcription toward the adaptation/resolution process. SIRT1 was the first sirtuin whose anti-inflammatory activity was recognized and probably the best studied so far.
Nowadays, most of the anti-inflammatory effects performed by sirtuins are carried out by regulating the nuclear factor kappa B (NF-κB) complex. The NF-κB proteins are a family of ubiquitously expressed transcription factors representing a central mediator of proinflammatory gene induction and functions in both innate and adaptive immune cells. SIRT1, 2, 6, and 7 can deacetylate p65, a subunit of NF-κB, which repress NF-κB transcriptional activation and thus reduction of tumor necrosis factor α (TNF-α), IL-1β, IL-6, IL-12p40, and cyclooxygenase-2 (COX2) secretion (Wang et al., 2014; Yeung et al., 2004).
Lysine acetylation at the N-terminus tails of histones H3 and H4 is classically associated with increased gene expression, while deacetylation tends to increase chromatin compaction and gene silencing. As epigenetic regulators, sirtuins can downregulate (via lysine-deacetylation of histones) the activity of key transcription factors that govern the inflammatory response. During the adaptation stage of sepsis, SIRT1 binds in the promoter regions of TNF-α in THP-1 cells and enhances H4K16 deacetylation (Chen et al., 2016). Similarly, SIRT1 and SIRT6 attenuate NF-κB-driven expression of proinflammatory genes by deacetylating histone H3K9 in the promoters of NF-κB target genes (Kawahara et al., 2009; Wu et al., 2015; Zhang et al., 2019).
Besides NF-κB, sirtuins also regulate the activity of other multiple downstream nonhistone targets. For example, SIRT1 can deacetylate and downregulate the activity of tumor suppressor proteins such as p53 (Vaziri et al., 2001) and p73 (Dai et al., 2007), forkhead box proteins forkhead box protein O (FOXO) transcription factors (Motta et al., 2004), but also oncogenes such as signal transducer and activator of transcription 3 (STAT3) (Lu et al., 2014), survivin (Wang et al., 2008), and β-catenin (Firestein et al., 2008), among many others (Fig. 2).
A challenging aspect of these sirtuin-dependent mechanisms of modulation is to understand how a small population of posttranslationally modified proteins such as a deacylated transcription factor targeted by a given sirtuin leads to a maximal response of executive proteins at the cellular level. In other words, how a small fraction of activated proteins belonging to a larger population could account for a broad cellular response? A tentative explanation is that activation of a small number of molecules is sufficient to overcome a given threshold and generate significant responses. In this regard, it is widely known that cells can respond abruptly to gradually increasing signals by means of all or none or even sigmoidal-like responses, in which low concentrations of stimulus do not have much effect, while upon intermediate stimulus levels, the response rises steeply and continuously (Kochen et al., 2022).
For SIRT1, this may be the case, at least in some instances. In fact, measurement of the cellular levels of acetylated p65 in LPS-challenged cells where SIRT1 activity was modified by overexpression, knockdown, pharmacological activation or inhibition showed that TNF-α production correlates rather well with increasing levels of acetylated p65 in a sigmoidal resembling manner (Yang et al., 2012). However, there is a sort of redundancy in the control of p65 acetylation levels, since it can be simultaneously regulated also by sirtuins 2, 6 and 7 in response to a common stimulus (Chen et al., 2019a; Rothgiesser et al., 2010; Sun et al., 2014).
Thus, under these circumstances, the acetylation state of a given target such p65 will result from the simultaneous and convergent activity of several sirtuins, and so, the proportionality between a posttranslationally modified protein population and the full effect achieved by executive proteins will be quite different from that observed for a particular sirtuin-target pair. In addition, it should also be taken into account that a given sirtuin can simultaneously target different key metabolic regulators belonging to convergent but separate pathways. For example, in response to caloric restriction, SIRT1 exerts its known epigenetic regulation on gene transcription, but simultaneously is able to directly regulate the mammalian target of rapamycin (mTOR) activity (Ghosh et al., 2010).
Thus, the overall cellular response will be the consequence of the integration of two or more simultaneously activated parallel pathways. Probably the initially mentioned proportionality is more apparent than real and a more realistic scenario would arise when the cell is visualized as an integrator of diverse and simultaneous processes yielding a coherent output, where the dose–response ratio is closer to a hyperbolic graduate response than a biphasic one.
Finally, an interesting feature concerning the anti-inflammatory response exerted by oxidative stressors is the so-called hormetic response, which is a biphasic dose–response dynamic characterized by low-dose stimulation and high-dose inhibition (Calabrese et al., 2010). Hormetic response is a type of preconditioning and is mainly observed upon activation of nuclear factor erythroid 2-related factor 2 (NRF2), a transcription factor that regulates the expression of antioxidant proteins to protect against oxidative damage triggered by injury and inflammation (Calabrese and Kozumbo, 2021). Given the cross talk of NRF2 mainly with SIRT1 and SIRT6, it is plausible an involvement of these sirtuins in hormetic responses.
The observed effect of low concentrations of TNF-α in eliciting the basal redox signaling and antioxidative responses observed in cardiomyocytes (Shanmugam et al., 2016) is an example of that. Importantly, the features of the hormetic dose–response may have medical implications because it imposes constraints upon the doses of a drug to induce a desired effect.
Despite the consensus about the anti-inflammatory role played by sirtuins, some exemptions have been reported. As discussed, proinflammatory roles were observed in some specific instances, mainly for SIRT2, 6, and 7.
Sirtuin 1
The canonical pathway by which SIRT1 modulates the inflammatory response is through NF-κB inactivation by deacetylation on lysine 310 of RelA/p65 subunit (Yeung et al., 2004). Besides regulation of transcriptional activity of NF-κB, SIRT1 can also deacetylate the histone H3 at K9 position at the promoters of NF-κB target genes (Liu et al., 2011; Zhang et al., 2019) (Fig. 3). In addition, SIRT1 regulates several other intermediates of the inflammatory pathway. It was found that sirtuins, especially SIRT1 and SIRT3, play a protective role in the inflammation through the attenuation of Nod-like receptor family pyrin domain-containing 3 (NLRP3), which is the best-characterized inflammasome. In fact, NF-κB promotes the transcription of NF-κB-downstream target genes, such as NLRP3, Pro-IL-1β, and Pro-IL-18, which are necessary for inflammasome activation (Liu et al., 2017b).

Such activation may imply alternative pathways, as observed in a model of inflammatory-mediated demyelination in the central nervous system of mice (Liu et al., 2021), where the inflammatory response initially triggered by the activation of the vanilloid type 4 calcium channel (Trpv4) induced microglia activation, reduced peroxisome proliferator-activated receptor alpha coactivator 1α (PGC-1α), decreased mitochondrial function, and increased reactive oxygen species production. This response is mediated by the inflammasome, as NLRP3 knockdown inhibited glial activation and alleviated demyelination. Furthermore, in this inflammatory context, the SIRT1-mediated PGC-1α deacetylation ameliorates mitochondrial dysfunction and ROS production, in turn reducing NLRP3 inflammasome activation.
In an acute model of liver injury induced by acetaminophen, the Notch pathway activation in mesenchymal stromal/stem cells (MSCs) by its ligand Jagged1 increased COX2 production, which in turn activated the (AMP-activated protein kinase [AMPK]α/SIRT1) pathway in liver-resident macrophages. The last resulted in the deacetylation of spliced X-box-binding protein 1 (XBP1) transcription factor and the subsequent inhibition of NLRP3 inflammasome (Yu et al., 2022). Given the relevance of MSCs as a source of tissue-specific cells bearing unique properties, such as enhanced proliferation, multilineage differentiation, and their immunomodulatory properties, this type of cell represents an interesting target for treating various inflammatory diseases in animals and humans, including acute and chronic liver diseases.
Neuroinflammation has emerged as a key contributor during the pathogenesis of several neurodegenerative diseases, including Alzheimer's disease (AD). Increased mTOR activity has been detected in AD-affected brains. It has been recently suggested that the elevated mTOR expression in brain mice can be downregulated by all trans-retinoic acid (ATRA), an active metabolite of vitamin A, via an SIRT1-dependent mechanism (Guo et al., 2021). ATRA can also attenuate neuroinflammation due to abusive alcohol consumption in an SIRT1-dependent manner (Priyanka et al., 2019), apparently through NF-κB inactivation since supplementation with ATRA reduced NF-κB expression and translocation to the nucleus. The latter suggests that the mechanism of action of ATRA may be mediated through NF-κB. Moreover, the decreased SIRT1 activity due to alcohol exposure could be reverted upon ATRA supplementation.
However, given the ability of SIRT1 to modulate ATRA-dependent transcriptional activity (Tang et al., 2014) as well as to directly regulate mTOR signaling pathway proteins (Ghosh et al., 2010), the ultimate mechanism responsible for this effect remains unclear.
The HuR protein regulates messenger RNA (mRNA) stability and translation through RNA-recognition motifs present in the protein. Thus, SIRT1 mRNA stabilization is achieved by HuR binding to its 3′ UTR (untranslated region). For example, in senescent cells, checkpoint kinase 2 (CHK2)-mediated phosphorylation of HuR dampened the interaction between HuR and SIRT1 mRNA, leading to decreased SIRT1 protein levels (Abdelmohsen et al., 2007). Moreover, a decay of SIRT1 mRNA stability due to calcium-induced HuR-SIRT1 dissociation has been proposed as a mechanism responsible for neuronal damage after glutamatergic hyperexcitatory stimulation (Yang et al., 2020). A similar posttranscriptional regulation of SIRT1 has been recently proposed in the context of alcoholic hepatosis (Zhang et al., 2021), where ethanol-dependent phosphorylation of HuR triggers the dissociation of HuR-SIRT1 mRNA complex, again promoting SIRT1 mRNA decay.
Interestingly, genetic deletion of Fas-activated serine/threonine kinase (FASTK) diminished ethanol-induced HuR phosphorylation and HuR-SIRT1 mRNA complex dissociation, thereby enhancing SIRT1 mRNA stability. Given the outstanding role of SIRT1 in protection against NAFLD and alcoholic fatty liver diseases, FASTK may represent an important hub during the pathogenesis of inflammatory-based liver diseases. The main regulatory mechanisms of inflammation involving SIRT1 are summarized in Table 1.
Regulatory Mechanisms of Inflammation Involving Sirtuin1
LPS, lipopolysaccharide; mRNA, messenger RNA; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa B; NLRP3, nod-like receptor family pyrin domain-containing 3; PGC-1α, peroxisome proliferator activated receptor-gamma coactivator 1 alpha; SIRT, sirtuin; TNF-α, tumor necrosis factor α; UTR, untranslated region; XBP1, X-box-binding protein 1.
Targeting of SIRT1 by noncoding microRNAs (miRNAs) is a mechanism of inflammatory regulation that is increasingly explored. Through SIRT1 downregulation, miRNAs usually delay the transition from acute to adaptation stage of inflammation. Since every single miRNA targets hundreds of transcripts, many of which are regulated by multiple miRNAs, the list of miRNAs that regulate or can potentially regulate SIRT1 is continuously growing. The first detected miRNA that modulates SIRT1 was miR-34a, inducing apoptosis under genotoxic stress conditions in human colon cancer HCT116 cells. MiR-34a binds directly to the 3′ UTR of SIRT1 mRNA and declines its expression. The latter leads to an increase in acetylated p53 and expression of p21 and PUMA, two transcriptional targets of p53 that regulate the cell cycle and apoptosis, respectively (Yamakuchi et al., 2008). However, miR-34a-mediated regulation of SIRT1 was also reported in hepatocytes and macrophages.
In hepatocytes, SIRT1 is key for the regulation of the transcriptional coactivator PGC-1α and the adipogenic transcription factor peroxisome proliferator-activated receptor (PPAR)γ. In fact, miR-34a is one of the most increased miRNAs in hepatocytes in both genetic and diet-induced obese (DIO) mice (Choi et al., 2013), and remarkably increased in patients suffering hepatic steatosis (Purushotham et al., 2009). Thus, miR-34a has emerged as a candidate to play a key role during the pathogenesis of hepatic steatosis. Moreover, miR-34a can also suppress the expression of NAMPT, the rate-limiting enzyme for NAD+ biosynthesis, via binding directly to the 3′ UTRs of NAMPT mRNA (Choi et al., 2013). As discussed later, the control of NAD+ concentration is key to governing the interplay among SIRT1, 3, and 6 and achieving a successful inflammatory response during sepsis in macrophages.
MiR-138 also targets SIRT1 and impacts the macrophage inflammatory response by activating NF-κB and involving the AKT signaling pathway (Bai et al., 2018). Consistently, the protective effect exerted by microRNA-138 antagomir in LPS-treated macrophages disappeared when SIRT1 was knocked down with small interfering RNA (siRNA) and in challenged SIRT1 knockout (KO) mice. SIRT1 is also targeted by microRNA-217/138-5p to elicit the inflammatory response, oxidative stress, and the induction of cell apoptosis in a cell model of Parkinson's disease (Wang et al., 2019). Given that not only inflammation but also apoptosis and oxidative stress response are affected by the activation of microRNA-217/138-5p axis, these miRNAs may be affecting additional important regulatory players besides SIRT1. In this regard, the downregulation of miRNAs miR-217 and miR-543 mitigated the inflammatory response in in vivo and in vitro models of viral myocarditis by regulating the SIRT1/AMPK/NF-κB signaling pathway (Xia et al., 2020).
Interestingly, as observed for microRNA-217/138-5p, increased apoptosis of cardiomyocytes and decreased superoxide dismutase (SOD) activity were paralleled with enhanced synthesis of inflammatory factors.
The levels of circulating monocyte chemoattractant protein-1 (MCP-1) and IL-8 in obese individuals are higher than in lean persons and are linked with characteristics of metabolic syndrome such as atherosclerosis, insulin resistance, and cardiovascular disease (Kim et al., 2006). Interestingly, miR-132 was reported to be upregulated in LPS-challenged THP-1 macrophages (Taganov et al., 2006) and associated with the proinflammatory response via the release of IL-8 and MCP-1 in human adipocytes in vitro (Strum et al., 2009). It was shown that miR-132 can bind SIRT1 mRNA and downregulate SIRT1 translation. Overexpression of miR-132 is sufficient to induce NF-κB translocation, p65 acetylation, and production of MCP-1 and IL-8 in stimulated adipocytes.
Worthy of note, miR-132 inhibition by means of serum deprivation reduced p65 acetylation and partially prevented the generation of MCP-1 and IL-8, pointing to the importance of the nutrient availability as a regulator of the miR-132/SIRT1/NF-κB axis on adipose tissue (Strum et al., 2009). A novel regulatory pathway involving miR132/212, NF-κB, SIRT1, and a circular RNA (circRNA) form derived from the SIRT1 host gene was reported in primary cultures of vascular cells (Kong et al., 2019). circRNAs are a novel class of noncoding RNAs formed by back-splicing of exons, characterized by covalently closed-loop structures with neither 5′ to 3′ polarity nor a polyadenylated tail. They are highly stable in vivo compared with linear ones and can be found in exosomes. It has been shown that circRNAs generally regulate homologous mRNA expression by acting as cytoplasmic miRNA “sponges,” RNA-binding proteins, or nuclear transcriptional regulators (Li et al., 2018).
In the cytoplasm, circ-SIRT1 sequesters NF-κB and precludes p65 from entering the nucleus to drive the inflammatory response. Complementarily, circ-SIRT1 binds to miR-132/212, which interferes with SIRT1 mRNA, facilitating the expression of host SIRT1 gene. As a result, increased SIRT1 promotes deacetylation and inactivation of p65, switching vascular smooth muscle cells to the noninflammatory phenotype (Kong et al., 2019). These findings open new venues of research on inflammatory regulation as it broadens the variety of target options available for therapeutic purposes.
MiR-22 interaction with SIRT1 has been mainly studied in the context of cancer, where it is considered a tumor suppressor (Centomo et al., 2022). Because direct targeting of the 3′ UTR of SIRT1 mRNA by this miRNA was observed in several kinds of cells, both in vitro and in vivo, the overexpression of this miR-22 can potentially impact on a wide array of cell functions such as apoptosis and senescence (Hu et al., 2019; Li et al., 2021; Zhao et al., 2019) and chronic inflammation-based diseases. Highly abundant levels of miR-22 in the serum of NAFLD patients have been observed (López-Riera et al., 2017), and miR-22 expression has been clearly shown to be inversely correlated to that of SIRT1 in the liver (Yang et al., 2020). The latter suggests a crucial role of miR-22 during the pathogenesis of liver steatosis.
However, given that miR-22 can simultaneously target other genes involved in the control of inflammation, metabolism, and obesity such as PGC-1α/PPARα, fibroblast growth factor receptor 1, or the hepatic cannabinoid receptor (Azar et al., 2020; Gurha et al., 2013; Hu et al., 2020), the actual contribution of the miR-22/SIRT1 axis to the inflammatory phenotype is difficult to evaluate. A recent article showing that increased free fatty acids present in the serum of mice fed with a choline-deficient amino acid diet inhibited gluconeogenesis and induced NAFLD via miR-22 targeting of SIRT1 seems to confirm a direct involvement of this miRNA, at least in the liver energetic metabolism (Yadav et al., 2023). More recently, the targeting of SIRT1 by miR-22 was proposed as a mechanism responsible for inflammation and increased apoptosis during septic cardiomyopathy (Wang et al., 2021).
Finally, in an in vitro model of rheumatoid arthritis, miR-22 was reported to inhibit synovial fibroblast proliferation and proinflammatory cytokine production via targeting SIRT1 (Zhang et al., 2020a). Although it seems contradictory with most of the evidence gathered so far, given that miR-22 takes part of complex signaling networks implying positive or negative feedback loops, this type of finding should not be surprising. Targeting of SIRT1 by miRs under several experimental and clinical conditions concerning inflammation is depicted in Figure 4.

In conclusion, SIRT1 is considered to have strong anti-inflammatory properties due to the regulatory effects on transcription factors, including NF-κB and their downstream proinflammatory effectors. Moreover, SIRT1 also exerts modulatory roles on the inflammasome. However, although SIRT1 is probably the most studied sirtuin and whose anti-inflammatory mechanisms are better understood, this does not mean that SIRT1 anti-inflammatory roles are more prominent than those of other sirtuins. As further discussed, SIRT2, 6, and 7 can also target NF-κB and SIRT2 can also modulate NLRP3 inflammasome activation. In this regard, available data suggest that sirtuins form a functional network leading a cellular response that represents more than the sum of each individual contribution. In this scenario, it is hard to determine the importance of each specific sirtuin to the anti-inflammatory response.
Sirtuin 2
SIRT2 has been well characterized as the main cytoplasmic sirtuin, being the only one that colocalizes with tubulin in the cytosol (North et al., 2003). This unique colocalization, together with its function as tubulin deacetylase, suggested a role in the dynamics of microtubules, the control of the microtubule-based intracellular transport of organelles, and in the progression of cell cycle.
More recent studies have increasingly shown that SIRT2 can participate in the regulation of gene expression and signal transduction, mainly through posttranslational modification of target genes and proteins, pointing SIRT2 as an emerging player in several metabolic pathways. Nuclear resident proteins p53, p300, and histones (H3 and H4) have been identified as substrates of SIRT2 (Black et al., 2008; Heltweg et al., 2006; Vaquero et al., 2006). The apparent contradiction of a protein being in the cytoplasm but exhibiting specificity for nuclear substrates was resolved when SIRT2 localization was monitored during cell cycle progression. SIRT2 localizes to the cytoplasm throughout the cell cycle, but during the G2–M transition it translocates and deacetylates H4K16 in the nucleus (Pereira et al., 2018; Vaquero et al., 2006). Localization of SIRT2 has also been observed in mitochondria (Liu et al., 2017a).
Therefore, SIRT2 can be involved in metabolic pathways and disorders related to inflammation and oxidative stress. Although its role in some diseases is still controversial, given the multiple functions regulating physiological and pathological signal transduction, SIRT2 is currently regarded as a potential target for disease treatment.
As mentioned, systemic low-grade chronic inflammation is relevant in the pathogenesis of metabolic disorders being the main regulators of the NF-κB complex, the mitogen-activated protein kinase (MAPK) and the inflammasome (NLRP3). Similar to SIRT1, a canonical role for SIRT2 is binding and deacetylation of p65 subunit of NF-κB at Lys 310, therefore downregulating the NF-κB pathway and yielding an anti-inflammatory effect (Rothgiesser et al., 2010) (Fig. 3). The downregulation of the NF-κB pathway by SIRT2 has been extensively reported in diverse experimental models and pathological conditions (Lin et al., 2013; Zhang & Chi, 2018). More recently, two novel substrates of SIRT2 were proposed, the Hsp90 chaperone, whose deacetylation reduces the extent of LPS-induced inflammation by suppressing the expression of inflammatory factors via the SIRT2-Hsp90-glucocorticoid receptor axis (Sun et al., 2020a), and the hepatocyte nuclear factor 4 alpha (HNF4α) protein, a master regulator of hepatic differentiation.
SIRT2 binding and deacetylating of HNF4α on Lys 458 seem to be sufficient to revert high-fat diet-fed-induced insulin resistance, hepatic steatosis, and systemic inflammation in mice, whereas SIRT2 liver-specific ablation exacerbated these metabolic dysfunctions (Ren et al., 2021).
In the central nervous system, SIRT2 counteracts microglia activation upon LPS treatment. In these cells, the TLR activation generates two opposite effects: a proinflammatory response through NF-κB activation and simultaneously an SIRT2 dephosphorylation and activation that in turn downregulate NF-κB and moderate the inflammatory response (Pais et al., 2013). Noteworthy, other authors have reported the proinflammatory roles of SIRT2, also using microglia as a model of neuroinflammation (Wang et al., 2016). Thus, the role played by SIRT2 in inflammation seems to be more complex and its consequences are less predictable than those observed for SIRT1. For example, SIRT2 deficiency impairs bone marrow-derived macrophage polarization to M1 and inhibits LPS-induced iNOS mRNA and protein expression. This cell phenotype is paralleled by a diminished phosphorylation of p65 at Ser536 and therefore its nuclear translocation to drive the expression of downstream target genes such as IL-1β, IL-6, MCP-1, MMP-9, and MMP-13.
Mechanistically, the deficiency of SIRT2 reduces the phosphorylation and degradation of IκBα, the protein that normally keeps sequestered NF-κB proteins in the cytosol, resulting in impaired NF-κB activation (Lee et al., 2014).
Pharmacological inhibition of SIRT2 further supports the putative proinflammatory role of this sirtuin in LPS-activated microglia and suggests a regulation through the MAPK signaling pathway. The SIRT2 inhibitor AGK2 decreased the phosphorylation of MAPK and moderated the increase of phosphorylation of p38, JNK, and ERK, after LPS challenge. The last was paralleled with diminished production of inflammatory cytokines (iNOS, TNF-α, and IL-1β) in vitro, as well as iNOS in brain mice (Jiao et al., 2020). Consistently, AGK2 inhibition of SIRT2 increased the acetylated form of mitogen-activated protein kinase 1 (MAPK-1), which dephosphorylates and inhibits all three MAPKs in the nucleus. Of note, pharmacological inhibition of SIRT2 also increased tubulin deacetylation both in vivo and in vitro, suggesting that the need of tubulin acetylation for a strong inflammatory response (see discussion below) is not a rule and may depend on the context where the inflammation takes place.
The upregulation of MAPK-1 in SIRT2 KO mice was also observed in the context of cisplatin-induced acute kidney injury (Jung et al., 2020) and the acetylation of MAPK-1 was found significantly increased in SIRT2 knockdown tubular epithelial cells. Given that MAPK-1 is currently introduced as a new protein in anti-inflammation, particularly for monocyte and macrophage activation (Kim and Asmis, 2017; Moosavi et al., 2017), SIRT2 emerges as a relevant player in the MAPK pathway during inflammation.
NLRP3 activity is dependent on the cytoskeleton. Microtubules are important for the assembly of the NLRP3 inflammasome, specifically for the association of NLRP3 with its adaptor apoptosis-associated speck-like protein containing a CARD (ASC) (Misawa et al., 2013). In fact, inhibition of the tubulin-based transport of mitochondria-bearing ASC toward the sites where NLRP3 locates suppressed IL-1β production in macrophages treated with an inducer of the NLRP3 inflammasome. Because mitochondria are preferentially transported by acetylated microtubules, removing this acetylation by SIRT2 was proposed as a control step of NLRP3 activation (Misawa et al., 2013). Consistently, SIRT2 KO mice were found to be protected from blood–brain disruption, edema, and cell death after traumatic brain injury, such protection being paralleled by diminished NLRP3 inflammasome expression (Wang et al., 2023b).
Interestingly, depletion of NAD+ levels in macrophages by FK866, a specific inhibitor of the NAD+ salvage pathway, mediated mitochondrial re-localization toward perinuclear regions, NLRP3 activation, cleavage of caspase-1, and production of IL-1β (Shim et al., 2021). Given that a drop of NAD+ levels below a critical value may inhibit sirtuin activity and considering that microtubule-based transport of mitochondria depends on tubulin acetylation, this observation suggests a pivotal role for NAD+ during the NLRP3-based inflammatory response in macrophages.
On the contrary, acetylation of NLRP3 facilitates the assembly and activation of the NLRP3 inflammasome. In this regard, it was recently proposed that SIRT2 can govern the acetylation state of NLRP3 in a switch on/off manner (He et al., 2020). This switch was shown to be age-dependent and physiologically relevant since it prevents aging, overnutrition-associated chronic inflammation, and insulin resistance in mice. This may represent a novel mechanism to understand aging-associated inflammation and glucose homeostasis in rodents. Also, it provides a link between the age-dependent decline of NAD+ levels in tissues and the chronic inflammation developed during aging. However, the activation of NLRP3 as a consequence of increased caloric intake can simultaneously occur through an SIRT3-dependent mechanism (Traba et al., 2015). As further discussed, SIRT3 downregulation by a high-calorie diet leads to increased production of ROS, a well-known inducer of NLRP3 assembly and activation (Traba et al., 2017).
Thus, such switch on/off modulation of inflammasome activation is likely the result of at least two coordinated mechanisms running simultaneously and pointing to the same target, rather than the sum up of two independent pathways.
In summary, although SIRT2 was initially described as exclusively cytoplasmic, the role of SIRT2 in the nucleus and even in the mitochondria is currently increasingly studied. Of note, the emerging role of SIRT2 in the regulation of the inflammasome adds a promising target for the therapeutic control of inflammation. Finally, SIRT2 could be related to the decline of NAD+ levels, associated with aging.
Sirtuin 3
SIRT3 is a soluble 44-kDa protein whose last 25 amino acids at the NH2-terminal are responsible for localization at the mitochondrial matrix. A further proteolytic process by matrix-processing peptidases yields a 28-kDa protein bearing enzymatic activity (Onyango et al., 2002; Schwer et al., 2002). Among mitochondrial sirtuins, SIRT3 appears to have the most potent deacetylase activity, although it exhibits also demyristoylase activity (Madsen et al., 2016; McGinnis et al., 2022). Similar to other sirtuins, SIRT3 possesses a hydrophobic pocket that accommodates -N acetyl lysine groups (and -N myristoyl groups) and other different pockets for NAD+ binding. The presence of a substrate group stabilizes NAD+ binding that is specific and essential for deacetylation activity (Madsen et al., 2016).
It is known that NAD+ levels decline in several metabolic-associated diseases, including aging (Chini et al., 2017), however, SIRT3 expression and activities also fluctuate between fed/fasting states. Because SIRT3 deacylates several mitochondrial enzymes related to energetic metabolism such as those involved in fatty acid oxidation (FAO), tricarboxylic acid (TCA) cycle, and oxidative phosphorylation (OXPHOS), this sirtuin is considered a sensor of nutrient stress. For this reason, SIRT3 was proposed as a therapeutic target for metabolic syndrome, hepatic steatosis, and NAFLD (Friedman et al., 2018; Nassir and Ibdah, 2016).
Prolonged fed state and accumulation of ATP and NADH lead to a reduction in the TCA cycle and accumulation of citrate, in turn reconverting to acetyl CoA by ATP citrate lyase. The last represents an increase in cellular levels of the main substrate of lysine acetyltransferases for protein acetylation (Jennings et al., 2022). However, due to a high turnover of metabolites and pH, acetylation or acylation in mitochondria may even occur nonenzymatically (Goodman et al., 2018; McGinnis et al., 2022). For example, in HFD-fed mice, hyperacetylation of mitochondrial enzymes correlates with a decrease in NAD+ levels and a reduction in SIRT3 activity, without affecting SIRT1 (Kendrick et al., 2011). Under fasted conditions, SIRT3 deacetylates long-chain acyl coenzyme A dehydrogenase and improves FAO (Fig. 5).
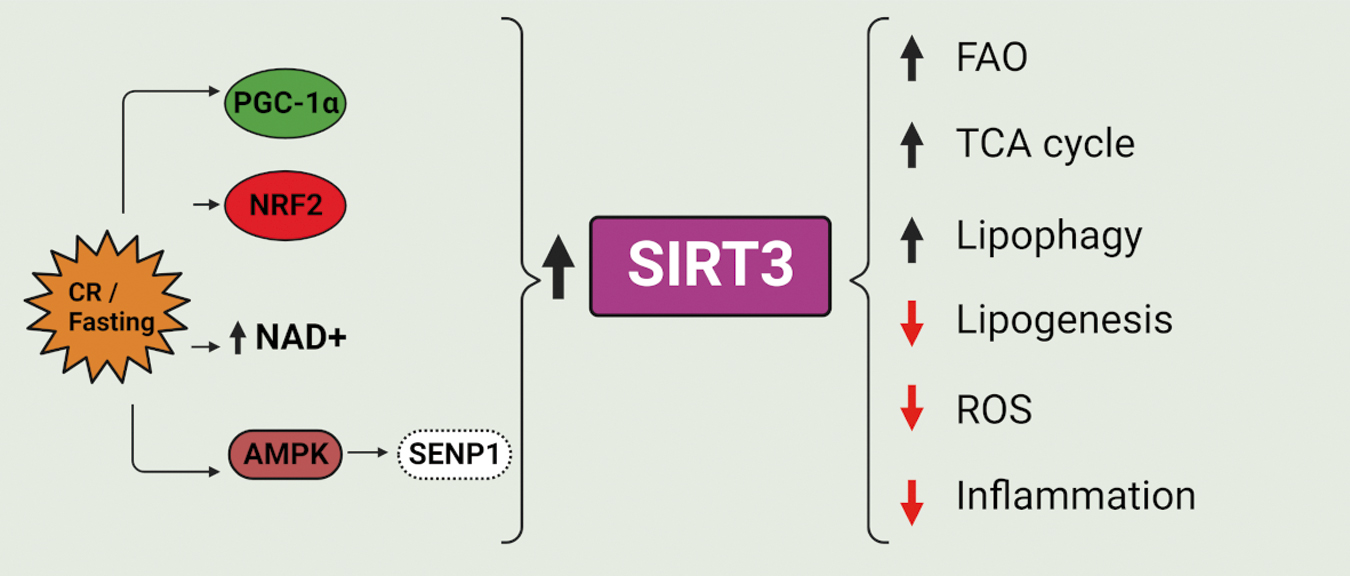
Many other enzymes belonging to FAO, OXPHOS, urea cycle, and ketogenesis are also targeted for deacetylation and activation by SIRT3 (Chen et al., 2011; Finley et al., 2011; Hallows et al., 2011; Hallows et al., 2006; Hirschey et al., 2010; Yu et al., 2012). Therefore, SIRT3 activation accelerates FAO, bursts the mitochondrial TCA cycle and OXPHOS to catabolize acetyl-CoA, but also controls ROS levels. Dysregulation of lipid metabolism due to increased fatty acid synthesis and reduced FAO leads to lipid droplet (LD) accumulation and diseases associated with obesity, such as metabolic syndrome, and liver steatosis associated with NAFLD and nonalcoholic steatohepatitis (NASH), all of them positively correlated with hyperacetylation of mitochondrial proteins and SIRT3 decrease (Zhang et al., 2022; Zhang et al., 2020b; Zheng et al., 2018).
Accumulation of LDs can also occur by impaired lipophagy, a lysosomal lipolytic pathway also associated with NAFDL development (González-Rodríguez et al., 2014; Singh et al., 2009). Lipophagy occurs when LDs are recognized by the macroautophagy machinery and incorporated into autophagosomes to release free fatty acids and alleviate lipotoxicity (Singh et al., 2009). Low levels of SIRT3 and the inhibited macroautophagy flux present in HFD-fed mice can be reverted by overexpression of SIRT3. It appears that SIRT3 is essential for the formation of a complex with -perilipins, heat shock proteins, and lysosome to destabilize LDs and promote LD dispersion (Zhang et al., 2019). This process is mediated by activating the AMPK and perilipin phosphorylation (Zhang et al., 2020b).
In summary, SIRT3 activation can promote a shift in metabolism toward lipid degradation by three different metabolic approaches as follows: (1) inducing LD mobilization to release fatty acids; (2) increase in FAO by enhancing mitochondrial beta oxidation/OXPHOS; and (3) inhibiting lipogenesis.
SIRT3 transcription and expression can be increased by the transcription factor estrogen receptor-related α (ERRα), activated by PGC-1α, but also by other different factors (Giralt et al., 2011). An interesting regulation in SIRT3 is the SUMOylation promoted by sentrin-specific protease 1 (SENP1). This is an important mechanism for the adaptive immune system, especially in T cell memory development. In this study, low glucose was shown to activate the AMPK-SENP1-SIRT3 axis promoting mitochondrial fusion, OXPHOS fueled by FAO substrates, and T cell survival (Fig. 5). Also, the SENP1-SIRT3 axis is involved in M2 macrophage polarization via deacetylation and increased activity of glutamate dehydrogenase (GDH) 1 and α-ketoglutarate production (Zhou et al., 2022). In fact, mitochondria have a central role in the metabolic reprogramming of macrophage polarization (Liu et al., 2015; Liu et al., 2012; Van den Bossche et al., 2017) and inflammation is often associated with SIRT3 reduced activity.
As discussed later, increased expression of SIRT3 because of rising in NAD+ levels occurs during the adaptation stage of inflammation in monocytes (Liu et al., 2015). Of note, reprogramming from glucose to FAO metabolism is a necessary condition to shift from the acute to the adaptation phase. In such a context, SIRT3 is important to buster the TCA cycle and drive an increased mitochondrial oxygen consumption rate and ATP production. This effect is achieved by deacetylation and activation of key targets such as isocitrate dehydrogenase 2 (IDH2) and superoxide dismutase 2, concomitant with increases in citrate synthase activity (Liu et al., 2015). DIO mice have reduced SIRT3 expression in adipose tissue, bone marrow-derived macrophages (Zhou et al., 2023) and SIRT3 KO DIO mice are prone to accelerated body weight gain and severe inflammation (Zhou et al., 2023).
SIRT3 knockdown in macrophages led to increased expression of inflammatory cytokines, iNOS and COX2 (Boniakowski et al., 2019), which may be explained by hyperacetylation of mitochondrial succinate dehydrogenase (SDH) with succinate accumulation that results in Krüppel-like factor 4 suppression and activation of proinflammatory macrophage phenotype (Zhou et al., 2023). On the contrary, increased SIRT3 expression is associated with decreased inflammation, lower intracellular levels of ROS, and improved mitochondrial function. SIRT3 expression increases during the M1-M2 transition of wound macrophages. SIRT3 KO mice have a significant increase in proinflammatory cytokines and delayed wound healing following injury, similar to DIO mice. Consistently, the transfer of SIRT3+/+ macrophages to SIRT3−/− mice reduces the expression of proinflammatory cytokines and improves wound healing. This is confirmed by the ability of an FABP4 inhibitor to elevate SIRT3 expression in DIO wound macrophages.
Therefore, SIRT3 and FABP4, its upstream regulator, are targets for the regulation of macrophage-mediated inflammation in obesity and diabetic wound healing (Boniakowski et al., 2019).
The SIRT3-dependent modulation of inflammation observed in obesity is also associated with inflammasome priming. NLRP3 complex amplifies inflammation by activating caspase 1 and proinflammatory cytokines in obesity-induced inflammation, however, in fasting and caloric restriction, NLRP3 formation is reduced (Strowig et al., 2012). Fasting diminished NLRP3 inflammasome activity in peripheral blood mononuclear cells and monocytes (Traba et al., 2015). One of the possible mechanisms of SIRT3-dependent NLPR3 suppression in fasting is mediated by the deacetylation of SOD2 and reduction of mitochondrial ROS, a signal that usually triggers NLRP3 assembly (Traba et al., 2017). SIRT3 overexpression also seems to decrease palmitate-induced mitochondrial ROS and reverts autophagy-related 5 (ATG5) acetylation in autophagosome that otherwise triggers inflammasome activation (Liu et al., 2018). In addition, dependent SIRT3 deacetylation of pyruvate dehydrogenase (PDH) is also related to the assembly of NLRP3.
SIRT3 ablation results in the induction of IL-1β in bone marrow-derived macrophages and shifts the metabolic phenotype from OXPHOS to glycolysis in perivascular adipose tissue (Wei et al., 2021). However, the ability of SIRT3 to modulate other inflammatory pathways such as MAPK and NF-κB cannot be ruled out and needs further investigation. Therefore, it seems that SIRT3 modulation of mitochondrial enzymes such as SOD to control ROS signaling and others such as GDH, PDH, and SDH to control the metabolic shifting can alter the macrophage inflammatory phenotype, inflammatory tissue status, and development of NAFLD-related conditions.
Another important regulator of SIRT3 is NRF2 was predicted to bind the SIRT3 promoter and regulate its expression in several tissues (Satterstrom et al., 2015), being important as a nutrient sensor during metabolic reprogramming and inflammation. Interestingly, NRF2 is implicated in liver inflammation induced by salt. NAFLD can be promoted also by a higher salt intake diet (HSD) and the role of SIRT3 in hepatic inflammation was demonstrated by an HSD-induced cardiovascular damage model (Gao et al., 2022). The expression of SIRT3 in the liver was significantly inhibited by HSD. Specific liver SIRT3 overexpression with AAV8-TBG-SIRT in the HSD-NAFLD mouse model reduced aspartate aminotransferase and proinflammatory cytokines, together with lower status of lipid accumulation, hepatocyte steatosis, and inflammatory cell infiltration.
Moreover, acetylation of H3K27 and inhibition of Nrf2 binding to SIRT3 promoter are the main reason for the continuous decline of SIRT3 expression. Thus, the hepatoprotective role of Nrf2 seems to be SIRT3-dependent.
In conclusion, SIRT3 is a relevant player in the control of inflammation processes. The role of SIRT3 during inflammation can be better understood taking into consideration its ability to modulate mitochondrial metabolism as well as the importance of mitochondria during inflammation. Through deacetylating key enzymes belonging to the TCA cycle and main anaplerotic reactions, SIRT3 governs the dynamics of fatty acid accumulation or catabolism, thus participating in the pathogenesis of NAFLD and related conditions. Similarly, SIRT3 contribution to the immunometabolic adaptation to acute inflammation in monocytes relies, in part, on its ability to coordinate together with sirtuins 1 and 6, fast switches of mitochondrial bioenergetics.
Sirtuin 4
The mitochondrial SIRT4 is a rather understudied member of the sirtuin family. One key function of SIRT4 is related to metabolism regulation. Although initially classified as a mono-ADP-ribosyltransferase (Haigis et al., 2006) with a weak deacetylase activity compared with other sirtuins, it was later shown to have a more robust deacylase activity toward 3-hydroxy-3-methyl-glutarylated (HMG) lysine residues (Pannek et al., 2017), as well as lipoamidase and deacetylase substrate specificity (Laurent et al., 2013b; Mathias et al., 2014).
Many cellular metabolic activities, including insulin secretion, glutamine metabolism, and fatty acid metabolism, are connected to SIRT4 (van de Ven et al., 2017). It has been shown that SIRT4 inhibits PDH activity, hydrolyzing the lipoamide cofactors from the dihydrolipoylysine acetyltransferase (DLAT), a component of PDH complex (Mathias et al., 2014). Laurent et al., (2013b) demonstrate that malonyl CoA decarboxylase (MCD) is deacetylated by SIRT4, promoting lipogenesis and inhibiting β-oxidation. SIRT4 also regulates the expression of PPARα and consequently inhibits downstream genes associated with fatty acid catabolism (Laurent et al., 2013a).
SIRT4 negatively regulates insulin secretion in pancreatic β cells, ADP-ribosylating, and inhibiting mitochondrial GDH. In turn, GDH controls amino acid-stimulated insulin secretion by regulating glutamine and glutamate oxidative metabolism. Specifically, due to its ADP-ribosyl transferase activity, SIRT4 can downregulate GDH activity in β cells, thereby reducing insulin secretion in response to glutamine and leucine (Haigis et al., 2006). A decreased NAD/NADH ratio countered these effects when calorie intake was restricted.
Consistently, depletion of SIRT4 from insulin-producing cells results in increased insulin secretion in response to glucose. Mass spectrometry analysis showed that SIRT4 coimmunoprecipitated with the insulin-degrading enzyme (IDE) and the ADP/ATP translocases 2 and 3 (ANT2 and ANT3), reinforcing the hypothesis of a role in the regulation of insulin secretion in response to glucose (Ahuja et al., 2007) and supporting the evidence suggesting a protective effect on Diabetes Mellitus (DM). The ADP/ATP translocase ANT2a catalyzes the exchange of ATP generated in the mitochondria by ATP synthase against ADP produced in the cytosol, and so, SIRT4 overexpression is associated with increased ATP levels. Moreover, ANT2/3 is more active when SIRT4 activity is preserved (Ho et al., 2013).
SIRT4 is downregulated during inflammation (Wu et al., 2022), and currently available data increasingly suggest that this sirtuin has an active role in inflammation. For example, SIRT4 is downregulated during osteoarthritis in human patients; moreover, upon overexpression in primary human chondrocytes, three critical players in osteoarthritis progression such as MMP-13, IL-6, and TNF-α were reduced (Dai et al., 2020). Similarly, human umbilical vein endothelial cells stimulated with LPS decreased the expression of SIRT4, whereas silencing of SIRT4 exacerbated the expression of proinflammatory cytokines (IL-1β, IL-6, and IL-8), COX-prostaglandin system (COX-2), extracellular matrix remodeling enzymes MMP-9, and the adhesion molecule ICAM-1 (Tao et al., 2015). Mechanistically, SIRT4 interferes with the NF-κB signaling pathway by preventing NF-κB nuclear translocation into the nucleus (Fig. 3), however, the mechanism by which this mitochondrial sirtuin can control NF-κB nuclear-cytoplasmic shuttling remains to be studied.
SIRT4 was observed to be upregulated in infiltrating T-reg cells after spinal cord injury in mice. Conversely to the above discussed, SIRT4 expressed in infiltrating T-reg cells impaired the anti-inflammatory modulation otherwise exerted by such cells (Lin et al., 2019). In fact, SIRT4 overexpression in isolated splenic CD4+ CD25+ T cells downregulated FOXP3 and immunoregulatory factors such as IL-10 and TGF-β. Moreover, the authors showed that the modulation of FOXP3 by SIRT4 is mediated through AMPK. Mechanistically, SIRT4 decreased the phosphorylation of AMPK, so given that activated AMPK can drive naive T cells to differentiate toward T-reg cells both in vitro and in vivo (Sun et al., 2017), then the suppression of the anti-inflammatory phenotype of T-reg cells could be better explained because of AMPK downregulation.
Finally, in vivo models have shown that SIRT4 is downregulated in cardiomyocytes after myocardial ischemia/reperfusion (I/R) injury, and that SIRT4 overexpression decreases myocardial infarct size. This protective effect of SIRT4 against I/R injury has been reported to be associated with preserved mitochondrial function and reduced myocardial apoptosis (Zeng et al., 2018). The association of SIRT4 with I/R injury in other organs such as the liver, brain, lung, and kidney is still unexplored.
The regulation of glutamate metabolism by SIRT4 is key to control the glucose homeostasis but also to account for its role as a tumor suppressor, mainly explained through the inhibition of glutamine production (Chen et al., 2019b). Moreover, SIRT4 works as a molecular switch mediating colorectal cancer cell proliferation through glutaminase-mediated activation of the Akt/GSK3β/CyclinD1 pathway (Cui et al., 2021). Although the precise function of SIRT4 in inflammatory processes still requires further exploration, current findings suggest that it may play a significant role in modulating the inflammatory response. Given the importance that mitochondrial metabolism is gaining in the scenery of inflammation, SIRT4 represents an interesting prospect for future research in this field.
Sirtuin 5
Among mammal sirtuins, the mitochondrial SIRT5 is the only well-accepted lysine desuccinylase (Du et al., 2011), and is involved in regulating energy metabolism, metabolic homeostasis, and inflammation. It does so through the regulation of mitophagy, autophagy, apoptosis, and cell response against oxidative stress (Li et al., 2019; Wang et al., 2019; Wu et al., 2022).
The current evidence supporting a role for SIRT5 in regulating autophagy is contradictory. When SIRT5 is overexpressed, the mitochondrial size is increased and mitophagy decreases, whereas the opposite effect is observed in SIRT5-silenced cells or upon treatment with the SIRT5 inhibitor MC3482 (Polletta et al., 2015). However, its overexpression in HCG27 gastric cancer cell lines could enhance autophagy and apoptosis via the AMPK/mTOR pathway (Gu et al., 2021). In addition, SIRT5-induced deacetylation of lactate dehydrogenase B and its consequent activation trigger autophagy and growth of colorectal cancer cells (Shi et al., 2019). Finally, the mitochondrial uncoupling protein UCP1 was found to be a new target of SIRT5 desuccinylation and elevated succinylation of this mitochondrial protein in SIRT5 KO brown adipose tissue, resulted in altered metabolic flexibility and mitophagy (Wang et al., 2019).
SIRT5 participates in the regulation of apoptosis through lysine deacetylation and may influence apoptosis-related proteins. For example, SIRT5 deacetylates cytochrome C, a mitochondrial protein with a central function in oxidative metabolism and in apoptosis initiation (Schlicker et al., 2008). SIRT5 overexpression ameliorated cytochrome C leakage and activation of caspase 3 to alleviate apoptosis (Li et al., 2019), thus suggesting for SIRT5 a protective role against apoptosis. However, impaired cell viability and proliferation were observed upon SIRT5 overexpression in epidermal keratinocytes stimulated with IL-17 to recreate an in vitro model of psoriasis. This phenotype was paralleled by diminished production of IL-8, IL-1β, IL-6, and phospho p65 protein.
The levels of p-ERK and STAT3 were significantly decreased after SIRT5 overexpression, indicating that the anti-inflammatory effect of SIRT5 in keratinocytes is mediated by ERK/STAT3 (Wang et al., 2023a). The last observation is consistent with a recent report showing that the expression levels of SIRT1-5 were diminished in human and mouse skin with psoriasis lesions, while SIRT6 and 7 were increased (Fan et al., 2019). The role of SIRT5 as a suppressor of proliferation has been reported in pancreatic β cells in vitro by downregulating transcription of pancreatic and duodenal homeobox 1 (PDX1) through deacetylating H4K16 (Ma and Fei, 2018). In pancreatic ductal adenocarcinoma, deletion of SIRT5 promotes the noncanonical use of glutamine to supply building blocks for macromolecular biosynthesis and cell proliferation (Hu et al., 2021).
Similar to SIRT4, the downregulation of SIRT5 promoted the secretion of insulin, suggesting that besides SIRT4, SIRT5 may also serve a relevant role in the pathogenesis of type 2 DM.
On the contrary, there is also abundant literature describing SIRT5 as a promoter of cell proliferation in the context of cancer. For instance, SIRT5 promotes proliferation in prostate cancer cells by increasing the activity of the MAPK pathway through acetyl-CoA acetyltransferase 1 (Guan et al., 2020). Moreover, citrate synthase desuccinylation by SIRT5 promotes colon cancer cell proliferation (Ren et al., 2020). Similarly, SIRT5 desuccinylation of serine hydroxy methyltransferase 2, a mitochondrial enzyme needed for glycine synthesis, controls proliferation in cancer-derived cell lines (Yang et al., 2018). Finally, SIRT5 regulates cell proliferation directly or indirectly by influencing the expression of transcription factors, such as E2F1 in hepatocellular carcinoma (Chang et al., 2018). The current data available suggest a dual role for SIRT5 in regulating proliferation, depending on cell type and the pathological context. This is worth further exploration.
The improvement in mitochondrial function and increased FAO could account for the protective effect of SIRT5 against age-related cardiac alterations. In fact, SIRT5 KO mice exhibit a reduced cardiac function and display signs of hypertrophic cardiomyopathy upon aging (Sadhukhan et al., 2016). Moreover, these mice have also shown increased cardiac fibrosis, compared with age-matched wild-type mice. In whole-body SIRT5 KO mice, impaired mitochondrial medium-chain FAO drove periportal macrovesicular steatosis (Goetzman et al., 2020). Consistently, a decreased expression of SIRT1-3, SIRT5, and SIRT6 in patients with NAFLD or fibrosis has been observed in several studies (Wu et al., 2014).
The role of SIRT5 during macrophage activation is still unclear. In M1 polarized macrophages upon LPS challenge, SIRT5 is split from HAUSP, a deubiquitinating enzyme that stabilizes SIRT5, prompting it to ubiquitination by TRIM21 and further degradation by the proteasome. In a feedback loop, SIRT5 degradation sustains the acetylation of TRIM21 at Lys351, thereby increasing its E3 ligase activity, thus enhancing IL-1β production (Yao et al., 2022). KO of TRIM21 in SIRT5-deficient mice protects against colitis. Conversely, a proinflammatory role of SIRT5 in macrophages was suggested by Zhu et al. (2022), who observed that the anti-inflammatory effect exerted by glutamine in burn-driven sepsis was due to SIRT5 repression. Glutamine controls metabolic remodeling and switches macrophage to OXPHOS by replenishing essential intermediate metabolites. Thus, glutamine supports M2 polarization in IL-4-treated murine macrophages by sustaining the activity of PDH, a checkpoint enzyme in the TCA cycle.
This effect is mediated by its metabolite α-ketoglutarate, which inhibits SIRT5 expression and SIRT5-dependent desuccinylation on PDHA1, thereby maintaining PDH activity and allowing an entire OXPHOS process that supports M2 polarization. The role of SIRT5 in I/R injury is also controversial and probably tissue-specific, restricting I/R progression in the heart (Hershberger et al., 2017) while deteriorating injury in the kidney and brain (Diaz-Cañestro et al., 2018). Its effect on hepatic I/R remains still unclear. In this regard, SIRT5 overexpression alleviated liver I/R injury and inflammation in mice in a hypoxia/reoxygenation in vitro model (Zheng et al., 2022). Moreover, SOD1 and IDH2 knockdown in cells abolished the effect of SIRT5 on restraining oxidative stress and inflammation, suggesting that both SOD1 and IDH2 are downstream effectors of SIRT5.
As mitochondrial sirtuin, SIRT5 is involved in metabolic homeostasis, inflammation, and cell response against oxidative stress through regulating mitochondrial metabolism, mitophagy, and apoptosis. In this regard, its effects on either stimulating or suppressing cell proliferation can in principle be understood due to the ability of SIRT5 to control mitochondrial anaplerosis to replenishing TCA cycle intermediates and key molecules able to switch the metabolic program between growth and quiescence. Thus, SIRT5 can either promote or prevent specific metabolic pathways depending on cell type and availability of nutrients.
Sirtuin 6
SIRT6 was initially postulated as an important regulator of life span in mice (Kanfi et al., 2012). Curiously, SIRT6 is the unique sirtuin that could bind NAD+ without an acetylated substrate, raising the possibility of SIRT6 as an NAD+ sensor (Pan et al., 2011). SIRT6 enzymatic activities include deacetylation, fatty acid deacylation, and mono-ADP-ribosylation, thus allowing it to participate in several cellular pathways. As such, SIRT6 is involved in DNA damage repair, transcriptional repression, telomeric maintenance, metabolic homeostasis, senescence, cell cycle, and immune regulation, among other functions (Michishita et al., 2008; Mostoslavsky et al., 2006; Rezazadeh et al., 2019; Roichman et al., 2021; Tasselli et al., 2016; Tennen et al., 2011; Toiber et al., 2013; Xu et al., 2015).
A severe decrease in lymphocyte number was initially observed in the context of systemic alterations caused by SIRT6 deficiency in mice (Mostoslavsky et al., 2006). This observation represented a starting point to understanding the implications of SIRT6 in immune regulation. The first studies suggested an anti-inflammatory role through NF-κB signaling inhibition (Fig. 3). SIRT6 interacts with the RelA/p65 subunit and is recruited to the chromatin to deacetylate H3K9 at promoter regions and silencing NF-κB target genes (Kawahara et al., 2009). In addition, similar to SIRT1 and SIRT2, SIRT6 affects the acetylation levels on p65 (Lys310), attenuating NF-κB signaling in several situations and experimental models (Cheng et al., 2023; Sun et al., 2014; Wu et al., 2015).
Moreover, an additional mechanism of NF-κB regulation by SIRT6 was proposed in HEK293F cells where SIRT6 indirectly promotes IkBa expression (NF-κB repressor), which restores cytoplasmic relocalization of NF-κB and prevents its overactivation (Santos-Barriopedro et al., 2018). SIRT6 ablation leads to hyperacetylation of H3K9 and increased occupancy of c-JUN at promoters of proinflammatory genes for MCP-1, IL-6, and TNF-α (Xiao et al., 2012). The functional link between SIRT6, NF-κB, and c-JUN seems relevant in vivo and was suggested after observing that the proinflammatory phenotype developed by SIRT6 KO mice was at least partially due to the upregulation of inflammatory genes and hyperactivation of the NF-κB pathway (Kawahara et al., 2009).
In agreement, SIRT6 deficiency in the immune cells of mice results in liver inflammation and fibrosis through overexpression of c-JUN/AP-1 signaling (Xiao et al., 2012), whereas in bone marrow-derived macrophages, SIRT6 deletion promoted NF-κB activation and concomitant IL6, TNF-α, and INF-β increased expression, leading to STAT3 activation and M1 polarization. As a result, SIRT6 myeloid-specific deficient mice subjected to HFD are more susceptible to developing diet-induced inflammation and insulin resistance (Lee et al., 2017). Interestingly, an augmented response to HFD was described in the Fabp4-Cre (Sirt6-FKO) mouse model, where deletion of SIRT6 in preadipocytes and mature adipocytes leads to overweight, insulin resistance, glucose intolerance, and adipose inflammation compared with wild-type mice (Kuang et al., 2017; Xiong et al., 2017). In addition, adipocyte-specific SIRT6 KO mice showed increased infiltration of macrophages and an up-regulation of M1 macrophage genes in adipose tissue (Song et al., 2019).
In addition, SIRT6 protection was also demonstrated during vascular inflammation. Using an experimental model of atherosclerosis, endothelium-specific SIRT6 KO mice showed increased production of MCP-1, IL-6, and IL-1 in response to hyperlipidemia. This mechanism was postulated to be NRF2-dependent since overexpression of SIRT6 increased NRF2 and expression of target genes, whereas knockdown of SIRT6 produced the opposite effect (He et al., 2021). Similar results were obtained in endothelial-to-mesenchymal transition (EndMT) during vascular inflammation, where SIRT6 suppressed transition by attenuating the endothelial inflammatory response (Chen et al., 2021). NRF2 activation by means of SIRT6 overexpression or increased NAD+ levels upregulated antioxidant proteins HO-1 and SRXN1 in neurotoxic astrocytes obtained from amyotrophic lateral sclerosis rat models (Harlan et al., 2019). In turn, this antioxidant response protects cocultured motor neurons from apoptosis.
Given the inflammatory component of ALS, this observation highlights the potential role of the SIRT6/NRF2 axis in the pathogenesis of inflammatory-based conditions triggered by increased oxidative stress.
In this regard, Liu et al. (2015, 2012) shed light on how intracellular NAD+ fluctuations modulate inflammatory response in a sirtuin-dependent manner. In macrophages, SIRT1 and SIRT6 simultaneously sense changes in NAD+ levels to reprogram cellular metabolism during acute inflammatory responses. The transition from early to late inflammation relies on an important metabolic switch facilitated by SIRT1 and SIRT6. This switch involves a shift from enhanced glycolysis to increased FAO (Liu et al., 2012). Upon TLR4 receptor stimulation, there is an induction of Nampt expression, leading to elevated NAD+ levels. This rise in NAD+ concentration plays a crucial role in activating SIRT6, which in turn decreases glycolysis, and SIRT1, which promotes FAO. Worthy of note, such a sharp change in cell metabolism can be better explained as a consequence of the coordinate signaling of these two sirtuins that, in turn, get activated once a critical threshold of NAD+ concentration is achieved.
Importantly, increased NAD+ levels trigger two additional SIRT1-dependent effects: first, an upregulation of SIRT3 expression that improves mitochondrial respiration and decreases ROS production; second, stimulation of mitochondrial biogenesis. Remarkable, mitochondrial dysfunction during adaptation may adversely influence sepsis outcome, while accelerating mitochondrial biogenesis during early sepsis can increase survival in septic animals (Singer, 2014). In the context of acute systemic infection, this NAD+- triggered mechanism ensures a transition to a lower energy restorative state that balances immunity and inflammation, and restores homeostasis. Importantly, the functional relationship between nuclear SIRT1 and mitochondrial SIRT3 suggests that NAD+ generation in the two subcellular compartments is coordinated during adaptation.
A role for SIRT6 in lineage differentiation and function of immune cells was proposed. SIRT6 deletion impairs spontaneous maturation, interferes with adaptive immune responses (Lasigliè et al., 2016), and with M1 to M2 transition of macrophages. Consistently, myeloid-specific SIRT6-deficient mice exhibited increased infiltration of M1 and decreased M2 macrophages due to impaired Akt signaling. Similar results were observed in the context of obesity, finding increased M1 polarization of bone marrow macrophages and migration toward adipose-derived chemoattractants (Lee et al., 2017). Conversely, SIRT6 overexpression promotes macrophage M2 transformation (Ji et al., 2019). SIRT6 deficiency contributes to delayed wound healing with reduced collagen deposition, suppressed angiogenesis, and reduced expression of wound healing-related genes, compared with wild-type mice (Koo et al., 2019).
Finally, a role for SIRT6 in IFN-γ synthesis by T cells was suggested since its expression was reduced upon SIRT6 knockdown with siRNA. Similarly, splenocytes from SIRT6 KO mice secreted less IFN-γ compared with wild-type mice (Bruzzone et al., 2009).
On the contrary, an important number of publications highlight the proinflammatory roles that SIRT6 can exert under specific circumstances (Fig. 3). The first evidence regarding the proinflammatory role of SIRT6 was observed in activated immune cells, where an increase of NAD+ concentration promoted TNF-α mRNA translation in an SIRT6-dependent manner (Van Gool et al., 2009). Consistently, SIRT6 was reported to catalyze the hydrolysis of myristoylated lysine 19 and 20 of TNF-α, allowing TNF-α secretion in cultured cells (Jiang et al., 2016; Jiang et al., 2013), whereas in vivo pharmacological inhibition of SIRT6 decreases TNF-α secretion after LPS stimulation in mice peritoneum (Bresque et al., 2022) as well as improves oral glucose tolerance and reduced levels of insulin, triglycerides, and cholesterol in a mouse model of type 2 DM (Sociali et al., 2017).
A reduction of systemic inflammation and hyperglycemia was also observed upon an SIRT6 myeloid-specific ablation, in an obesity-induced inflammation mouse model (Bresque et al., 2022). These findings suggest a scenario where SIRT6 can perform either as a pro- or as an anti-inflammatory player, probably depending on the stage of the inflammatory process or upon a combination of metabolic clues simultaneously present.
Finally, in pancreatic cancer cells, SIRT6 enhanced the expression of the proinflammatory cytokines IL-1α, IL-6, IL-8, and TNF-α and indirectly promoted cell migration by modulating the intracellular ADPr levels, through a Ca2+-NFAT-dependent signaling (Bauer et al., 2012). Moreover, in an EAE (experimental autoimmune encephalomyelitis) mouse model, SIRT6 inhibition impairs dendritic cell migration and downregulates pathogenic T cell infiltration and inflammatory responses, leading to a delay in EAE onset (Ferrara et al., 2020). Figure 6 summarizes the data discussed above.

The evidence available so far depicts SIRT6 as a multitasking enzyme involved in the regulation of multiple pathways of different hierarchies. Apparently, depending on the context and cell type involved, SIRT6 can switch from a proinflammatory to an anti-inflammatory phenotype. This plasticity could be assigned, in part, due to SIRT6's capacity to regulate and reprogram the metabolic requirements to drive the transition from an early inflammatory response to a late-stage and resolution phase (Liu et al., 2015; Medzhitov et al., 2012). In addition, SIRT6-dependent transcriptional repression/activation allows it to regulate immune genes and determine specific cell phenotypes such as macrophage polarization, dendritic cell migration, or T cell infiltration. In the present context, SIRT6 rises as a candidate target for pharmacological intervention.
Sirtuin 7
Sirtuins 6 and 7 were the two last mammalian sirtuins to be described (Frye, 2000). SIRT7 is a widely expressed nucleolar protein that is associated with active rRNA genes where it interacts with RNA polymerase I (Pol I) as well as with histones. Overexpression of SIRT7 increases Pol I-mediated transcription, whereas knockdown of SIRT7 results in decreased association of Pol I with recombinant DNA (rDNA) and a reduction of Pol I transcription. Depletion of SIRT7 stops cell proliferation and triggers apoptosis (Ford et al., 2006). In addition, SIRT7 has been shown to function in aging-related processes. SIRT7 KO mice had shorter life spans, showed decreased stress resistance, and developed age-dependent inflammatory cardiomyopathy, implying that SIRT7 is involved in inflammatory processes (Vakhrusheva et al., 2008).
However, the evidence concerning the role of SIRT7 in inflammation suggests a complex landscape, where experimental observations are sometimes inconsistent or difficult to interpret. For example, the knockdown of SIRT7 in primary breast cells promoted the phosphorylation and translocation of p65 to the nucleus and consequently increased the secretion of downstream inflammatory cytokines. Conversely, SIRT7 overexpression had the opposite effect (Chen et al., 2019a) (Fig. 3). In addition, in a murine model of accelerated aging, the decline of SIRT7 resulted in overexpression of progerin and upregulation of proinflammatory cytokines IL-1β and IL-6 in human umbilical vein endothelial cells; overexpression of SIRT7 effectively alleviated the inflammatory response, rejuvenates blood vessels and extends life span (Sun et al., 2020b). Interestingly, a novel mechanism involving SIRT7 in inflammation was recently proposed to understand the persistent inflammation in renal tissue leading to nephropathy during DM (Li et al., 2022).
The authors proposed that SIRT7 modulates hyperglycemic memory in DM, where glucose-mediated mutual inhibition between SIRT7 and a transcription factor of the ETS oncogene family (ELK1) induced death associated protein kinase 3 (DAPK3) transcription and inflammation despite normoglycemia, thus forming a vicious cycle and accounting for the hyperglycemic memory during diabetes.
Published evidence in favor of a proinflammatory role of SIRT7 is also abundant (Fig. 3). Observations in fibroblast cells suggested that loss of SIRT7 promoted the translocation of NF-κB p65 to the cytoplasm (Sobuz et al., 2019). These authors demonstrated that SIRT7 interacts with a small GTPase named Ras-related nuclear antigen (Ran) and deacetylates Lys37, thus suppressing the export of p65 from the nucleus. Similarly, SIRT7 silencing in human pulmonary artery endothelial cell (HPAEC) cultures decreased total NF-κB levels in nuclear fractions in basal conditions and in LPS-stimulated cultures. SIRT7 silencing in HPAEC also attenuated a basal and an LPS-induced increase in phosphorylated NF-κB and the expression of NF-κB downstream genes, IL-6, and IL-8 (Wyman et al., 2020).
Besides the anti-inflammatory effect achieved by SIRT7 silencing, HPAEC also underwent EndoMT with decreases in VE-cadherin and platelet and endothelial cell adhesion molecule 1 (PECAM1) and increases in collagen, alpha-smooth muscle actin, and TGFβ receptor 1. Similar results were also obtained in a model of acute kidney injury, where SIRT7 KO mice reduced the inflammation and tubular damage after bilateral I/R (Sánchez-Navarro et al., 2022). Notably, increased fibrosis seems to be a common feature in several experimental models where SIRT7 is downregulated, suggesting the activation of common pathways upon SIRT7 inhibition, beyond the cell model used.
These contradictory results imply that the regulatory effects of SIRT7 on the inflammatory process may be variable under diverse circumstances, that is, experimental models or even specific pathologies. Further studies will be necessary before claiming SIRT7 as a therapeutic target aiming to modulate the inflammatory response in diverse scenarios.
Conclusions and Perspectives
The available evidence supporting a close causal relationship between sirtuins and inflammation is significant and still growing. Initially regarded as anti-inflammatory regulators, sirtuins are currently emerging as multifaceted proteins, able to stimulate or suppress inflammatory pathways, apparently in a context-dependent manner. This behavior is far to be understood, and contradictory observations are abundant in the specialized literature. Such contradictions may be mostly apparent and probably reflects the ignorance that science still has about the regulation of such a complex process. Nevertheless, a wide range of regulatory mechanisms used by sirtuins to modulate the inflammatory status are currently known and others are being subject of intense research. In addition, the use of cell-tissue-specific KO models as well as the availability of new and specific inhibitors and activators will allow us to deeply tease apart the complex function of each sirtuin in different inflammatory contexts.
This review highlighted the translational potential of sirtuin targeting to regulate inflammatory responses, hoping that taking advantage of their potentiality as key metabolic regulators will enable further therapeutic interventions on inflammation-related disorders. Future challenges concerning the use of sirtuins as therapeutic targets will require determining the exact role that each sirtuin may have in specific time frames of the inflammatory process, aiming to design more specific and efficient interventions.
Footnotes
Authors' Contributions
C.E. and A.C. designed and conceived the review. All authors wrote and reviewed the article. L.S., A.C., and M.B. conceived and designed the figures and tables.
Author Disclosure Statement
No financial or commercial conflict of interest to disclose.
Funding Information
This work was funded by the CSIC I + D program (Grant ID 498-2020, CSIC-Udelar), PEDECIBA, and FOCEM.