
Abstract
Significance:
Stringent regulation of protein homeostasis pathways, under both physiological and pathological conditions, is necessary for the maintenance of proteome fidelity and optimal cell functioning. However, when challenged by endogenous or exogenous stressors, these proteostasis pathways can become dysregulated with detrimental consequences for protein fate, cell survival, and overall organism health. Most notably, there are numerous somatic pathologies associated with a loss of proteostatic regulation, including neurodegenerative disorders, type 2 diabetes, and some cancers.
Recent Advances:
Lipid oxidation-derived reactive carbonyl species (RCS), such as 4-hydroxynonenal (4HNE) and malondialdehyde, are relatively underappreciated purveyors of proteostatic dysregulation, which elicit their effects via the nonenzymatic post-translational modification of proteins. Emerging evidence suggests that a subset of germline proteins can serve as substrates for 4HNE modification. Among these, prevalent targets include succinate dehydrogenase, heat shock protein A2 and A-kinase anchor protein 4, all of which are intrinsically associated with fertility.
Critical Issues:
Despite growing knowledge in this field, the RCS adductomes of spermatozoa and oocytes are yet to be comprehensively investigated. Furthermore, the manner by which RCS-mediated adduction impacts protein fate and drives cellular responses, such as protein aggregation, requires further examination in the germline. Given that RCS-protein adduction has been attributed a role in infertility, there has been sparked research investment into strategies to prevent lipid peroxidation in germ cells.
Future Directions:
An increased depth of knowledge regarding the mechanisms and substrates of RCS-mediated protein modification in reproductive cells may reveal important targets for the development of novel therapies to improve fertility and pregnancy outcomes for future generations.
Introduction
In healthy cells, a complex network of protein homeostasis or “proteostasis” machinery acts in concert to maintain proteome fidelity and ensure optimal functioning of somatic and reproductive cells alike. The currently curated proteostasis network of human somatic cells comprises more than 2000 proteins (more than 10% of the human proteome), including molecular chaperones and proteolytic enzymes, that are responsible for the coordination of three interconnected arms of proteostasis (Klaips et al., 2017). These proteostatic arms can be broadly classified as follows: (i) polypeptide synthesis and folding, (ii) maintenance of protein conformation and solubility, and (iii) protein degradation. The mechanisms and key effectors associated with each of these processes have been extensively reviewed by Hipp et al. (2019) and Jayaraj et al. (2020). A summary of proteostatic regulation under both physiological and pathological conditions is provided in Figure 1.
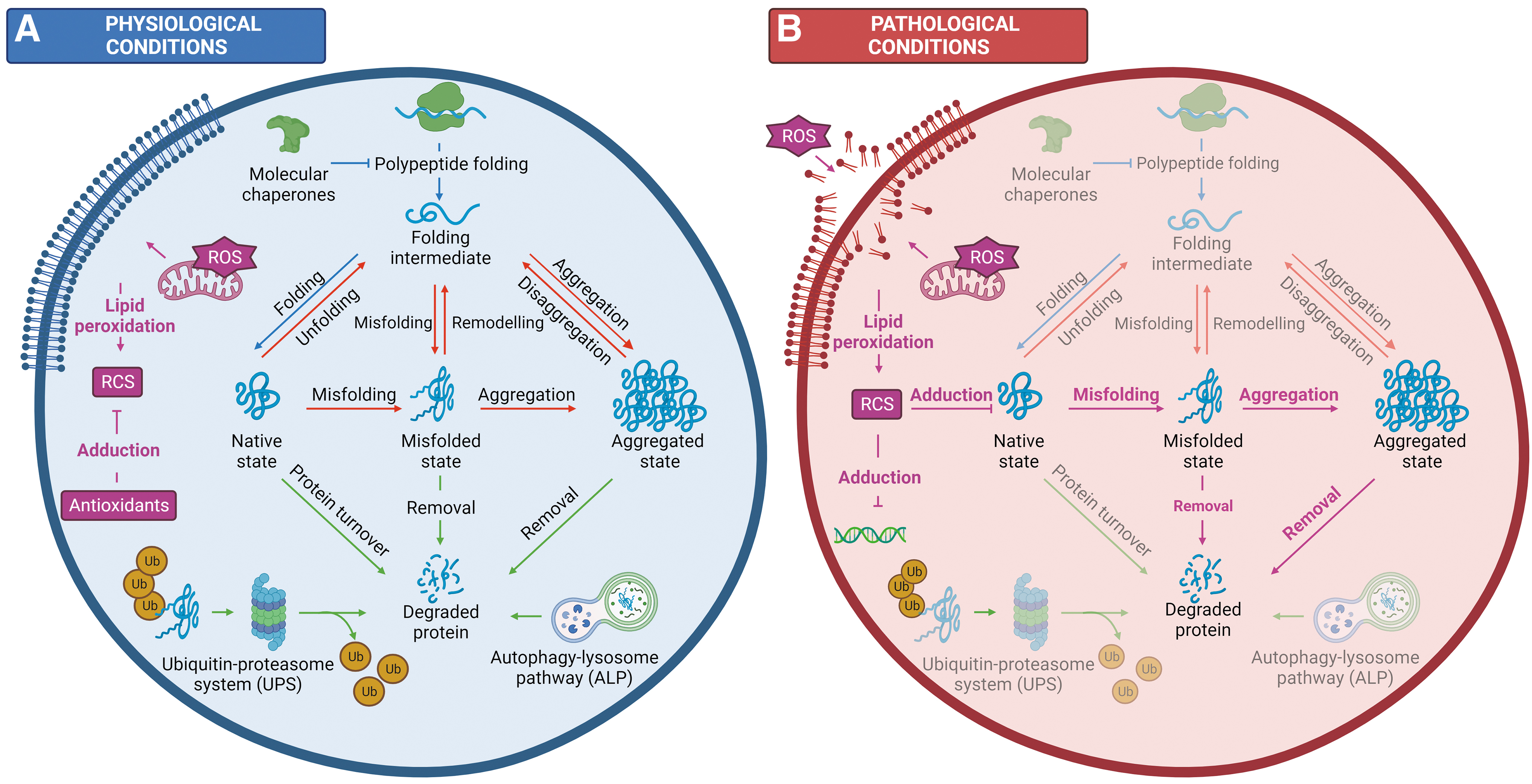
The proteostasis network ensures that proteins are generated at the correct time, cellular location, and abundance to support cell function (arm 1) and orchestrates adaptation in response to diverse stress conditions (Arnsburg and Kirstein-Miles, 2014). Most of these proteins must adopt a unique, thermodynamically stable three-dimensional structure to gain functionality (Anfinsen, 1973). This de novo folding (arm 1) is mediated by protein–protein interactions with molecular chaperones, namely members of the heat shock protein (HSP) family (Fink, 1999). Once folded, proteins must maintain their native structure, which is complicated by the fact that functionality often requires considerable conformational flexibility. As a result, the biologically active conformation of many proteins is often only marginally stable or metastable (Jayaraj et al., 2020).
These metastable proteins tend to populate misfolded species that are prone to forming toxic aggregates, including soluble oligomers and fibrillar amyloid deposits (Hartl, 2017). This is further compounded by underlying genetic mutations, translational errors, abnormal protein modifications, and/or physiological stressors that consistently produce a basal level of misfolded proteins (Sweeney et al., 2017). To prevent deleterious consequences, misfolded and/or aggregated protein species must be remodeled (arm 2), or alternatively, removed by degradation through one of two key proteolytic pathways (arm 3): the ubiquitin–proteasome system (UPS) or the autophagy–lysosome pathway (ALP). Together, the three interconnected arms of proteostasis prevent the accumulation of potentially cytotoxic protein aggregates and maintain proteome fidelity (Fig. 1A) (Hipp et al., 2019).
Despite stringent regulation of proteostasis pathways, maintaining a balanced proteome becomes a challenge in the face of exogenous and endogenous stressors with the consequences of proteostatic dysregulation ranging from protein misfolding to cellular degeneration and tissue damage (Bromfield et al., 2019; Bromfield et al., 2017a; Bromfield et al., 2015; Cafe et al., 2020; Kurtishi et al., 2019; Nixon et al., 2019; Peters et al., 2020). This is particularly challenging for reproductive cells, including sperm and developing germ cells, as they conduct their final maturation steps and complete the fertilization of oocytes in the absence of de novo transcription and translation. As such, sperm are known to be among the most sensitive cell types to the extracellular environment (Dutta et al., 2021; Gharagozloo and Aitken, 2011).
Among the most pervasive of these cellular stressors are those that induce a state of oxidative stress (i.e., thermal stress (Houston et al., 2018)), which can severely compromise cellular function and drive cells toward a synonymous pathway of pathology and demise. For example, the detrimental consequences of proteostatic dysregulation during periods of oxidative stress can be gleaned from immunological studies. In response to injury or infection, an array of immune cells, including neutrophils, are recruited to the affected tissue where they deploy a variety of highly microbicidal weapons (Ulfig and Leichert, 2021). As part of their arsenal against pathogens, neutrophils produce a suite of oxidizing agents, namely hypochlorous acid (HOCl) and hypobromous acid, that are capable of modifying protein (Winter et al., 2008), DNA (Henderson et al., 2001; Prütz, 1996), and lipid substrates (Winterbourn et al., 1992).
Despite the ability to target and damage several components of the pathogen, HOCl primarily serves as a strong inducer of oxidative unfolding and protein aggregation (Winter et al., 2008), which, if left unchecked, can lead to widespread loss of function, cytotoxicity, and cell death (Ulfig and Leichert, 2021). In this way, neutrophils have been shown to mediate pathogen clearance through the targeted attack of proteostatic pathways. Unfortunately, to the detriment of the host, hypohalous acids (i.e., HOCl) have low specificity and are in no way selective for pathogens and/or their macromolecular substrates. As a result, excessive generation and accumulation of these products can induce unfolding and aggregation of host proteins, ultimately leading to cell death (Ulfig and Leichert, 2021) and collateral tissue damage (Aratani, 2018; Davies and Hawkins, 2020; Kisic et al., 2016; Ndrepepa, 2019; Pravalika et al., 2018).
An often-overlooked tier of proteostatic regulation, particularly during periods of oxidative stress, is the nonenzymatic post-translational modification of proteins by reactive carbonyl species (RCS) (Fig. 1B). This review discusses RCS with respect to their generation, reactivity, and ability to adduct to macromolecular substrates, with a particular focus on RCS-mediated post-translational modifications and their consequences at both the protein and cellular levels. While critical research within this area has been forged by those in the field of neurodegeneration, we aim to provide insight into the impact of RCS in the male germline, namely the role of RCS in directing protein fate and how remodeling of the proteome by RCS may be linked to the dysregulation of proteostasis pathways.
Reactive Carbonyl Species
RCS, historically termed electrophilic lipid aldehydes, are a physiologically relevant and highly reactive group of compounds that are generated under conditions of oxidative stress (Mano, 2012). In the absence of adequate antioxidant defenses, reactive oxygen species (ROS) produced by either intracellular metabolic changes or extracellular sources such as ionizing radiation or thermal stress (Gaschler and Stockwell, 2017) can cause extensive damage to a range of nucleic acid, protein, and lipid substrates (He et al., 2017; Lundgren et al., 2018; Su et al., 2019). Of particular interest to this review, ROS including superoxide (O2 •−), hydroxyl radical (•OH), and hydrogen peroxide (H2O2) can attack the lipidic carbon–carbon double bond(s) in unsaturated fatty acids, consequently initiating a cascade of lipid peroxidation (Ayala et al., 2014).
Lipid peroxidation
Polar lipids (i.e., glycolipids and phospholipids) and neutral lipids (i.e., sterols) are all vulnerable to the damaging and potentially lethal process of electron scavenging (Girotti, 1998; Kanner et al., 1987; Yin et al., 2011), with omega-3 and omega-6 (hereafter, ω-3 and ω-6) polyunsaturated fatty acids (PUFAs) serving as primary targets for peroxidative modification (Ayala et al., 2014; Mihalas et al., 2017). Notably, PUFAs that are constituents of structural phospholipids within the cellular membrane including docosahexaenoic acid, eicosapentaenoic acid, γ-linolenic acid, arachidonic acid (AA), and linoleic acid (LA) act as substrates for oxidative attack (Fig. 2) (Ayala et al., 2014; Esterbauer et al., 1991; Tallima and El Ridi, 2018). The process of lipid peroxidation can be divided into the following three distinct phases: (i) initiation, (ii) propagation, and (iii) termination (Girotti, 1998; Kanner et al., 1987; Yin et al., 2011).
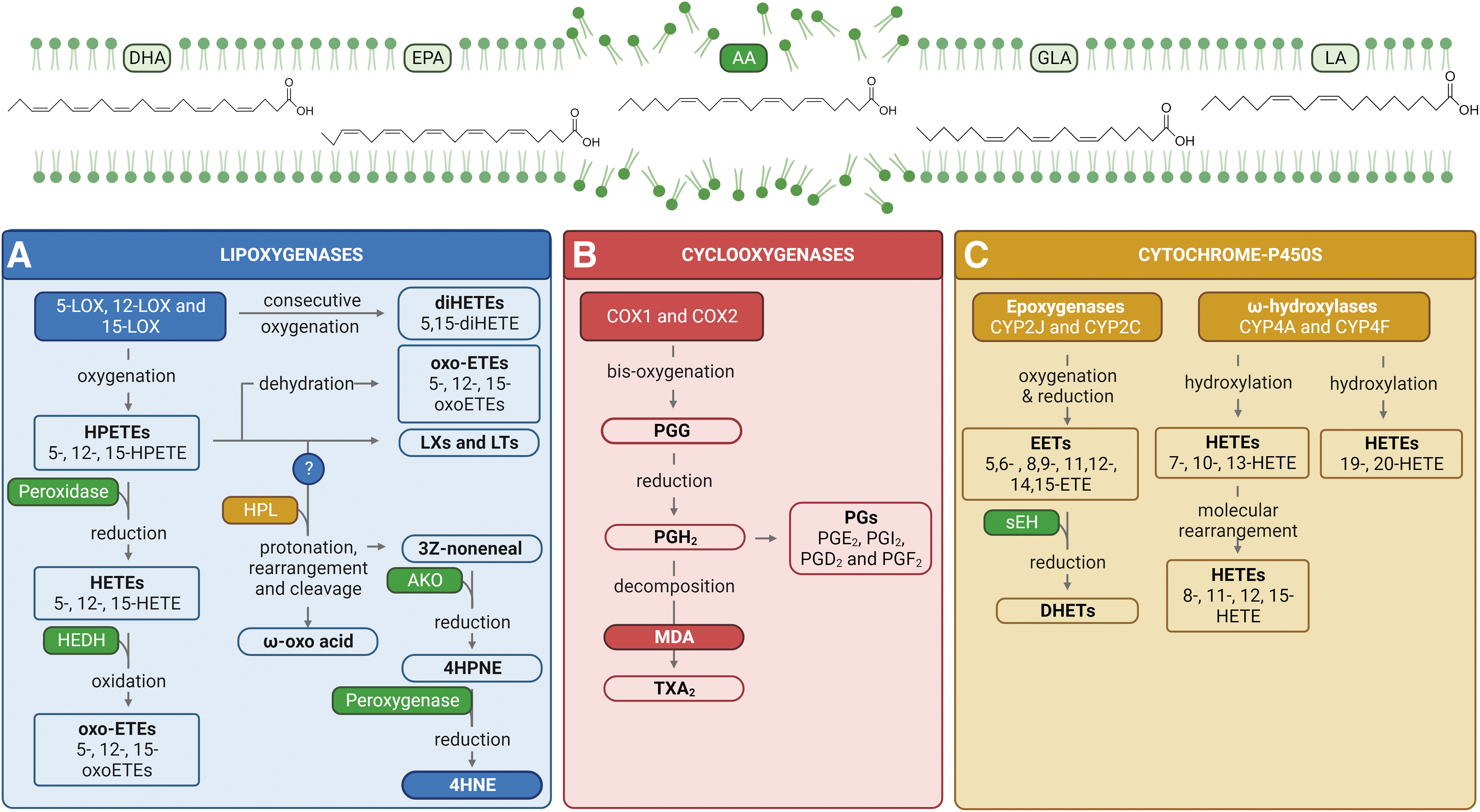
During the initiation stage, PUFA substrates undergo a series of reactions including allylic hydrogen atom abstraction by ROS (i.e., O2 •−, •OH, and H2O2) and molecular rearrangement that results in the formation of unsaturated lipid radicals. In the propagation phase, these lipid radicals rapidly react with oxygen to form lipid peroxyl radicals, which in turn abstract a hydrogen from additional PUFAs to generate a new lipid radical (thus perpetuating the lipid peroxidation cycle) and a lipid hydroperoxide. In this way, a single oxidation event can trigger an auto-oxidative chain reaction that will continue until a termination product is produced. Alternatively, these lipid peroxyl radicals can be further metabolized or enzymatically converted into a suite of secondary lipid peroxidation products including RCS.
Among these, the most abundant are 4-hydroxynonenal (4HNE) and malondialdehyde (MDA), while acrolein (ACR) is the most reactive (Esterbauer et al., 1991; Fuloria et al., 2020; Pizzimenti et al., 2013). Termination of this lipid peroxidation cascade occurs when antioxidant molecules such as glutathione (GSH), superoxide dismutase, and some vitamins (C and E) donate a hydrogen atom to the lipid peroxyl species resulting in the formation of non-radical products. For additional information regarding these mechanisms and the chemistry associated with lipid peroxidation pathways, the reader is referred to excellent reviews by Volinsky and Kinnunen (2013) and Ayala et al. (2014).
A long prevailing paradigm within the carbonyl stress field suggests that nonenzymatic lipid peroxidation is a major contributor to RCS formation in vivo. Evidence of this can be drawn from several studies, including one notable investigation conducted by Kadiiska et al. (2005), which sought to define the contribution of cyclooxygenase (COX) enzymes to the production of RCS and their intermediates. Key results from this study revealed an acute elevation of MDA and isoprostane levels (an index of exogenous lipid peroxidation) in the urine and plasma of rodents poisoned with carbon tetrachloride, an oxidative stressor. Although reduced slightly, MDA and isoprostane levels were not significantly inhibited in the presence of nonsteroidal anti-inflammatory drugs (NSAIDs), which, among other functions, primarily serve as competitive inhibitors to suppress COX activity. While these data clearly suggest that nonenzymatic pathways are a major contributor to RCS generation in vivo, there are some limitations to this study that potentially underestimate the contributions of enzymatic routes.
Most notably, this study aims to define the contribution of COX enzymes independent of other oxidative enzymes (i.e., lipoxygenases [LOXs] and cytochrome P-450s [CYPs]); however, there is likely some level of redundancy between these pathways. While LOX and CYP enzymes are not currently known to play a role in the catalytic generation of MDA or isoprostanes, it is a possibility that novel, noncanonical oxidation pathways become activated in the absence of sufficient COX activity. Furthermore, the authors highlight that the efforts to define the contributions of enzymatic versus nonenzymatic RCS sources are complicated by the antioxidant actions of conventional NSAIDs (Kadiiska et al., 2005). Taken together, these limitations do not diminish nor negate these findings; however, they do highlight the importance of further investigation into the production of RCS via enzymatic pathways.
While comparatively less is understood about the contribution of enzymatic pathways in vivo, compelling evidence gleaned from in vitro germ cell models suggest that enzymatic pathways are a central mechanism through which RCS are generated in the germline. Importantly, data support that selective inhibition of arachidonate 15-lipoxygenase (ALOX15) using PD146176 (6,11-dihydro[1]benzothiopyrano[4,3-b]indole) is successful in reducing oxidative stress, lipid peroxidation, and the production of RCS, namely 4HNE, in both mouse and human germ cells (Bromfield et al., 2017b; Walters et al., 2018). In this way, inhibition of ALOX15 can afford protection to human sperm during conditions of oxidative stress, with notable improvements in motility, acrosome reaction rates, and zona pellucida binding (Walters et al., 2018). Considering the critical role of these oxidative enzymes in reproductive cells, this review herein will focus on the enzymatic generation of RCS.
RCS production by enzymatic processes
Literature pertaining to somatic cells has documented several enzymes capable of catalyzing the lipid oxygenation and hydrogen abstraction processes that lead to the production of RCS. These primarily include LOX enzymes, COXs, and to a lesser extent, CYP enzymes. The molecular mechanisms of lipid substrate oxidation by each of these enzymes have been extensively described by Wang et al. (2019) and Hajeyah et al. (2020). Here, we provide a brief overview of the products generated by each of these enzyme families (Fig. 2).
LOX enzymatic activity
LOXs are a family of non-heme iron dioxygenases that catalyze the oxidation of PUFA (and occasionally sterol) substrates into an array of metabolites including hydroxyeicosatetraenoic acids (HETEs) derived from AA, leukotrienes (LTs), and lipoxins (LXs) (Fig. 2A). LOX isoforms are often classified based on their positional specificity for oxygenation (Shi et al., 2020). In mammalian cells, carbon atoms at the 5-, 12-, and 15-positions are thought to serve as the primary oxidation sites of AA, with these reactions catalyzed by 5-LOX, 12-LOX, and 15-LOX enzymes, respectively (Wang et al., 2019). Here, we will solely focus on the products of 5-LOX-catalyzed AA metabolism; however, the same general principles apply for 12- and 15-LOX enzymes. The insertion of molecular oxygen at carbon atom 5 in AA generates 5-hydroperoxyeicosatetraenoic acid (5-HPETE), which is either reduced by peroxidase enzymes into 5-HETE or, alternatively, converted to bioactive lipid mediators such as LTs and LXs (Wang et al., 2021).
This insertion of oxygen at one end of AA does not impinge on the reactive pentadiene system at the other end, thus consecutive oxygenation at the 5- and 15-carbons can yield 5,15-diHETEs (Tejera et al., 2012). Downstream of these events, 5-oxo-eicosatetraenoic acid can also be synthesized via (i) enzymatic or nonenzymatic dehydration of 5-HPETE or (ii) 5-hydroxyeicosanoid dehydrogenase-catalyzed oxidation of 5-HETE (Powell and Rokach, 2015). In a similar manner, LOX enzymes can metabolize LA into hydroperoxyoctadecadienoic acids (HPODEs), hydroxyoctadecadienoic acids, dihydroxyoctadecadienoic acids, and oxooctadecadienoic acids (Kirpich et al., 2016).
The mechanisms through which these oxidative intermediates are metabolized into RCS are not yet fully understood. One proposed mechanism is the lipoxygenase–hydroperoxide lyase (LOX-HPL) pathway in which lipid hydroperoxides (i.e., LA-derived 9- and 13-HPODEs) serve as the primary substrates for RCS formation. In plants, for instance, 9-HPODE is subjected to protonation, rearrangement (C-C to C-O), and subsequent cleavage by 9-HPL (CYP74). This reaction yields two products: (i) 9-oxonanoic acid (an ω-oxo acid) and (ii) 3Z-nononeal (a volatile aldehyde). 3Z-nononeal is rapidly oxidized by alkenal oxygenase to form 4-hydroperoxynonenal (4HPNE), an immediate precursor of 4HNE. Analogous reactions are theorized to facilitate the conversion of hydroperoxides from other ω-6 PUFAs, specifically AA-derived 11- and 15-HPETEs to 4HPNE, which can be further reduced to 4HNE by cellular peroxygenases (Fig. 2) (Ayala et al., 2014; Noordermeer et al., 2001; Noordermeer et al., 2000; Schneider et al., 2001; Spickett, 2013).
COX enzymatic activity
COXs, also known as prostaglandin G/H synthases, are rate-limiting enzymes involved in the conversion of AA into prostaglandins (PGs) and later, through additional mechanisms, thromboxanes (TXs) (Fig. 2B). There are two distinct COX isoforms: COX1 is constitutively expressed in most cells and is the dominant source of prostanoids (active lipid mediators; i.e., PGs and TXs) that maintain tissue homeostasis (Ding et al., 2003). On the contrary, COX2 is an inducible isoform with its expression stimulated by growth factors, cytokines, and tumor promoters (Wang et al., 2021). Given this, COX2 is hypothesized to play a central role in prostanoid formation in inflammation and various pathological disease states (Rouzer and Marnett, 2009; Wang et al., 2021).
Possessing two separate but linked active sites, COX enzymes catalyze the bis-dioxygenation and subsequent reduction of AA to PGG2 and PGH2 (Rouzer and Marnett, 2009). These PGs serve as substrates for metabolism by a series of downstream enzymes to generate a family of prostanoids (Ding et al., 2003; Wang et al., 2021), including PGE2, PGI2, PGD2, PGF2, and TXA2 (Cao and Prescott, 2002; FitzGerald and Patrono, 2001; Funk, 2001; Hata and Breyer, 2004; Samuelsson et al., 1978; Smith et al., 2000; Wlodawer and Samuelsson, 1973). MDA can be generated as a by-product of thromboxane synthase-catalyzed decomposition of PGH2 to TXA2 (Hecker et al., 1987).
CYP enzymatic activity
CYP enzymes are a superfamily of heme-containing monooxygenases that catalyze a diverse range of oxidative reactions, including the metabolism of AA (Fig. 2C). The CYP family can be divided into two subgroups according to their mechanism of action, those being CYP epoxygenases (i.e., CYP2J and 2C) and CYP ω-hydroxylases (i.e., CYP4A and 4F) (Spector et al., 2004; Wang et al., 2021). The epoxygenase activity of CYP enzymes catalyzes insertion of an oxygen atom on a carbon attached to one of the double bonds of AA. This double bond is subsequently reduced to form AA epoxides, also referred to as epoxyeicosatrienoic acids (EETs). Epoxidation can occur at any of the four double bonds of AA, giving rise to four unique regioisomers: 5,6-EET, 8,9-EET, 11,12-EET, and, the dominant form, 14,15-EET (Spector, 2009). The resultant EETs are further metabolized, mainly by soluble epoxide hydrolase, into corresponding dihydroxyepoxyeicosatrienoic acids or diols (Chacos et al., 1983; Moghaddam et al., 1996; Zeldin et al., 1995; Zeldin et al., 1993).
Alternatively, CYP ω-hydroxylases mediate the metabolism of fatty acids via the addition of a hydroxyl group to the ω or ω-1 carbon atom. Notably, ω-hydroxylation of AA yields several metabolic products, including 19- and 20-HETEs (Ni and Liu, 2021). In a similar manner, CYPs can hydroxylate the three bisallylic carbons of AA to generate 7-, 10-, and 13-HETEs. The latter compounds are unstable in acidic conditions and can rearrange to give conjugated diene HETEs. 13-HETE undergoes molecular rearrangement into 11- and 15-HETEs, whereas 10-HETE can be converted into 8- and 12-HETEs (Brash et al., 1995; Powell and Rokach, 2015).
Ultimately, members of the LOX, COX, and CYP families function both independently and in combination to generate short-chain RCS, such as 4HNE, MDA, and ACR. While the male germline is known to possess many of the same peroxidative enzymes detected in somatic cells, including COX1 and COX2 (Matzkin et al., 2010; Perrotta et al., 2012), their role in germ cells is yet to be fully elucidated. Current knowledge surrounding the role of RCS in the male germline will be discussed later in this review (see the RCS in the Context of Reproduction section). In addition to the enzymatic mechanisms discussed previously, RCS may also be generated via nonenzymatic pathways, as reviewed by Ayala et al. (2014) and Spickett (2013).
RCS structure and reactivity
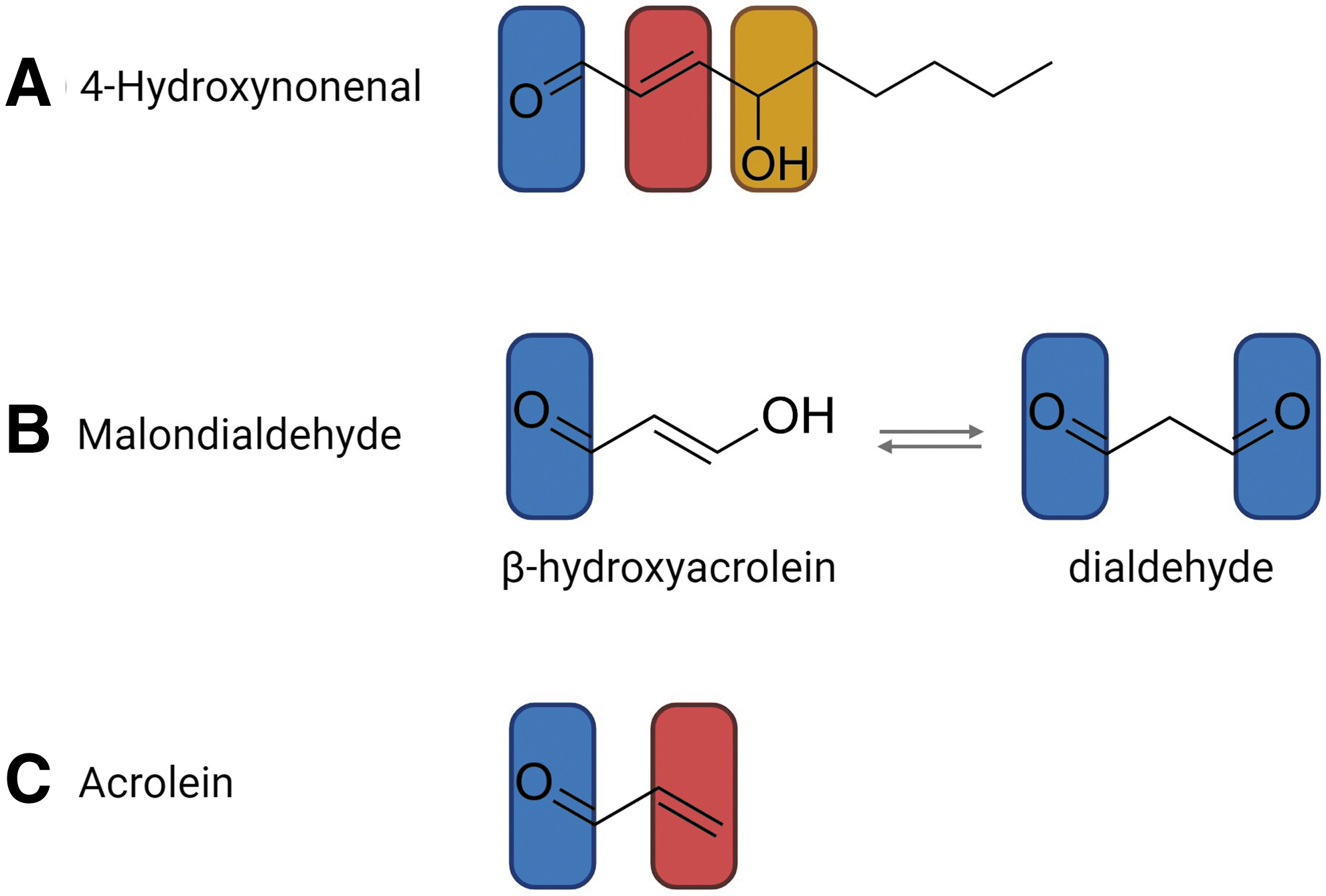
Lipid peroxidation-derived RCS are a large class of electrophilic (electron-deficient) carbonyl compounds that contain at least one hydrogen atom substituent on the carbonyl carbon atom (LoPachin and Gavin, 2014). RCS can be divided into three subclasses based on the incorporation of additional functional moieties: (i) α,β-unsaturated aldehydes such as 4HNE and ACR, (ii) keto-aldehydes such as 4-oxononenal, and (iii) dialdehydes including MDA (Hwang et al., 2016; Vistoli et al., 2013). Here, we briefly outline the structure and reactivity of 4HNE, MDA, and ACR (Fig. 3).
4HNE reactivity
4HNE is a highly toxic molecule containing three functional groups that contribute to its high reactivity (Fig. 3A). The first of these is a carbon–carbon double bond (alkene; C = C, C2/C3) that can be a target for irreversible Michael additions (protein carbonylation) or can undergo reduction or epoxidation. The second is an aldehyde (carbonyl; C = O) group, which can react with thiol substrates (organosulfur compound; R–SH) to yield an acetal/thioacetal or can be a target for Schiff base formation, oxidation or reduction. Finally, 4HNE harbors a secondary alcohol (hydroxyl group attached to a saturated carbon atom, which is bonded to two alkyl groups; R–CHOH–R′) that can be oxidized to a ketone (Ayala et al., 2014; Bilska-Wilkosz et al., 2022; Schaur, 2003; Schaur et al., 2015).
MDA reactivity
MDA is a small and highly abundant three-carbon molecule that, due to its pH-dependent tautomeric properties, exists in several different forms in aqueous solution (Esterbauer et al., 1991). At neutral pH, the dominant form is the relatively unreactive enolate anion. At lower pH, such as that induced under stress conditions, MDA exists in equilibrium between the highly reactive protonated enol aldehyde (β-hydroxyacrolein) and dialdehyde forms (Fig. 3B). The β-hydroxyacrolein form contains an α,β-unsaturated carbonyl group that can be targeted to Michael addition and Schiff base reactions (Ayala et al., 2014; Morales and Munné-Bosch, 2019; Weber et al., 2004). Contrastingly, the dialdehyde form is characterized by two carbonyl groups, joined by the common group, R–CHO, consisting of a carbonyl center bonded to hydrogen. These carbonyl groups are able to form two Schiff bases (Jové et al., 2020).
ACR reactivity
ACR (2-propen-1-al) is the simplest and most reactive of the α,β-unsaturated aldehydes (Esterbauer et al., 1991; Witz, 1989). Chemically, ACR is a type-2 alkene composed of a polarizable alkene bond (C1/C2, C = C) in close proximity to a carbonyl group (C = O) (Fig. 3C). The resultant structure forms a conjugated system that contains mobile outer shell π-electrons. Typically, alkene functional groups are electron rich. In ACR, however, the combination of π-electron mobility and electron-withdrawing capacity of the carbonyl group creates a strong electron deficiency at the terminal β-carbon (LoPachin et al., 2009). In a similar manner to the previously discussed RCS, these alkene and carbonyl functional groups can be targeted for Michael additions and Schiff base reactions, respectively.
Ultimately, these functional groups afford RCS the ability to form covalent bonds, via Michael additions and Schiff bases, with nucleophilic (electron-rich) sites on biological targets, such as DNA, lipids, and proteins. The resultant RCS-macromolecule adducts can elicit a variety of physiological and pathological effects, either through direct (impaired substrate function) or indirect (cell signaling) mechanisms. Most commonly, RCS adduction leads to the inhibition of cellular processes and eventual cytotoxicity (Beauchamp et al., 1985; Esterbauer et al., 1991; Kehrer and Biswal, 2000; LoPachin and Gavin, 2014; Witz, 1989).
Regulation of Protein Fate
Traditionally, RCS were investigated for their contributions to DNA damage and genotoxicity. However, more recently, RCS have been attributed a central role in protein dysregulation and proteotoxicity (Ayala et al., 2014; Mihalas et al., 2017). This is largely mediated by protein lipoxidation, a form of nonenzymatic post-translational modification characterized by the covalent adduction of reactive lipid species (i.e., RCS) to protein substrates (Jové et al., 2020; Pamplona, 2011; Thorpe and Baynes, 2003; Viedma-Poyatos et al., 2021). The resulting RCS-protein adducts, also termed advanced lipoxidation end products, have been shown to disrupt proteostasis through the induction of polypeptide cross-linking, structural perturbation (i.e., misfolding), and protein aggregation (Dalleau et al., 2013; Doorn and Petersen, 2002; Esterbauer et al., 1991; Mihalas et al., 2017; Pizzimenti et al., 2013; Sayre et al., 2006). Herein, this review outlines the chemistry and mechanisms underpinning 4HNE-, MDA-, and ACR-mediated protein adduction and the consequences of these modifications for protein fate.
RCS-mediated protein adduction and cross-linking
RCS preferentially attack proteins that contain strong nucleophilic sites, with the most common targets being the sulfur atom of cysteine (Cys), imidazole nitrogen of histidine (His), and the amine nitrogen of lysine (Lys) (Casini et al., 2002; Grimsrud et al., 2008; Guengerich et al., 2001; Labenski et al., 2009). It is important to note that a number of factors, including overall protein structure, cellular context, and the local pH, all contribute to the intrinsic reactivity of these amino acids. During conditions of oxidative stress and subsequent elevated lipid peroxidation, the alteration of these factors, combined with a surge in RCS production, drives an increase in the number of proteins susceptible to RCS adduction (Andringa et al., 2014).
4HNE-protein adducts
4HNE is able to modify proteins through the covalent adduction of nucleophilic amino acid residues, specifically deprotonated side chains of Cys, His, and Lys (Andringa et al., 2014; Ayala et al., 2014; Dalleau et al., 2013; Doorn and Petersen, 2002; Mihalas et al., 2017; Peters et al., 2020; Pizzimenti et al., 2013; Sayre et al., 2006; Wall et al., 2014). Depending on the functional group involved in this reaction, 4HNE-mediated protein modification can yield a variety of adducts. For instance, the addition of thiol or amino compounds on the electrophilic β-carbon (carbon of the double bond) of 4HNE produces the corresponding Michael adduct. The preference of substrates for Michael addition (from highest to lowest) is Cys>>His>Lys (Fig. 4A) (Doorn and Petersen, 2002).
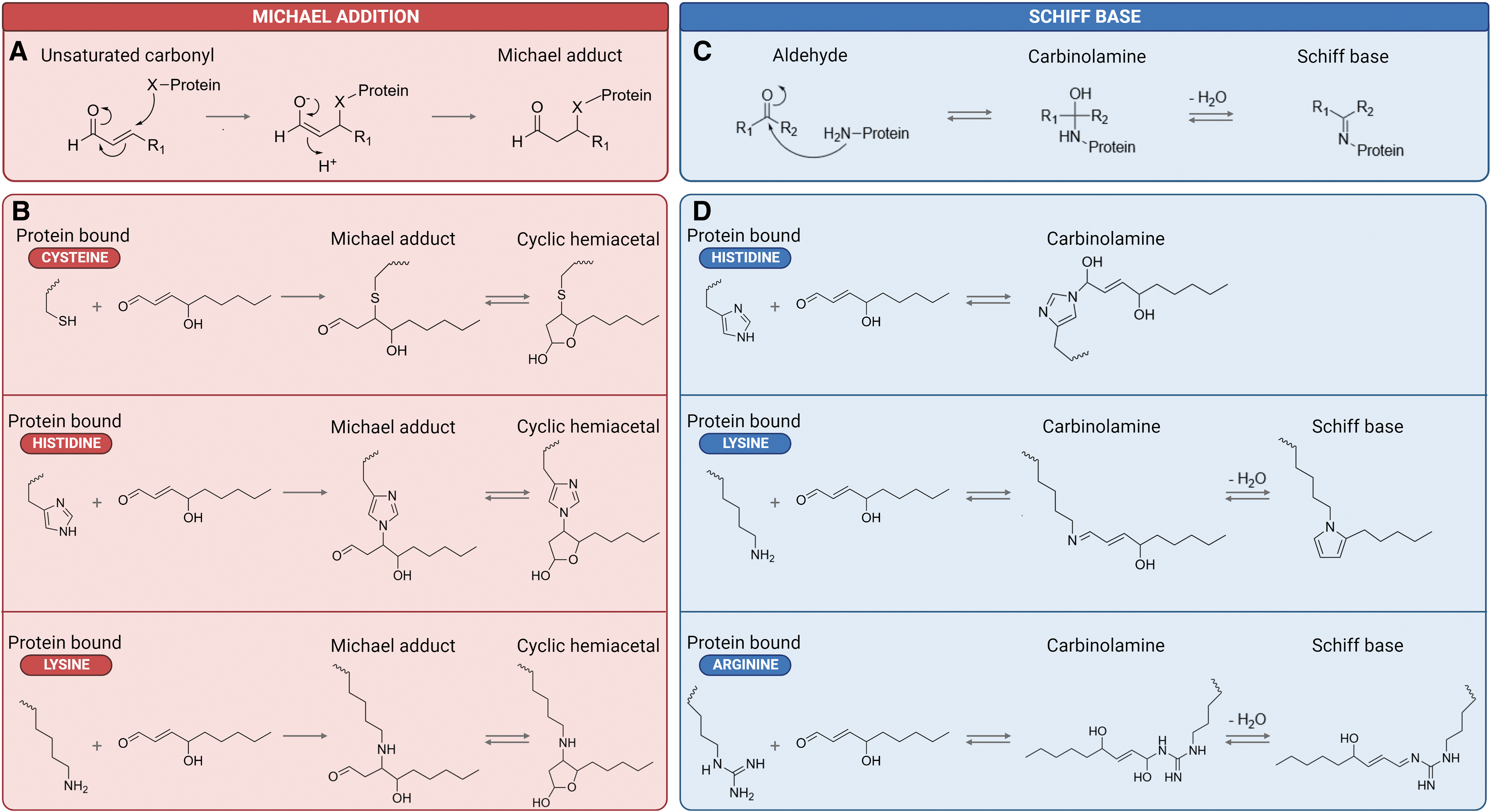
Following this primary reaction, which confers rotational freedom to the C2–C3 bond, secondary reactions involving the carbonyl and hydroxyl group may occur (Schaur, 2003). Notably, the reaction between the nitrogen atom of primary amines (i.e., amino acids) and the aldehyde group of 4HNE yields a reversible Schiff base (Fig. 4B) (Ayala et al., 2014). His, Lys, and, less commonly, arginine (Arg) act as primary targets for Schiff base formation (Andringa et al., 2014; Isom et al., 2004). Broadly, Michael adducts account for >99% of HNE protein modifications, whereas Schiff base adducts are less prevalent even in the presence of excess HNE (Bruenner et al., 1995; Rauniyar and Prokai, 2009; Uchida and Stadtman, 1992).
The formation of RCS-protein adducts is biologically significant given these compounds can participate in secondary deleterious reactions, notably, intramolecular and/or intermolecular protein cross-linking (Ayala et al., 2014; Bilska-Wilkosz et al., 2022; Petersen and Doorn, 2004). Mechanistically, 4HNE covalently cross-links proteins through sequential 1,4 Michael addition and 1,2 Schiff base reactions between residues on the same or two independent polypeptide chains (Carini et al., 2004; Cohn et al., 1996; Siegel et al., 2007). These protein cross-linking events can have detrimental consequences for protein function, cell survival, and overall organism health (see the Biological consequences of protein carbonylation section).
As mentioned previously, RCS are constantly being produced at basal concentrations as a by-product of lipid peroxidation. At these physiological levels, three major detoxification pathways are responsible for the enzymatic conversion of 4HNE into less reactive chemical species and the maintenance of steady-state concentrations (Zhong and Yin, 2015). These mechanisms involve (i) conjugation with GSH, which occurs spontaneously or can be catalyzed by glutathione S-transferases, (ii) oxidation by aldehyde dehydrogenase (ALDH), and (iii) reduction of ALDH intermediates by aldo-keto reductases (AKRs) such as alcohol dehydrogenase. The intermediate products generated by these reactions; 4HNE-GSH, hydroxynonenoic acid, and 1,4-dihydroxynonene, respectively, may be further metabolized or excreted by the cell (Fig. 6A) (Ayala et al., 2014; Castro et al., 2017; Zhong and Yin, 2015). While this detoxification initially limits the availability of 4HNE for macromolecular substrate adduction, these mechanisms become rapidly overwhelmed as the concentration of 4HNE increases. At this point, the cell enters a state of carbonyl stress.
MDA-protein adducts
Despite a relatively low reactivity toward free amino acids, MDA possesses a high affinity for the modification of peptide chains/protein-bound amino acids (Esterbauer et al., 1991). In a similar manner to 4HNE, the two distinct isomeric structures of MDA can modify proteins by (i) Michael addition at the β-carbon of the α,β-unsaturated carbonyl group in the β-hydroxyacrolein isomer or (ii) Schiff base formation at the carbonyl carbon in this isomer or in the dialdehyde (Weber et al., 2004). While the free ɛ-amine group of Lys acts as the primary target of these reactions (Esterbauer et al., 1991; Kikugawa and Beppu, 1987), other amino acid substrates including Arg, His, tyrosine, and methionine may also be altered to some extent (Chio and Tappel, 1969; Jové et al., 2020; Requena et al., 1997).
In addition to modifying nucleophilic groups, MDA can also react with acetaldehyde (a product of MDA metabolism) under conditions of oxidative stress. This reaction yields hybrid protein conjugates termed MDA acetaldehyde adducts (Tuma, 2002; Tuma et al., 2001) that have been shown to be highly immunogenic (Ayala et al., 2014; Duryee et al., 2008; Wållberg et al., 2007; Wang et al., 2012; Wang et al., 2007; Weismann and Binder, 2012).
As mentioned previously, MDA readily forms a reversible Schiff base with the side chain of Lys residues to generate the MDALys adduct; however, Lys-MDA-Lys cross-links may also be produced (Chio and Tappel, 1969; Domingues et al., 2013; Jové et al., 2020; Kikugawa and Beppu, 1987; Miyata et al., 1998; Refsgaard et al., 2000; Requena et al., 1997; Slatter et al., 1998). The molecular consequences of MDALys adduct formation mostly include alterations in physicochemical properties, such as conformation (Thorpe and Baynes, 2003), charge (Mol et al., 2019), hydrophobicity and solubility (Dalfó and Ferrer, 2008), aggregation (Dalfó and Ferrer, 2008; Requena et al., 1997; Vay et al., 2001), loss of enzymatic activity (Domingues et al., 2013; Yarian et al., 2005), and accelerated rate or resistance (in the case of Lys-MDA-Lys cross-links) to proteolysis (Mahmoodi et al., 1995). The mechanisms involved in MDALys adduct and Lys-MDA-Lys cross-link formation, as well as the cytotoxic effects of these molecules have been thoroughly reviewed by Jové et al. (2020).
ACR-protein adducts
In a similar manner to 4HNE and MDA, ACR preferentially targets the nucleophilic side chains of Cys, His, and Lys residues, as well as free N-terminal amino groups (Esterbauer et al., 1991). ACR readily reacts with these amino acid groups via two distinct mechanisms: (i) Schiff base formation at the carbonyl group or (ii) Michael addition at the terminal β-carbon (Cai et al., 2009). The retained aldehyde group in ACR-amino acid Michael addition adducts can further react with additional nucleophilic groups to form intramolecular and/or intermolecular cross-links (Burcham and Pyke, 2006).
Furthermore, evidence suggests that several molecules of ACR can react with the same residue, thus forming complex adducts such as Nɛ-(3-formyl-3,4-dehydropiperidino)lysine (FDP-Lys) (Uchida et al., 1998) and Nɛ-(3-methylpyridinium)lysine (Furuhata et al., 2003). These complex and highly stable adducts can further perpetuate the damaging effects of ACR. By way of example, FDP-Lys adducts generated in the ACR-modified protein can function as thiol-reactive electrophiles that covalently bind to the free sulfhydryl group (Cys residues) in key substrates such as GSH. Together, these data indicate that elevated production of ACR and its protein adducts during conditions of oxidative stress can further potentiate a state of redox imbalance via the depletion of GSH (Furuhata et al., 2002).
Biological consequences of protein carbonylation
RCS-mediated modification, specifically the irreversible process of protein carbonylation via Michael addition, can direct proteins toward a range of fates that have been well described in somatic cells, the most common of which is protein misfolding. There are three major fates for these misfolded proteins: (i) refolding and rescue of native structure by chaperones, (ii) degradation (clearance) by proteases leading to a loss of function, or (iii) aggregation leading to cytotoxicity and the development of disease states (Yadav et al., 2019; Yerbury et al., 2005). Broadly speaking, these cell fate decisions are determined by concentration-dependent exposure to RCS and typically depend on the substrate as well as the degree of oxidative damage it sustains (Ayala et al., 2014; Castro et al., 2017; Dalleau et al., 2013; Gonzales, 2001; Smerjac and Bizzozero, 2008). Here, we outline the dose-dependent effects of 4HNE as a driver of physiological and pathological protein fate decisions.
4HNE as a signaling molecule
Initially, proteotoxic stress, defined as the accumulation of misfolded protein species, elicits adaptive responses in cells aimed at restoring proteostasis, thus promoting cell survival during conditions of elevated stress (Brancolini and Iuliano, 2020). At low levels, 4HNE can play a physiologically significant role in the regulation of central signaling pathways (Forman et al., 2008; Jaganjac et al., 2012) and cellular processes (Shoeb et al., 2014). This is largely attributed to the activation of proteins involved in signal transduction and gene expression, including transcription factors, receptors, kinases, and phosphatases (Ayala et al., 2014; Ullery and Marnett, 2012). By way of example, nuclear factor erythroid 2-related factor 2 (NRF2) is a central transcription factor involved in the regulation of several genes that contain antioxidant-responsive elements.
Under physiological conditions, NRF2 is sequestered in the cytoplasm by the repressor protein, Kelch-like ECH-associated protein 1 (KEAP1). In the absence of excess oxidant molecules, NRF2 expression is maintained at low levels through a process of ubiquitination and proteasomal degradation. Conversely, under conditions of oxidative stress, KEAP1 is adducted by 4HNE at Cys513 and Cys518, resulting in disrupted binding of the KEAP1-NRF2 complex and reduced degradation of NRF2 (Gao et al., 2020). Adduction of additional cysteine residues in KEAP1, including Cys151, Cys288, Cys226, and Cys368, is similarly thought to be involved in 4HNE-induced activation of the NRF2 signaling pathway (Dinkova-Kostova et al., 2002; McMahon et al., 2010; Parvez et al., 2015).
Free from the inhibition of KEAP1, accumulated cytoplasmic NRF2 translocates into the nucleus and activates the expression of antioxidant-responsive element-containing genes to stimulate antioxidant (i.e., glutathione reductase, glutathione peroxidase, glutaredoxin, peroxiredoxin, and thioredoxin) and cytoprotective defenses (Fig. 5) (Chorley et al., 2012; Cyran and Zhitkovich, 2022; Schmidlin et al., 2019; Thimmulappa et al., 2002). Moreover, NRF2 activation is associated with upregulated expression of AKR and ALDH enzymes, which, as discussed earlier, are capable of detoxifying sublethal levels of 4HNE (Breitzig et al., 2016; Dalleau et al., 2013; Schaur et al., 2015).
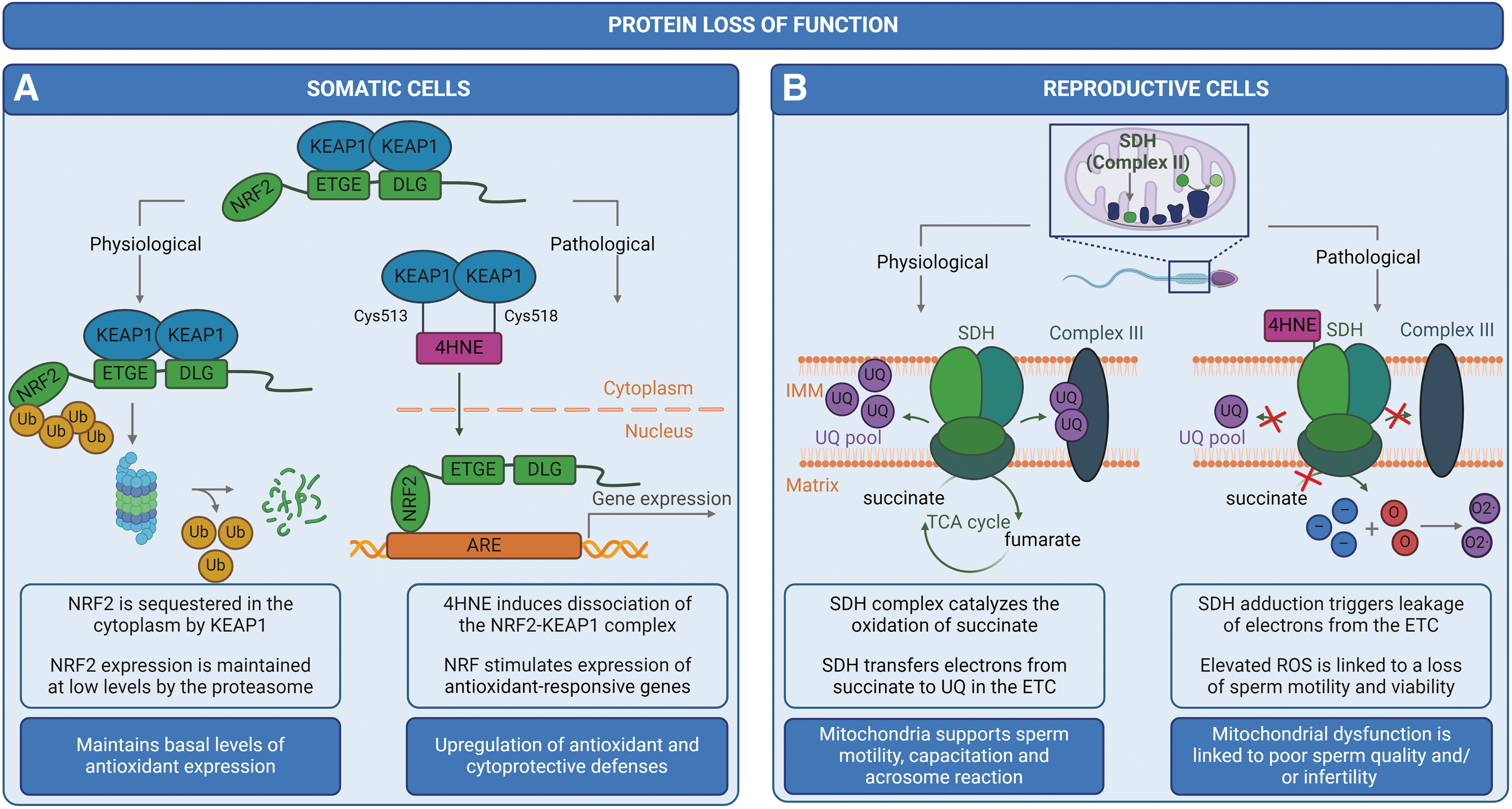
There is also evidence to suggest that NRF2 facilitates the clearance of potentially cytotoxic misfolded and aggregated proteins species through the targeted activation of ALP (Qin et al., 2019; Wang et al., 2014) and/or UPS-mediated (Pajares et al., 2017) degradation pathways. In this way, 4HNE can positively regulate metabolism and restore proteostasis (Ayala et al., 2014; Dalleau et al., 2013; Gan and Johnson, 2014; Grimsrud et al., 2008; Huang et al., 2012; Ishii et al., 2004; Levonen et al., 2004; Siow et al., 2007; Tanito et al., 2007; Zhang et al., 2010). Unfortunately, the antioxidant capacity of the cell can be rapidly depleted as the concentration and activity of 4HNE increases, thus propagating a state of proteotoxic stress.
Protein misfolding and loss of function
At moderate levels, 4HNE induces protein denaturation with consequences for secondary and tertiary structures, stability, and function (Alviz-Amador et al., 2019; Fritz et al., 2012; Perluigi et al., 2012; Smathers et al., 2012). Examples of 4HNE-induced loss of protein function in the context of somatic biology are presented in Figure 5A. These intrinsic changes typically manifest in the form of misfolded and/or nonfunctional proteins. Even a low level of misfolded proteins can be detrimental to the cell, with some studies suggesting that the presence of one metastable or misfolded protein can destabilize additional unrelated proteins within the proteome (Brettschneider et al., 2015; Gidalevitz et al., 2009; Gidalevitz et al., 2006; Taylor and Dillin, 2011). This phenomenon, termed as “bystander” mechanism, has been observed as a consequence of the high amyloid burden in neurodegenerative disorders, such as Alzheimer's disease (Xu et al., 2013).
The accumulation and subsequent aggregation of these misfolded protein species not only impair normal cellular function but may elicit cytotoxicity, ultimately leading to the induction of cell death via apoptotic, necrotic, or ferroptotic pathways (Ayala et al., 2014; Haeri and Knox, 2012). To combat the accumulation of these potentially cytotoxic non-native species, key effectors of the proteostasis network are responsible for the inhibition or reversal of protein misfolding/aggregation, or alternatively, direct terminally misfolded proteins toward a range of degradation pathways (Klaips et al., 2017; Tittelmeier et al., 2020). Most notably, a diverse and ubiquitous class of proteins, termed molecular chaperones, recognizes and transiently stabilizes hydrophobic motifs exposed on the surface of misfolded proteins (Stirling et al., 2003). A mere binding activity is considered a holdase function that does not require ATP. This holdase activity temporarily stabilizes misfolded protein species, thereby preventing (i) their association with other unstable proteins, (ii) protein aggregation, and/or (iii) aberrant degradation.
However, protein folding and refolding often rely on an ATP-dependent cycle that facilitates repeated binding and release of the same or different chaperones and/or co-chaperones (Brehme et al., 2014; Bukau et al., 2000; Cho et al., 2015; Liberek et al., 2008; Mayer and Bukau, 2005; Preissler and Deuerling, 2012; Reichmann and Suss, 2015; Tittelmeier et al., 2020). Despite these investments into the maintenance of proteostasis, the accumulation of misfolded protein species is further compounded by the fact that molecular chaperones, including heat shock protein 70 (HSP70), heat shock protein 90 (HSP90), and the protein disulfide isomerase family, are directly targeted by 4HNE (Castro et al., 2017).
For example, 4HNE-mediated modification of heat shock protein A2 (HSPA2), the inducible variant of HSP70, significantly inhibits the protein refolding ability of this chaperone in vitro. This inhibitory effect has been indirectly linked to the covalent adduction of Cys267 in the ATPase domain of HSPA2 (Carbone et al., 2004). Given that the affinity of the HSP70 family for protein substrates is a function of nucleotide (i.e., ATP or ADP) presence at the ATPase domain (Fink, 1999), it follows that modification of this site can alter the state of nucleotide binding and, ultimately, reduces the affinity of HSPA2 for its substrate (Carbone et al., 2004).
In addition to directly modifying proteins, carbonyl-induced oxidative stress may also lead to ATP depletion as observed in bacterial cells subjected to HOCl stress (Hannum et al., 1995; Ulfig and Leichert, 2021). Ultimately, depletion of cellular ATP leads to the inactivation of essential proteases and ATP-dependent chaperone systems (i.e., HSP70/HSP40) that typically protect against oxidative stress-induced unfolding and protein aggregation (Khor et al., 2004; Winter et al., 2005). To any extent, the incapacitation of these remodeling pathways contributes significantly to the accumulation of misfolded and potentially cytotoxic protein species that are either degraded or go on to form aggregates.
Protein degradation
The targeted removal of carbonylated proteins can occur through several proteolytic mechanisms. While we exclusively discuss pathways associated with the UPS here, the proteostasis network also employs the ALP, which digests modified and/or misfolded proteins. The 20S ubiquitin-independent proteasome is known to catalyze the degradation of misfolded proteins based on its ability to detect exposed hydrophobic amino acids and aromatic residues (Grimsrud et al., 2008; Grune and Davies, 2003; Grune et al., 1997).
As mentioned previously, RCS adduction induces several physiochemical changes within the target protein, one of those being an increase in surface hydrophobicity (Thorpe and Baynes, 2003). This occurs when hydrophobic moieties from within the protein core are exposed on the surface via oxidative rearrangement (partial unfolding) of secondary and tertiary protein structure (Grune and Davies, 2003; Grune et al., 1997). Such changes render RCS-modified proteins more susceptible to degradation than their non-oxidized counterparts (Castro et al., 2017; Dukan et al., 2000; Grune et al., 2004; Grune et al., 2003; Huang et al., 1995; Maisonneuve et al., 2008).
Alternatively, RCS-modified proteins may also serve as targets for preferential ubiquitination (compared with their native counterparts), with this modification tagging them for degradation either by the 26S ubiquitin-dependent proteasome (Botzen and Grune, 2007) or by a lysosome and ubiquitin-dependent but proteasome-independent pathway (Marques et al., 2004). In a similar way to ubiquitination, it has been postulated that carbonylation itself may tag proteins for degradation (Dukan et al., 2000; Grune et al., 2003; Maisonneuve et al., 2008). Illustrative of this phenomenon, key examples of somatic and reproductive-related protein degradation pathways induced by 4HNE adduction are depicted in Figure 6.

To the detriment of the cell, the efficiency of proteolytic machinery rapidly declines during aging as the concentration of intracellular RCS increases, thus contributing to the accumulation of misfolded proteins and the perpetuation of proteotoxicity. This decline in proteolytic function may result from the direct modification of proteases by RCS, or, alternatively, the indirect inhibition of proteolytic enzymes by the carbonylated proteins they intend to degrade (see the Protein aggregation section) (Smerjac and Bizzozero, 2008). For instance, several studies suggest that members of the UPS, including the 20S and 26S proteosome, serve as targets for modification by 4HNE (Bardag-Gorce et al., 2005; Castro et al., 2017; Ferrington and Kapphahn, 2004; Grune and Davies, 2003; Kessova and Cederbaum, 2005; Okada et al., 1999; Vieira et al., 2000). In this way, RCS-mediated inhibition of protein degradation machinery increases the number of misfolded species available to participate in cross-linking and/or aggregation events.
Protein aggregation
While moderately carbonylated proteins are susceptible to misfolding and degradation, heavily carbonylated proteins have a propensity to form high-molecular-weight aggregates (Gonos et al., 2018; Smerjac and Bizzozero, 2008). Protein aggregates are a continuum of misfolded structures that range from cytotoxic globular oligomers, which retain some solubility, to more advanced amyloid fibrils or diffuse amorphous aggregates, which are largely insoluble (Adamcik and Mezzenga, 2018; Breydo and Uversky, 2015; Chen et al., 2017; Luo et al., 2014; Wu et al., 2022; Wu et al., 2021; Wu et al., 2020). As mentioned earlier, aggregates are capable of inhibiting the proteasome whose function is to degrade them, thus conferring resistance to proteolysis and further accelerating the rate of aggregate formation (Castro et al., 2017).
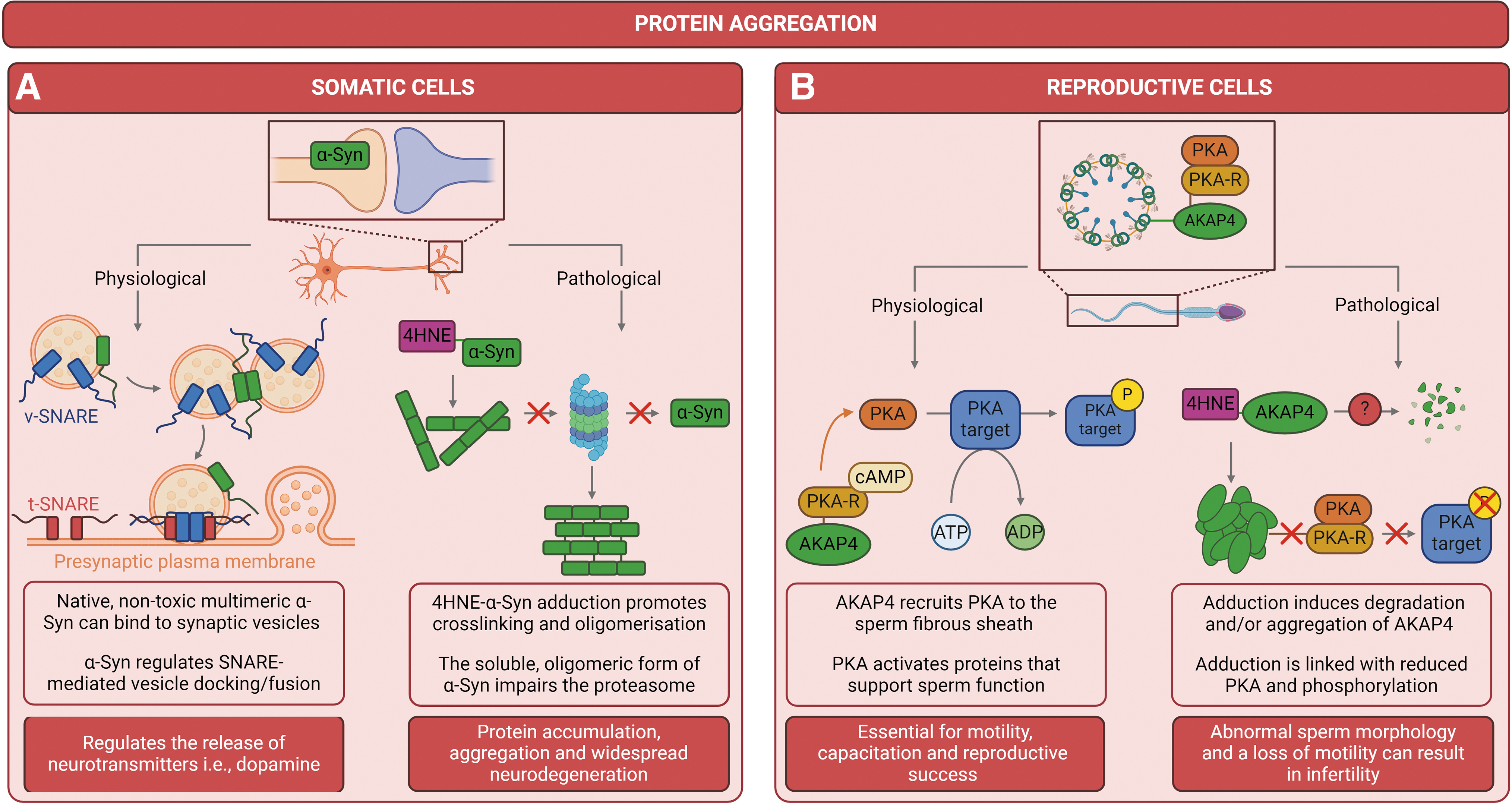
For instance, 4HNE modification of neuronal proteins, such as amyloid beta peptide (Aβ) and α-synuclein (α-Syn), can promote intermolecular cross-linking, oligomerization, and the subsequent formation of protofibrillar aggregates that can inhibit the proteasome (Bae et al., 2013; Shringarpure et al., 2000). Mechanistically, the soluble oligomeric forms of Aβ and α-Syn have been shown to inhibit the 20S proteasome through allosteric stabilization of a closed substrate gate conformation, consequently blocking substrate translocation into the catalytic degradation chamber. While the ability to degrade proteins is reduced as oligomers begin to accumulate, the level of proteasomal impairment continues to increase exponentially as more proteins accumulate and oligomerize (Fig. 7A) (Thibaudeau et al., 2018). The accumulation of protein oligomers and/or aggregates can perturb organelle and cellular functions, trigger cell death pathways, and ultimately, lead to a decline in the overall well-being of an organism (Iuchi et al., 2021).
In the context of human health, aberrant protein aggregation is a common feature associated with the development and/or progression of serious pathological conditions, collectively termed protein misfolding diseases (PMDs). Upward of 30 PMDs have been identified to date, including diverse systemic disorders such as type 2 diabetes mellitus (Mukherjee et al., 2015), cystic fibrosis (Fraser-Pitt and O'Neil, 2015), and certain types of cancer (Li et al., 2022). The most prevalent and increasingly common classes of PMDs are neurodegenerative proteinopathies (i.e., Alzheimer's disease, Parkinson's disease, and Creutzfeldt–Jakob disease) that arise from protein misfolding/aggregation in the central nervous system.
It is important to note that PMDs are typically defined by the misfolding of one or several distinct aggregation-prone protein(s). For example, neuropathological protein deposits are predominantly composed of Aβ and tau in Alzheimer's disease (Hardy and Higgins, 1992; Iqbal et al., 2005; Tiwari et al., 2019), α-Syn in Parkinson's disease (Atik et al., 2016; Stefanis, 2012; Yoo et al., 2023), and TAR DNA-binding protein 43 in multiple disorders such as frontotemporal dementia and amyotrophic lateral sclerosis (Jo et al., 2020; Steinacker et al., 2019). Identification of the individual protein(s) responsible for these disorders makes it easier to track disease and develop target-based treatments.
RCS in the Context of Reproduction
While the contribution of RCS to disrupted proteostasis is a relatively new concept in biology, critical research in this area has been forged by those in the field of neurodegeneration. Neurons are extremely long-lived and nondividing cells that are required to maintain proteostasis over the entire life course. This renders neurons inherently vulnerable to damage by RCS during aging and redox stress, and accordingly, neuronal cell lines have been central to the study of stress-induced protein aggregation (Barnham et al., 2004; Kurtishi et al., 2019). The importance of maintaining neuronal proteome integrity is eloquently illustrated by the fact that most neurodegenerative disorders, as mentioned earlier, are characterized by the accumulation of abnormal aggregates in the form of soluble oligomers, fibrils, and large insoluble protein inclusions (Hetz, 2021; Petrovic et al., 2020; Soto and Pritzkow, 2018; Yerbury et al., 2016).
In addition to neurons, reproductive cells also provide a unique cellular context to build fundamental knowledge of RCS-induced protein damage. Considering that the mature male gamete possesses a severely limited antioxidant capacity and has no ability to enact transcription or translation, it must solely rely on the proteostasis network for survival and the maintenance of protein quality (Cafe et al., 2021; Sala and Morimoto, 2022). Similarly, the female gamete is a long-lived cell that is subjected to prolonged periods of arrest (up to decades) and phases of transcriptional and translational quiescence that place immense pressure on protein networks (Peters et al., 2020; Sala and Morimoto, 2022). Distinctively, in the case of both gametes, the maintenance of proteostasis throughout germ cell life is central to the preservation of genomic fidelity and the fitness of the next generation.
Within this cellular context, a growing body of research suggests that RCS, specifically 4HNE, can directly and indirectly regulate protein homeostasis in the male germline (Aitken et al., 2012b; Baker et al., 2015; Bromfield et al., 2017a; Bromfield et al., 2015; Cafe et al., 2020; Mihalas et al., 2017; Moazamian et al., 2015; Nixon et al., 2019). This is further impactful in mature spermatozoa that are particularly vulnerable to RCS-induced modification as they (i) rely heavily on post-translational modifications (i.e., tyrosine phosphorylation) to acquire functional competence during maturation within the epididymis and the female reproductive tract and (ii) contain a high proportion of ω-3 and ω-6 PUFAs that serve as substrates for oxidative attack, thus yielding high concentrations of 4HNE (Aitken et al., 2012b; Bromfield et al., 2017a; Gautier and Aurich, 2022; Jones et al., 1979; Rao et al., 1989; Skerrett-Byrne et al., 2022). Here, we summarize current data surrounding 4HNE-induced protein dysfunction in the male germline.
4HNE-mediated protein modification
As in somatic cells, the generation of excess 4HNE during aging and conditions of oxidative stress can have detrimental consequences for male germline proteins. Most notably, 4HNE-mediated protein adduction has been shown to alter sperm function, with an observable decline in motility (Nowicka-Bauer and Nixon, 2020) and normal morphology, reduced capacity to undergo the acrosome reaction, and an impaired ability to interact with the zona pellucida of an oocyte (Aitken et al., 2012a, 2012b; Bromfield et al., 2015). This is largely attributed to the ability of 4HNE to modify a subset of vulnerable targets within the germ cell and/or sperm proteome. Among these, prevalent targets include succinate dehydrogenase (SDH) (Aitken et al., 2012b), HSPA2 (Bromfield et al., 2015), and A-kinase anchor protein 4 (AKAP4) (Nixon et al., 2019). With many potential targets of 4HNE modification in the male and female germlines still unexplored, the subsequent sections discuss the impact of 4HNE on its well-characterized targets within male reproductive cells.
SDH-4HNE adduction
Mitochondria play a fundamental role in energy production, redox equilibrium, calcium regulation, and apoptotic pathways, all of which are necessary to support sperm motility, capacitation, hyperactivation, and the acrosome reaction, as well as oocyte fusion and penetration (Boguenet et al., 2021). Given the aforementioned functions, it follows that mitochondrial dysfunction is linked to a decline in sperm quality and/or infertility (Durairajanayagam et al., 2021). In fact, recent comparative proteomic studies indicate that proteins associated with mitochondrial oxidative phosphorylation (OXPHOS) and the tricarboxylic acid (TCA) cycle are dysregulated in sperm samples from asthenozoospermia patients (characterized by a reduced or complete absence of motility) compared with fertile men (Amaral et al., 2014; Nowicka-Bauer et al., 2018).
Among the identified mitochondrial proteins, SDH activity has been positively correlated with sperm function and fertility (Woodhouse et al., 2022). Located in the inner mitochondrial membrane, the SDH enzyme complex plays a unique dual role in respiration, (i) catalyzing the oxidation of succinate into fumarate in the TCA cycle (Cecchini, 2003; Miyadera et al., 2003) and (ii) transferring electrons from succinate to ubiquinone in the electron transport chain as part of the OXPHOS pathway (Cecchini, 2003; Miyadera et al., 2003; Sun et al., 2005).
During conditions of elevated oxidative stress, 4HNE adducts to SDH proteins localized within the mitochondria-rich midpiece of the sperm flagellum (Aitken et al., 2012b). This finding is consistent with previous studies (in rodents) that have identified mitochondrial SDH as a target for 4HNE modification in the central nervous system (Picklo et al., 1999) and the diabetic heart (Lashin et al., 2006). This indicates that SDH proteins may be conserved targets of 4HNE adduction in the body, and their adduction may be an early indication of pathology. In human spermatozoa, 4HNE-SDH adduction results in protein loss of function and widespread mitochondrial dysfunction.
Notably, 4HNE-mediated disruption of SDH activity facilitates the leakage of electrons from the electron transport chain, subsequently increasing ROS production and activating apoptotic pathways, thus perpetuating a state of oxidative stress (Fig. 5B). The cascade of events that follow 4HNE-SDH adduction can be characterized by a rapid loss of mitochondrial membrane potential, impaired motility, oxidative DNA damage and fragmentation, as well as a profound loss of cell viability (Aitken et al., 2012b), all of which are associated with infertility phenotypes and poor offspring outcomes (Aitken and De Iuliis, 2010; Aitken et al., 2011; Espinoza et al., 2009; Koppers et al., 2008; Mitchell et al., 2011).
HSPA2-4HNE adduction
HSPA2 is a testis-enriched member of the HSP70 chaperone family implicated in a variety of cellular processes including protection of the proteome from stress (Filipczak et al., 2012), protein (re)folding and disaggregation (Mayer and Bukau, 2005), transmembrane transport of client proteins (Dun et al., 2012, Huszar et al., 2000) and the assembly/dissociation of multimeric protein complexes (Redgrove et al., 2011). Within the human testis, HSPA2 is expressed at two discrete stages; (1) during meiosis as a component of the synaptonemal complex, and (2) during spermiogenesis/sperm maturation, where it is considered to provide a robust discriminative index of fertilizing potential (Ergur et al., 2002, Huszar et al., 2006, Huszar et al., 2000, Motiei et al., 2013). In addition to its generic chaperoning roles, HSPA2 is also involved in several testis-specific functions, including regulation of germ cell differentiation (Eddy, 1999), the replacement of histones by protamines during nuclear compaction (Govin et al., 2006) and priming of the sperm surface for zona pellucida recognition and binding (Bromfield et al., 2015, Nixon et al., 2015, Redgrove et al., 2011, Redgrove et al., 2013, Redgrove et al., 2012).
Defective sperm-zona pellucida interaction is a major contributor to male infertility (Liu and Baker, 2003). Studies by Redgrove et al. (2012) revealed that in human spermatozoa these interactions are largely mediated by the regulated expression of multimeric zona pellucida-receptor complexes on the sperm surface. The most extensively studied of these complexes is constituted by; (1) sperm adhesion molecule 1 (SPAM1), a hyaluronidase primarily involved in cumulus cell matrix dispersal (Kimura et al., 2009, Lathrop et al., 1990), (2) arylsulfatase A (ARSA), an enzyme implicated in adhesion to sulphated ligands adorning the zona pellucida (Carmona et al., 2002, Tantibhedhyangkul et al., 2002), and (3) HSPA2 which coordinates receptor complex formation and remodeling during capacitation (Redgrove et al., 2012).
Given this regulatory role, it follows that 4HNE-mediated modification of HSPA2 causes a reduction in human sperm–egg interaction in vitro, likely due to the attenuation of HSPA2 chaperoning activity and the resultant failure to correctly position oocyte receptors (i.e. SPAM1 and ARSA) on the anterior surface of the sperm head (Bromfield et al., 2015). Furthermore, 4HNE-adduction is correlated with HSPA2 ubiquitination and subsequent degradation through proteasomal pathways, thus contributing to a significant reduction in HSPA2 abundance in developing germ cells (Fig. 6B). Ultimately, dysregulation of HSPA2 is associated with oligozoospermia (low sperm count) (Cedenho et al., 2006, Lima et al., 2006), poor sperm quality (Ayaz et al., 2018) and reduced levels of zona binding (Huszar et al., 2000), as well as impaired fertility and adverse pregnancy outcomes following in vitro fertilisation (IVF) or intracytoplasmic sperm injection (ICSI) (Ergur et al., 2002, Huszar et al., 1992).
AKAP4-4HNE adduction
AKAP4 is the most abundant constitutive protein of the sperm fibrous sheath (Brown et al., 2003), a unique cytoskeletal structure surrounding the axoneme and outer dense fibers that extend along the principle-piece of the flagellum (Eddy et al., 2003). Functionally, AKAP4 is responsible for recruiting cAMP-dependent protein kinase A (PKA) to the fibrous sheath (Miki et al., 2002) and acts as a subcellular scaffold to tether PKA within the immediate proximity of its enzymatic substrates (Brown et al., 2003). In this way, AKAP4-coordinated compartmentalization of PKA serves to regulate the specificity of signal transduction pathways and metabolic processes that support the development, activation, and maintenance of sperm motility and capacitation (Luconi et al., 2011; Sergeant et al., 2019).
The importance of AKAP4 for reproductive success is eloquently highlighted by gene ablation studies in which male mice lacking precursor (proAKAP4) and mature AKAP4 expression are rendered infertile due to abnormal sperm morphology (i.e., aberrant fibrous sheath formation and a shorted flagella) and a loss of progressive motility (Fang et al., 2019; Miki et al., 2002). Recent studies have reported the application of affinity-based isolation techniques coupled with mass spectrometry to characterize the 4HNE adductome of oxidatively stressed human spermatozoa (Baker et al., 2015). Among the dominant targets identified was AKAP4, with these data revealing at least three AKAP4 peptides that harbor either one or two 4HNE-modified residues following exogenous treatment with the aldehyde (Baker et al., 2015). Further validation of these data confirmed that both the precursor and mature forms of AKAP4 are conserved targets for 4HNE adduction in post-meiotic germ cells (round spermatids) and mature mouse and human spermatozoa (Nixon et al., 2019).
Evidently, 4HNE modification is associated with a substantial reduction in proAKAP4 and AKAP4 levels, although this loss appears to be independent of conventional proteolytic degradation pathways. A concomitant increase in the degree of proAKAP4 and AKAP4 aggregation was similarly observed in germ cells following 4HNE treatment. While the mechanisms are not yet fully understood, it has been theorized that 4HNE-induced aggregation of these proteins may reduce their activity and thus affect the efficacy with which they are recovered from cell lysates (Nixon et al., 2019).
As in somatic cells, RCS-mediated protein modification in the germline can have detrimental consequences for both protein and cell function. Taking proAKAP4 and AKAP4 for example, Nixon et al. (2019) observed that 4HNE-induced aggregation and/or nonclassical degradation of these proteins is correlated with reduced levels of PKA and tyrosine kinase phosphorylation in mature human spermatozoa. These data suggest that 4HNE can dysregulate capacitation-associated phosphorylation, potentially through attenuation of the signaling framework assembled around the AKAP4 scaffold.
Downstream of these signaling events, 4HNE negatively impacts the motility profile of capacitated human spermatozoa (i.e., altered velocity parameters and reduced progressive motility) as well as the ability of these cells to undergo the acrosome reaction (Fig. 7B) (Nixon et al., 2019). As a caveat for the interpretation of these data, it is important to consider that alternate proteins involved in capacitation signaling (PKA), motility (dynein, outer dense fiber protein 1), and membrane remodeling (HSPA2) can also serve as primary targets for 4HNE modification (Baker et al., 2015). The broad-spectrum of 4HNE targets makes it difficult to differentiate the downstream consequences of proAKAP4 and AKAP4 adduction from those elicited by the modification of other proteins.
Future Directions and Perspectives
Despite growing knowledge in this field, we are yet to fully characterize the complete RCS adductome in the male germline, and there are consequently several key questions remaining (Fig. 8), including: (i) Which proteins comprise the proteostasis network of germline cells and how do they compare to somatic cell proteostasis components? (ii) Which germline proteins are targeted for modification by RCS? What function do these proteins possess and why are they targeted? (iii) Are RCS-modified germline proteins preferentially degraded or aggregated? (iv) Is there potential to mitigate RCS-induced proteotoxicity using novel therapies? Regarding the latter question, developments in our understanding of RCS production and metabolism have led to recent innovations for their mitigation. Notably, several RCS-trapping drugs are under development for the treatment of somatic conditions, such as dry eye disease, that might also be applicable for preventing RCS-induced damage in the germline.

Reproxalap (previously termed ADX 102 or NS-2) is a small molecular inhibitor [2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol] now being developed by Aldeyra Therapeutics to prevent the production of reactive aldehyde species (RASP; including many RCS). Patients with dry eye disease have been found to have elevated levels of MDA and downstream inflammation (Augustin et al., 1995; Choi et al., 2016). As such, RASP have been named as an objective sign of dry eye disease since 2020. Moreover, aqueous-deficient dry eye is likely to be a protein conformational disease with a significant fraction of the tear proteome impacted structurally (Azharuddin et al., 2015; Ramos-Llorca et al., 2022). Thus, inhibition of RASP may improve outcomes for patients through the mitigation of multiple pathways featuring RCS. Reproxalap has demonstrated activity in phase 2/3 trials in noninfectious anterior uveitis, dry eye disease, and allergic conjunctivitis (Cavanagh et al., 2022; Clark et al., 2022; Clark et al., 2021a; Clark et al., 2021b; Clark et al., 2021c; Mandell et al., 2020) and has recently received acceptance as a new drug application by the Food and Drug Administration (February 2023).
While this drug has so far been successful when applied topically to the eye, proof-of-concept studies for its use as a cellular inhibitor of RASP were supplied in a model of lung injury derived from cigarette smoke. Specifically, it was demonstrated that reproxalap (termed here as ADX-102) dose dependently protected against smoke- and alcohol-induced cilia slowing, decreases in bronchial epithelial wound repair, and decreases in epithelial monolayer resistance without showing cellular toxicity (up to 100 μM) (Ochoa et al., 2022). These data indicate that reproxalap and other drugs that prevent the production and/or action of RASP may find widespread applications in RCS-specific conditions, such as male infertility.
Concluding Remarks
Taken together, the data discussed in this review highlight the susceptibility of male germline proteins to damage by 4HNE and the detrimental consequences of RCS-mediated post-translational protein modification for overall sperm quality and/or male fertility. While we exclusively discuss protein damage in this review, it is important to mention that 4HNE-DNA adduction has also been shown to contribute to poor fertility outcomes (Aitken, 2020; Aitken et al., 2013), and it is likely that these mechanisms work in unison. In a similar manner to being able to track specific disease-causing proteins in neurons that precede neurological disorders, increased depth of knowledge regarding proteinopathies in the male germline may reveal a subset of putative targets for the development of novel therapies to improve fertility and pregnancy outcomes for future generations.
Footnotes
Acknowledgments
The authors gratefully acknowledge Dr. Jessica Walters, Distinguished Emeritus Professor John Aitken, and Ms. Shenae Cafe for their contributions to the ideas discussed within this article.
Authors' Contributions
S.P.S.: Conceptualization, data curation, visualization, writing—original draft preparation. B.N.: Supervision, writing—reviewing and editing. D.A.S-B.: Supervision, writing—reviewing and editing. N.D.B.: Writing—reviewing and editing. E.G.B.: Conceptualization, writing—reviewing and editing, visualization, supervision, funding acquisition.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by an Australian Research Council Discovery Early Career Researcher Award to Elizabeth G. Bromfield (DE210100103), and funding support to Elizabeth G. Bromfield from the College of Engineering, Science and Environment, The University of Newcastle, Australia.