
Abstract
Aims:
Increasing nicotinamide adenine dinucleotide (NAD+) availability has been proposed as a therapeutic approach to prevent neurodegeneration in amyotrophic lateral sclerosis (ALS). Accordingly, NAD+ precursor supplementation appears to exert neuroprotective effects in ALS patients and mouse models. The mechanisms mediating neuroprotection remain uncertain but could involve changes in multiple cell types. We investigated a potential direct effect of the NAD+ precursor nicotinamide mononucleotide (NMN) on the health of cultured induced pluripotent stem cell (iPSC)-derived human motor neurons and in motor neurons isolated from two ALS mouse models, that is, mice overexpressing wild-type transactive response DNA binding protein-43 (TDP-43) or the ALS-linked human superoxide dismutase 1 with the G93A mutation (hSOD1G93A).
Results:
NMN treatment increased the complexity of neuronal processes in motor neurons isolated from both mouse models and in iPSC-derived human motor neurons. In addition, NMN prevented neuronal death induced by trophic factor deprivation. In mouse and human motor neurons expressing ALS-linked mutant superoxide dismutase 1, NMN induced an increase in glutathione levels, but this effect was not observed in nontransgenic or TDP-43 overexpressing motor neurons. In contrast, NMN treatment normalized the TDP-43 cytoplasmic mislocalization induced by its overexpression.
Innovation:
NMN can directly act on motor neurons to increase the growth and complexity of neuronal processes and prevent the death induced by trophic factor deprivation.
Conclusion:
Our results support a direct beneficial effect of NAD+ precursor supplementation on the maintenance of the neuritic arbor in motor neurons. Importantly, this was observed in motor neurons isolated from two different ALS models, with and without involvement of TDP-43 pathology, supporting its therapeutic potential in sporadic and familial ALS. Antioxid. Redox Signal. 41, 573–589.
Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration of both upper and lower motor neurons, leading to muscle atrophy, weakness, fasciculations, and spasticity (van Es et al., 2017). However, the disease presents a high degree of heterogeneity with up to 50% of patients with ALS revealing a more extensive involvement of the central nervous system (CNS), including the development of behavioral changes and cognitive impairment (van Es et al., 2017). The clinical heterogeneity of the disease is also evident regarding the site of onset (bulbar or spinal), degree of upper and lower motor neuron involvement, and progression rate (Goyal et al., 2020). Most ALS cases are sporadic, often referred to as SALS, and only about 10% of the cases present with a familial history (familial ALS, FALS) (Ghasemi and Brown, 2018). Multiple ALS-associated genes have been identified, but mutations in C9orf72 (open reading frame 72 in chromosome 9), SOD1 (superoxide dismutase 1), TARDBP (transactive response DNA binding protein-43; TDP-43), and FUS (fused in sarcoma) account for most familial cases in populations of European ancestry (Hardiman et al., 2017, Renton et al., 2014). Although mutations in TDP-43 account for about 4% of FALS cases, TDP-43 pathology is observed in most SALS and FALS cases, except for FUS- or SOD1-linked FALS cases (Blair et al., 2010, Mackenzie et al., 2010, Mackenzie et al., 2007). SOD1 was the first ALS-linked gene identified (Rosen et al., 1993). TDP-43 pathology is also observed in more than half of patients with fronto-temporal dementia (FTD), and it has been shown that about 10%–15% of FTD patients develop motor neuron disease (FTD/ALS), whereas 20%–50% of patients with ALS are diagnosed with probable or definite FTD (Kiernan et al., 2011). Most of the mechanistic understanding of ALS pFathology emerged from the study of rodent models overexpressing ALS-linked mutant human SOD1s, including hSOD1G93A, which develop a progressive motor neuron disease that recapitulates key pathological hallmarks observed in patients with ALS (Vinsant et al., 2013a, Vinsant et al., 2013b).
Innovation
Increasing NAD+ availability has been proposed as a therapeutic approach for ALS. The mechanisms mediating neuroprotection remain elusive. We show that the NAD+ precursor NMN can directly act on motor neurons to increase the growth and complexity of neuronal processes and prevent the death induced by trophic factor deprivation. NMN supplementation exerts neuroprotection in iPSCderived human motor neurons and in motor neurons isolated from two models of ALS, with and without involvement of TDP-43 pathology, indicating that NMN neuroprotection is not restricted to FALS cases and could be observed in SALS cases displaying TDP-43 pathology (Fig. 1).
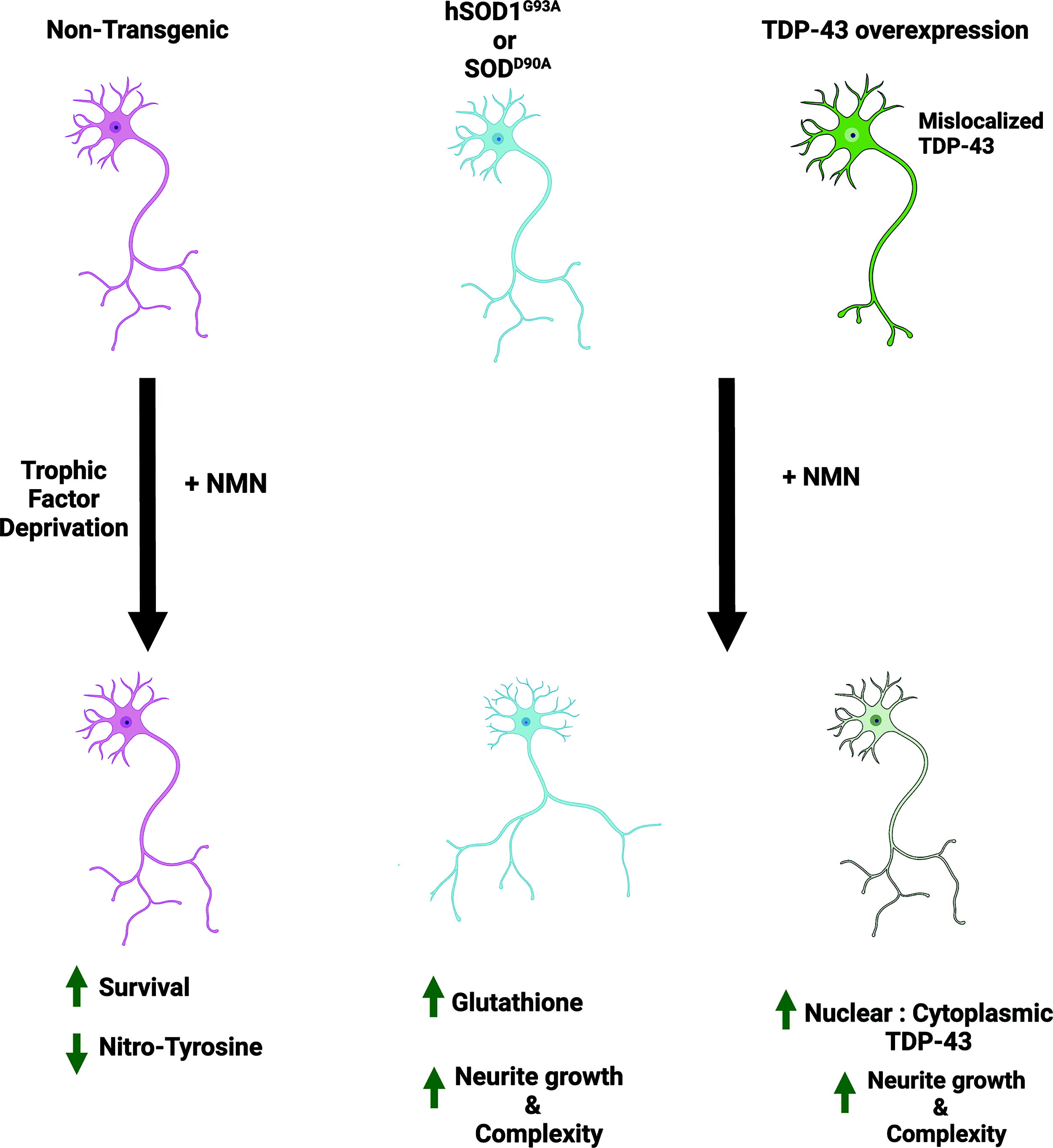
Various molecular mechanisms have been implicated in the motor neuron degeneration observed in ALS, including protein misfolding and aggregation, impaired RNA trafficking/metabolism, and altered cytoskeletal dynamics. These mechanisms lead to mitochondrial dysfunction, oxidative stress, altered proteostasis, axonal transport defects, altered dendrite morphology, and excitotoxicity (Ghasemi and Brown, 2018). In addition, although ALS-linked genes are expressed by motor neurons, the disease involves a non-cell-autonomous process that requires dysfunction of neighboring glial cells and affects disease progression (Ditsworth et al., 2017, Ilieva et al., 2009, Pehar et al., 2017).
No cure or effective treatment is currently available for ALS, although three drugs (riluzole, edaravone, and a combination of sodium phenylbutyrate/taurursodiol) slightly slow disease progression (Bensimon et al., 1994, Paganoni et al., 2020, Sawada, 2017). Thus, the development of novel therapeutic approaches is of utmost relevance. Increasing the availability of nicotinamide adenine dinucleotide (NAD+) has been proposed as a therapeutic approach to prevent neurodegeneration in aging and pathological conditions, including ALS (Hou et al., 2019, Pehar et al., 2018). In addition to its participation in redox reactions, NAD+ is part of key signaling pathways. Although redox reactions do not involve a net consumption of the dinucleotide, just a switch between the reduced (NADH) and the oxidized (NAD+) form, NAD+-signaling processes lead to the degradation of the dinucleotide (Canto et al., 2015, Pehar et al., 2018). Three different families of enzymes participate in NAD+-signaling processes, including poly (adenosine diphosphate-ribose) polymerases (PARPs), adenosine diphosphate (ADP)-ribosyl cyclases, and sirtuins. The consumption of NAD+ by these enzymes is coupled to alterations in posttranslational modifications of proteins (PARPs and sirtuins) or the production of second messengers (ADP-ribosyl cyclases) (Pehar et al., 2018). Different approaches have been used to increase cellular NAD+ availability in the CNS, including (i) inhibition of NAD+ consuming enzymes (PARPs and CD38) or (ii) increasing NAD+ biosynthesis, by supplementation with NAD+ precursors (nicotinamide [NAM], nicotinamide mononucleotide (NMN) or nicotinamide riboside [NR]) or by upregulating the rate-limiting enzyme in the salvage biosynthetic pathway (nicotinamide phosphoribosyl transferase; NAMPT) (Canto, 2022, Pehar et al., 2018). Importantly, increasing NAD+ availability by treatment with NMN or NR, or by NAMPT overexpression, reverts the toxicity of ALS astrocytes toward co-cultured motor neurons (Harlan et al., 2016). In addition, in hSOD1G93A overexpressing mice, NR supplementation delays motor neuron degeneration and decreases gliosis and inflammatory markers in the spinal cord, which is reflected in a modest extension in life span (Harlan et al., 2020). The mechanisms mediating the observed neuroprotection in vivo remain uncertain. Although the neuroprotective effects of NR supplementation could be explained by its ability to revert astrocyte-mediated neurotoxicity (Harlan et al., 2016), a direct neuroprotective effect of increasing NAD+ availability in the neuronal compartment is also plausible. We hypothesize that NAD+ precursor supplementation could have a direct neuroprotective effect in motor neurons. Thus, we investigated the effect of NMN supplementation on the health of cultured motor neurons isolated from the spinal cord of two different ALS mouse models, that is, mice overexpressing wild-type TDP-43 and mice overexpressing the ALS-linked mutant hSOD1G93A. We observed that NMN supplementation increases the growth and complexity of neuronal processes in motor neurons isolated from both mouse models. A similar result was observed in induced pluripotent stem cell (iPSC)-derived human motor neurons. In addition, NMN treatment supports motor neuron survival in the absence of trophic factors and corrects the cytoplasmic mislocalization of TDP-43 induced by its overexpression. Together, our results support a direct neuroprotective effect of increasing NAD+ availability in motor neurons, providing insight into the potential mechanisms responsible for the beneficial effects of this therapeutic approach in ALS.
Results
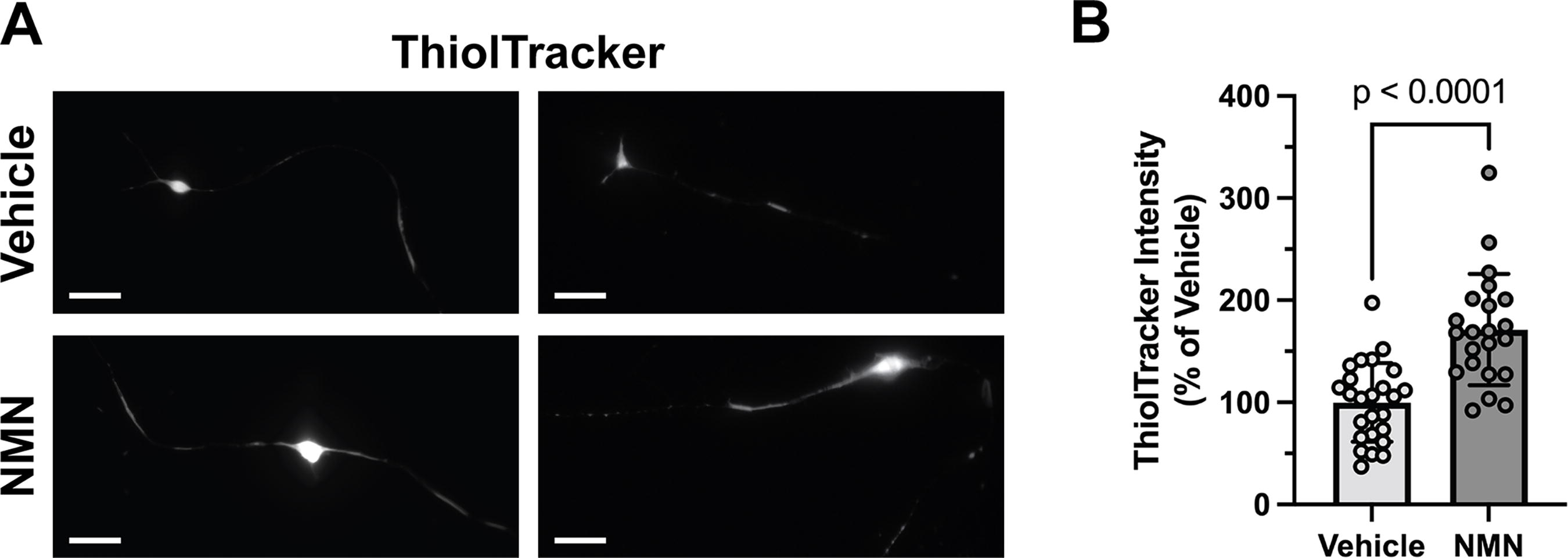
It has been shown that in different cell types and through multiple mechanisms, NAD+ availability significantly impacts the overall antioxidant defenses of the cell (reviewed in (Pehar et al., 2018)). Glutathione (γ-l-glutamyl-l-cysteinyl-glycine, GSH) is the main non-protein thiol and the most abundant antioxidant in mammalian cells (Dringen et al., 2000). Maintenance of GSH levels in its reduced form regulates the sensitivity of motor neurons to apoptotic stimuli and alters disease progression in SOD1-linked ALS mouse models (Pehar et al., 2007, Vargas et al., 2011, Vargas et al., 2008). We observed that treatment with the NAD+ precursor NMN increases the levels of reduced GSH in motor neurons isolated from mice overexpressing mutant hSOD1G93A. Owing to the limited yield of primary motor neuron culture preparations, we estimated the levels of reduced GSH using the thiol-reactive fluorescent probe, ThiolTracker™ Violet. Reduced GSH constitutes the vast majority of free thiols in the cell; thus, this probe has been commonly used as an indicator of intracellular reduced GSH (Mandavilli and Janes, 2010). Confirmation of the probe’s ability to detect changes in GSH content in motor neurons is shown in Supplementary Figure S1. We observed an increase of ∼71% in the intensity of the thiol-reactive probe in hSOD1G93A motor neurons 48 h after NMN treatment (Fig. 2).
Treatment with NMN also appears to affect the morphology of cultured hSOD1G93A motor neurons. Although we did not observe differences between nontransgenic and hSOD1G93A motor neurons treated with vehicle, after treatment with NMN, hSOD1G93A motor neurons displayed an increase in total neurite length and in the complexity of neuronal processes, as determined by Sholl analysis (Fig. 3). This effect of NMN treatment was not observed in nontransgenic motor neurons (Fig. 3). Importantly, no difference in soma size were observed after treatment. Thus, a similar result was obtained after correcting total neurite length by soma size (Supplementary Fig. S2).

Motor neuron survival in vivo and in cell culture depends on the presence of trophic factors (Oppenheim, 1996). Multiple growth factors, neurotrophins, and cytokines have been shown to support motor neuron survival in vitro and in vivo, including glial cell-line-derived neurotrophic factor (GDNF) (Henderson et al., 1994) and brain-derived neurotrophic factor (BDNF) (Henderson et al., 1993). In the absence of exogenously added trophic factors, about 45% of the motor neurons cultured in B27-supplemented Neurobasal media (see Methods section) are lost during the first 2 days in vitro (Fig. 4). Trophic factor deprivation induces motor neuron apoptosis through a pathway involving increased production of nitric oxide and the formation of peroxynitrite and can be prevented by increasing antioxidant defenses in motor neurons (Estevez et al., 2000, Estevez et al., 1998). GSH plays a key role in limiting the deleterious effects of nitric oxide in the nervous system (Heales et al., 1999). Thus, we evaluated whether the increase in GSH levels induced by NMN treatment was sufficient to prevent motor neuron death in cultures maintained without exogenously added trophic factors. We observed that NMN treatment prevented the death of hSOD1G93A motor neurons induced by trophic factor deprivation (Fig. 4A). Although treatment with 2 mM NMN did not affect the survival of hSOD1G93A motor neurons cultured in the presence of GDNF (1 ng/mL), it significantly increased the survival in cultures maintained in the absence of trophic factors, to a value not statistically different from control conditions. NMN still exerted a protective effect at concentrations of 1 mM, but motor neuron survival was significantly lower when compared with cultures maintained with GDNF. At lower concentrations (0.1 mM), the neuroprotection was lost (Fig. 4A). A similar neuroprotective effect was observed in motor neurons isolated from nontransgenic animals, although in this case, concentrations of 1 mM were sufficient to increase motor neuron survival to a level not significantly different from control cultures maintained in the presence of GDNF (Fig. 4B). Again, this neuroprotection was not observed at lower concentrations (Fig. 4B). This is in agreement with previous reports showing that NMN requires millimolar concentrations to exert a maximal effect at increasing NAD+ levels in different mammalian cells in culture (Harlan et al., 2016, Yang et al., 2019).

As mentioned above, the death of nontransgenic motor neurons in response to trophic factor deprivation occurs through a pathway involving increased production of nitric oxide and superoxide, the formation of peroxynitrite, and the subsequent increase in protein tyrosine nitration (Estevez et al., 1998). Treatment with NMN (2 mM) prevented the increase in nitrotyrosine immunoreactivity induced by trophic factor deprivation (Fig. 5).

To further confirm the relevance of the results obtained in primary mouse motor neurons overexpressing mutant ALS-linked hSOD1, we evaluated the effect of NMN on human motor neurons differentiated from iPSCs derived from a patient with ALS carrying a mutation in the SOD1 gene (SOD1D90A). The efficiency of motor neuron differentiation in our cultures was evaluated by immunofluorescence with antibodies against β-III-Tubulin, and the motor neuron markers Islet-1 homeoprotein, Choline O-acetyltransferase (ChAT), and the homeobox protein HB9 (motor neuron and pancreas homeobox protein 1). Representative images of the immunostaining analysis are shown in Supplementary Figure S3: 60.0 ± 10.7% of the cells were β-III-Tubulin positive; of these, 99.5 ± 1.2% were ChAT positive, 100% were Islet-1 positive, and 96.5 ± 4.7% were HB9 positive. No glial fibrillary acidic protein (GFAP) expression was observed in the cultures. We confirmed that NMN (2 mM) treatment induced an approximately 2-fold increase in total NAD+ levels in these cultures (Fig. 6A). In addition, we observed that NMN treatment increased GSH content in iPSC-derived human motor neurons, as estimated by ThiolTracker Violet staining (Fig. 6B) or biochemical analysis (Tietze method; Fig. 6C). Further validating the ability of ThiolTracker Violet staining to detect changes in GSH content in motor neurons, the same magnitude of change in GSH levels was determined by both methods (∼26% increase by probe staining and ∼23% increase by biochemical analysis). Lastly, as observed in primary mouse motor neuron cultures, Sholl analysis revealed an increase in the complexity of neuronal processes in iPSC-derived human motor neurons after NMN treatment (Fig. 6D).

We also explored the effect of NMN treatment on motor neurons from another ALS mouse model not linked to SOD1 mutations. Since, as indicated above, TDP-43 pathology is observed in most SALS and FALS cases (except for SOD1-linked FALS), we decided to use a model of TDP-43 induced neurotoxicity. We isolated motor neurons from mice overexpressing wild-type TDP-43, a mouse model that develops progressive degeneration of cortical and spinal motor neurons (Wils et al., 2010). We observed that motor neurons overexpressing TDP-43 displayed decreased total neurite length and neurite complexity when compared with motor neurons isolated from nontransgenic littermates (Fig. 7A–C). NMN treatment completely reverted the effect of TDP-43 overexpression, increasing total neurite length and the complexity of neuronal processes to a level similar to that observed in nontransgenic motor neurons. It is worth noting that neurite complexity and length in nontransgenic motor neurons were not affected by NMN treatment (Fig. 7A–C).

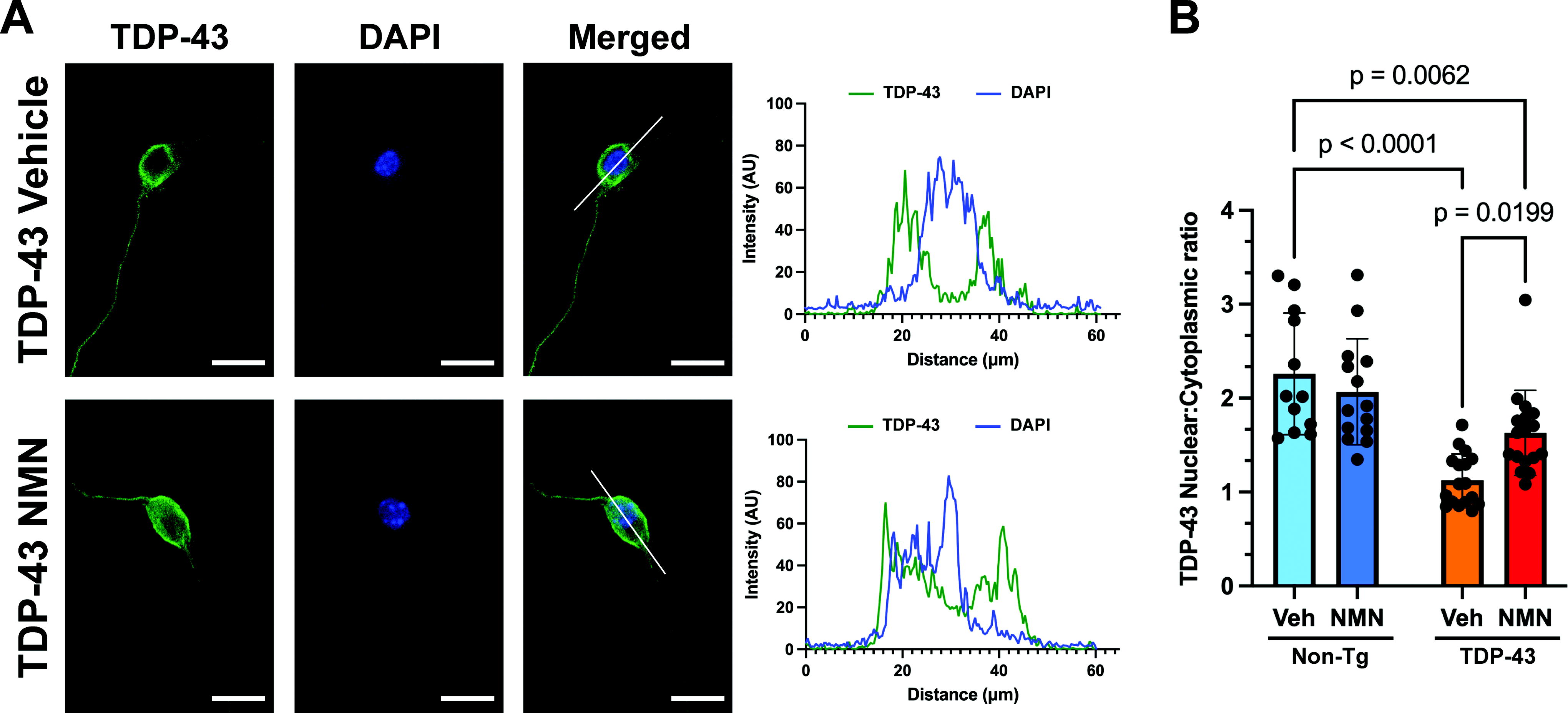
We also evaluated whether NMN was also able to increase the levels of reduced GSH in motor neurons overexpressing TDP-43, as we observed in motor neurons isolated from hSOD1G93A mice (Fig. 3). However, we were not able to detect significant changes in the staining intensity of the thiol-reactive fluorescent probe after NMN treatment in nontransgenic or TDP-43 overexpressing motor neurons (Fig. 7D). Therefore, we evaluated the potential effect of NMN treatment on TDP-43 subcellular localization, which has been linked to its toxicity in ALS (Barmada et al., 2010, Suk and Rousseaux, 2020). After immunostaining with specific antibodies against total TDP-43 or phosphorylated TDP-43 at serine residues 409/410 (pS409/410), we observed that motor neurons overexpressing TDP-43 displayed a decrease in the nuclear:cytoplasmic staining intensity ratio, indicative of TDP-43 mislocalization into the cytoplasm after its overexpression. However, in agreement with another report (Herzog et al., 2017), we did not observe the formation of TDP-43 aggregates. Remarkably, 48 h after NMN treatment, we observed an increase in the nuclear localization signal of TDP-43 and pS409/410 TDP-43, compared with vehicle-treated cultures (Figs. 8 and 9). Although motor neurons isolated from nontransgenic litter mates displayed about twice as much TDP-43 staining intensity in the nucleus than in the cytoplasm (nuclear:cytoplasmic ratio = 2.26 ± 0.65), neurons isolated from TDP-43 overexpressing mice displayed similar TDP-43 staining intensity in both cellular compartments (nuclear:cytoplasmic ratio = 1.13 ± 0.28). In contrast, NMN treatment significantly increased the nuclear:cytoplasmic ratio to 1.63 ± 0.45 (Fig. 8B). Importantly, the effect of NMN was restricted to TDP-43 overexpressing motor neurons, as we did not observe a significant change in the subcellular localization of the protein after NMN treatment in nontransgenic motor neurons (Fig. 8B). A similar result was obtained after pS409/410 TDP-43 immunostaining. In this case, the phosphorylated form of the protein was again preferentially located in the nucleus of nontransgenic motor neurons with a nuclear:cytoplasmic ratio of 4.2 ± 0.7. TDP-43 overexpression induced an increase in the cytoplasmic localization of the phosphorylated protein, with neurons displaying a nuclear:cytoplasmic ratio of 1.56 ± 0.53. NMN treatment completely reversed this change in the subcellular localization of the phosphorylated protein, increasing the nuclear:cytoplasmic ratio to 3.73 ± 1.71 (Fig. 9B). Again, we did not observe a significant change in the subcellular localization of the phosphorylated protein after NMN treatment in nontransgenic motor neurons (Fig. 9B).

Discussion
Increasing NAD+ availability by precursor supplementation was shown to exert neuroprotective effects in hSOD1G93A ALS mice, modifying the metabolic plasticity of skeletal muscles, and improving motor neuron survival while decreasing neuroinflammation in the spinal cord (Harlan et al., 2020). In addition, a clinical trial in a small cohort of patients with ALS showed that treatment with a combination of nicotinamide riboside and pterostilbene significantly improved the revised ALS functional rating scale (ALSFRS-R) score, pulmonary function, muscular strength, and skeletal muscle/fat weight ratio compared to patients receiving a placebo control (de la Rubia et al., 2019). The mechanisms mediating neuroprotection were not defined but could involve changes in the biology of multiple cell types. Indeed, increasing NAD+ availability by supplementation of the culture media with NAD+ precursors reverts the neurotoxicity of ALS astrocytes toward co-cultured motor neurons (Harlan et al., 2016). Our results show that NMN supplementation also has the potential to directly modify the biology of motor neurons to exert neuroprotection in the context of ALS pathology.
NMN treatment increased neurite length and complexity in cultured motor neurons isolated from two different ALS mouse models, overexpressing hSOD1G93A or wild-type TDP-43. However, we observed a differential effect of NMN treatment depending on the genotype of the motor neurons. Although NMN supplementation induced an increase in GSH levels in motor neurons expressing ALS-linked mutant SOD1, both in mouse and in iPSC-derived human motor neurons, this effect was not observed in nontransgenic or in TDP-43 overexpressing motor neurons. This observation could be explained by the mechanism by which ALS-linked mutant hSOD1s induce toxicity. SOD1 functions as a homodimer, with each monomer containing a catalytic copper ion bound to the active site, and a structurally important zinc ion bound to a conserved region of the protein (Kayatekin et al., 2008). ALS-linked mutant proteins are less stable and prone to losing zinc ions, which induces a conformational change that exposes the copper ion and facilitates aberrant redox biology (Crow et al., 1997a). The altered coordination of copper makes zinc-deficient SOD1 a more efficient oxidant, rapidly oxidizing intracellular antioxidants including GSH (Estevez et al., 1999). This allows the protein to operate in reverse—that is, the copper is reoxidized by oxygen leading to the production of superoxide at the expense of cellular antioxidants such as GSH (Estevez et al., 1999). In the presence of increased nitric oxide production, the enzyme can then catalyze the formation of peroxynitrite, a strong oxidant involved in a motor neuron-specific cell death pathway activated by multiple toxic stimuli, including trophic factor deprivation (Estevez et al., 1998, Pehar et al., 2007, Raoul et al., 2002). Thus, the effect of NMN treatment on GSH levels may be particularly evident in motor neurons overexpressing ALS-linked mutant SOD1s, which depend on intracellular antioxidants to counteract the toxicity of the mutant protein. Although NMN did not increase GSH levels in nontransgenic motor neurons in basal conditions, it completely prevented the death induced by trophic factor deprivation. The death pathway activated by trophic factor deprivation involves increased production of nitric oxide and the formation of peroxynitrite and can be blocked by increasing antioxidant defenses in motor neurons (Estevez et al., 2000, Estevez et al., 1998). Accordingly, we observed that NMN supplementation prevented the increase in nitrotyrosine immunostaining induced by the increased production of peroxynitrite after trophic factor deprivation in nontransgenic motor neurons. Together, our results suggest that under oxidative stress conditions imposed by trophic factor deprivation or mutant ALS-linked SOD1 expression, NMN supplementation exerts a protective effect in motor neurons.
Morphological abnormalities, including decreased dendritic length and spine density, have been reported in spinal and cortical motor neurons of ALS mice overexpressing hSOD1G93A (Fogarty et al., 2016, Martin et al., 2013). Moreover, degeneration of apical dendrites in corticospinal motor neurons was observed in familiar and sporadic cases of ALS (Genc et al., 2017). It has been proposed that alterations in neurofilament organization can contribute to motor neuron degeneration in ALS (Cleveland, 1996). Neurofilaments, composed of three different subunits, that is, light (NF-L), medium (NF-M), and heavy (NF-H), constitute a major cytoskeletal component in neurons and are the primary determinants of axonal diameter and normal dendritic arborization in large motor neurons (Cleveland, 1996, Zhang et al., 2002). It was previously shown that nitration of tyrosine residues in the NF-L subunit inhibits neurofilament assembly in vitro (Crow et al., 1997b) and that the toxicity of ALS-linked mutant SOD1 can, at least in part, be explained by its ability to catalyze peroxynitrite formation and tyrosine nitration (Crow et al., 1997a, Estevez et al., 1999). Thus, it is plausible that the effect of NMN on the growth and maintenance of the neuritic arbor in motor neurons could be explained, at least in part, by the ability of NMN to increase GSH levels and prevent tyrosine nitration.
We observed an increase in neurite length and complexity after NMN treatment in both hSOD1G93A and TDP-43 motor neurons. This likely occurs through different molecular mechanisms because GSH levels did not appear to be affected by NMN supplementation in TDP-43 overexpressing motor neurons. Indeed, we observed that in TDP-43 overexpressing motor neurons, the effect of NMN treatment includes alterations in the subcellular localization of the protein. TDP-43 is a predominantly nuclear ribonucleoprotein that shuttles into the cytoplasm and neuronal processes while performing its functions. In healthy cells, TDP-43 is mainly localized in the nucleus, where it regulates transcription, microRNA (mRNA) splicing, and mRNA processing, whereas in the cytoplasm, it regulates the subcellular localization, translation, and degradation of mRNAs (Lagier-Tourenne et al., 2010). TDP-43 pathology is observed in most SALS and FALS cases, except for FUS- or SOD1-linked FALS cases (Blair et al., 2010, Mackenzie et al., 2010, Mackenzie et al., 2007), and is characterized by its clearance from the nucleus and by accumulation of insoluble cytoplasmic inclusions containing hyperphosphorylated and ubiquitinated C-terminal fragments of the protein (Neumann et al., 2009, Neumann et al., 2006). There is still no consensus regarding whether it is the nuclear depletion of TDP-43, its cytoplasmic accumulation, and/or the formation of insoluble aggregates that induces motor neuron toxicity in ALS (Da Cruz and Cleveland, 2011). In cortical neuron cultures, overexpression of wild-type or mutant TDP-43 induces neurotoxicity by a mechanism directly linked to the cytoplasmic mislocalization of the protein and independent of the formation of cytoplasmic inclusions (Barmada et al., 2010). Accordingly, although we did not observe the overt formation of cytoplasmic TDP-43 aggregates in motor neurons isolated from TDP-43 overexpressing mice, we observed an increase in the cytoplasmic localization of the protein and a decrease in total neurite length and complexity. These results agree with the report by Fallini et al. (2012) showing that TDP-43 is actively recruited to motor neuron processes and, when overexpressed, negatively regulates neurite outgrowth. We observed that supplementation with the NAD+ precursor NMN corrected the subcellular localization of TDP-43, favoring its nuclear localization, and averted the detrimental effects of TDP-43 overexpression on neurite length and complexity.
The shuttling of TDP-43 from the nucleus to the cytoplasm and neuronal processes plays a key role in the regulation of its biological functions, and both knockdown and upregulation of the protein significantly alter neuronal function. Although Fallini et al. (2012) reported an increase in axonal arbor length after silencing TDP-43 expression in motor neurons, other reports have shown opposite results, with the silencing of TDP-43 expression significantly reducing axon growth and neurite complexity in human embryonic stem cell-derived motor neurons and in mouse primary motor neurons (Briese et al., 2020, Klim et al., 2019). A similar effect of dysregulated TDP-43 expression was observed in cortical and hippocampal neurons, with overexpression or knockdown of TDP-43 expression inducing a similar decrease in the complexity of neuronal processes (Herzog et al., 2017). In primary mouse motor neurons, the defects in axon growth induced by silencing of TDP-43 were associated with reduced axonal protein synthesis due to dysregulated RNA transport and disturbed mitochondrial function (Briese et al., 2020). Strikingly, the defects in axon growth induced by TDP-43 knockdown appear to be rescued by treatment with NAM, an NAD+ precursor that is converted to NMN in a reaction catalyzed by NAMPT, the rate-limiting step in the biosynthetic NAD+ salvage pathway (Briese et al., 2020). The report indicates that NAM treatment partially reverses the defects in protein synthesis observed in axons of TDP-43 depleted motor neurons (Briese et al., 2020). Because NAM can inhibit sirtuin activity (end-product inhibition) (Canto, 2022), it is not clear if the effects of NAM treatment are a consequence of increased NAD+ availability or potential effects on sirtuin activity. TDP-43 associates and regulates maturation and splicing of thousands of pre-mRNAs (Da Cruz and Cleveland, 2011), and multiple pathways have been shown to be dysregulated in neurons with altered TDP-43 expression and subcellular mislocalization (Briese et al., 2020, Herzog et al., 2020, Klim et al., 2019). Accordingly, the neuroprotective effects induced by NAD+ precursor supplementation on the different phenotypes induced by dysregulated TDP-43 expression may involve the engagement of multiple mechanisms that may act in concert, including an increase in antioxidant defenses, improved mitochondrial function, and/or improved proteostasis, all of which can be dependent or independent of sirtuin activity regulation (Lautrup et al., 2019). Our results indicate that NMN supplementation exerts its neuroprotective effects in cells overexpressing TDP-43 by restoring normal subcellular localization of the protein.
Wallerian degeneration observed after axotomy or in cases of chemotherapy-induced neuropathies has been directly linked to an accumulation of NMN in axons because of the loss of NMNAT2, the enzyme that synthesizes NAD+ from NMN and ATP (Di Stefano et al., 2017, Di Stefano et al., 2015). However, manipulations that increase NMN levels do not induce axonal degeneration and can actually prevent axonal degeneration (Liu et al., 2018, Sasaki et al., 2016, Sasaki et al., 2006). Attempts to prevent axonal degeneration in ALS by targeting the pathways that control Wallerian degeneration have generated inconsistent results (Ferri et al., 2003, Fischer et al., 2005, Peters et al., 2018, Vande Velde et al., 2004, Veriepe et al., 2015, White et al., 2019). Regardless of the involvement of Wallerian degeneration in ALS pathology, our data show that NMN supplementation can confer axonal protection in motor neurons isolated from two dissimilar models of ALS, with and without involvement of TDP-43 pathology. In SOD1-linked ALS mouse models, preventing motor neuron cell death alone does not appear to be sufficient to prevent muscle denervation and change the course of the disease, whereas the extent of axonal degeneration directly correlates with disease severity (Gould et al., 2006, Parone et al., 2013). Therefore, axonal protection remains an important therapeutic target in ALS. Our results support a beneficial effect of NAD+ precursor supplementation on the growth and maintenance of motor neuron processes, further supporting its therapeutic potential in the context of ALS pathology.
In summary, we observed a direct neuroprotective effect of increasing NAD+ availability in motor neurons. Our results suggest that the neuroprotection conferred by NMN supplementation in motor neurons expressing ALS-linked mutations, or exposed to oxidative stress conditions imposed by trophic factor deprivation, is mediated by a mechanism that involves an increase in GSH content. Accordingly, we observed that NMN treatment prevented the increase in nitrotyrosine immunostaining induced by trophic factor deprivation. However, it is important to mention that our results do not provide a definitive mechanistic confirmation of a link between these observations. Moreover, we found that the neuroprotection conferred by NMN treatment in TDP-43 overexpressing motor neurons did not appear to involve alterations in GSH levels, although we did observe changes in the subcellular localization of the protein. Further research is needed to clearly outline the mechanism by which NMN treatment regulates the nuclear-cytoplasmic trafficking of TDP-43.
The fact that millimolar concentrations of NMN are required to exert neuroprotection in motor neuron cultures highlights the translational challenges that strategies to modify NAD+ availability face. NMN concentrations in blood are estimated to be in the micromolar range, likely because of the instability of this intermediate in blood (Camacho-Pereira et al., 2016, Chen et al., 2022). NMN levels appear to be even lower than NR levels, because NMN is degraded to the nucleoside (NR) in order to enter the cell (Nikiforov et al., 2011). Despite these observations, both precursors have been shown to increase NAD+ levels in vivo and to exert protective effects (Pehar et al., 2018). In multiple experimental setups in mammalian cell cultures, NMN and NR supplementation in the millimolar range is required to reach a maximal biological effect (Canto et al., 2012, Harlan et al., 2016, Yang et al., 2019). Importantly, even when pharmacokinetic studies are not able to detect millimolar concentrations of NAD+ precursors in vivo, the main observations obtained in cell culture models have been reproduced in preclinical animal studies (Covarrubias et al., 2021, Pehar et al., 2018), arguing for the relevance of the results obtained in cell culture models using these concentrations of NAD+ precursors.
Materials and Methods
Reagents
Unless otherwise specified, cell culture media, serum, and supplements were obtained from ThermoFisher Scientific-Gibco, whereas other reagents were from Sigma-Aldrich.
Mice
Transgenic mice overexpressing hSOD1G93A, strain B6.Cg-Tg(SOD1*G93A)1Gur/J (Gurney et al., 1994), were obtained from the Jackson Laboratory (stock #004435) and were maintained as hemizygous animals in a C57BL/6J background. Transgenic mice overexpressing wild-type TDP-43 under the control of the mouse Thy1 promoter, strain B6;SJL-Tg(Thy1-TARDBP)4Singh/J (Wils et al., 2010), were also obtained from the Jackson Laboratory (stock #012836). This transgenic line was maintained in hemizygosis, and carriers were mated to produce nontransgenic and homozygous animals used in cell culture experiments. All animal procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Animal Care and Use Committee of University of Wisconsin–Madison (Animal Welfare Assurance number A3368-01) approved the animal protocols pertinent to the experiments reported in this manuscript.
Primary motor neuron cultures
Primary motor neuron cultures were prepared from 12.5 embryonic day mouse spinal cords using an OptiPrep-gradient centrifugation protocol as previously described (Liu et al., 2020). Genotyping of transgenic embryos was performed by real-time polymerase chain reaction during tissue dissection, and all embryos of the same genotype were pooled together for motor neuron isolation. Motor neurons were plated on 4-well multidishes (Nunclon) or 4-well cell culture slides (Mattek) precoated with polyornithine–laminin. Cultures were maintained in Neurobasal medium supplemented with 2% B-27 supplement, 25 μM L-glutamate, 25 μM 2-mercaptoethanol, 0.5 mM L-glutamine, and 2% horse serum. Neuronal survival was supported by the addition of GDNF (1 ng/mL; Sigma-Aldrich), or a combination of GDNF (1 ng/mL) and BDNF (0.1 ng/mL; R&D Systems). NMN (Sigma-Aldrich) was added 2 h after motor neuron plating. Motor neuron survival was determined after 48 h in vitro by direct counting, in a prefixed area of the dish, of all cells displaying intact neurites longer than four cell bodies in diameter, which typically express the motor neuron markers Islet-1/2 homeoprotein, ChAT, and the homeobox protein HB9 (motor neuron and pancreas homeobox protein 1), as determined by immunofluorescence. The purity of the cultures was confirmed by immunofluorescence with antibodies against β-III-Tubulin, ChAT, Islet-1, and HB9: 99.5 ± 2.0% of the cells were β-III-Tubulin positive; of these, 100% were ChAT or Islet-1 positive, and 98.6 ± 3.4% were HB9 positive.
iPSC-derived human motor neuron cultures
An iPSCs line derived from a patient with ALS carrying the mutation D90A in the SOD1 gene was obtained from the WiCell Research Institute (iPSC ID# WC034i-SOD1-D90A). iPSCs were differentiated into motor neurons using the STEMdiff™ Motor Neuron Differentiation protocol from Stemcell Technologies (Catalog #100–0871) following the manufacturer’s instructions. Briefly, iPSCs were differentiated from day 0 to day 9 into motor neuron progenitors using an embryoid body formation protocol in AggreWell™ 400 plates. On day 9, the embryoid bodies were dissociated, and motor neuron progenitors were plated on poly-L-ornithine and laminin-coated wells for differentiation to postmitotic motor neurons. Motor neuron progenitors were plated at a density of 2600 cells/cm2 in 4-well cell culture slides (MatTek) for cell morphology analysis, ThiolTracker™ Violet staining, and immunostaining analysis of motor neuron markers. Motor neuron progenitors were plated on 24-well plates (TPP Techno Plastic Products) at a density 21,000 cells/cm2 for total GSH and NAD+ assay. Motor neuron differentiation efficiency was evaluated at day 16 by immunofluorescence with antibodies against β-III-Tubulin, ChAT, Islet-1, HB9, and the astrocyte marker GFAP.
Cell morphology analysis
Images of primary motor neurons at 20× magnification were imported into the National Institute of Health Fiji (Image J) software (Schindelin et al., 2012) for morphology analyses. To measure total neurite length, motor neurons were traced in two dimensions semi-automatically using the Fiji plugin SNT (Arshadi et al., 2021). The reconstructed neuronal arbors were then analyzed using the Sholl Analysis module within SNT (Ferreira et al., 2014). Ten concentric circles with a radius step size of 20 μm starting from the center of the soma were generated to a final radius of 200 μm. The number of times the arbors intersect each concentric circle was recorded and expressed graphically as the number of crossings. The same analysis was conducted on iPSC-derived human motor neuron cultures in images acquired using a 10× objective and using a concentric circle radius step size of 20 μm starting from the center of the soma to a final radius of 400 μm.
Intracellular glutathione detection
To estimate GSH content in mouse primary motor neuron cultures, we used the ThiolTracker Violet probe (GSH detection reagent, ThermoFisher Scientific; #T10095). The ability of the probe to detect changes in intracellular thiols in motor neurons was confirmed in a series of control experiments in which nontransgenic motor neuron cultures were treated with buthionine–sulfoximine (BSO; 10 nM), an inhibitor of GSH synthesis (Griffith and Meister, 1979), or tert-butylhydroquinone (tBHQ; 500 nM), an activator of Nrf2-driven transcription in neurons, which regulates the expression of both subunits of the enzyme catalyzing the rate-limiting step in GSH synthesis (glutamate-cysteine ligase catalytic and modifier subunits; GCLC and GCLM) (Johnson et al., 2002, Lee et al., 2003). Accordingly, ThiolTracker Violet stain intensity significantly decreased or increased after treatment with BSO or tBHQ, respectively (Supplementary Fig. S1). ThiolTracker Violet staining was performed as previously described (Mandavilli and Janes, 2010). Briefly, live motor neuron cultures were incubated at 37°C with 10 μM ThiolTracker Violet diluted in Dulbecco’s phosphate buffered saline (DPBS) containing calcium and magnesium (Corning). After 30 min, the solution was discarded, and the cells were fixed with 4% paraformaldehyde (PFA) and 0.1% glutaraldehyde in PBS for 30 min at room temperature. After washing, cells were permeabilized with 0.1% Triton™ X-100 for 15 min at room temperature and stained for 1 h at room temperature with Alexa Fluor 488 phalloidin probe (1:400, ThermoFisher Scientific, #A12379). Slides were mounted using Gel/Mount aqueous mounting medium (Electron Microscopy Sciences), and images were acquired using an Axio Observer 5 microscope (Carl Zeiss Microscopy) with identical settings for all treatment groups. Quantification of reduced GSH levels was performed using the Fiji (Image J) software. A mask was generated from the phalloidin probe channel to outline the soma of iPSC-derived human motor neurons or soma and neurites of primary mouse motor neurons. This mask was then overlaid onto the ThiolTracker Violet channel to measure integrated density.
GSH assay
Total GSH levels (GSH and GSSG) were determined using the Tietze method as previously described (Vargas et al., 2006). Total glutathione content was normalized by the total protein concentration in the sample, as determined with the bicinchoninic acid (BCA) protein assay (ThermoFisher Scientific).
NAD+ quantification
After treatment, cells were lysed in 0.5M perchloric acid and maintained on ice. Following centrifugation at 14,000 g for 10 min at 4°C, the acid-soluble fraction was neutralized with an equal volume of 0.8M KHCO3, and NAD+ levels were determined by an enzymatic cycling assay (Li and Sauve, 2015). NAD+ content was corrected for the total protein concentration in the sample, as determined with the BCA protein assay (ThermoFisher Scientific).
Immunostaining
Primary spinal cord motor neuron or iPSC-derived human motor neuron cultures were pre-fixed by replacing half of the cell culture media with fixation solution consisting of 4% PFA and 0.1% glutaraldehyde in DPBS for 15 min at 37°C. For complete fixation, all the media was replaced with fixation solution and incubated for an additional 20 min on ice. After washing with PBS, slides were permeabilized with 0.1% Triton X-100 in PBS and blocked for at least 1 h with 10% goat serum, 2% bovine serum albumin, and 0.1% Triton X-100 in PBS. Slides were then incubated overnight at 4°C with primary antibodies diluted in blocking solution. The following primary antibodies were used: rabbit anti-TDP-43 (1:200, Proteintech, #10782-2-AP), rabbit anti-Phospho-TDP43 (Ser 409/410) (1:500, Proteintech, #22309-1-AP), and mouse anti-β-Tubuin III (neuronal) (1:400, Sigma-Aldrich, #T8578). An additional set of immunostaining was conducted using blocking solution containing 5% donkey serum and 0.1% Triton X-100 in PBS. The following primary antibodies were used: rabbit anti-ChAT (1:200, Proteintech, 20747-1-AP), goat anti-ChAT (1:100, Novus Biologicals, NBP1-30052), goat anti-GFAP (1:450, Novus Biologicals, NB100-53809), rabbit anti-HB9 (1:300, Invitrogen, PA5-23407), rabbit anti-Islet 1 (1:200, Abcam, ab109517), chicken anti-β-Tubuin III (neuronal) (1:300, Novusbio, NB100-1612), and rabbit anti-nitrotyrosine (1:150, ThermoFisher Scientific, A-21285). After incubation with primary antibodies, slides were incubated for 1 h at room temperature with fluorophore-conjugated secondary antibodies in the appropriate blocking solution. The secondary antibodies used were goat anti-mouse Alexa Fluor 594 (1:1000, ThermoFisher-Invitrogen, #A-11032), goat anti-rabbit Alexa Fluor 488 (1:1000, ThermoFisher-Invitrogen, #A-11034), biotin-SP-conjugated AffiniPure Donkey Anti-Rabbit IgG (1:800, Jackson ImmunoResearch) followed by Alexa Fluor 594-conjugated Streptavidin, Alexa Fluor 488-conjugated Donkey Anti-Goat IgG (1:800, Jackson ImmunoResearch), Alexa Fluor 647-conjugated Donkey Anti-Goat IgG (1:800, Jackson ImmunoResearch), Alexa Fluor 488-conjugated Donkey Anti-Mouse IgG (1:800, Jackson ImmunoResearch), and Alexa Fluor 647-conjugated Donkey Anti-Chicken IgG (1:800, Jackson ImmunoResearch). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; ThermoFisher-Invitrogen). Slides were mounted using Gel/Mount aqueous mounting medium (Electron Microscopy Sciences). As a negative control, the primary antibodies were omitted. Image acquisition was performed on a Nikon A1RS HD Confocal Microscope at the University of Wisconsin-Madison Optical Imaging Core (OIC). Images were acquired with identical settings using a 60× oil-immersion 1.4 NA objective at 1024 × 1024 pixels, with a z-stack step size of 0.5 μm. Human motor neuron images used for morphology analyses were acquired with a Nikon A1R Confocal Microscope (OIC), using identical settings, with a 10 × 0.45 NA objective at 1024 × 1024 pixels.
Immunofluorescence image quantification
Quantification of Phospho-TDP-43 (P-TDP-43) and TDP-43 subcellular localization was performed using the National Institute of Health Fiji (ImageJ) software (Schindelin et al., 2012), as previously described (Kelley and Paschal, 2019). Briefly, the z-stack corresponding to the center of the nucleus was chosen for analysis. A mask of the nucleus was created using the DAPI channel, and a mask of the soma was generated using the β-Tubulin-III channel. Each mask was then overlaid onto the P-TDP-43 or TDP-43 channel, and the integrated density and mean fluorescent intensity were measured. The cytoplasmic intensity was calculated by subtracting the nuclear integrated density from the soma integrated density. The mean fluorescence intensity of the cytoplasm was calculated by dividing the cytoplasmic intensity by the region of interest (ROI) area. Finally, the nuclear-to-cytoplasmic ratio was calculated by dividing the mean cytoplasmic intensity by the mean nuclear intensity.
Nitrotyrosine immunostaining intensity was quantified using the National Institute of Health Fiji software. A mask of the soma was created using the β-Tubulin-III channel. The mask was then overlaid onto the nitrotyrosine channel to measure the integrated density.
Statistical analysis
Comparisons between two groups were performed using unpaired t-tests with Welch’s correction. The D’Agostino-Pearson normality test (omnibus K2 test) was used to evaluate if data samples were consistent with a Gaussian distribution. Multiple group comparisons were performed by one-way analysis of variance (ANOVA) followed by Tukey’ post-test. The Brown–Forsythe test for equal variances was used to confirm equal variances in data groups prior to performing ordinary one-way ANOVA. When comparing the effect of genotype and treatments, two-way ANOVA was used followed by Tukey’s or Dunnett’s post-tests. Sholl analysis data in iPSC-derived human motor neuron cultures treated with vehicle or NMN were analyzed by multiple t-tests. Differences were declared statistically significant if p ≤ 0.05. All statistical computations were performed using Prism 9.0 (GraphPad Software).
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke (grants R01NS100835 and R01NS089640). This work used resources and facilities of the William S. Middleton Memorial Veterans Hospital (Madison, WI, USA). The contents of this article do not represent the views of Veterans Affairs or the U.S. government. The authors thank the University of Wisconsin–Madison OIC for the use of its equipment and facilities. The summary graphic illustration was created with BioRender.com.
Authors’ Contributions
H.L.H., M.A., S.A., C.B.C., M.R.V., and M.P. performed experiments and analyzed data. H.L.H. and M.P. wrote the article. All authors reviewed and approved the content of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke grants R01NS100835 and R01NS089640.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.