
Abstract
Significance:
Aberrant redox homeostasis, characterized by the enhancement of intracellular reactive oxygen species (ROS) and antioxidant defenses, is among the well-known cancer hallmarks. Understanding the regulatory mechanisms of redox homeostasis in cancer cells has become the focus of many studies. Epigenetic and post-translational modifications (PTMs), as pivotal regulators of multiple biological processes, play critical roles in tumorigenesis and development.
Recent Advances:
DNA and RNA methylation are important forms of epigenetic modifications. Recent evidence suggests that DNA/RNA methylation and PTMs can modulate redox homeostasis in multiple manners including affecting key molecules in ROS production, elimination, and redox-related signaling, thereby participating in tumor progression.
Critical Issues:
The regulatory effects of DNA/RNA methylation and PTMs on ROS are of crucial importance for tumor progression. In this review, we introduce the dual role of ROS in cancer, and then focus on the mechanistic role of DNA/RNA methylation and PTMs, especially ubiquitination and acetylation, in regulating redox homeostasis to involve in cancer progression.
Future Directions:
A complete understanding of how epigenetics and PTMs function in the regulation of redox homeostasis in cancer progression might expand a new direction for the progression mechanisms and therapeutic targets of cancer. Antioxid. Redox Signal. 39, 531–550.
Introduction
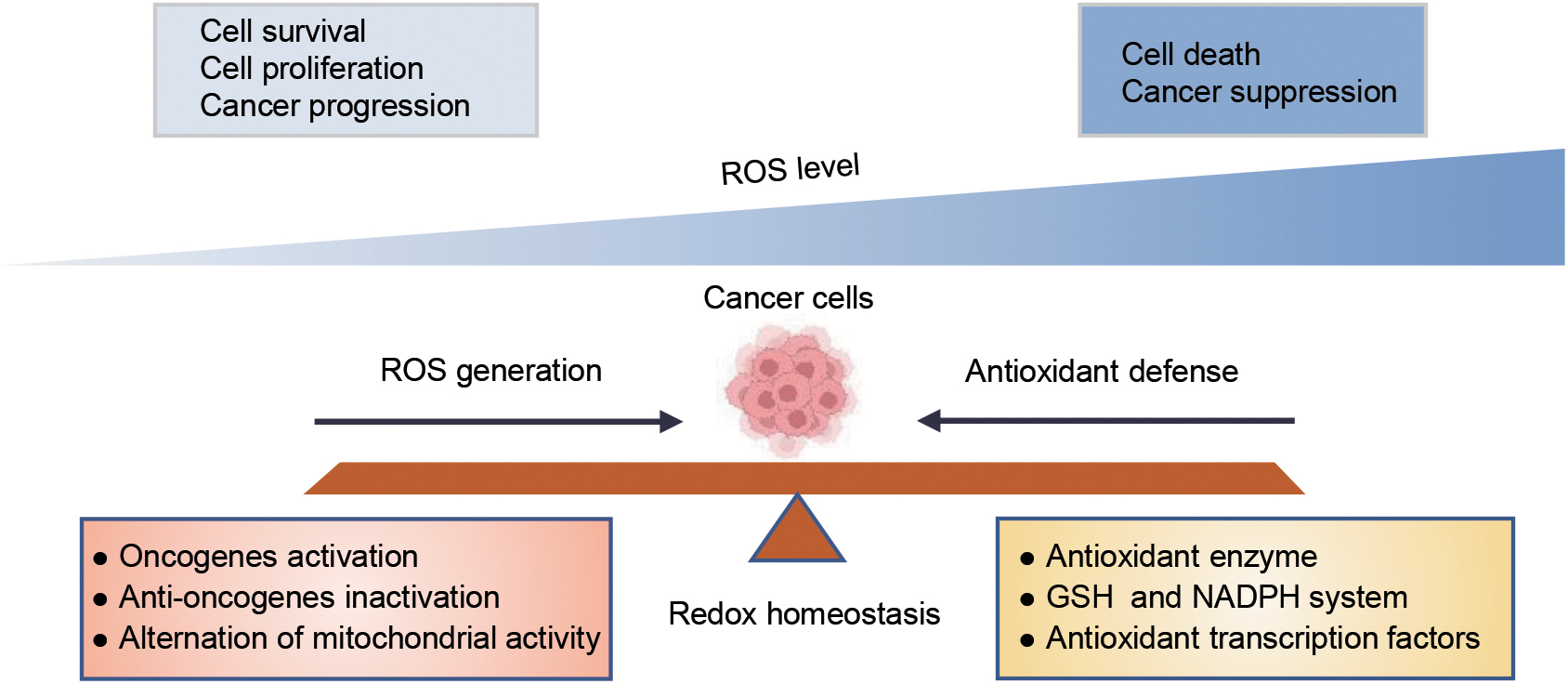
Intracellular redox homeostasis mainly refers to the balance between reactive oxygen species (ROS) generation and antioxidant systems. Aberrant redox homeostasis, one of the major features of cancer, exhibits increased abundance of ROS and antioxidant defenses (Hayes et al., 2020; Reczek et al., 2017). Extensive evidence has indicated that ROS play a paradoxical role in cancer depending on the intracellular ROS concentration. Low ROS levels can actually promote cancer cell proliferation, survival, and metastasis, while high ROS levels induce oxidative damage leading to various forms of cell death such as apoptosis, autophagy, and ferroptosis (Hayes et al., 2020; Zhang et al., 2021a).
The proliferation and metastasis of tumor cells are accompanied by the increase of ROS production, while cancer cells increase their antioxidant defense systems to control the ROS homeostasis for maintaining their malignant phenotypes and simultaneously avoiding excessive ROS-induced cell death (Dong et al., 2019; Harris and DeNicola, 2020). Therefore, the regulation of redox homeostasis plays a complicated and important role in tumorigenesis and progression, and it is urgent to search for pivotal regulators involved in maintaining redox homeostasis in cancer cells.
DNA and RNA methylation are important forms of epigenetic modifications, which can regulate gene expression independent of DNA/RNA sequence changes at the level of transcription and post-transcription, respectively, and play vital roles in tumor development (Klutstein et al., 2016; Michalak et al., 2019).
In addition, post-translational modifications (PTMs) act as a molecular switch to quickly change the cell state mainly including ubiquitination, phosphorylation, acetylation, and methylation. These PTMs regulate the activity, stabilization, localization, and degradation of target proteins at the post-translation level (Chen et al., 2020a). DNA/RNA methylation and PTMs as important regulators are involved in several cellular processes, including the cell proliferation, differentiation, apoptosis, and DNA repair in cancer (Dawson and Kouzarides, 2012; He et al., 2019; Pan and Chen, 2022).
Importantly, accumulative evidence has supported that the role of DNA/RNA methylation and PTMs in tumor progression is also closely associated with redox regulation (Fig. 1), and their regulatory effects are very complex that has not been reviewed. In this review, we introduce the dual role of ROS in cancer, and then aim to provide an overview of recent evidence on the mechanistic role of DNA/RNA methylation and PTMs, especially ubiquitination and acetylation, in regulating redox homeostasis to involve in cancer progression, which may provide a positive outlook for the progression mechanisms and therapeutic targets of cancer.

Sources of ROS
ROS are intracellular chemical species produced by oxygen that mainly contain superoxide anion (O2 •−), hydrogen peroxide (H2O2), and hydroxyl radical. ROS are produced mainly in a variety of metabolic reactions mediated by mitochondrial electron transport chain (ETC), lipoxygenases, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX), and cyclooxygenases (Marengo et al., 2016). ETC is composed of four protein complexes I–IV, which supports the operation of oxidative phosphorylation (OXPHOS) system to produce ROS as by-products. Free electrons, which are taken by coenzyme Q and cytochrome c (Cyt c), and released by NADH and FAD, reduce O2 to water at complex IV.
NOX, another important source of ROS, consists of membrane subunits, including NOX1, NOX3–5, dual oxidase 1 and dual oxidase 2, cytosolic proteins, and the small guanosine triphosphate (GTP)-binding protein Rac 1 and 2. O2 •− is formed via single electron reduction of O2 in the course of the combination of diverse NOX subunits (Tochhawng et al., 2013). ROS can also ascend in emergency condition and in the process of defense after exogenous stimuli. For instance, ultraviolet (UV) radiation promotes the mitochondria to produce O2 •−, and pattern recognition patterns on pathogens make phagocytic cells manufacture O2 •− (Helfinger and Schröder, 2018).
ROS and Cancer
From a previous study, it appears that cancer cells exhibit aberrant redox homeostasis with elevated ROS levels compared with normal cells (Gorrini et al., 2013). ROS play a dual role in cancer (Reczek et al., 2017).
On the one hand, low levels of increased ROS drive cellular signaling pathways supporting cancer cell proliferation and survival to accelerate tumor development. However, excessive high levels of ROS can lead to cell death and limit tumor progression by inducing detrimental oxidative stress (Perillo et al., 2020). Therefore, cancer cells adopt multiple antioxidative defense mechanisms to control ROS levels (Harris and DeNicola, 2020), resulting in a relative balance between ROS production and elimination in cancer cells, hence preventing high levels of ROS induced various forms of cell death and promoting tumor development (Fig. 2). A detailed description of the role of ROS and antioxidant defense mechanisms in cancer is provided below.
Tumorigenic effects of ROS
Gene mutation is an important molecular basis for tumorigenesis (de Sa Junior et al., 2017). ROS can directly act on DNA hydrogen bonds to make the DNA double strands open and break. Then, the internal base-pairs are fully exposed to ROS, which stimulates the oxidative modification of base-pairs and causes the mutation of base-pairs in the nucleus (Srinivas et al., 2019).
ROS can also inhibit DNA damage repair by targeting key signal molecules, such as ataxia telangiectasia mutated, ataxia telangiectasia and Rad3-related, checkpoint kinase 1 and 2, and key enzymes required for DNA damage repair, such as humans oxoguanine DNA glycosylase 1, and breast cancer gene 2. ROS hamper DNA damage repair and augment gene mutations accumulation, leading to further expansion in genomic instability and transformation into malignant cells (Huang et al., 2021).
ROS also promote the proliferation of cancer cells by involving in the activation of growth factors. Signals of growth factor receptors (GFRs) and integrins accelerate the accumulation of O2 •− and the conversion of O2 •− to H2O2. GFRs are stimulated through specific growth hormones or ligation of integrins, then trigging the Ras-Raf-Erk and the PI3K-Akt signaling pathways. These signaling pathways enhance the production of NOXs. Integrins facilitate lipoxygenases to generate lipid hydroperoxides as well. Hydroperoxides from these sources activate receptor tyrosine kinases and suppress protein tyrosine phosphatases, thereby improving the proliferation of cancer cells (Hegedus et al., 2018).
After a period of proliferation, cancer cells begin to metastasize through epithelial–mesenchymal transition (EMT), which destroys the cellular matrix and promotes angiogenesis. ROS-induced oxidative stress can activate the transforming growth factor-β (TGF-β) and WNT signaling pathway, which play a critical role in promoting EMT through SMAD and β-catenin (Dongre and Weinberg, 2019).
In addition, ROS can accelerate the activation of downstream matrix metalloproteinases (MMPs) and serine proteases by activating MAPK and AP-1, and facilitate the degradation of extracellular matrix (Sosa et al., 2013). Besides, ROS can drive angiogenesis by regulating Ang1 and p44/42 MAPK signaling pathways as well as p300, vascular endothelial growth factor-α, hypoxia inducible factor 1 (HIF1), p53, and MMPs (Kirtonia et al., 2020).
Tumor-suppressive effects of ROS
Apoptosis
Apoptosis is a kind of programmed cell death that is the main mode of cell death induced by oxidative stress. Cyt c is distributed outside of mitochondrial inner membrane, and its anchoring and release is the key to mitochondria-mediated apoptosis (Babbitt et al., 2015). ROS induce the release of Cyt c and propel cancer cells apoptosis by activating p38MAPK signaling pathway (Wang et al., 2013).
In addition, ROS improve the abnormal release of Ca2+ in the endoplasmic reticulum (ER), induce ER stress, and lead to the accumulation of misfolded proteins in ER. Mitochondrial protein oxidation exalts the transcription of C/EBP-homologous protein (CHOP), which activates mitochondrial unfolded protein response (UPRmt) and eventually leads to ER stress-dependent apoptosis of tumor cells (O'Malley et al., 2020).
Ferroptosis
Ferroptosis is an iron-dependent form of programmed cell death induced by excessive ROS, especially lipid hydroperoxides (Jiang et al., 2021). GPX4 is the core enzyme of antioxidant metabolism in the process of ferroptosis and plays an essential role in reducing intracellular oxidative stress. The glutamate/cystine antiporter (system XC −) is also an important intracellular antioxidant system, which transports cysteine required for glutathione (GSH) synthesis to scavenge ROS. And glutamate metabolism is necessary for ferroptosis caused by cysteine depletion. Suppression of system XC −-mediated cystine uptake disrupts the oxidative defense system and triggers ferroptosis of cancer cells (Yang and Stockwell, 2016).
Autophagy
Autophagy is an autolytic process in which damaged proteins or organelles are encapsulated in vesicles and fused with lysosomes to form autophagosomes (Zhang et al., 2021a). Recent studies have shown that ROS are capable to accelerate autophagy in cancer, while autophagy relieves oxidative damage by removing protein aggregates and damaged organelles (Chen et al., 2009). Specifically, ROS could regulate autophagy of tumor cells through TIGAR, HIF1-BNIP3/NIX, Nrf2-p62, FOXO3-LC3/BNIP3, Atg4 signaling pathways. Tumor cells are able to resist oxidative stress by scavenging misfolded proteins through autophagy with the help of chaperone-mediated autophagy pathway, mitosis pathway, and p62 delivery pathway (Li et al., 2015).
Antioxidant defense in cancer
To avoid excessive ROS-induced cell death, tumor cells adopt various antioxidant defense mechanisms to maintain a relative balance between ROS production and elimination. One of the important mechanisms is the overexpression of antioxidant enzymes such as superoxide dismutase (SOD), catalase, GPXs, and peroxiredoxin to adaptively enhance the antioxidant capacity. SOD can rapidly turn superoxide into H2O2, which is subsequently converted to H2O by peroxiredoxin, GPX, and catalase. Tumor cells have highly activated antioxidant systems, including NADPH-dependent GSH reductase and TRXR systems.
Among them, GPXs convert H2O2 to H2O, and GSH reductase reduces oxidized glutathione disulfide (GSSG) to GSH using NADPH as the electron donor (Forman and Zhang, 2021). Furthermore, tumor cells alter cell metabolism to regulate the production of antioxidants NADPH and GSH in response to oxidative stress. NADPH plays an important role in the cellular antioxidant system.
On the one hand, GSH reductases use NADPH as an important cofactor to convert GSSG to GSH, which then acts as an auxiliary substrate of GPX to decrease peroxides to H2O. On the other hand, TRXR takes advantage of NADPH as an electron donor to keep the reduced form of TRX, which facilitates O2 scavenging and DNA synthesis by reducing nucleotide reductase (Ju et al., 2020). The metabolic glycolysis pathway can be shifted to the pentose phosphate pathway (PPP) to produce NADPH in many cancer cells. Most GSH is present in reduced form.
After reacting with oxidizing substances such as ROS, it is oxidized to its oxidized form, GSSG. Loss of GSH will disrupt redox homeostasis, leading to ROS accumulation, and eventually triggering cell dysfunction and death. The glutamine metabolic pathway to generate GSH is the most fundamental pathway to control ROS levels. Therefore, glutamine uptake and glutaminolysis are dramatically increased in cancer cells (Niu et al., 2021).
Tumor cells maintain antioxidant capacity by regulating key redox-related transcription factors such as nuclear factor E2-related factor 2 (Nrf2), HIF1, p53, nuclear factor kB (NF-κB), and forkhead box protein O (FOXO) as well. Among them, Nrf2 function is controlled by Kelch-like ECH–associated protein 1 (Keap1), and the increased ROS in cells induces a protective antioxidant response by disrupting Keap1 degradation of Nrf2. Nrf2 acts as a transcription factor that binds to antioxidant response elements (AREs) to activate the expression of several antioxidant genes such as GCLC, NQO1, GSTM1, GPX4, and PRDX1 (Forman and Zhang, 2021). HIF1 reduces the formation of ROS mainly by improving the efficiency of electron transfer to O2, upregulating the expression of NADH dehydrogenase NDUFA4L2 and increasing glycolytic factors (Cyran and Zhitkovich, 2022).
The antioxidant effect of p53 is vital for tumor growth and survival. p53 interferes with DNA oxidation and mutation by inhibiting ROS. In addition, p53 can induce a series of antioxidant enzymes, such as MnSOD, Sestrins, TIGAR, and GLS2 (Budanov, 2014). These antioxidant systems promote tumor proliferation, invasion, metastasis, and drug resistance. Consequently, these systems may act as important targets for antitumor research.
Drugs or agents affecting the redox balance of tumor cells
With the in-depth study of the redox mechanism of tumor cells, a number of drugs or agents of manipulating redox status have been developed. Most of the traditional chemotherapy drugs, including anthracyclines, platinum complexes, alkylating agents, camptothecin compounds, arsenics, and topoisomerase inhibitors, have oxidative function, and their clinical therapeutic effects are closely related to the production of extremely high ROS in a short period of time (Gorrini et al., 2013).
In addition to drugs that induce ROS production such as cisplatin, doxorubicin, fenretinide, and bleomycin, there are drugs that target antioxidant systems for tumor therapy (Wang et al., 2021). Buthionine sulfoximine (BSO) is an effective inhibitor of GSH synthesis, and has anticancer effects on melanoma, ovarian and breast cancer (Poprac et al., 2017).
In addition, Telcyta (TLK-286), disulfiram, NOV-002, sulfasalazine, and menadione all target the inhibition of GSH antioxidant system for tumor therapy (Kirtonia et al., 2020). The TRX/TRXR inhibitor PX-12 has been shown to inhibit the growth and induce the apoptosis of a variety of tumor cells, including lung, liver, gastric, and colorectal cancer (CRC), by increasing ROS levels (Baker et al., 2006). Dimesna (BNP7787), targeting TRX and GRX, has been used to treat a variety of solid tumors, including ovarian cancer and nonsmall cell lung cancer (NSCLC) (Lu et al., 2007).
2-Methoxyestradio and ATN-224 can be used to treat tumors by inhibiting the activity of SOD and inducing the imbalance of oxidative homeostasis of tumor cells (Forman and Zhang, 2021). In addition, a number of drugs of inhibitors for induction of ferroptosism in tumors have been found, such as erastin, sulfasalazine, sorafenib, artesunate, temozolomide, BSO, lapatinib, altretamine, ML-162, RSL-3, ML-210, and ATRA (Niu et al., 2021). Glutaminase 1 (GLS1) has also been shown to be a viable drug target for manipulating redox status in tumor cells.
The small molecule CB839 is a potent inhibitor of GLS1, and its clinical trials in lung cancer are underway (Romero et al., 2017). Also, ivosidenib and enasidenib are specific inhibitors for isocitrate dehydrogenase 1 (IDH1)/2 mutant and target mitochondrial ROS for antitumor therapy (Lu et al., 2007). More research is needed to gain a deeper and better understanding of clinical-grade ROS scavengers and inducers, and will have significance for tumor treatment.
Role of DNA Methylation in the Regulation of Redox Homeostasis in Cancer
DNA methylation, a type of epigenetic modification, is the covalent binding of methyl groups to the cytosine carbon position 5 of genomic CpG dinucleotides by DNA methyltransferase to form 5-methylcytosine (5mC). DNA methylation has a great significance for silence of retrovirus elements, modulating tissue-specific gene expression, genomic imprinting, and X chromosome inactivation (Moore et al., 2013). Genome-wide DNA hypomethylation has been reported in tumors, which bring about genomic instability and may cause proto-oncogene activation and tumor suppressor gene inactivation.
In addition, CpG islands in tumor-specific gene promoters have been found to be hypermethylated in DNA, which is a radical hallmark of many cancer cells (Kulis and Esteller, 2010). Recent evidence suggests that the role of DNA methylation in tumor progression is closely related to redox regulation (Lin et al., 2021; Szczepaniak et al., 2022; Zhang et al., 2013).
Regulation of ROS production by DNA methylation
Most intracellular ROS are by-products of OXPHOS. As the central pathway of cell metabolism, OXPHOS plays a pivotal role in almost all cells (Papa et al., 2012). Recent studies have shown that OXPHOS can be regulated by DNA methylation of transcription factor, antioncogene, and mitochondrial DNA to influence tumor progression (Damal Villivalam et al., 2021; Li et al., 2019; Ozturk et al., 2022). ISL2, a nuclear and chromatin-associated transcription factor, is a putative tumor suppressor. ISL2 is silenced by high DNA methylation in pancreatic ductal adenocarcinoma (PDAC), leading to increased mitochondrial OXPHOS in tumor cells, promoting production of large amounts of ATP and ROS accordingly (Ozturk et al., 2022).
The hypermethylation of the tumor suppressor gene PTEN promoter confines its expression, thereby inhibiting OXPHOS and triggering dysfunctional oxidative metabolism in esophageal cancer (EC) cells (Li et al., 2019). The changes in the CpG methylation of mitochondrial DNA can lead to various abnormalities of mitochondrial function, which promotes OXPHOS inducing ROS accumulation to inhibit hepatocellular carcinoma (HCC) progression (Zhao et al., 2021).
In addition, NOX, an imperative enzyme responsible for ROS production, has been reported to be regulated by DNA methylation. Both DNA methylation inhibitor 5-azacytidine and DNA methyltransferase 3A (DNMT3A) have been found to obstruct NOX methylation and reduce production of ROS, thereby allowing tumor cell survival (Damal Villivalam et al., 2021; Ma et al., 2018). Docetaxel, a commonly used clinical chemotherapy drug, has also been shown to preclude NOX promoter methylation and upgrade NOX levels, thereby inducing superoxide production and destroying vascular function of breast cancer cells (Szczepaniak et al., 2022).
Regulation of ROS elimination by DNA methylation
The hypermethylation of several enzymes related to ROS elimination has been investigated in various tumors. For example, DNMT3A mediates the hypermethylation in CpG islands upstream of SOD2 and silences SOD2 expression, resulting in mitochondrial oxidative stress in human mesenchymal stem cells (Jung et al., 2020). Promoter hypermethylation and suppression of GPX3 are associated with inflammatory breast carcinogenesis (Mohamed et al., 2014).
In addition, the promoter hypermethylation and expression silencing of NQO1, GSTpi, GPX, and MnSOD have been observed in cancer (Guo et al., 2015). In a high oxidative stress environment, tumor cells clean ROS mainly by increasing NADPH and GSH (Liu et al., 2022b). Recent studies have shown that GSH-related enzymes are principally influenced by DNA methylation or demethylation. For instance, demethylation of termed glutamine synthetase promoter enhances glutamine, which catalyzes the de novo synthesis of glutamate and ammonia by GS, sparking off ROS scavenging and tumor cells survival as well as drug resistance (Bott et al., 2015; Guo et al., 2021).
Jou et al. (2021) also have demonstrated that glutathione S-transferase Mu-5 (GSTM5) is hypermethylated in bladder cancer, which assists cancer cells invasion and metastasis by increasing GSH expression. PPP, a branch of glycolysis, is essential for ribonucleotides synthesis and an essential source of NADPH (Patra and Hay, 2014). The promoter hypermethylation and silencing of zinc finger DHHC-type containing 1 (ZDHHC1) have been found in multiple tumor cells, and reduction of ZDHHC1 precludes tumor growth by regulating PPP to induce oxidative stress (Le et al., 2020).
Fructose-1,6-bisphosphatase 1 (FBP1), a key metabolic enzyme, hydrolyses fructose 1, 6-bisphosphate to fructose 6-phosphate during gluconeogenesis. DNA demethylase TET2 activates FBP1 expression to promote clear cell renal cell carcinoma (ccRCC) growth via inhibition of glycolysis and PPP pathways (Zhang et al., 2022a). Moreover, cysteine metabolism is involved in the GSH synthesis and regulation of the ROS level. DNMT3B induces the promoter hypermethylation and suppression of cystathionine γ-lyase, a gene related to cysteine metabolism, contributing to the decrease of cysteine levels and increases of ROS levels, which is essential for promoting HCC proliferation and metastasis (Lin et al., 2021).
As previously mentioned, Keap1-Nrf2 signaling pathway is central to the cellular antioxidant defense mechanism (Guo et al., 2015). The promoter hypermethylation of Nrf2 and Keap1 has been detected in various types of tumors. Past research has shown that Keap1 gene promoter is frequently methylated in breast cancer (51%), CRC (53%), thyroid cancers (70.6%), and NSCLC (47%) (Guo et al., 2015), which is the main regulatory mechanism of Keap1 silencing in cancer. DNMT inhibitor genistein increases Keap1 expression through demethylation of CpG promoter, which leads to overexpression of Nrf2 and promotes lung adenocarcinoma (LUAD) cells proliferation (Liu et al., 2016).
Zhang et al. (2021b) reported that Nrf2 is decreased due to its promoter hypermethylation, which inhibits the expression of GPX4 and decreases the expression of ROS and lipid peroxides, thereby constraining ferroptosis caused by oxidative stress. In addition, there is evidence that DNMT1 and DNMT3A lead to the promoter methylation and silencing of Nrf2 to promote prostate cancer cells apoptosis (Zhang et al., 2013).
Regulatory Effects of RNA N6-Methyladenosine Modification on Redox Homeostasis in Cancer
N6-methyladenosine (m6A) has received increasing attention as a novel epigenetic modification, and is the most abundant post-transcriptional modification of internal RNA in eukaryote identified in the 1970s (Desrosiers et al., 1974). m6A modification is dynamic and reversible, mainly mediated by m6A-related regulators, including m6A methyltransferases (“writers,” such as METTL3, METTL14, and WTAP), demethylases (“erasers,” such as FTO and ALKBH5), and m6A-binding proteins (“readers,” such as YTHDF1/2/3, YTHDC1/2, and IGF2BP1/2/3).
m6A is the most prevalent RNA epigenetic regulator involved in multiple aspects of RNA metabolism through affecting messenger RNA (mRNA) splicing, nuclear export, translation efficiency, RNA stability, and microRNA (miRNA) processing (Chen et al., 2019). In recent years, the role of m6A modification in cancer has been extensively investigated. It is now clear that the expression of m6A modification and m6A-related regulators is dysregulated in various cancers, such as acute myeloid leukemia (AML), HCC, lung cancer, CRC, and breast cancer (Huang et al., 2020).
And m6A plays a dual role in carcinogenesis and tumor suppression. m6A regulators can change the m6A status of oncogenes or suppressor genes causing their up/downregulation, thus affecting tumor growth, differentiation, and metastasis (Wang et al., 2020). Emerging studies have reported that m6A modification can also affect antioxidant responses in cancer via multiple pathways, including regulating the generation of ROS, influencing antioxidant systems or affecting redox signaling, thus affecting tumorigenesis and tumor progression (Yang and Chen, 2021).
Regulation of ROS generation by m6A
m6A can directly affect ROS production by regulating key players in the mitochondrial respiration. Drosha and DGCR8 are two constituent proteins of the miRNA processing microprocessor complex, which are required for the processing of primary miRNAs into precursor miRNAs (Han et al., 2009). RALY is a fresh RNA-binding protein identified as a key constituent part of the Drosha complex. Overexpressed RALY-mediated miRNA processing is modulated by the m6A switch, and the upregulated miRNAs systematically downregulate the expression of metabolism-related genes and inhibit OXPHOS, resulting in CRC cells proliferation (Sun et al., 2021).
PGC-1α transcriptional coactivator is a master regulator of mitochondrial biogenesis and plays a complex role in redox regulation by decreasing mitochondrial production of ROS (Guo et al., 2018; Kaarniranta et al., 2018). FTO plays an antitumor role in ccRCC by inducing oxidative stress. Mechanically, FTO increases PGC-1α expression by reducing the m6A level of PGC-1α mRNA transcription, thereby restoring mitochondrial activity and inducing ROS production (Zhuang et al., 2019).
Adenylate kinase 4 (AK4) is localized in the mitochondrial matrix and modulates the cell energy metabolism. Tamoxifen-resistant breast cancer cells exhibit upregulation of m6A modification at the 5′ untranslated region (UTR) of AK4 mRNA. METTL3 can induce ROS production and p38 phosphorylation by increasing the expression of AK4, and inhibit mitochondrial apoptosis, thus promoting breast cancer resistance to tamoxifen (Liu et al., 2020).
Regulation of antioxidant systems by m6A
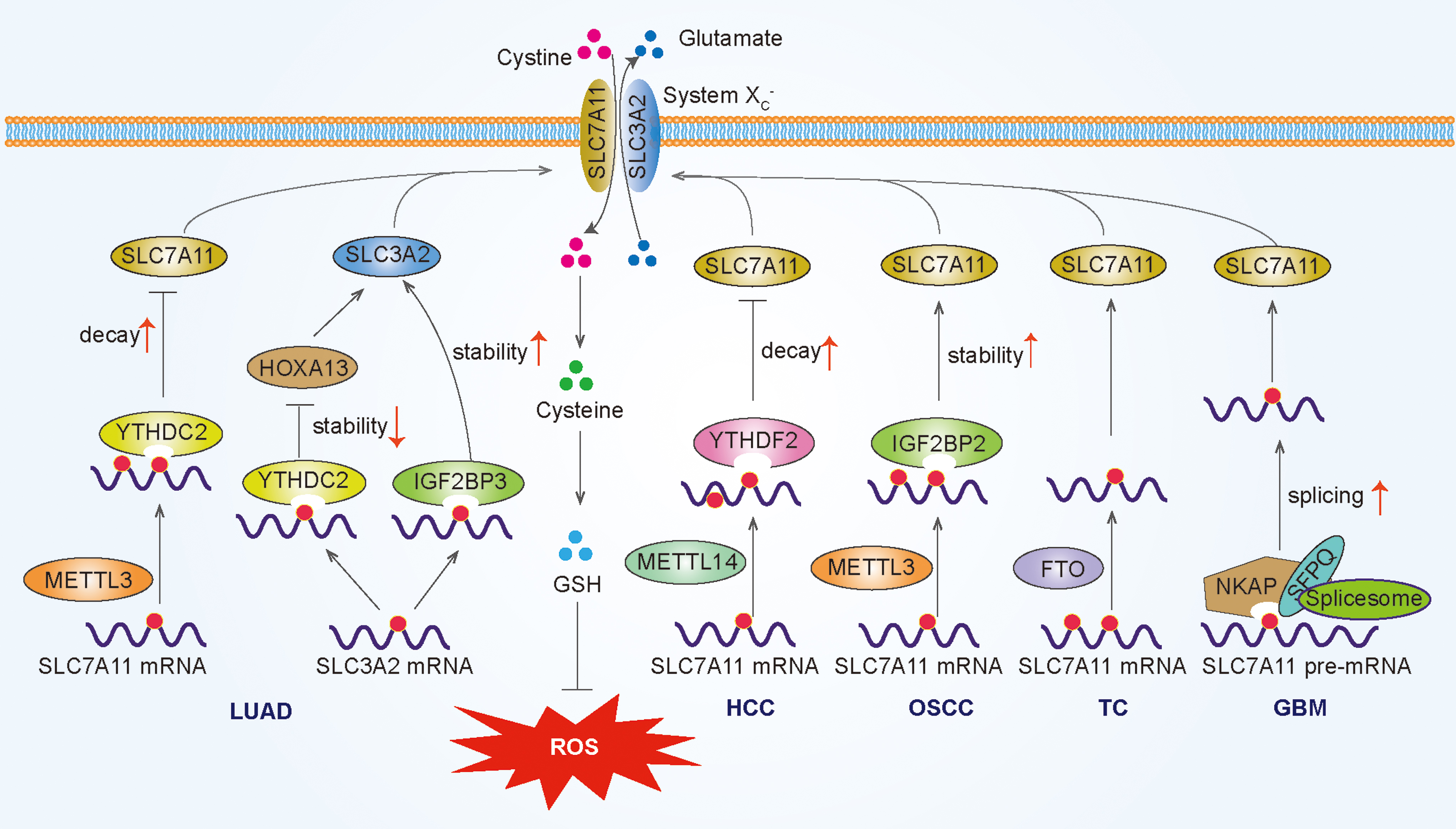
The system XC − is an important antioxidant transporter importing cystine for GSH biosynthesis to maintain cellular ROS homeostasis, composed of two subunits, solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2) (Dixon et al., 2014). Notably, SLC7A11 mRNA exhibits aberrant m6A modification in a variety of human tumors, and m6A plays a dual role in antioxidant response by mediating SLC7A11 up/downregulation. On the one hand, YTHDC2 exhibits antitumor activity via suppressing antioxidant system XC − in LUAD.
Mechanistically, m6A-methylated SLC7A11 transcripts are recognized by YTHDC2 to promote SLC7A11 mRNA degradation (Ma et al., 2021a). Hypoxia blocks ferroptosis of HCC cells via suppression of METTL14 triggered YTHDF2-mediated degradation of SLC7A11 mRNA (Fan et al., 2021). On the other hand, METTL3 promotes oral squamous cell carcinoma progression by enhancing IGF2BP2-dependent stabilization of SLC7A11 mRNA (Xu et al., 2021a).
Similarly, METTL3-mediated m6A modification enhances the stability of SLC7A11 mRNA to suppress ferroptosis in hepatoblastoma (Liu et al., 2022a). Another study showed that the downregulation of SLC7A11 m6A methylation mediated by FTO can prevent thyroid cancer progression (Ji et al., 2022). Furthermore, the RNA-binding protein NKAP acts as a new m6A-binding protein and promotes SLC7A11 mRNA splicing and maturation, thereby inhibiting glioblastoma cells ferroptosis (Sun et al., 2022). SLC3A2 is also regulated by m6A modification. Qiao and colleagues found that IGF2BP3 inhibits ferroptosis in LUAD cells by enhancing SLC3A2 mRNA stability in an m6A-dependent manner (Xu et al., 2022).
In another study, YTHDC2 suppresses SLC3A2 via inhibiting homeobox A13 (HOXA13) in an m6A-indirect manner inducing ferroptosis in LUAD cells (Ma et al., 2021b). In conclusion, m6A modification plays a dual role in redox homeostasis by mediating SLC7A11 up/downregulation to affect the system XC −, thereby affecting cancer progression mainly through ferroptosis (Fig. 3).
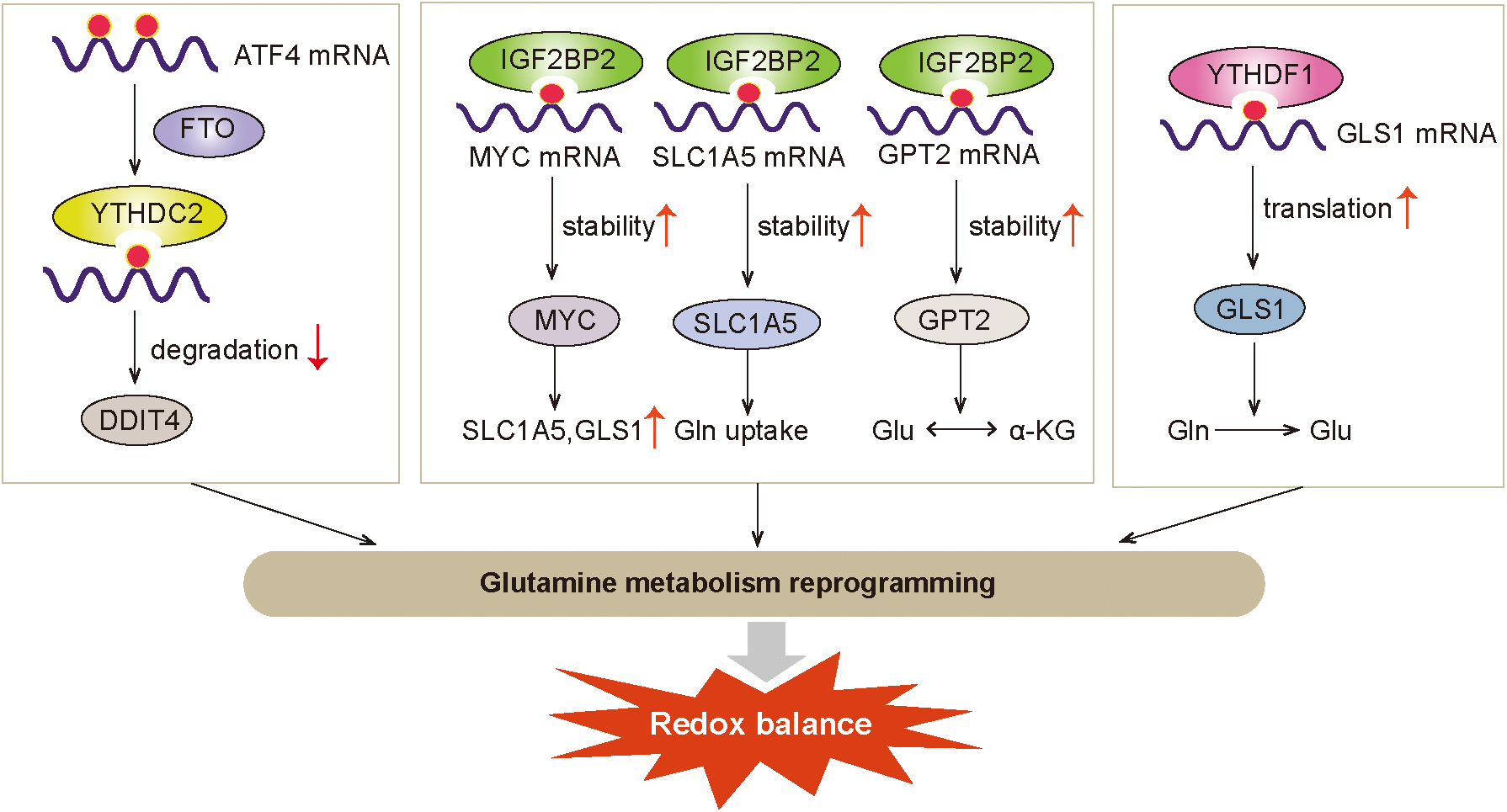
In cancer cells, glutaminolysis is an important nitrogen and carbon source for tumor cells to support tumor cell biosynthesis, and regulates intracellular redox homeostasis by producing antioxidants such as GSH and NADPH (Daye and Wellen, 2012; DeBerardinis and Cheng, 2010; DeBerardinis et al., 2007). Alanine-serine-cysteine transporter 2 (ASCT2 or SLC1A5 [solute carrier family 1 member 5]), a cell surface Na+-dependent neutral amino acid transporter, is primarily responsible for glutamine uptake (Scalise et al., 2018).
The key regulators of glutamine uptake and glutaminolysis have shown to be regulated by m6A modification. Chen and colleagues reported that IGF2BP2 is highly expressed in AML, playing an oncogenic role. IGF2BP2 promotes AML development and self-renewal of leukemia stem/initiating cells by upregulating the expression of key targets in the glutamine metabolic pathway such as MYC, glutamic pyruvate transaminase 2 (GPT2), and SLC1A5 in an m6A-dependent manner (Weng et al., 2022).
Another study has found that YTHDF1 is significantly upregulated in colon tumors and promotes protein synthesis of glutaminase GLS1 by binding to the 3’ UTR of GLS1 to modulate the GLS1-glutamine metabolism axis-mediated cisplatin resistance in CRC (Chen et al., 2021). In addition, glutaminolysis inhibition promoted activating transcription factor 4 (ATF4) mRNA expression by abrogating m6A modification and YTHDF2-mediated RNA decay in CRC cells to induce prosurvival autophagy during glutaminolysis inhibition (Han et al., 2021). These studies suggest that m6A modification promotes antioxidant response and cancer progression via regulating the expression of key targets in the glutamine metabolic pathway (Fig. 4).
Regulation of redox signaling by m6A
A large number of studies have found that the antioxidant response mediated by Nrf2 plays an important role in promoting the occurrence and development of tumors. m6A modification has been reported to alter redox state by influencing Nrf2 signaling (Fig. 5). In nonsmall cell lung carcinoma cells, the depletion of YTHDF1 mediates a decrease in the translation efficiency of Keap1, leading to the upregulation of Nrf2 and its downstream ARE response factor antioxidant AKR1C1, which render cancerous cells resistant to cisplatin treatment (Shi et al., 2019).
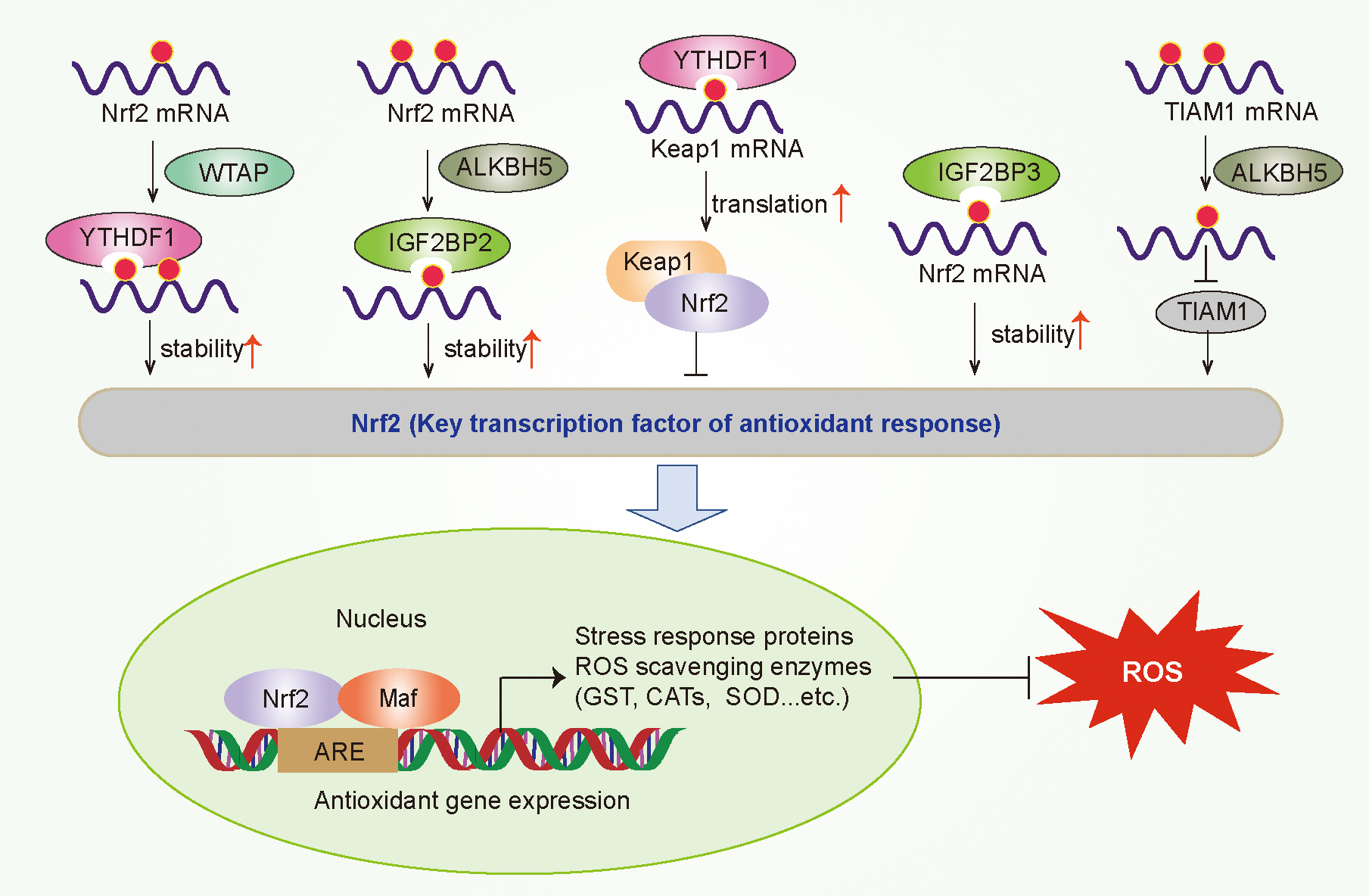
IGF2BP3 negatively regulates sorafenib-induced ROS and ferroptosis in HCC cells by promoting the stability of Nrf2 mRNA through identifying its m6A modification (Lu et al., 2022). WTAP has been reported to be upregulated in bladder cancer tissues and cells, associated with poor prognosis. WTAP can promote the m6A modification of Nrf2 mRNA at the 3′-UTR, which is recognized by YTHDF1 and enhances its mRNA stability to promote the viability of bladder cancer cells and inhibits erastin-induced ferroptosis (Wang et al., 2023).
ALKBH5 can increase the resistance of hypopharyngeal squamous cell carcinoma to ferroptosis via dysregulating NFE2L2/Nrf2 expression in an m6A-IGF2BP2–dependent mechanism (Ye et al., 2022). In addition, ALKBH5 inhibited thyroid cancer progression by inducing ferroptosis through m6A-TIAM1-Nrf2/HO-1 axis (Li et al., 2023c).
In addition to affecting Nrf2 signaling, m6A can regulate redox-related KRAS and MYC signal pathway. KRAS promotes ROS generation and metabolic reprogramming of cancer cells by regulating mitochondrial function, and has been reported to be abnormally activated in a variety of tumors such as NSCLC and CRC (Hu et al., 2012). In NSCLC, the high expression of FTO is positively correlated with the activation of KRAS signal transduction pathway (Shi et al., 2020).
MYC increases antioxidant capacity by activating Nrf2 transcription, and decreases mitochondrial production of ROS by downregulating PGC-1α and suppressing mitochondrial biogenesis (Chong et al., 2018; DeNicola et al., 2011). YTHDF2 recognizes m6A modification of MYC mRNA to increase the stability and expression of MYC, thereby mediating the viability of glioblastoma stem cells (Dixit et al., 2021).
Similarly, IGF2BP1 promotes tumorigenesis by upregulating MYC stability and expression in an m6A-dependent manner (Zhu et al., 2020). Downregulation of FTO increased m6A modifications of MYC mRNA, thereby enhancing YTHDF1-dependent translation of MYC mRNA and promoting proliferation and tumorigenesis in LUAD by metabolic reprogramming (Yang et al., 2021).
Collectively, these findings suggest that m6A affects tumorigenesis and tumor development by participating in redox regulation. However, the regulation of m6A modification on redox homeostasis in cancer is complex and dynamic, and its regulatory mechanism needs to be systematically and deeply studied.
Regulation of PTMs on Redox Homeostasis in Cancer
PTMs are covalent additions appended to the side chains of amino acids (Pan and Chen, 2022), mainly composed of methylation, phosphorylation, acetylation, ubiquitylation, SUMOylation, glycosylation, and ADP-ribosylation (Zhao and Shilatifard, 2019). In tumor cells, proteins undergone different PTMs are involved in the regulation of cell survival, cell cycle, and proliferation to provide a continuous proliferation signal, conducing to abnormally rapid proliferation of cancer cells (Han et al., 2018). Importantly, accumulating evidence has suggested that PTMs can also regulate redox homeostasis to participate in tumor development. Here, we focus on the effects of the major PTMs such as ubiquitination and acetylation on redox regulation.
Regulatory effects of ubiquitination on redox homeostasis in cancer
Ubiquitination refers to the binding of ubiquitin molecules to proteins through an isopeptide bond formed between the C terminus of ubiquitin and the side chain lysine of the target protein (Ramazi et al., 2020). Ubiquitination is essential for many physiological processes, including cell survival, differentiation, and innate and acquired immunity.
Like kinases, components of the ubiquitination system are frequently deregulated, leading to various diseases such as cancer (Mansour, 2018). Components of the ubiquitin system associated with cancer include E3 ligases, E1 and E2 enzymes, DUBS, and the proteasome. These members of the ubiquitin can regulate redox homeostasis through affecting redox-related transcription factors, antioxidant effectors, and ROS production to play a vital role in tumor progression (Pellegrino et al., 2022).
Ubiquitination regulates redox-related transcription factors
Previous studies have shown that many transcription factors including Nrf2, heat shock factor 1 (HSF1), FOXO, NF-κβ, and p53 regulate intracellular redox status, and have all been considered to be involved in tumor development (Hayes et al., 2020). These redox-related transcription factors have been reported to be regulated by ubiquitination in different tumors, and participate in tumorigenesis and progression.
Ubiquitination of P53
P53, an important tumor suppressor, can increase antioxidant capacity by activating the key regulator genes involved in GSH synthesis and NADPH production (Nguyen et al., 2018; Pan and Chen, 2022). Ubiquitination of P53 also plays an essential role in tumor progression, and its main forms of ubiquitination are monoubiquitination, K48-linked, and K63-linked polyubiquitination (Sun et al., 2020). Enzymes that mediate p53 ubiquitination and degradation mainly include COP1, CHIP, F-box, and WD-repeat-domain–containing 7 (FBXW7), HUWE1, RING1, RCHY1, RNF115, TRIM23, TRIM24, TRIM28, TRIM69, TRIM39, and TRIM71.
While p53 deubiquitylation is mediated by various deubiquitinating enzymes, such as OTUD1, OTUD3, OTUD5, ubiquitin-specific protease 7 (USP7), USP11, USP15, USP29, USP24, and USP42 (Brooks et al., 2007; Hock and Vousden, 2010; Hong et al., 2014; Sun et al., 2020). For example, Fan et al. (2013) found that USP7 inhibitor P22077 induced tumor cell apoptosis in neuroblastoma cells with intact USP7-HDM2-p53 axis. Besides, Yang et al. (2005) have identified a family of small molecules (HLI98) that can prohibit the E3 activity of HDM2 to suppress the degradation of P53 enhancing its tumor suppressor function.
In addition, Zeng et al. found that SHARPIN could promote ubiquitination and degradation of p53, thereby withholding the expression of antioxidant proteins GPX4, SOD-1, and SOD-2, inducing excessive oxidative stress and restraining the proliferation of cholangiocarcinoma cells (Zeng et al., 2022).
Ubiquitination of Nrf2
Nrf2 is the most critical antioxidant transcription factor maintaining cellular redox homeostasis (Wang et al., 2021). The stability and transcriptional activity of Nrf2 are tightly regulated by the ubiquitination-mediated proteasome pathway. Ubiquitination of Nrf2 is mediated by E3 ubiquitin ligases Keap1-Cullin3-RBX1 and β-TrCP-SKP-CUL1-RBX1 (Tebay et al., 2015). Nrf2 can also be deubiquitinated by deubiquitinase 3 (DUB3). DUB3 can suppress the K48-linked ubiquitination of Nrf2, and increase the stability and transcriptional activity of Nrf2, thereby inducing drug resistance of CRC (Zhang et al., 2019a).
In NSCLC, USP11 is positively correlated with Nrf2 expression, and USP11 promotes cell proliferation and ferroptotic resistance by deubiquitinating and stabilizing Nrf2 (Meng et al., 2021). Ge et al. (2017) also found that iASPP, a known p53 inhibitor, can compete with Nrf2 for Keap1 binding, conducing to Nrf2 accumulation by reducing Nrf2 ubiquitination, which contribute to cancer growth and drug resistance.
Ubiquitination of NF-κB
NF-κB represents a family of transcription factors including heterodimeric or homodimer combinations of p50, p52, p65/RelA, RelB, and c-Rel subunits (Morgan and Liu, 2011). NF-κB is recognized as a master switch for inflammation, and regulates redox through the expression of pro-oxidative genes and widespread H2O2 production (Oliveira-Marques et al., 2009). NF-κB is also regulated by the ubiquitination-mediated proteasome pathway. In breast cancer, F-box and WD-repeat-domain–containing 2 (FBXW2), as an E3 ligase, mediates the ubiquitination and degradation of NF-κβ p65 to suppress tumorigenesis and paclitaxel resistance (Ren et al., 2022).
Tripartite motif (TRIM) family proteins play a critical role as E3 ubiquitin ligases in cancer progression. TRIM22 promotes glioblastoma cells proliferation by activating NF-κB. The mechanism is that TRIM22 catalyzes the K48-linked ubiquitination and degradation of IκBα, a negative regulator of NF-κB, leading to the increase of NF-κB (Ji et al., 2021). NF-κβ can also be activated by TRIM47. NF-κB–activating protein kinases, such as protein kinase C epsilon (PKC-ɛ) and protein kinase D3 (PKD3), are directly ubiquitinated and stabilized by TRIM47 to promote the proliferation and tamoxifen resistance of breast cancer cells (Azuma et al., 2021).
Ubiquitination of FOXO
FOXO transcription factors are fatal tumor suppressors, and participate in the maintenance of intracellular redox homeostasis by inducing genes associated with ROS elimination and mitochondrial redox improvement (Klotz et al., 2015). FOXO1, FOXO3a, and FOXO4 are unstable proteins that are destroyed by ubiquitin-mediated proteolysis. E3 ligases SKP2 and NEDD4-like E3 ubiquitin ligase (NEDD4L) catalyze ubiquitination and degradation of FOXO1 to suppress breast cancer stemness via downregulating SOX2 transcription (Yu et al., 2019).
In addition, SKP2 facilitates ubiquitin-mediated FOXO1 degradation to suppress tumor in a mouse lymphoma model (Huang et al., 2005). FOXO1 is also modulated by ubiquitin-conjugating enzyme E2 T (UBE2T). UBE2T motivates cell proliferation and precludes apoptosis in NSCLC by the ubiquitination-mediated FOXO1 degradation (Yin et al., 2020). E3 ligase MDM2 also plays a role in the polyubiquitination and degradation of FOXO3 (Hu et al., 2017; Jiramongkol and Lam, 2020).
Human telomerase reverse transcriptase (hTERT) promotes cell invasion in gastric cancer by enhancing E3 ligase MDM2-mediated FOXO3a ubiquitination and degradation (Hu et al., 2017). TRIM33 protects ROS-induced apoptosis by inhibiting FOXO3a ubiquitylation and degradation (Zheng et al., 2014). FOXO3a is also a target of deubiquitinase USP9x (Zheng et al., 2014).
Ubiquitination affects antioxidant systems
In addition to regulating redox-related transcription factors, ubiquitination can affect tumor progression by affecting the activities of antioxidant systems, including NADPH and GSH (Meng et al., 2022; Yang et al., 2022b; Zhang et al., 2023). Recent evidence suggests that ubiquitination affects the production of GSH through regulating the system XC − with glutamine metabolism, and regulates NADPH production via the regulation of key enzymes such as glucose-6-phosphate dehydrogenase (G6PD), malic enzyme 1 (ME1), and IDH1 (Fig. 6).
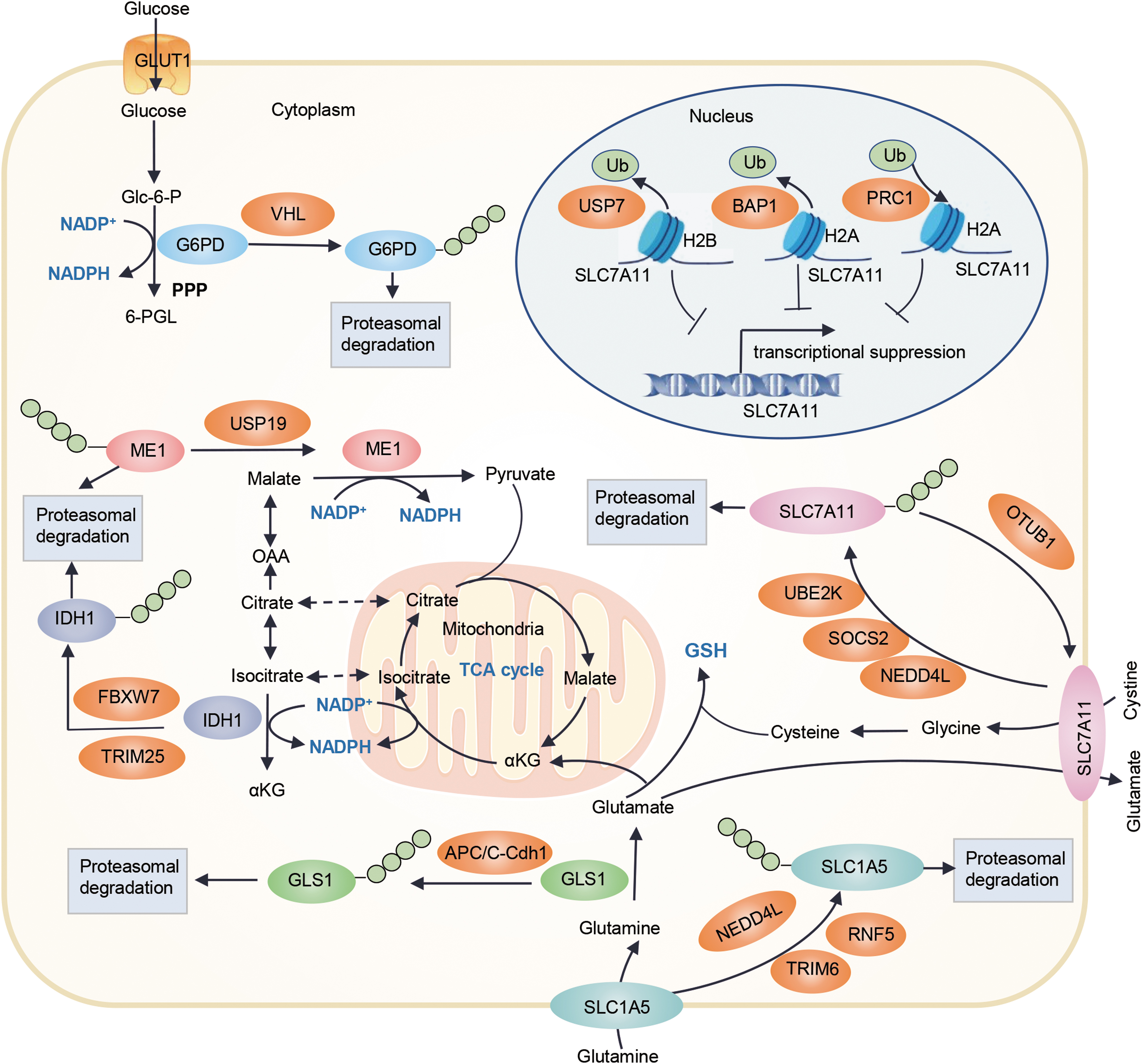
Regulation of antioxidant system Xc − by ubiquitination
As previously described, SLC7A11, an essential component of the antioxidant system Xc−, can promote antioxidant defense by importing cystine for GSH biosynthesis. At present, both ubiquitination- and deubiquitination-mediated SLC7A11 protein stability in tumors have been reported. E3 ubiquitin ligases NEDD4L and SOCS2 have shown to regulate SLC7A11 ubiquitination in breast cancer and HCC. Shao and colleagues demonstrated for the first time that SOCS2 can enhance the ubiquitination and degradation of SLC7A11, which ultimately leads to the occurrence of ferroptosis and the radiosensitization of HCC (Chen et al., 2023).
Similarly, NEDD4L-mediated SLC7A11 ubiquitination has also been demonstrated in breast cancer radiotherapy (Liu et al., 2021). In addition, OTU deubiquitinase, ubiquitin aldehyde binding 1 (OTUB1), obstructs the proteasomal-dependent degradation of SLC7A11 by removing SLC7A11 ubiquitination and enhances the sensitivity of cells to erastin-induced ferroptosis (Liu et al., 2019).
Zhou and colleagues also found that ubiquitin-conjugating enzyme E2K (UBE2K) prohibits SLC7A11 ubiquitination to augment its expression by directly combining with LINC00578, thereby inhibiting ROS generation and ferroptosis to promote pancreatic cancer progression (Li et al., 2023a). In addition, histone ubiquitination can regulate SLC7A11 expression by mediating its transcription. A study has shown that deficiency of nuclear deubiquitinase BRCA1-associated protein-1 (BAP1) can upregulate SLC7A11 expression by suppressing histone 2A ubiquitination (H2Aub) on the SLC7A11 promoter and lead to cancer cells resistance to ferroptosis (Zhang et al., 2018).
H2Aub modified by the ubiquitin ligase polycomb repressive complex 1 (PRC1) at the SLC7A11 promoter also inhibits SLC7A11 (Zhang et al., 2019b). Wang et al. (2019a) demonstrated that histone 2B monoubiquitylation (H2Bub) mediated by USP7 can also mediate transcriptional activation of SLC7A11 to suppress ferroptosis sensitivity. Together, these recent findings indicate the critical role of the histone ubiquitination and ubiquitination-mediated proteasomal degradation in regulating SLC7A11 expression to affect antioxidant regulation and ferroptosis in cancer.
Regulation of glutamine metabolism by ubiquitination
As described above, glutamine metabolism can produce antioxidants such as GSH and NADPH to maintain redox homeostasis. E3 ubiquitin ligase NEDD4L can suppress mitochondrial metabolism by promoting ubiquitin proteasomal degradation of SLC1A5 and inhibiting glutamine uptake in pancreatic cancer cells (Lee et al., 2020).
In addition, NEDD4L inhibits esophageal squamous cell carcinoma progression and glutamine metabolism by ubiquitination of c-Myc to reduce the expressions of SLC1A5. E3-ubiquitin ligase tripartite motif-containing 6 (TRIM6) hampers glutamine uptake and ferroptosis in lung cancer cells by promoting SLC1A5 ubiquitination and degradation (Zhang et al., 2023).
Jeon et al. (2015) found that the E3 ubiquitin ligase ring finger protein 5 (RNF5) can modulate glutamine uptake, tricarboxylic acid cycle component levels, simultaneously increasing autophagy and cell death by ubiquitinating the L-glutamine carrier proteins SLC1A5 and SLC38A2. GLS1, the first enzyme in glutaminolysis, is also targeted for destruction by anaphase-promoting complex/cyclosome-Cdh1 (APC/C-Cdh1) (Colombo et al., 2011). Together, these results suggest that ubiquitination may play an important role in redox regulation by modulating glutaminolysis.
Regulation of NADPH production by ubiquitination
The G6PD-mediated PPP is the main source of cytosolic NADPH production, and G6PD can also be involved in tumor development by regulating the antioxidant system (Zhang et al., 2022b). Interestingly, G6PD expression has been confirmed to be significantly reduced under hypoxic conditions and reversed by the proteasome inhibitor MG132 (Chettimada et al., 2015). Recently, Wang and colleagues found that E3 ubiquitin ligase von Hippel–Lindau (VHL) can directly bind and ubiquitinate G6PD at the K366 and K403 to regulate the stability of G6PD (Meng et al., 2022).
In addition to NADPH production via PPP, IDHs and MEs are involved in the NADPH pools in the cytoplasm and mitochondria. FBXW7, as an essential recognition factor of the ubiquitin proteasome degradation pathway, has been confirmed to be negatively correlated with the expression level of IDH1 in glioma. Loss of FBXW7 leads to NADPH homeostasis disruption in IDH1 mutant glioma cells by blocking sterol regulatory element-binding protein 1 (SREBP1) degradation to significantly increase the expression of neomorphic mutant IDH1, which increases PPP activity and NADPH consumption, conferring high sensitivity to radiotherapy (Yang et al., 2022b).
Li and colleagues demonstrated that ubiquitin-specific peptidase 19 (USP19) antagonizes RNF-1–mediated ME1 degradation by deubiquitination, thereby promoting NADPH generation and ROS elimination, which plays an important role in colorectal carcinogenesis (Zhu et al., 2021). In addition, Zhao et al. (2022) showed that ME2 can increase the production of 2-hydroxyglutarate by regulating the breakdown of glutamine and the reaction of pyruvate and NADPH, thereby reducing MDM2-mediated ubiquitination of mutant p53, finally leading to tumorigenesis.
Ubiquitination modulates ROS production
Ubiquitination plays an important role in maintaining ROS homeostasis by regulating oncogenes that promote ROS generation such as KRAS and Rac1 (Chong et al., 2018). Monoubiquitination of KRAS at lysine147 can increase GTP loading and its affinity for specific downstream effectors such as PI3K and Raf (Sasaki et al., 2011). Cullin3-based E3 ubiquitin ligase complex facilitates the polyubiquitination and ubiquitin-proteasome–mediated degradation of KRAS (Abe et al., 2020). SAG is part of the SCF (Skp1, Cullins, and F-box proteins) E3 ligases, the largest member of the E3 ubiquitin ligase family. It can inhibit KRAS activation through the Ras-Erk pathway, thereby preventing ROS production and inhibiting papilloma proliferation (Xie et al., 2015).
The small RhoGTPase Rac1 mediates ROS production through activating NOX, and participates in the progression of breast cancer, melanoma, liver cancer, and other tumors by affecting cell proliferation, adhesion, and apoptosis (Abdrabou and Wang, 2018; Svensmark and Brakebusch, 2019). The degradation of Rac1 is regulated via their ubiquitination-mediated proteasomal pathway. E3 ubiquitin ligases HACE, XIAP, and c-IAP1 promote ubiquitylation of Rac1 at K147 (Abdrabou and Wang, 2018; Woolery et al., 2014). It has been shown that loss of HACE1 can confine the ubiquitylation and degradation of Rac1, leading to an increase of Rac1 activity, which accelerates ROS production contributing to breast cancer progression (Kumar et al., 2019).
The NOX family of enzymes is responsible for the generation of ROS (Harrison and Selemidis, 2014). Recent evidence suggests that ubiquitination is a novel mechanism of NOX enzymes in cancer, and the mechanism varies significantly depending on the NOX isoform. Heat shock protein 90 (Hsp90) has been identified as a novel regulator of NOX1, NOX2, and NOX5, and the role of Hsp90 is opposed by Hsp70 and CHIP, which motivate the ubiquitination-mediated degradation of NOX proteins and reduce ROS production (Chen et al., 2012).
Silencing deubiquitinating enzymes CYLD accelerates the degradation of NOXO1 protein by promoting ubiquitination and regulates prostate cancer progression (Haq et al., 2022). NOX4 is polyubiquitinated at K48 by Connexin32 to promote its degradation and reduction of H2O2 (Chen et al., 2020b). In CRC, Xu et al. (2021b) reported that PRDX1 can mediate NOXA degradation via UBE2F-Neddylated cullin-5 (CUL5). Another study also found that NOXO1 activity is regulated by E3 ligase CBl-mediated ubiquitination in CRC cells (Joo et al., 2016). Overall, enzymes in the ubiquitin system are involved in cancer regulation through a novel mechanism regulating NOX enzymes and may be potential therapeutic targets.
Regulatory effects of acetylation on redox homeostasis in cancer
Protein acetylation modification is the process by which an acetyl group is added to a protein lysine residue with the action of acetyltransferase, which can be reversed by deacetylase. Abnormal acetylation levels may cause initiation and progression of tumors, and are closely related to the malignant phenotype of tumors (Yang et al., 2022a). Lysine deacetylases are divided into two subgroups: Zn2+-dependent histone deacetylases (HDACs) and NAD+-dependent deacetylases (sirtuins) (Wu et al., 2022).
In this review, we emphasize on the role of sirtuins in redox regulation. We reviewed that sirtuins regulate oxidative stress in cancer through regulating redox pathways, including AMPK, Nrf-2, FOXO, and NF-κβ.
Acetylation affecting redox homeostasis in cancer by regulating the AMPK signaling pathway
AMPK is an important pathway associated with redox regulation, which is often activated under conditions of oxidative stress. Activated AMPK plays an antioxidant role through various pathways such as promoting insulin sensitivity and fatty acid oxidation. SIRT1, SIRT3, SIRT4, and SIRT6 have been investigated to regulate AMPK pathway. Deacetylation of liver kinase B1 (LKB1), an upstream regulator of AMPK, can be regulated by SIRT1 and SIRT3, and concurrently increases its activity leading to activation of AMPK (Li et al., 2020; Vancura et al., 2018). SIRT1 can also reverse chemoresistance and cancer stemness properties of gastric cancer by suppressing the AMPK pathway (An et al., 2020).
In addition, SIRT6 can prompt the expression of AMPK and upregulate the expression of antioxidant genes to inhibit oxidative stress (Wang et al., 2016). Our research studies have shown that SIRT4 is also involved in the regulation of AMPK. On the one hand, SIRT4 inhibits pancreatic tumorigenesis by activating AMPKα and inducing autophagy. On the other hand, SIRT4 deletion promotes HCC progression and metastasis by inactivating AMPKα and promoting glutamine catabolism (Li et al., 2023b; Wang et al., 2019b).
Acetylation affecting redox homeostasis in cancer by regulating the Nrf2 signaling pathway
Nrf2 is an intracellular transcription factor that can protect cells from oxidation by regulating the expression of antioxidant genes. SIRT1 activates Nrf2 via transforming the structure of Keap1, leading to Nrf2 translocation into the nucleus and upregulating the expression of GSH S-transferase and glucuronosyltransferase to resist oxidative stress (Tang et al., 2014).
In addition, Cao et al. (2016) found that SIRT2 could regulate Nrf2 levels by regulating Akt phosphorylation, and thus changing the levels of glutamate cysteine ligase (GCL) and GSH. Cai et al. (2021) also reported that SIRT6 restraint could inactivate Keap1/Nrf2 signaling pathway and downregulate GPX4, thereby triggering ferroptosis and overcoming drug resistance in gastric cancer cells. All this evidence suggests that SIRT1, SIRT 2, and SIRT 6 play an antioxidant role in tumor regulation through Nrf2.
Acetylation affecting redox homeostasis in cancer by regulating the FOXO signaling pathway
As mentioned earlier, FOXO is an important antioxidant tumor suppressor gene, which is regulated by SIRT1 and SIRT3. For example, SIRT3 can activate the expression of FOXO3 and subsequently promote the transcription of antioxidant genes such as CAT and MnSOD, to eliminate ROS (Kwon et al., 2015). Huo et al. (2017) also found that when ROS increased, the expression of SIRT1 decreased, causing increase in the expression and acetylation of FOXO3a.
Besides, it has been shown that SIRT1 downregulates FOXO-mediated apoptotic pathways under oxidative stress conditions, thereby favoring the survival of cancer cells under stress (Ford et al., 2005). SIRT1 has also been reported to regulate FOXO in gastric cancer and breast cancer (An et al., 2020). In breast cancer, SIRT1 can also directly co-operate with FOXO and regulate FOXO pathway involved in tumor metastasis (Dilmac et al., 2022). The above studies indicate that SIRT1 and SIRT3 can regulate FOXO to enhance antioxidant defense and promote tumor progression.
Acetylation affecting redox homeostasis in cancer by regulating the NF-κB signaling pathway
NF-κB is a nuclear transcription factor that can enhance the production of ROS and promote oxidative stress. Recent studies have shown that SIRT1-mediated deacetylation can reduce ROS level through inhibiting NF-κB activity in cancer. For example, SIRT1 promotes HCC growth by inducing the deacetylation of NF-κB p62 and suppressing the NF-κB activity (Feng et al., 2021). Zheng et al. (2020) have reported that metformin can enhance the SIRT1 pathway, which further increase the expression of NF-κB p65 and finally promote cancer cell pyroptosis.
Another study has found that SIRT1 reduces the sensitivity to radiotherapy, and increases the drug resistance of lung cancer cells by restraining the expression of NF-κB and its downstream regulators (Ji et al., 2018). Unlike SIRT1, SIRT3 can be activated by the MTDH-NFκB-p65 complex to enhance MnSOD levels and promote EMT in triple negative breast cancer cells (Neeli et al., 2020). In addition, icariin has been investigated to inhibit the activation of the NF-κB/EMT pathway, resulting in the upregulation of SIRT6 expression and leading to the inhibition of breast cancer cell migration and invasion (Song et al., 2020).
Conclusion
Given that ROS play a dual role in tumorigenesis and development, cancer cells exhibit aberrant redox homeostasis, which promotes tumor malignant phenotypes while avoiding various forms of oxidative stress-induced cell death. Consequently, there is an urgent need to understand the mechanisms of maintaining redox homeostasis in cancer cells. With the extensive study of DNA/RNA methylation and PTMs in tumors, the understanding of their functional mechanism in tumor progression has become increasingly comprehensive and profound.
Based on the above studies, it provides strong evidence that DNA/RNA methylation and PTMs as key regulators of redox homeostasis play critical roles in tumor progression. Therefore, exploring the potential mechanisms of these modifications in the redox regulation will provide new research directions for cancer progression mechanisms and therapeutic targets. The studies summarized herein suggest that DNA/RNA methylation and PTMs can regulate redox homeostasis in multiple manners including affecting key regulators in ROS production, elimination, and redox-related signaling, thereby participating in tumor progression.
In summary, expanding knowledge of the mechanistic role of epigenetic modifications and PTMs in regulating ROS homeostasis would contribute to the development of new strategies based on redox regulation for cancer treatment.
Footnotes
Acknowledgment
The authors would like to extend their gratitude to the Department of Clinical Laboratory of the Second Hospital of Shandong University for supporting this work.
Authors' Contributions
Q.Q. and S.Y. surveyed the literature, collected information, and wrote the article. J.L. collected the related references and prepared the figures. L.D. and P.L. devised the idea and revised the article. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This work was supported by the Natural Science Foundation of Shandong Province (ZR2020QH197) and the National Natural Science Foundation of China (82130067 and 82002228).