
Abstract
Aims:
Scavenger receptor class B type I (SRBI) promotes cell cholesterol efflux and the clearance of plasma cholesterol. Thus, SRBI deficiency causes abnormal cholesterol metabolism and hyperlipidemia. Studies have suggested that ferroptosis is involved in lipotoxicity; however, whether SRBI deficiency could induce ferroptosis remains to be investigated.
Results:
We knocked down or knocked out SRBI in renal HK-2 cells and C57BL/6 mice to determine the expression levels of ferroptosis-related regulators. Our results demonstrated that SRBI deficiency upregulates transferrin receptor 1 (TFR1) expression and downregulates ferroportin expression, which induces iron overload and subsequent ferroptosis in renal tubular epithelial cells. TFR1 is known to be regulated by hypoxia-inducible factor-1α (HIF-1α). Next, we investigated whether SRBI deletion affected HIF-1α. SRBI deletion upregulated the mRNA and protein expression of HIF-1α, and promoted its translocation to the nucleus. To determine whether HIF-1α plays a key role in SRBI-deficiency–induced ferroptosis, we used HIF-1α inhibitor and siHIF-1α in HK-2 cells, and found that downregulation of HIF-1α prevented SRBI-silencing–induced TFR1 upregulation and iron overload, and eventually reduced ferroptosis. The underlying mechanism of HIF-1α activation was explored next, and the results showed that SRBI knockout or knockdown may upregulate the expression of HIF-1α, and promote HIF-1α translocation from the cytoplasm into the nucleus via the PKC-β/NF-κB signaling pathway.
Innovation and Conclusion:
Our study showed, for the first time, that SRBI deficiency induces iron overload and subsequent ferroptosis via the HIF-1α/TFR1 pathway.
Introduction
Lipids are essential for cell survival and play important roles in cells (Li et al., 2022b; Slatter et al., 2016). However, in nonadipocytes, excessive accumulation of lipid intermediates can result in changes in the intracellular environment, cellular dysfunction, and even cell death, which is known as lipotoxicity (Li et al., 2017). The kidney is one of the organs most vulnerable to lipotoxicity (Liu et al., 2022). Several studies have shown that serum lipid abnormalities and renal lipid accumulation are closely associated with renal disease (Opazo-Rios et al., 2020; Vaziri, 2016).
Scavenger receptor class B type I (SRBI)-deficient and apolipoprotein E (ApoE)-deficient mice are two types of models commonly used for hyperlipidemia. SRBI is a transporter that promotes cell cholesterol efflux from the plasma membrane to high-density lipoprotein (HDL), which plays an important role in reverse cholesterol transport (Chistiakov et al., 2017; Wang et al., 2007).
Innovation
The findings of this study indicate that scavenger receptor class B type I (SRBI) is associated with not only cholesterol metabolism but also iron metabolism and ferroptosis, thereby expanding its functional scope. In summary, this investigation demonstrated for the first time that SRBI deficiency induces ferroptosis via the hypoxia-inducible factor-1α/transferrin receptor 1 signaling pathway in renal tubular epithelial cells. These results provide insights into the involvement of SRBI in ferroptosis and suggest it as a potential therapeutic target for the prevention and treatment of organ and cellular damage resulting from ferroptosis.
Clearance of plasma cholesterol is mediated by hepatic SRBI. SRBI mutations can cause the loss of SRBI function, resulting in reduced cholesterol efflux. Unlike hyperlipidemia caused by ApoE deficiency, which mainly results in elevated plasma triglycerides, SRBI-deficient mice have higher total and free cholesterol in the plasma.
Lipotoxicity is closely related to inflammation, oxidative stress, apoptosis, and ferroptosis (Bai et al., 2020; Katsoulieris et al., 2010). Ferroptosis is a new type of regulated cell death (Dixon et al., 2012). It refers to iron-dependent cell death, and is mainly characterized by glutathione (GSH) depletion, increased reactive oxygen species (ROS), and lipid peroxidation (Bai et al., 2020). Glutathione peroxidase 4 (GPX4) is a GSH-dependent enzyme, and its inactivation leads to ROS accumulation and accelerates lipid peroxidation, thus inducing cells to be more susceptible to ferroptosis (Friedmann Angeli et al., 2014). Ferroptosis has been linked to hyperlipidemia-related diseases (Bai et al., 2020; Wang et al., 2022; Yu et al., 2022); however, the underlying mechanisms remain unclear.
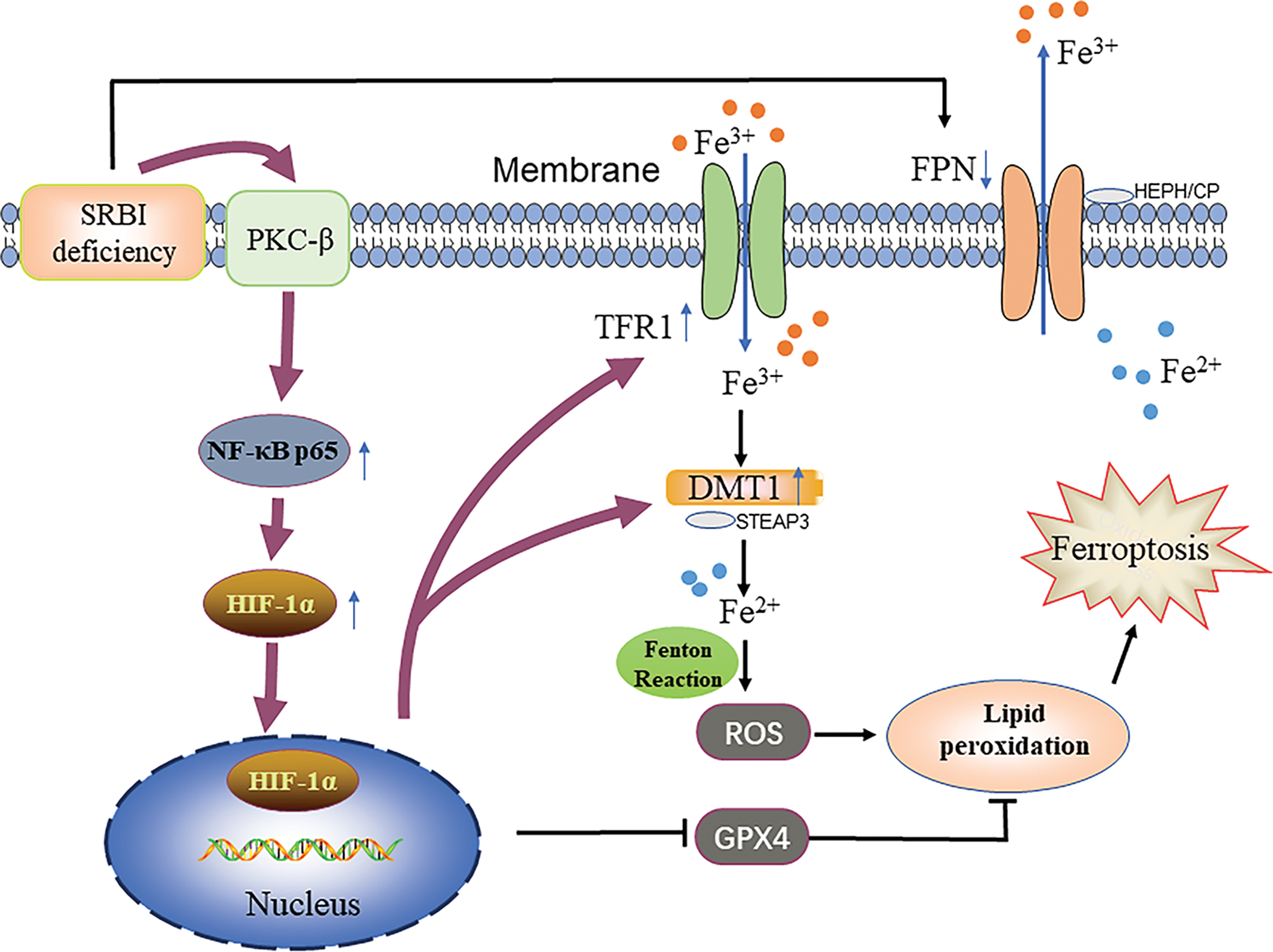
Interestingly, this study found that SRBI knockout mice developed iron overload significantly in their kidneys, especially in renal tubular cells, which was more obvious than that in ApoE-deficient mice, indicating that iron overload is probably related to SRBI gene deletion, not solely explained by lipid deposition. Therefore, we hypothesized that SRBI deficiency can induce iron overload and subsequent ferroptosis in renal tubular epithelial cells, and further explored the underlying mechanism (Fig. 1).
Results
SRBI deficiency induced oxidative stress and inflammation in the mouse kidney
SRBI deficiency is accompanied by dyslipidemia. First, we assessed the serum lipid profiles of the SRBI-deficient mice. Compared with wild-type (WT) mice, there were significantly elevated plasma total and free cholesterol levels in SRBI-deficient mice, whereas plasma triglyceride levels did not increase (Fig. 2A). Hematoxylin and eosin (HE) and periodic acid-Schiff (PAS) staining were performed on kidney sections to evaluate renal histology, and the results showed that tubular damage was significantly increased in SRBI-deficient mice, including tubular necrosis, vacuolization, and loss of brush borders (Fig. 2B, C).

In addition, indicators of oxidative stress and inflammation were assessed in both the groups. As shown in Figure 2D, SRBI-deficient mice had higher ROS levels than WT mice. Dihydroethidium (DHE) staining of frozen sections of the mouse kidney also showed increased DHE staining in SRBI-deficient mice (Supplementary Fig. S1A, B). Western blot analysis showed that the protein expression of p47phox, a crucial regulator of NADPH oxidase (Nox) activation, was higher in SRBI-deficient mice than that in WT mice (Fig. 2E, F).
Quantitative real-time polymerase chain reaction (PCR) results showed that the expression of renal tubule injury markers, kidney injury molecule 1 (Kim1) and neutrophil gelatinase-associated lipocalin (Ngal), were upregulated in SRBI-deficient mice, indicating that SRBI-deficient mice exhibit severe kidney injury. In addition, the mRNA expression of inflammatory cytokine Il-6 significantly increased and anti-inflammatory cytokine Il-10 decreased in SRBI-deficient mice compared with WT mice, while mRNA levels of tumor necrosis factor Tnf-α also increased and Il-1β tended to increase, indicating that SRBI deficiency evoked an inflammatory response in the mouse kidney.
Quantitative real-time PCR also showed that the mRNA levels of Nox1 and Nox4 were upregulated, indicating considerably higher levels of oxidative stress in SRBI-deficient mice (Fig. 2G). As shown in Figure 2H, sections from SRBI-deficient mice showed increased F4/80-positive areas. Moreover, Ly6G-positive areas were slightly increased in SRBI-deficient mice (Fig. 2I), indicating neutrophil infiltration and macrophage activation in the kidneys of SRBI-deficient mice. Overall, SRBI deficiency causes hypercholesterolemia, pathological kidney injury, oxidative stress, and inflammation.
SRBI-deficient mice exhibit significant iron overload in renal tubular epithelial cells
Hyperlipidemia and oxidative stress are intimately associated with ferroptosis (Bai et al., 2020; Zhou et al., 2022). It has been reported that ferroptosis is involved in lipid peroxidation in aortic endothelial cells of ApoE-deficient mice (Bai et al., 2020). However, whether ferroptosis is involved in renal lipotoxicity in ApoE- and SRBI-deficient mice remains to be investigated. To comprehensively analyze changes in iron levels and distribution in ApoE- and SRBI-deficient mice, Prussian blue staining of the kidney sections was performed.
It suggests that SRBI-deficient mice exhibit significant iron overload in their renal tubular epithelial cells compared with WT mice, while there were no obvious differences between ApoE-deficient and WT mice (Fig. 3A, B). Quantitative determination of iron content also showed that iron concentrations were significantly increased in the kidneys of SRBI-deficient mice (Fig. 3C). Taken together, these results suggested that iron overload in renal tubular epithelial cells of SRBI-deficient mice may be caused by SRBI knockout.

Due to the higher iron concentrations in renal tubular epithelial cells in SRBI-deficient mice, other tubular pathological changes were further evaluated. Masson's trichrome staining indicated that SRBI-deficient mice had increased collagen deposition in the renal tubular interstitium (Fig. 3D). The expression levels of fibrosis-related proteins were assessed by Western blot. The results showed that the protein level of α-smooth muscle actin tended to increase, and the expression of fibronectin was upregulated in SRBI-deficient mice (Fig. 3E), indicating that SRBI-deficient mice were more susceptible to renal fibrosis.
Subsequently, the expression levels of autophagy-related proteins were detected using Western blot. Compared with WT mice, SRBI-deficient mice showed a decrease in the ratio of LC3II/LC3I and a significant increase in the level of SQSTM1/P62 protein (Fig. 3F), indicating impaired autophagic flux in the mouse kidney. The expression level of Beclin1 showed no significant change in SRBI-deficient mice (Supplementary Fig. S2A, B).
TUNEL staining revealed more dead cells in kidney tissues of SRBI-deficient mice (Fig. 3G, H), indicating that SRBI deficiency induces cell death. As shown in Supplementary Figure S3, flow cytometry analysis showed that SRBI silencing promoted apoptosis in HK-2 cells, which was consistent with the TUNEL staining results. Taken together, these results demonstrated that SRBI deficiency has multiple effects on mouse kidneys, including iron overload, fibrosis, impaired autophagy, and apoptosis.
SRBI silencing induced ferroptosis in HK-2 cells
SRBI-deficient mice exhibit significant iron overload and oxidative stress in the renal tubular epithelial cells, which may be due to lipotoxicity or SRBI deletion. To further determine the role of SRBI in ferroptosis progression, small-interfering RNA (siRNA) against SRBI was transfected into tubular epithelial HK-2 cells (Fig. 4A). SRBI-deficient mice have higher plasma total and free cholesterol levels. Therefore, HK-2 cells were stimulated with 50 μg/mL cholesterol to simulate hypercholesterolemia. Intracellular ROS production was detected by 2′,7′-dichloro-dihydro fluorescein diacetate (DCFH-DA) staining.
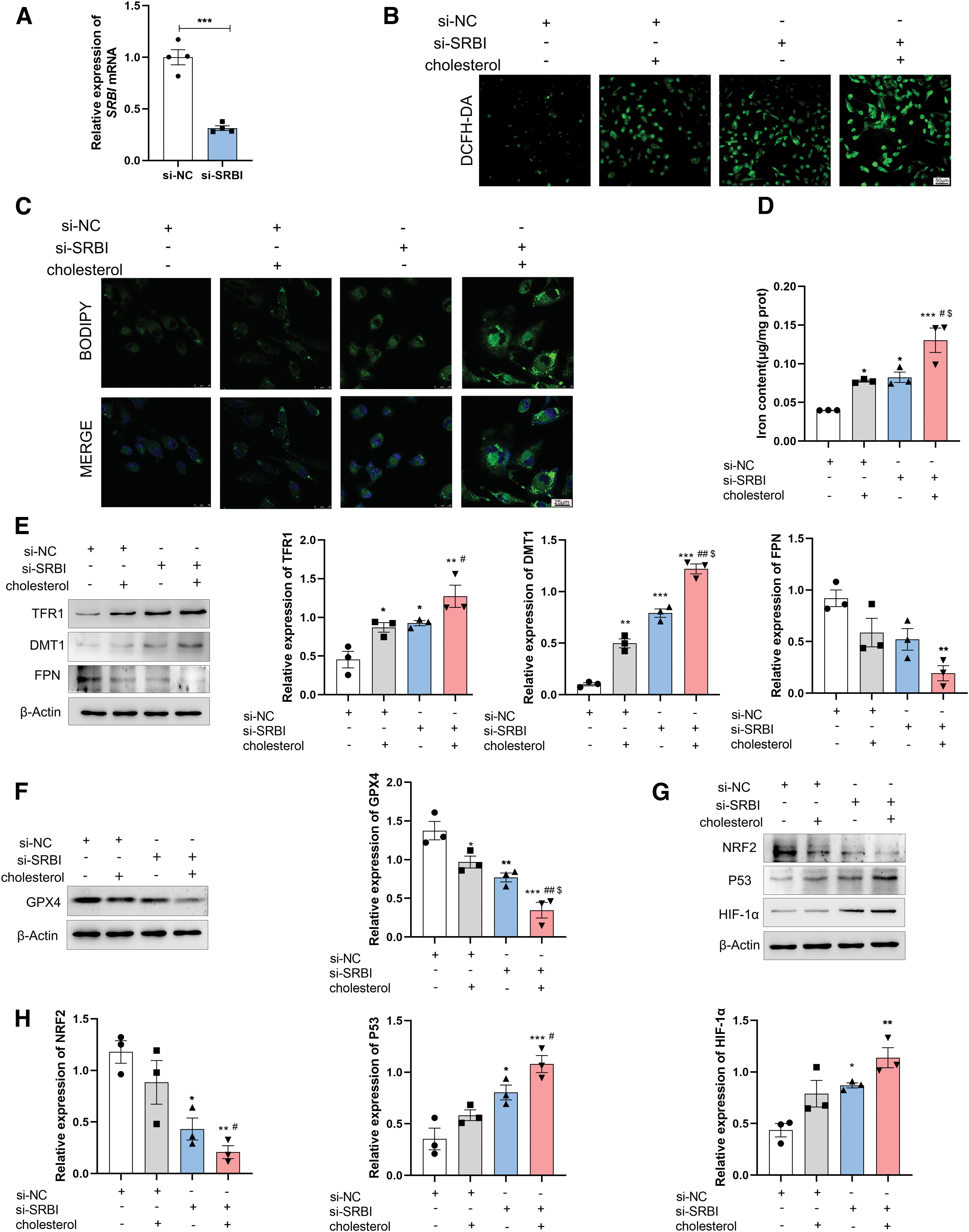
Compared with the control group, high-cholesterol treatment induced excessive ROS production in HK-2 cells. Interestingly, SRBI silencing alone increased ROS production in the HK-2 cells. More importantly, SRBI silencing accelerated the generation of ROS induced by high cholesterol (Fig. 4B), indicating that HK-2 cells had higher ROS levels when both stimuli were applied together. Mitochondrial ROS production was also evaluated using MitoSOX Red, and the results were similar to those for cellular ROS (Supplementary Fig. S4).
In addition, BODIPY staining was performed to assess lipid peroxidation. SRBI silencing increased lipid peroxidation compared with that in the control group. Moreover, SRBI knockdown aggravated lipid peroxidation compared with that in the high-cholesterol group (Fig. 4C).
As shown in Figure 4D, the intracellular iron levels were upregulated in the SRBI-silenced group. SRBI knockdown exacerbated the iron overload induced by high-cholesterol levels. Therefore, the protein levels of iron metabolism- and ferroptosis-related regulators were measured. The transferrin receptor 1 (TFR1) is required for iron uptake (Daniels et al., 2010), divalent metal transporter 1 (DMT1) mediates iron transport (Sandberg et al., 2018), and ferroportin (FPN) mediates iron efflux (De Domenico et al., 2010). Compared with the control group, the protein levels of TFR1 and DMT1 were upregulated, whereas the protein level of FPN was downregulated in the SRBI-silencing group.
The high-cholesterol group showed similar changes, which were aggravated by SRBI silencing (Fig. 4E). In addition, GPX4 plays an important role in ferroptosis. GPX4 converts toxic lipid peroxides to nontoxic alcohol lipids via GSH, which provides cytoprotection against ferroptosis (Fang et al., 2021; Fernandez-Mendivil et al., 2021). As shown in Figure 4F, SRBI silencing contributed to the downregulation of GPX4, and the knockdown of SRBI aggravated the high-cholesterol–induced GPX4 reduction.
Transcription factors, such as hypoxia-inducible factor-1α (HIF-1α), nuclear factor erythroid 2-related factor 2 (NRF2), and tumor protein p53 (P53), have been reported to play important roles in ferroptosis (Lovatt et al., 2020; Zhang et al., 2022; Zou et al., 2019), but the mechanisms remain unclear. Our results showed that both high-cholesterol and SRBI silencing upregulated the protein levels of P53 and HIF-1α, while the expression of NRF2 was downregulated. Moreover, the loss of SRBI exacerbated the high-cholesterol–induced upregulation of P53 and HIF-1α and the downregulation of NRF2 (Fig. 4G, H). These results clearly indicated that SRBI silencing could induce ferroptosis in HK-2 cells, which may be achieved through transcriptional regulation.
SRBI deficiency induced ferroptosis in the mouse kidney
It has previously been shown that iron overload can induce ferroptosis (Deng et al., 2023). To provide further evidence for the role of SRBI in ferroptosis, ferroptosis-related proteins were detected in SRBI-deficient mice. In this study, SRBI deficiency increased the expression of TFR1 and DMT1, and decreased the expression of FPN (Fig. 5A), indicating that the loss of SRBI was closely related to iron metabolism.
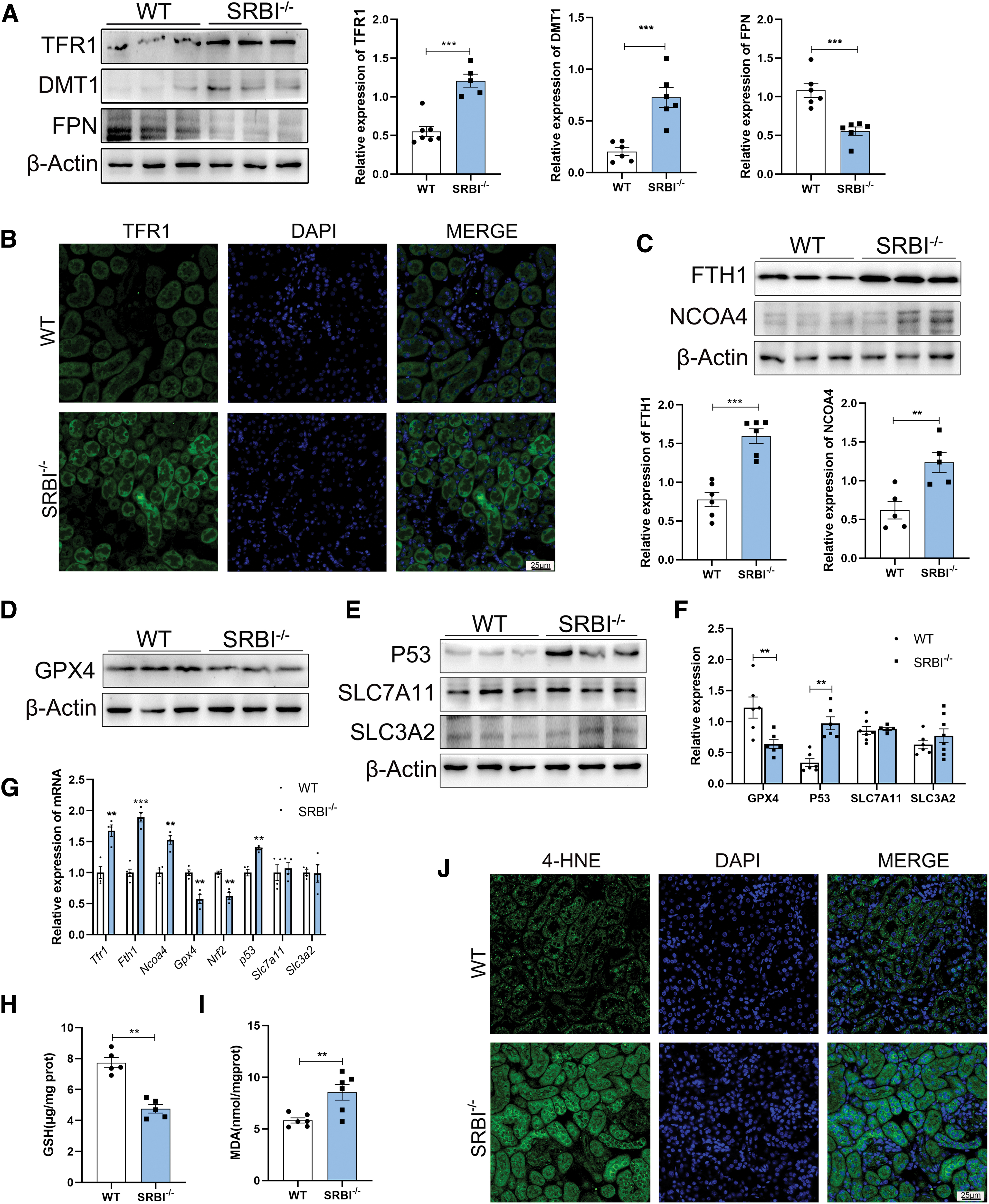
In addition, the immunofluorescence (IF) assay showed that SRBI deletion upregulated TFR1 protein in the renal tubules (Fig. 5B). The expression levels of the autophagy-related proteins ferritin heavy chain 1 (FTH1) and nuclear receptor coactivator 4 (NCOA4) were upregulated in the kidneys of SRBI-deficient mice (Fig. 5C). SRBI deficiency also downregulated the protein level of GPX4 (Fig. 5D, F), which led to increased susceptibility of cells to ferroptosis.
System Xc- is a cystine–glutamate antiporter that consists of the light chain subunit solute carrier family 7 member 11 (SLC7A11) and the heavy chain subunit solute carrier family 3 member 2 (SLC3A2) (Shin et al., 2017). SLC7A11 and SLC3A2 mediate cystine uptake to synthesize GSH (Dodson et al., 2019; Li et al., 2023; Zhang et al., 2022), and downregulation of SLC7A11 contributed to ferroptosis (Chen et al., 2020).
Compared with the WT group, the expression level of P53 was upregulated in the SRBI-deficient mouse kidney, whereas the expression levels of SLC7A11 and SLC3A2 showed no significant change (Fig. 5E, F), indicating that SRBI might not be linked to the cystine/glutamate antiporter system Xc-. Slc7a11 and Slc3a2 mRNA levels showed no significant changes in SRBI-deficient mice (Fig. 5G), which was consistent with the Western blot results.
In addition, the mRNA levels of Tfr1, Fth1, Ncoa4 and P53 were significantly elevated, and the expression of Gpx4 and Nrf2 was downregulated in SRBI-deficient mice (Fig. 5G). Moreover, GSH content was significantly reduced in SRBI-deficient mice compared with WT mice (Fig. 5H), indicating that SRBI deficiency triggers GSH depletion. Malondialdehyde (MDA) content (Fig. 5I) and 4-hydroxynonenal (4-HNE) IF (Fig. 5J) in SRBI-deficient mice were higher than those in WT mice, demonstrating that SRBI deficiency induces lipid peroxidation. Collectively, these results suggested that SRBI deficiency induces ferroptosis and lipid peroxidation in the mouse kidneys, which may be closely related to abnormal iron metabolism.
SRBI knockout or knockdown upregulated HIF-1α and promoted nuclear translocation
Our findings demonstrated that both SRBI silencing and knockout induced ferroptosis, and we next sought to explore the underlying molecular mechanisms. TFR1 is a key protein that facilitates cellular iron uptake, and elevated TFR1 levels result in increased cellular iron uptake (Daniels et al., 2010; Gammella et al., 2021). Studies have demonstrated that HIF-1 is involved in the transcriptional activation of TFR1, and that TFR1 expression is upregulated by HIF-1 (Bianchi et al., 1999; Cai et al., 2023; Tacchini et al., 1999).
To investigate whether SRBI silencing regulates TFR1 through HIF-1α, the expression level of HIF-1α was measured by IF (Fig. 6A). The results showed that HIF-1α was significantly upregulated in the kidneys of SRBI-deficient mice, which corresponded with its mRNA level (Fig. 6B) and protein expression (Fig. 6C) in vivo.
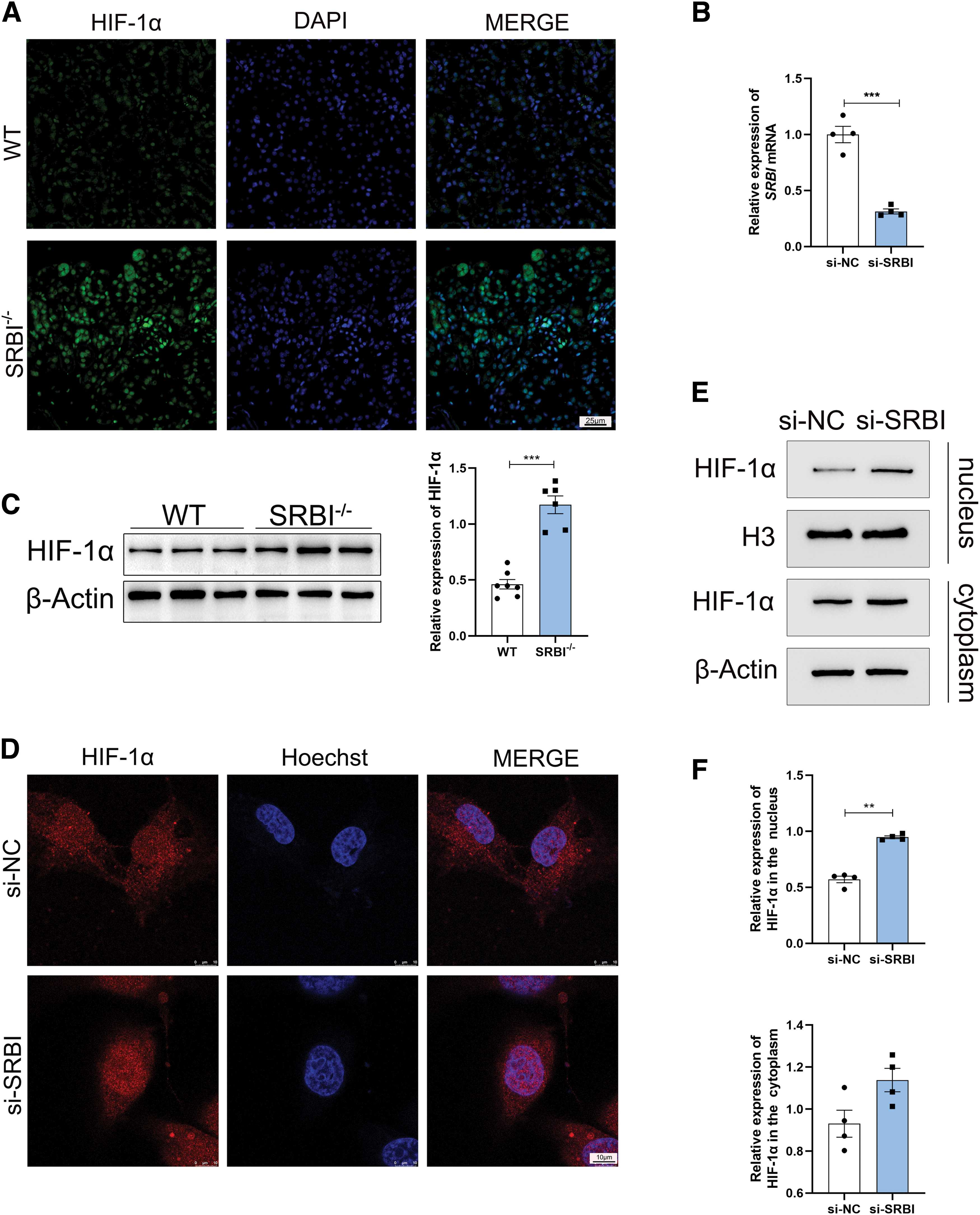
In addition, IF revealed that SRBI silencing induced translocation of HIF-1α to the nucleus of HK-2 cells (Fig. 6D). We separated nuclear and cytoplasmic proteins from HK-2 cells for further exploration. Compared with the control group, the expression of HIF-1α tended to increase in the cytoplasm of the SRBI-silencing group, whereas the expression of HIF-1α was significantly upregulated in the nucleus of the SRBI-silencing group, indicating that SRBI silencing promoted the nuclear translocation of HIF-1α (Fig. 6E, F).
SRBI silencing caused ferroptosis through the activation of HIF-1α/TFR1 pathway
To further investigate the role of HIF-1α in SRBI silencing, we inhibited the HIF-1α pathway using the HIF-1α inhibitor Oltipraz. The results showed that HIF-1α inhibition reversed the elevation of TFR1 and DMT1 protein levels (Fig. 7A) and reduced the depletion of GPX4 (Fig. 7B). The intracellular iron level was upregulated in the SRBI-silencing group compared with that in the control group, and HIF-1α inhibition alleviated this upregulation (Fig. 7C). Inhibition of HIF-1α also attenuated ROS generation initiated by SRBI silencing (Supplementary Fig. S5). Given the limited specificity of these inhibitors, oltipraz may also affect Nrf2 signaling.
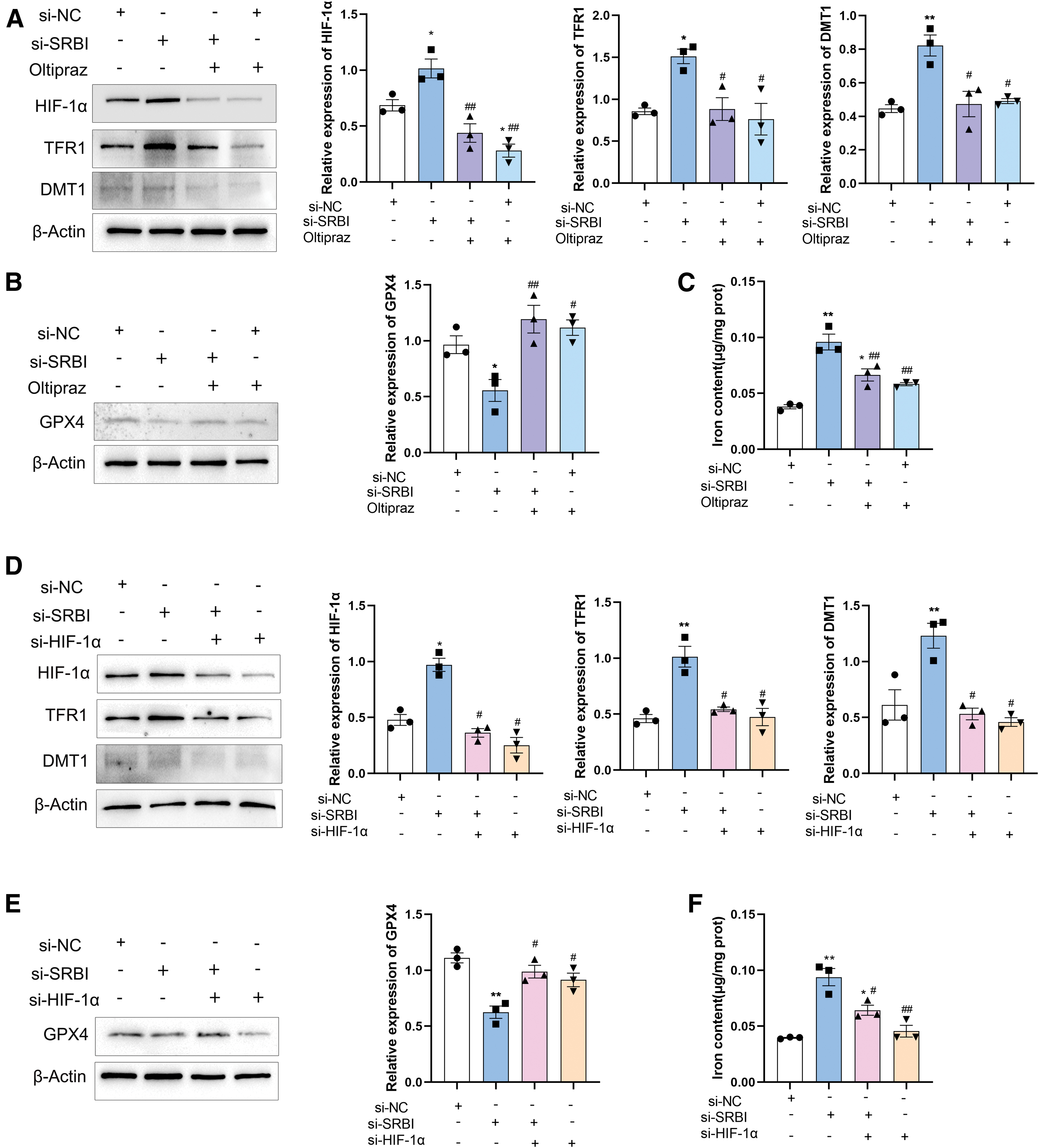
To further assess the involvement of HIF-1α in SRBI silencing, HK-2 cells were transfected with HIF-1α siRNA, and the results were similar to those obtained with the HIF-1α inhibitor (Fig. 7D, E). The results of iron content delineated that HIF-1α knockdown reduced the iron overload triggered by SRBI silencing (Fig. 7F). Taken together, we speculated that HIF-1α is involved in the upregulation of TFR1 caused by SRBI silencing. Previous studies have suggested that HIF-1α expression is upregulated by ROS (Kim et al., 2020).
To further examine whether ROS are involved in the upregulation of HIF-1α, the ROS scavenger, N-acetylcysteine (NAC), was used. The results suggest that HIF-1α expression remained unchanged after ROS scavenging (Fig. 8A), indicating that HIF-1α was not induced by ROS in this study. Interestingly, NAC attenuated the iron overload induced by SRBI silencing (Fig. 8B), suggesting that NAC rescued HK-2 cells from ferroptosis. It can be inferred that intracellular ROS production may be triggered by HIF-1α upregulation. Taken together, these results demonstrated that SRBI silencing induces ferroptosis via the HIF-1α/TFR1 pathway.
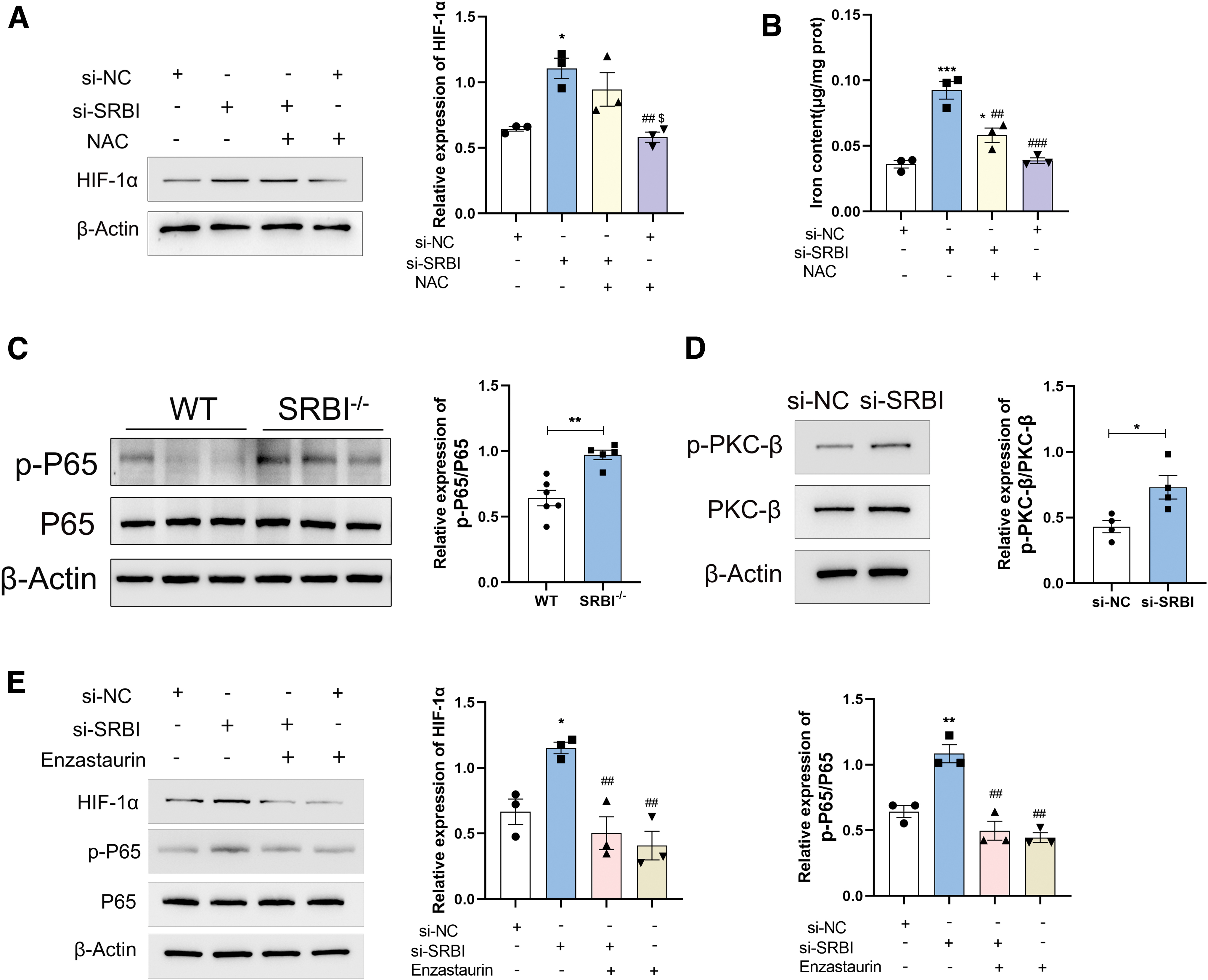
HIF-1α was activated via PKC-β/NF-κB signaling pathway
The mechanisms underlying HIF-1α activation were explored further. Previous studies have shown that NF-κB acts upstream of HIF-1α (Tacchini et al., 2008). Therefore, NF-κB expression was detected using Western blot. Compared with WT mice, the expression level of total NF-κB showed no significant change in the SRBI-deficient mouse kidney, whereas the expression level of phospho-NF-κB (p-NF-κB p65) was significantly upregulated, indicating that NF-κB was activated in the SRBI-deficient mouse kidney (Fig. 8C).
PKC-β is an upstream regulator of NF-κB (Basson et al., 2015; Bermejo et al., 2013), and we detected PKC-β activation in HK-2 cells. Compared with the control group, SRBI silencing upregulated the expression of p-PKC-β, indicating that SRBI silencing might promote the activation of PKC-β (Fig. 8D). Next, the PKC-β inhibitor enzastaurin (LY317615) was used to block PKC-β activation. The results showed that PKC-β inhibition reversed the elevation of HIF-1α and p-NF-κB (p-P65) protein levels induced by SRBI silencing, indicating that PKC-β is upstream of NF-κB and HIF-1α (Fig. 8E). Taken together, SRBI knockout or knockdown may increase the expression of HIF-1α through the PKC-β/NF-κB signaling pathway.
Discussion
Previous studies have shown that SRBI is involved in HDL metabolism, cholesterol homeostasis, and inflammation (Linton et al., 2017). However, the role of SRBI in ferroptosis remains unclear. In this study, we explored the role and underlying mechanism of SRBI in ferroptosis. The major findings were as follows: (i) SRBI knockout or knockdown caused iron overload in renal tubular epithelial cells, which contributed to ferroptosis. (ii) Iron overload and subsequent ferroptosis were induced by SRBI deficiency, not only by hypercholesterolemia. (iii) SRBI deficiency caused iron overload and ferroptosis through the HIF-1α/TFR1 signaling pathway (Fig. 1). This is the first study to show that SRBI deficiency initiates ferroptosis via the HIF-1α/TFR1 pathway.
Ferroptosis is an iron-dependent regulated cell death characterized by the destruction of cellular antioxidants and increased intracellular iron levels, which initiate detrimental lipid ROS deposition (Ni et al., 2022). Hyperlipidemia is closely related to ferroptosis, which has been observed in ApoE-deficient mice (Bai et al., 2020; Wang et al., 2022; Yu et al., 2022), a classical animal model of hyperlipidemia.
Notably, in this study, Prussian blue staining showed higher iron levels in the kidneys of SRBI-deficient mice than in ApoE-deficient mice. Thus, it is reasonable to speculate that ferroptosis is triggered by SRBI deficiency and not merely by lipid overload. Therefore, iron content and other ferroptosis-related indicators were detected under high-cholesterol and SRBI-silencing conditions in our study. These results revealed that SRBI silencing initiates iron overload and subsequent ferroptosis. This effect was even more exaggerated in a high-cholesterol environment, which further supported our speculation.
From a biochemical perspective, ferroptosis mainly involves the consumption of GSH and decreased GPX4 (Liang et al., 2022; Wang et al., 2020). GPX4 converts toxic lipid hydroperoxides to nontoxic lipid alcohols, thereby attenuating lipid peroxidation and ferroptosis (Feng et al., 2019; Yang et al., 2014). Numerous studies have shown that reduced GSH levels, decreased GPX4 activity, and increased lipid peroxidation are involved in ferroptosis (Sun et al., 2018; Wang et al., 2020; Xu et al., 2021). We focused on these indicators in this study. The results showed that GPX4 expression and GSH levels were significantly decreased in SRBI-deficient mouse kidneys. In vitro, the expression of GPX4 was also downregulated in SRBI-silenced HK-2 cells.
Ferroptosis is also linked to the metabolism of iron, amino acids, and ROS (Zheng and Conrad, 2020). Fe2+ can efficiently catalyze the Fenton reaction, in which hydroxyl radicals are generated from H2O2. Our results showed that the iron content increased in SRBI-knockdown HK-2 cells or knockout mice, which might partly trigger ROS production in renal tubular epithelial cells. Studies have demonstrated that ROS can increase the expression of p62 (Garcia et al., 2012; Liu et al., 2019). Elevated levels of p62 indicate impaired autophagic flux (Lai et al., 2020).
In addition, SRBI deficiency induced the downregulation of Nrf2. Recent studies have shown that a deficiency in Nrf2-GSH signaling enhances cell sensitivity to oxidants and Nrf-2 deficient mice have greater Nox-dependent ROS production (Kong et al., 2010; Reddy et al., 2007). It is possible that SRBI deficiency induced-Nrf2 downregulation results in upregulation of NOX, thereby leading to increased levels of ROS. Studies have shown that HIF-1α upregulates NOX (Jung et al., 2023).
In this study, SRBI knockout and knockdown upregulated the expression of HIF-1α. The upregulation of NOX might be attributable to the upregulation of HIF-1α induced by SRBI knockout. Lipid peroxidation is closely associated with increased ROS and reduced GPX4 (Ramosaj et al., 2021). In this regard, MDA and 4-HNE levels were elevated in SRBI-deficient mouse kidneys, and BODIPY staining indicated that SRBI silencing elicited lipid peroxidation in HK-2 cells.
In addition, it is known that the amino acid transporters SLC7A11 and SLC3A2 mediate cystine/glutamate transport, and SLC7A11 is vital for maintaining intracellular GSH levels (Li et al., 2022a). Our results showed that although loss of SRBI exacerbated the upregulation of P53, there was no difference in the mRNA and protein levels of SLC7A11 and SLC3A2 in the kidneys of SRBI-deficient and WT mice. It has already been revealed that P53 repressed the transcription of SLC7A11, which enhances ferroptosis (Chu et al., 2019). Therefore, the P53/SLC7A11 pathway may not play a dominant role in SRBI-deficiency–induced ferroptosis.
Based on our findings, we explored the mechanism underlying ferroptosis induced by SRBI deficiency. Iron metabolism is an important factor in ferroptosis (Ying et al., 2023). Cellular iron homeostasis is delicately regulated by various iron metabolism-related genes, including iron absorption, transport, storage, and efflux (Song et al., 2022). Most cells have the transferrin receptor TFR1 on their surface, which binds to iron-loaded transferrin.
Following the endocytosis of the complex, iron enters the cytoplasm via DMT1 in the endosomal membrane. In addition, FPN mediates cellular iron export (Anderson and Frazer, 2017; Zhou and Tan, 2017). To gain insight into these processes, the expression levels of iron homeostasis-related proteins were analyzed in vitro and in vivo. The results revealed that TFR1 and DMT1 were upregulated in SRBI-deficient mouse kidneys and SRBI-silenced HK-2 cells, whereas FPN was downregulated. This provides a plausible explanation for the increase in iron levels. Notably, TFR1 levels increased, suggesting that SRBI deficiency might increase intracellular iron levels via elevated TFR1 levels, thus eliciting abnormal iron metabolism and subsequent ferroptosis.
The mechanism of TFR1 upregulation was explored next. HIF-1α modulates the transcription of various genes (Guo et al., 2018), including iron metabolism-related genes, such as TFR1 (Fagundes et al., 2022; Tacchini et al., 1999). HIF-1α may play a biphasic regulatory role in ferroptosis. HIF-1α deletion reduces the sensitivity to ferroptosis in ES-2 cells (Zou et al., 2019). In contrast, the elevation and stabilization of HIF-1α can prevent ferroptosis in hepatic stellate cells (Yuan et al., 2022). Therefore, the expression of HIF-1α was measured in SRBI-deficient mouse kidneys and in SRBI-silenced HK-2 cells.
Western blot and IF results showed that SRBI deficiency upregulated HIF-1α in vitro and in vivo. To further validate the regulation of TFR1 by HIF-1α, HIF-1α inhibitor and siRNA were used. The results showed that TFR1 was downregulated after HIF-1α inhibition or knockdown, indicating that TFR1 is regulated by HIF-1α, consistent with previous studies (Tacchini et al., 2002; Tacchini et al., 1999; Tao et al., 2007).
In addition, HIF-1α inhibition or knockdown attenuated the elevation of DMT1 and the reduction of GPX4, and also reduced the iron content in HK-2 cells. Inhibition of HIF-1α attenuates ROS production. These results suggested that HIF-1α, as a transcription factor, could regulate the expression of TFR1, DMT1, and GPX4. Taken together, HIF-1α inhibition or knockdown attenuated SRBI-silencing–induced ferroptosis. Thus, it can be inferred that the HIF-1α/TFR1 pathway is involved in the ferroptosis initiated by SRBI silencing.
The activation of HIF-1α in this context was explored. In this study, both the mRNA and protein expression of HIF-1α were upregulated in SRBI-deficient mice, and HIF-1α was translocated from the cytosol to the nucleus in SRBI-silenced HK-2 cells. HIF-1α is known to be regulated by several factors. Previous studies have suggested that ROS increase the expression of HIF-1α (Chourasia et al., 2015). HIF-1α is also regulated by NF-κB (Frede et al., 2006).
To explore whether the upregulation of HIF-1α induced by SRBI silencing is mediated by ROS, ROS scavengers were used to eliminate intracellular ROS. Our results indicated that HIF-1α is not regulated by ROS, which is consistent with previous findings (Kim et al., 2013). In this study, HIF-1α inhibition attenuated ROS production, indicating that ROS may act downstream of HIF-1α. Upregulation and translocation of HIF-1α may be mediated by other pathways.
Next, we explored whether HIF-1α was upregulated by NF-κB, and found that NF-κB was activated in the kidneys of SRBI-deficient mice. Studies have shown that PKC-β is an upstream regulator of NF-κB (Basson et al., 2015; Bermejo et al., 2013). Our results also showed that PKC-β was activated in SRBI-silencing group. After PKC-β inhibition, the elevation of HIF-1α and p-NF-κB levels was reversed, indicating that SRBI knockout or knockdown might increase the expression of HIF-1α through the PKC-β/NF-κB signaling pathway.
In conclusion, this study showed for the first time that SRBI deficiency triggers ferroptosis through the HIF-1α/TFR1 signaling pathway in renal tubular epithelial cells.
Materials and Methods
Animal experiments
C57BL/6 mice and ApoE-deficient C57BL/6 mice were purchased from the Vital River Laboratory Animal Technology Company (Beijing, China). SRBI heterozygous mice were licensed for use by Professor Monty Krieger (Massachusetts Institute of Technology, Cambridge, MA). PCR analysis of mouse tail DNA was used to identify the SRBI homozygous mice, generated by crossing SRBI heterozygous female mice with SRBI homozygous male mice.
All mice were housed at Wuhan University Institute of Model Animal under controlled humidity (50% ± 5%), temperature (22°C ± 2°C), and light (12 h light/12 h dark cycle, lights on at 8:00 AM). The mice were housed under specific pathogen-free conditions with ad libitum access to food and water. Mice were fasted for 12 h before blood samples were collected by retro-orbital venous plexus puncture, and plasma was separated by centrifugation. All mice were anesthetized with sodium pentobarbital (30 mg/kg, i.p.). Kidneys of 12-month–old mice were collected immediately after cervical dislocation and snap-frozen in liquid nitrogen.
Animal maintenance and all experiments were approved by the Wuhan University Medical Animal Protection and Welfare Committee Animal Experimentation Program (No. 2018027; March 19, 2018). All experiments were performed in accordance with the guidelines of the China Animal Welfare Committee on the Protection and Use of Experimental Animals.
Cell culture and treatment
The human renal proximal tubular epithelial cell line HK-2 was cultured in Dulbecco's modified Eagle's medium/low-glucose medium (BL308A; Biosharp Co., Ltd., China) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin in a cell incubator (37°C, 5% CO2). Cells were treated with a final concentration of cholesterol (B20272; Shanghaiyuanye Bio-Technology Co., Ltd., Shanghai, China) of 50 μg/mL for 24 h. To suppress HIF-1α, Oltipraz (10 μM; MedChemExpress, Princeton, NJ) was used 24 h before treatment. NAC (5 mM, 616-91-1; Meilun Biotechnology Co., Ltd., Dalian, China) was used 24 h before treatment to remove ROS.
Lipid analysis
The plasma triglyceride and cholesterol levels were measured using a spectrophotometer. Free cholesterol, total cholesterol, and triglyceride levels were determined using commercial assay kits (E-BC-K 109-M, E-BC-K004-M; Elabscience Biotechnology Co., Ltd., Wuhan, China, and A110-1-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Histological, TUNEL staining, and IF
Kidney tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and sectioned at 4 μm. For histological evaluation, the kidney sections were stained with HE, PAS, and Masson trichrome (Masson) staining. To detect apoptotic cells, TUNEL staining of the kidney sections was performed (Promega, Madison, WI).
Mouse kidney sections and cells were fixed in 4% paraformaldehyde for 15 min, rinsed with phosphate-buffered saline (PBS), and permeabilized with 0.2% Triton-X100 in PBS for 3 min. After the samples were blocked with 5% bovine serum albumin, the primary antibodies were incubated at 4°C overnight. The primary antibodies used in this study were F4/80 (70076; Cell Signaling Technology, MA), Ly6G (87048; Cell Signaling Technology), TFR1 (A22161; Abclonal, Wuhan, China), HIF-1α (bs-20399R; Bioss Biotechnology, Beijing, China), and HIF-1α (20960-1-AP; Proteintech, Wuhan, China).
The following day, the sections and cells were washed three times in PBS and incubated with fluorescently labeled secondary antibodies (Cy3-coupled, 1:50; Boster, Wuhan, China) for 2 h at 37°C. Hoechst 33258 (Solarbio, Beijing, China) was used for the nuclear staining. The cells were viewed using a confocal microscope (Leica TCS SP8, Wetzlar, Germany).
GSH, MDA, iron content assay, and Prussian blue staining
Reduced GSH and MDA were measured using a GSH assay kit (BC1175; Solarbio) and MDA assay kit (A003-1; Nanjing Jiancheng), respectively, according to the manufacturer's protocol. Kidney tissues were placed in EP tubes on ice and ground. The samples were centrifuged (8000 rpm, 4°C, 10 min), and the supernatant was collected for substrate detection. Blank tubes, standard tubes, and sample tubes were prepared following the manufacturer's instructions. Optical density (OD) values were measured at 412 and 532 nm.
Iron content was measured using commercial kits (BC4355; Solarbio). Kidney tissue was homogenized and ground. The samples were centrifuged (10,000 g, 4°C, 10 min), and the supernatant was collected for substrate detection. The cells were scraped off the dish, and the lysates were collected. Blank, standard, and sample tubes were prepared according to the manufacturer's instructions. OD was measured at 520 nm.
To analyze iron overload, kidney sections were stained with Prussian blue. The sections were incubated with Prussian blue solution (G1422; Solarbio) for 60 min at room temperature, and then washed with distilled water. The slices were observed under a bright-field optical microscope.
Measurement of ROS production
OCT-embedded frozen kidney tissue was cryosectioned (3 μm-thick sections). ROS levels in the kidneys were measured by DHE staining. The fluorescence intensity of DHE staining was measured using ImageJ software. An ROS assay kit (BB-470512; BestBio Co., Shanghai, China) was used to detect ROS generation. We used the cortical fractions for further experiments according to the manufacturer's instructions. Intracellular ROS levels were measured using a Meilun ROS assay kit (MA0219; Meilun Biotechnology Co., Ltd.).
HK-2 cells were cultured at a density of 1 × 105 cells per well in six-well plates. DCFH-DA was diluted in serum-free medium at 1:1000 to a final concentration of 10 μM. After treatment, the working solution (1 mL) was added to each well and incubated for 30 min at 37°C before removing the culture medium. HK-2 cells were washed three times with serum-free medium to remove extracellular DCFH-DA.
A fluorescence microscope (IX73; Olympus, Tokyo, Japan) was used for the observation and photography. Mitochondrial ROS were detected using MitoSOX (HY-D1055; MedChemExpress), which was diluted in serum-free medium at 1:1000 to a final concentration of 5 μM. The working solution (1 mL) was added to each well and incubated for 15 min at 37°C before removing the culture medium. After washing three times with PBS or serum-free medium, the cells were viewed using a confocal microscope (Leica TCS SP8).
RNA interference
siRNA against SRBI (si-SRBI), HIF-1α (si-HIF-1α), and negative control (si-NC) were purchased from Tsingke Biotechnology Co., Ltd. (China). For siRNA transfection, Lipofectamine RNAiMax (Thermo Fisher, Waltham, MA) and OPTI-MEM medium (Thermo Fisher) were used according to the manufacturer's instructions. HK-2 cells were cultured in a normal medium for 24 h after transfection for subsequent experiments.
BODIPY staining
To assess the accumulation of lipid intermediates, HK-2 cells were cultured in confocal dishes. After intervention, HK-2 cells were incubated with 1 μM C11-BODIPY 495/503 nm (Thermo Fisher) for 15 min in the dark, and then washed with PBS three times (5 min each). Hoechst 33258 (1:100 dilution) was used for the nuclear counterstaining. HK-2 cells were incubated with Hoechst 33258 for 30 min in a cell incubator (37°C) and washed with PBS three times (3 min each). Lipid peroxidation was demonstrated by the green fluorescence of oxidized C11-BODIPY in the fluorescein isothiocyanate (FITC) channel. BODIPY staining was observed under a confocal microscope (Leica TCS SP8).
Quantitative real-time PCR
Total RNA was extracted from the kidneys using the TRIzol reagent (Thermo Fisher). RNA purity and concentration were measured using a NanoDrop spectrophotometer. The Vazyme reverse transcription kit was used to reverse transcribe RNA to cDNA. Real-time PCR was performed using SYBR Green (Vazyme Biotech Co., Ltd., Nanjing, China) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA). PCR conditions were as follows: initial denaturation at 95°C for 5 min, followed by 40 cycles of 15 s denaturation at 95°C, 30 s annealing at 60°C, and 30 s extension of primers at 72°C. Relative gene expression was analyzed using the 2−ΔΔCt method. The relative expression of each gene was normalized to that of β-actin and calculated according to the 2−ΔΔCt method. Primer sequences used are listed in Supplementary Table S1.
Western blot analysis
Kidney tissues (10 mg) and HK-2 cells were collected in a RIPA lysis buffer supplemented with protease inhibitors and homogenized. Protein concentrations were determined using a Coomassie Brilliant Blue assay kit (A045-2-2; Beyotime Biotechnology, Shanghai, China). Nuclear proteins were extracted using a nuclear protein extraction kit (R0050; Solarbio), in accordance with the manufacturer's instructions. Electrophoresis was conducted on 7.5%–15% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels before transfer to nitrocellulose membranes.
The membranes were incubated with primary antibodies overnight at 4°C, and then incubated with secondary antibodies for 1 h at 37°C. Electrochemiluminescence reagents (Beyotime Biotechnology) were used for detection. β-Actin was used as the internal reference. Blotted bands were quantified using the ImageJ software. The primary antibodies used are listed in Supplementary Table S2.
Flow cytometry
Apoptosis was analyzed by flow cytometry using an Annexin V-FITC/Propidium Iodide (PI) Apoptosis Kit (AT101; Multisciences, Hangzhou, China). According to the manufacturer's instructions, the cells were washed with PBS and collected in 1.5 mL EP tubes, and 5 μL Annexin V-FITC and 10 μL PI were added to each tube. The cells were incubated at room temperature in the dark for 5 min. Apoptotic and dead cells were detected using the Cytoflex S flow cytometer (Beckman, CA).
Statistical analysis
All data are presented as mean ± standard error of the mean from at least three independent experiments performed in parallel. All replicates represent biological replicates. Statistically significant differences were determined using an independent-sample t-test or one-way analysis of variance (ANOVA) using GraphPad Prism. p < 0.05 was considered statistically significant.
Data Access Statement
All relevant data are within the paper and its Supplementary Data.
Ethics Statement
The study was approved by the Wuhan University Medical Animal Protection and Welfare Committee Animal Experimentation Program (Number 2018027; March 19, 2018).
Footnotes
Authors' Contributions
L.-J.Y. and Q.L. performed the project and data analysis; Q.-Y.L. and J.-J.X. prepared figures; A.-Y.F., S.W., L.-H.N., and J.-W.H. collected the literature; X.-Y.W. edited the article; H.Y. conceived and designed part of the research; and B.-F.Z. designed the research and wrote the article. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by the National Natural Science Foundation of China 82370696 (XiaoYan Wu) and 82070471 (Hong Yu), Wuhan University Basic Medicine-Clinical Medicine Joint Project JCZN2022005 (Bai-Fang Zhang), and the Medical Sci-Tech Innovation Platform of Zhongnan Hospital, Wuhan University XKJS202035 (XiaoYan Wu).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.