
Abstract
Aims:
Preterm birth (PTB), recognized as delivery before 37 weeks of gestation, is a multifactorial syndrome characterizing as the main cause of neonatal mortality. Reactive oxygen species (ROS) have been identified as proinflammatory factors to cause placental inflammation, thereby resulting in several pregnancy outcomes. To date, limited knowledge regarding the underlying mechanisms of ROS-induced PTB has been reported. In this study, we aimed to investigate the role of oxidative stress in PTB and the protective effects of mitochondria-targeted antioxidant MitoTEMPO (MT) on preterm labor and offspring mice.
Results:
In this study, we found that preterm placentas had abnormal mitochondrial function, oxidative stress, and inflammatory response. In the lipopolysaccharide (LPS)-induced PTB mouse model, MT inhibited PTB by ameliorating maternal oxidative stress and inflammation, especially in placentas, thus improving placental function to maintain pregnancy. Antenatal administration of MT prevented LPS-induced fetal brain damage in acute phase and improved long-term neurodevelopmental impairments. Furthermore, our in vitro investigations validated that MT retarded the ROS accumulation and inflammatory response in LPS-stimulated trophoblast cells by promoting Kelch-like ECH-associated protein 1 (Keap1) degradation and subsequently activating nuclear factor erythroid 2-related factor 2 (Nrf2). By inhibiting Nrf2 activation, we discovered that the anti-inflammation and protective characteristics of MT were Nrf2/ARE pathway dependent.
Innovation and Conclusion:
MT inhibited PTB and fetal brain injury by inhibiting maternal inflammation and improving placental function through Keap1/Nrf2/antioxidant response element signaling pathway. Our findings provide a novel therapeutic strategy for PTB. Antioxid. Redox Signal. 41, 597–615.
Introduction
Preterm birth (PTB) is referred to delivery before 37 weeks of gestation. The average occurrence of PTB is about 11% worldwide, which is the leading cause of neonatal mortality (Harrison and Goldenberg, 2016). PTB also impairs neonatal development, causing higher prevalence of cerebral palsy, chronic lung disease, and visual and auditory deficits in preterm infants than in term infants. The severity of these poor outcomes is inversely related to gestational age and may persist into adulthood (Bell et al., 2022, Hagberg et al., 2012). Current clinical treatment for PTB is relatively limited, mainly tocolytics like magnesium sulfate, atosiban, ritodrine inhibiting uterine contractions, and glucocorticoids promoting fetal lung maturation. Antibiotics are not first-line clinical drugs for PTB. These drugs can prolong the gestation period and delay the onset of labor to some extent. But their efficacy is not reliable, and PTB rate has not been improved for decades. They are limited in suppressing intrauterine inflammation and improving the short-term and long-term sequelae of premature infants caused by intrauterine inflammation, and there are many side effects. Patra et al. (Patra et al., 2015) conclude that postnatal usage of glucocorticoids for more than 7 consecutive days in premature infants may affect fine motor and language development. In addition, studies have shown that atosiban as an oxytocin receptor antagonist may increase maternal oxidative stress and damage fetal brain function after 48 h of administration (Grzesiak et al., 2018). Ritodrine, as a beta agonist, is cardiotoxic to mother and fetus (Shigemi et al., 2019). Therefore, it is important and urgent to seek comprehensive and effective approaches for treating PTB and improving sequelae of preterm infants.
Innovation
The concrete pathogenesis of PTB remains unclear, and medical interventions to efficiently prevent preterm labor and fetal injury are lacking. This study explored the mechanism of mitochondrial oxidative stress and placental dysfunction in PTB. In inflammation-induced preterm mouse model, we confirmed the maternal use of mitochondria-targeted antioxidant.
PTB is a multifactorial syndrome and its etiology is complex, highly correlated with inflammation (Vrachnis et al., 2010). Owing to the stimuli such as microbial infection, sterile inflammation, and stress, the maternal–fetal immune tolerance and activation are disrupted, resulting in numerous immune cells infiltrating into maternal–fetal interface and overproducing inflammatory cytokines. Ultimately, the aberrantly expressed inflammatory factors trigger early initiation of labor (Gomez-Lopez et al., 2014, Wallenstein et al., 2016). However, the pathogenesis of inflammatory response–induced PTB is unclear. As a special structure during pregnancy, placenta is closely related to normal growth and development of fetus, maintenance of pregnancy, and initiation of labor. Current researchers have indicated that placental inflammation is correlated with PTB (Couture et al., 2023). Reactive oxygen species (ROS), as highly proinflammatory factors, are significantly elevated in preterm placentas, suggesting the involvement of oxidative stress in the pathogenesis of PTB (Millán et al., 2018). Under physiological conditions, ROS production and clearance in the placenta are in balance, which is conducive to placental function. ROS act as signaling molecules participating in regulating gene expression and downstream cellular activities, such as invasion, proliferation, and angiogenesis (Schieber and Chandel, 2014; Wong et al., 2017). We propose that placenta produces excessive ROS to induce placental inflammation and placental dysfunction under pathological conditions. To date, the pathophysiology process related to oxidative stress in preterm placentas is not completely understood. Placental mitochondria are working as the main energy source supporting mother and fetus development. Highly functional mitochondria are the major resource of placental ROS during late gestation. Furthermore, oxidative stress can be induced by mitochondrial structural damage, mitochondrial DNA mutation, or mitochondrial oxidative respiration alteration. Mitochondrial ROS (mtROS) can cause placental inflammation through activating redox-sensitive transcription factors to regulate inflammatory cytokines and directly activating the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway (Zhong et al., 2016). Consequently, targeting mtROS could be a viable therapeutic approach in PTB through improving placental inflammation.
MitoTEMPO (MT) is a selective mitochondria-targeted antioxidant, which is formed by the conjugation of the antioxidant piperidine nitroxide Tempol with the lipophilic positively charged triphenylphosphine cation. Since mitochondria is a negatively charged double-membrane chamber, MT can pass through the mitochondrial membrane structure driven by the membrane potential and accumulate hundreds of times in the mitochondria to remove mtROS. There is accumulating evidence suggesting that MT has positive effects on pregnancy complications such as fetal growth restriction and preeclampsia where mitochondrial oxidative damage performs a critical role throughout the pathology (Vaka et al., 2018, Zhu et al., 2022). Furthermore, MT is also proved to be effective in certain inflammatory disorders, including sepsis (Arulkumaran et al., 2021). Intrauterine inflammation activates fetal microglia to release excessive ROS, and microglia are highly exposed to ROS (Burd et al., 2012), which have a vital function in the pathogenesis of premature brain injury. It was reported that MT repaired neurological deficits and reduced neuronal apoptosis via activating nuclear factor erythroid 2-related factor 2/antioxidant response element (Nrf2/ARE) pathway (Thangaraj et al., 2020). The NF-E2–like 2 (Nrf2) transcription factor modulates numerous cytoprotective gene expression through binding to the ARE. Therefore, we suggested that MT may be an ideal treatment for PTB and fetal brain injury via suppressing oxidative stress and inflammation. However, the MT underlying molecular mechanism in treating PTB is still unknown. Throughout this investigation, we discovered the effects of MT on PTB in addition to the pathways through which MT accomplishes its functions.
Results
Altered placental function in human preterm placentas
We first sought to determine whether placental dysfunction is associated with PTB. Regarding placental vascularization, we observed substantially decreased vascular endothelial growth factor (VEGF) levels in placental villi of preterm patients (Fig. 1a). Ki67 and transferase dUTP nick end labeling (TUNEL) staining showed inhibited cell proliferation and increased apoptosis in trophoblasts of placental villi in preterm group (Fig. 1b, c). As for placental endocrine function and nutrient transport, decreased β-HCG, Glut1, and Glut3 levels were detected in preterm placentas (Fig. 1d, e), which may indicate reduced hormone production and glucose transfer in pathological preterm placentas. Except for the difference in expression of Glut 3 between TL (term labor) and TNL (term not in labor) placentas, no significant difference was observed in other placental functions between two groups.
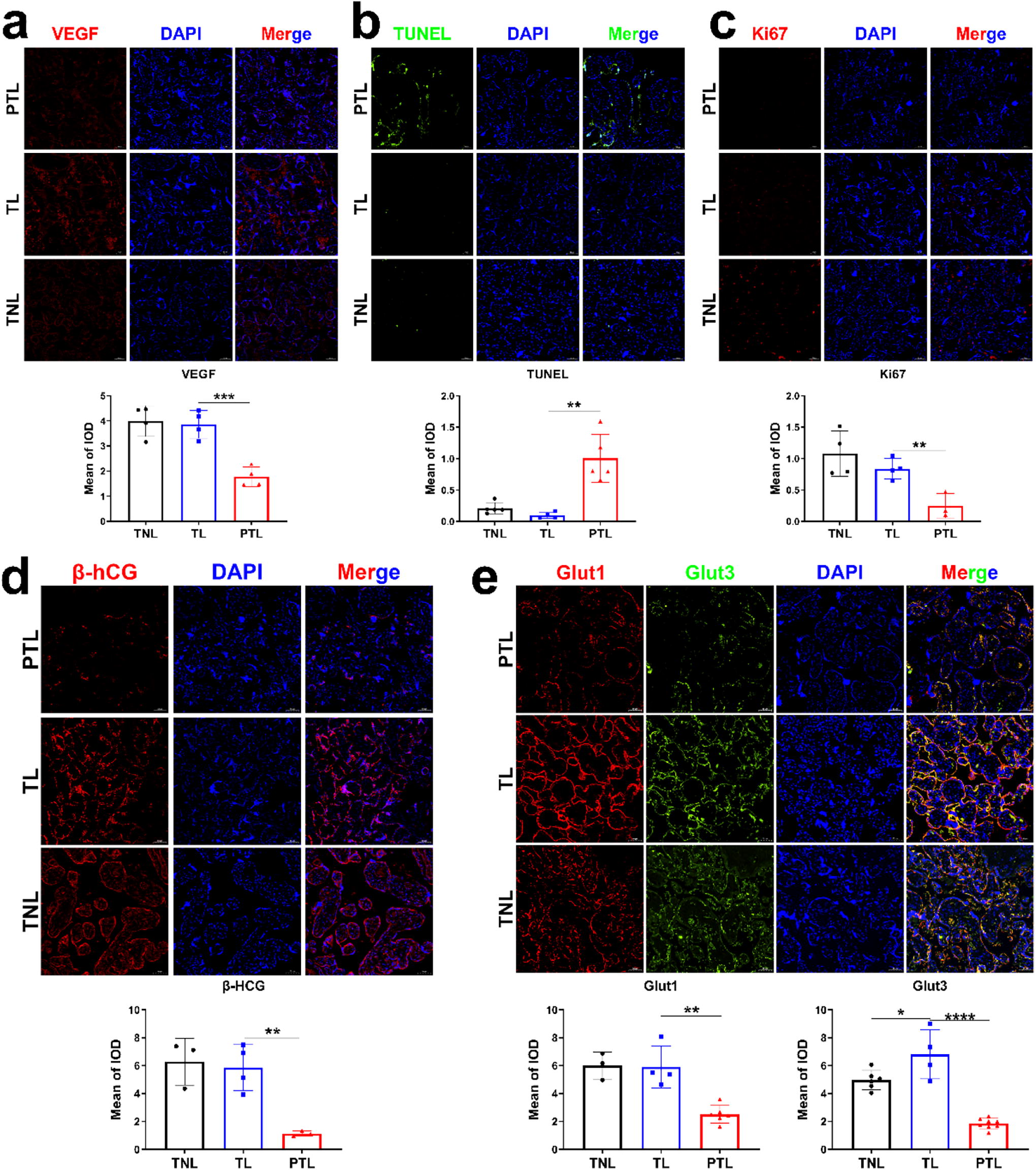
The alterations of placental functions in preterm birth.
Altered mitochondrial ultrastructure and function in human preterm placentas
Given the importance of mitochondria to placental function, we examined placental mitochondrial structure and function. We observed mitochondrial ultrastructure of placentas at magnified 30,000 times through transmission electron microscopy (TEM) (Fig. 2a). In preterm placenta, mitochondria were mostly located near the nucleus, and there was an asymmetric swollen translucent region at one end of the abnormal mitochondria, with few cristae not arranged in parallel. In term placenta, most mitochondria were oval in shape and contained intact mitochondrial outer membranes and parallel cristae, which extended throughout the mitochondria, with narrow gaps between the cristae. In order to analyze mitochondrial respiration and placental metabolic characteristics, we observed metabolites of TCA (tricarboxylic acid) cycle and glycolysis in the preterm and term placental villi and found that the intermediates in the TCA cycle, including aconitate, fumarate, and malate, were decreased in preterm placentas. Moreover, we found that glycolytic intermediates containing glucose 6-phosphate, fructose 6-phosphate, fructose 1, 6-bisphosphate, phosphoenolpyruvate, 3-phosphoglycerate, and lactate were all substantially elevated in preterm placentas. The abnormal activation of glycolysis may ensure energy supply for placental function to compensate for mitochondrial dysfunction in preterm placentas (Fig. 2b). Mitochondrial DNA (mtDNA) copy number, as a measure of mitochondrial function, was lowest in preterm placentas (Fig. 2c). The oxidized and reduced forms of nicotinamide adenine dinucleotide (NAD) is called the NAD+/NADH ratio. This ratio reflects both the mitochondrial redox state and the metabolic activities of cells. We detected NAD+ and NADH contents and found that NAD+ was highest in preterm placentas, with the ratio accordingly increased (Fig. 2d–f).
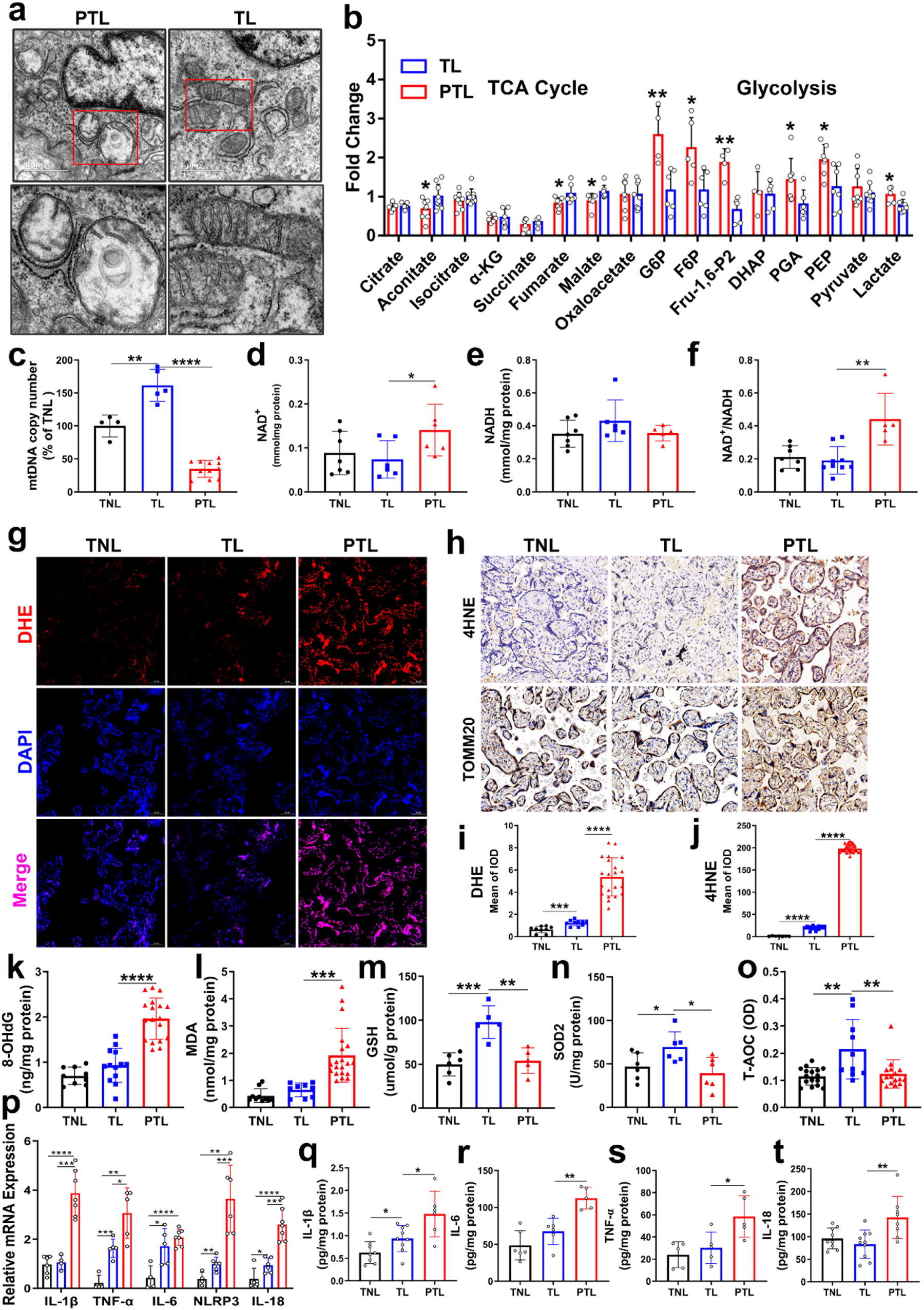
Mitochondrial dysfunction, oxidative stress, and inflammation in preterm placentas.
Oxidative stress and inflammation in human preterm placentas
To investigate whether placental oxidative stress and inflammation is associated with PTB, we tested the levels of oxidative stress parameters, including ROS, 4HNE, 8-OHdG, and MDA, and found that they were increased in preterm placentas compared with term placentas (Fig. 2g–l). We used TOMM20 as mitochondrial-specific biomarker and found that the expression site of the TOMM20 coincided with the 4HNE site, suggesting the role of mitochondria in placental oxidative stress response (Fig. 2h). The levels of antioxidant markers like GSH, SOD2 activities, and total antioxidant capacity (T-AOC) were attenuated in preterm placentas compared with term placentas (Fig. 2m–o). In term groups, the placentas in labor group showed increased oxidative stress and antioxidant levels compared with not in labor group. We assessed inflammation levels in placentas, and the results showed that the inflammatory gene expression, including IL-1β, TNF-α, IL-6, NLRP3, and IL-18, in the preterm placentas was higher compared with the other two groups (Fig. 2p). Meanwhile, enzyme-linked immunosorbent assay (ELISA) analysis showed increased inflammatory cytokines IL-1β, TNF-α, IL-6, and IL-18 in preterm placentas compared with term placentas. In term groups, the placentas in labor group showed higher expression of inflammatory factors than not in labor group (Fig. 2q–t).
MT prevents preterm labor and inhibits uterine contraction and pro-inflammatory mediators in amniotic fluid
To explore whether MT has protective effects in PTB, we calculated PTB rate of four groups (Table 1). All the pregnant mice in the lipopolysaccharide (LPS) group delivered within 24 h after LPS injection, and MT could effectively reduce the PTB rate of LPS-treated pregnant mice. Some of the pregnant mice were sacrificed 5 h after LPS or saline injection at D15 for anatomical observation. The uterine segmental space disappeared, and intrauterine bleeding occurred in the LPS-treated pregnant mice, indicating the onset of labor. Some fetuses of LPS-treated pregnant mice lacked cerebral perfusion or were even completely dead (Fig. 3a). The high expression of F4/80 indicates more infiltration of macrophages in the myometrium, causing strong inflammatory response, which promotes myometrial contraction to initiate labor. The results revealed that LPS injection substantially enhanced the positive expression of F4/80 and myometrial contraction-associated proteins in myometrium, and these impacts were noteworthily ameliorated in mice co-treated with MT (Fig. 3b, c). We measured the concentrations of 40 cytokines in amniotic fluid by quantitative inflammation array. In amniotic fluid, the inflammatory cytokines and chemokines expression levels in LPS group were greater than that in Control group (Fig. 3d). MT could effectively inhibit secretion of inflammation cytokines in amniotic fluid and meliorate inflammation response caused by LPS (Fig. 3e).

MitoTEMPO (MT) inhibited LPS-induced preterm labor and myometrial contraction, decreased pro-inflammatory mediators in amniotic fluid.
The Effect of MitoTEMPO on LPS-Induced Mouse Preterm Delivery
p < 0.001 showed significant difference in preterm rate between Control and LPS group.
p < 0.001 showed significant difference in preterm rate between LPS and LPS+MT group.
LPS, lipopolysaccharide; MT, MitoTEMPO.
MT ameliorates oxidative stress and inflammation in placenta and maternal circulation
To further investigate the influence of MT on oxidative stress, we examined oxidative status in mouse placenta and serum. We found that LPS significantly induced apoptosis and ROS production in placenta, as evidenced by increased staining of TUNEL and Dihydroethidium (DHE). Notably, these histological alterations were alleviated by MT (Fig. 4a–d). In addition, LPS greatly elevated the oxidative stress hallmarks of MDA in mice placenta and serum while being reduced by MT treatments (Fig. 4e, f). SOD activity representing antioxidant level was restored in placenta of MT-treated mice after LPS challenge (Fig. 4g).

MitoTEMPO (MT) ameliorated oxidative stress and inflammation in placenta and maternal circulation.
After that, inflammatory response was studied to determine whether it could be a therapeutic target for MT to exert its protective function. With ELISA analysis, we revealed that MT administration significantly decreased inflammatory cytokines in LPS-treated mice, as demonstrated by the reduced levels of IL-1b, IL-6, and TNF-α in placentas and maternal circulation (Fig. 4h–j, l–n). Progesterone is secreted by placenta in late pregnancy, which is important for maintaining pregnancy. LPS-reduced secretion of placental and serum progesterone was significantly increased in mice with MT treatment (Fig. 4k, o).
MT improves fetal brain injury and cognitive function in offspring mice
To investigate whether MT has protective effects on offspring, we first sought to evaluate cell apoptosis and neuroinflammation in fetal brain tissues. We found increased cell apoptosis in fetal brain tissues in LPS group by TUNEL staining, whereas MT decreased cell apoptosis in fetal brains and protected brain from LPS damage (Fig. 5a, b). The expression of neuroinflammation proteins, including iNOS, GFAP, and IBA1, was significantly increased in LPS group, which was reduced by MT prenatal treatment (Fig. 5c, d). Furthermore, we attempted to discover the long-term effects of MT on the brain function in offspring. For physical development of offspring mice, there was no substantial variation in the offspring mice weight from 1 to 7 weeks among three groups (Fig. 5e). Then we used offspring mice (5 weeks old) to carry out open field tests representing exploratory behavior and anxiety (Fig. 5f). No obvious difference of distance traveled, average speed, and time in center was found among three groups. We used offspring mice (8 weeks old) to conduct the Morris water maze experiment related to spatial learning and memory. In the training phase, the time required to the platform gradually decreased, and there was no substantial variation among three groups (Fig. 5g). In the test phase, no significant difference of distance traveled, average speed, time in target quadrant, and target crossings was found among three groups (Fig. 5h–l).

The protective role of MitoTEMPO (MT) on brain injury of offspring mice.
MT inhibits NF-κB and NLRP3 signaling pathway in trophoblast cells by suppressing ROS
Our in vivo findings demonstrated that the MT protected LPS-treated mice from inflammation and oxidative stress. To validate the MT function, in vitro studies were then performed with human trophoblast cells. At first, CCK-8 analysis examined whether MT was safe for cells. As shown in Figure 6a, MT administration at different concentrations had no notable effects on the survival of HTR-8/Svneo cells, indicating the noncytotoxicity of MT. Then the MT regulatory role on oxidative stress and inflammatory responses was further investigated in human trophoblast cells. Compared with LPS group, cells cotreated with MT revealed higher levels of antioxidant gene expression such as NAD(P)H quinone dehydrogenase (NQO), heme oxygenase 1 (HO-1), glutamate–cysteine ligase catalytic subunit (GCLC), and glutamate–cysteine ligase regulatory subunit (GCLM) (Fig. 6b). Immunofluorescence (IF) staining exhibited that LPS exposure increased cellular ROS and mitochondrial ROS levels in HTR-8/Svneo and BeWo cells, which were significantly abrogated by MT treatments (Fig. 6d, Supplementary Fig. S2a–c). We revealed that LPS strongly elevated IL-1a, IL-1β, IL-6, TNF-α, NLRP3, IL-8, and IL-18 expressions in cells, and this impact was notably reduced upon MT coincubation (Fig. 6c). Activating the NF-κB subunit performs a critical role in modulating inflammatory response. IF staining demonstrated that LPS induced a significant elevation of NF-κB subunit (p-p65) translocation into the nucleus and activated NF-κB pathway, whereas MT distinctly attenuated this translocation (Fig. 6e). Consistently, Western blot analysis showed that LPS stimulated inflammatory response, as evidenced by elevated expression levels of NF-κB (iNOS, p-p65, COX2, p-IκBα) and NLRP3 pathway (NLRP3, IL-1β, cleaved caspase 1, IL-18), which were efficiently reversed by MT (Fig. 6f, Supplementary Fig. S2d, e). We included uncropped raw images of all western blots as supplementary materials (Supplementary Data S4). We uploaded raw data of different expression of cytokines/chemokines in amniotic fluid from LPS and Control group (Figure 3d, Supplementary Table S3) and LPS and LPS+MT group (Figure 3e, Supplementary Table S4).

MitoTEMPO (MT) inhibited NF-κB and NLRP3 signaling pathway by reducing ROS in LPS-stimulated HTR-8/Svneo cells.
The protective effects of MT depends on Kelch-like ECH-associated protein 1/Nrf2 antioxidant pathway
As MT regulated Nrf2 induction in nerve cells, we examined if MT activated Nrf2 to perform protection to placental function and explore its mechanism. Nrf2 protein is continuously degraded in the cytosol by Kelch-like ECH-associated protein 1 (Keap1)-dependent ubiquitination pathway, hence we investigated the effect of MT on Keap1 and Nrf2 protein in HTR-8/Svneo cells. First, to explore the potential binding modes of MT with Keap1 or Nrf2, we performed molecular docking analysis. MT had docking scores of −6.605 with Keap1 and −3.248 with Nrf2 (Fig. 7a) and had binding modes with protein Keap1 at site 1 (Fig. S3), suggesting a strong interaction between MT and Keap1 at site 1 with the highest affinity, demonstrated in Figure 8b–d. MT resided in the hydrophobic pocket, enclosed by residues Val 418, Val 369, Val 608, Val 465, and Val 514, forming a stable hydrophobic association. Furthermore, we treated cells with CHX (Cycloheximide, protein synthesis inhibitor) to examine whether MT interacts with Keap1 to promote its degradation. When treated cells with LPS, Keap1 protein expression was relatively stable after treatment, but MT treatment stimulated a continuing degradation of Keap1 protein from 2 to 10 h (Fig. 7b, c). The results indicated that MT destabilized Keap1 protein through degradation pathway, thus dissociating Nrf2 from Keap1 and reducing the ubiquitylation degradation of Nrf2 to protect Nrf2 stability. Then, we knocked down Nrf2 expression by siRNA in vitro and inhibited Nrf2 activation by ML385 in vivo to explore if MT produces its protective function via Nrf2 activation. In in vitro experiments, Nrf2 knockdown inhibited expression of antioxidant proteins like NQO1 and HO-1 (Fig. 7d). MT treatment markedly decreased the protein expression of inflammatory molecules and activated Nrf2/HO-1 in trophoblast cells, but these effects were inhibited with Nrf2 knockdown (Fig. 7e, f). In in vivo experiments, compared with LPS+MT pregnant mice, the PTB rate of pregnant mice in LPS+MT+ML385 group was significantly increased (Fig. 7g). The fetal mice in LPS+MT+ML385 group were born prematurely, and some of them died in uterus with body structure completely disappeared (Fig. 7h). These outcomes proved that the protection role of MT against LPS-induced injury depended on the Nrf2 activation. Together, we hypothesized that MT might directly compete with Nrf2 for binding to Keap1 and limit Nrf2-Keap1 interaction, thereby preventing ubiquitination degradation of Nrf2 and leading to Nrf2 activation.

The protective function of MitoTEMPO (MT) depends on Nrf2 activation by promoting Keap1 degradation in LPS-stimulated HTR-8/Svneo cells.
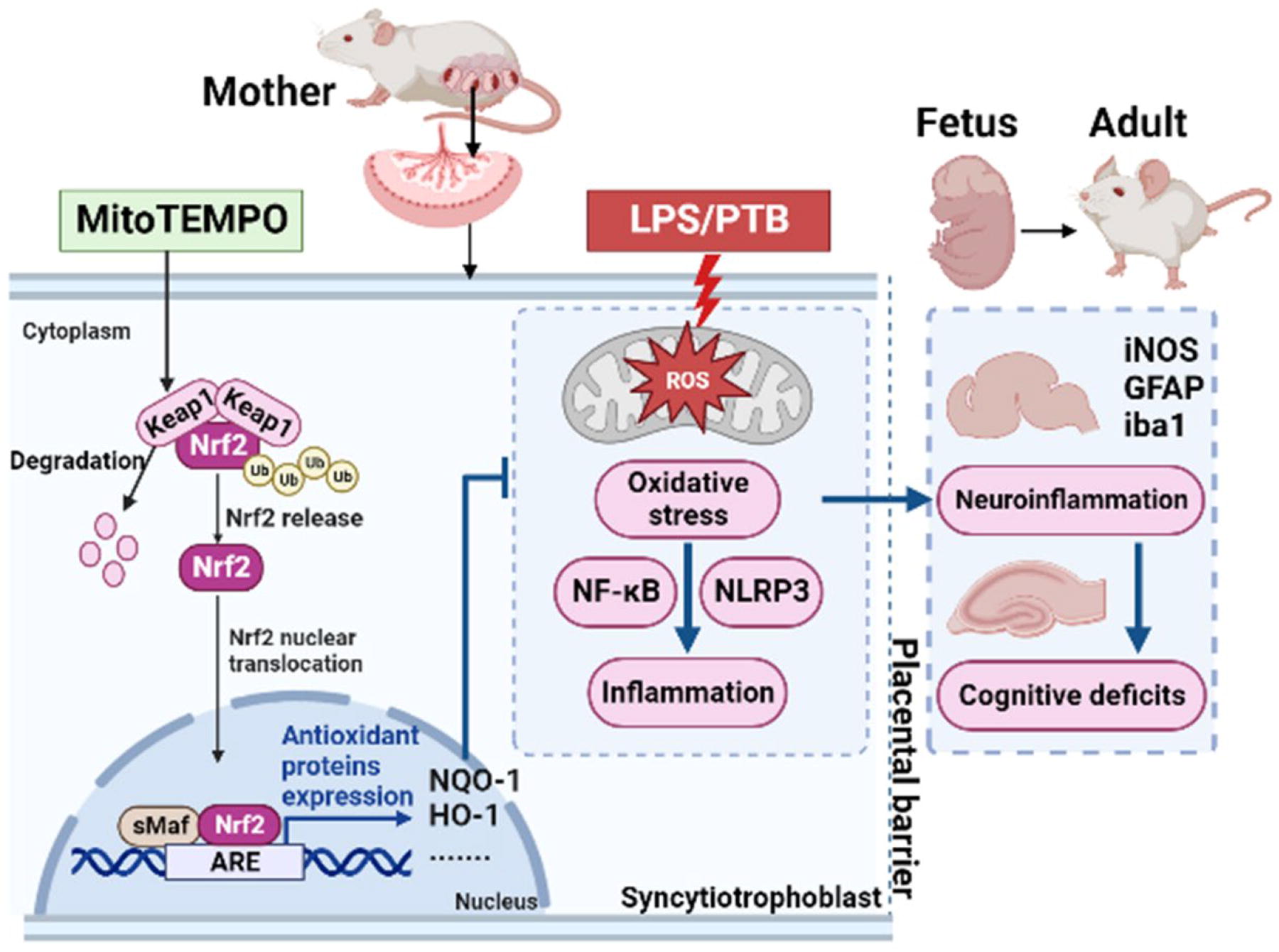
Schematic summary of MitoTEMPO (MT) inhibiting preterm birth and protecting offspring brain injury under the inflammatory condition. In simulation by LPS, MT promotes the degradation of Keap1 to inhibit the proteasome pathway degradation of Nrf2 and subsequently increases Nrf2 nuclear translocation, thus leading to Nrf2 nuclear accumulation and activation, which enhance the ARE transcription. The downstream products of ARE such as NQO1 and HO-1 greatly resist oxidative stress damage to improve intrauterine inflammatory circumstance, resulting in the inhibition of neuroinflammation in fetal brains and alleviation of cognitive deficits in offspring adults. The figure was created with BioRender (https://biorender.com/). ARE, antioxidant response element; Keap1, Kelch-like ECH-associated protein 1; LPS, lipopolysaccharide; Nrf2, nuclear factor erythroid 2-related factor 2.
Discussion
In this study, we discovered the mechanism of spontaneous PTB and found that mitochondrial dysfunction, oxidative stress, and inflammatory response impaired preterm placental function, thereby implicating the pathogenesis of PTB. We developed a mouse model of LPS-induced PTB and explored the therapeutic effects of MT on PTB. MT prevented PTB and protected offspring neurodevelopment by inhibiting placental oxidative stress and inflammatory response through Keap1/Nrf2/ARE pathway. The potential signaling pathways and underlying mechanisms that relied on our current experimental findings are illustrated in Figure 8.
The placenta is mainly composed of the chorionic plate, the villus lobules, the top plate, and the decidua basalis attached to the top plate. The villous lobule is the functional unit of placenta, and its main components are trophoblast cells (cytotrophoblast and syncytiotrophoblast) (Burton and Fowden, 2015). The placenta forms the maternal–fetal interface and participates in the labor initiation through various ways: (1) secretion of estrogen and corticosteroid-releasing hormone (CRH) diffuses into myometrium to transfer the myometrium from a resting state to an activated state (Wang et al., 2012) and (2) sterile inflammation in the placenta induces inflammation cascades involved in the onset of labor (Buhimschi et al., 2009). If placenta has abnormal hormone secretion, early activation of inflammatory response, and other pathological conditions, it will cause several pregnancy complications, such as intrauterine growth restriction, preeclampsia, and premature birth (Brien et al., 2019). Our study showed that preterm placenta had fewer angiogenesis, more cell apoptosis, suppressed hormone secretion, and glucose transport compared with term placenta. These pathological changes in preterm placenta are adverse to fetal development and pregnancy maintenance, suggesting that placental dysfunction is associated with PTB.
Mitochondria, as the main place for cell respiration and energy production, are involved in many placental physiological activities. Therefore, mitochondrial function is important for maintaining placental physiology. Accordingly, we observed the mitochondrial alterations in premature placenta. We found that premature placenta had decreased mtDNA copy number and abnormal mitochondrial structure, especially in cristae. Meanwhile, we found impaired mitochondrial function in preterm placenta, manifested as disrupted TCA cycle and aberrant activated glycolysis. The metabolic switch from TCA cycle to glycolysis may be a compensatory mechanism for mitochondrial damage to ensure energy supply. Mitochondrial dysfunction increases the release of mitochondrial damage–associated molecular patterns (mtDAMPs), such as mtROS, mtDNA, and adenosine triphosphate. Our results verified that preterm placenta overproduced ROS with decreased antioxidant capacity, resulting in the accumulation of ROS in the placenta. The excessive mtROS activates NF-κB pathway and cellular NLRP3 inflammasome, which subsequently stimulate the innate immune response to produce proinflammatory cytokines (Franchi et al., 2009). Recent studies have indicated that PTB is correlated with NF-κB and NLRP3 activation (Faro et al., 2019, Sheller-Miller et al., 2021). Coincidently, our data showed that these inflammatory cytokines significantly increased in preterm placenta. These data demonstrated that mitochondrial dysfunction induced excessive mtROS and triggered sterile inflammatory response, thus leading to placental inflammation and impaired placental function involved in PTB. Therefore, protecting placental mitochondrial function, especially reducing mtROS overproduction, may inhibit placental inflammation and prevent PTB.
As a mitochondria targeted antioxidant, MT was implicated in this study. As expected, MT treatment could inhibit PTB by decreasing oxidative stress and alleviating LPS-induced inflammation in maternal tissues, including myometrium, placentas, circulation, and amniotic fluid. Specifically, MT inhibited macrophage infiltration in the myometrium and also decreased the expression of uterine contractile proteins, thereby preventing premature labor onset. We also found that MT could increase placental progesterone synthesis. Progesterone performs a critical role in keeping myometrial quiescence and pregnancy, which is synthesized by the corpus luteum in early gestation and produced by the placenta during late gestation (Henson, 1998). Studies have shown that mitochondrial progesterone synthase containing steroidogenic acute regulatory protein, cholesterol side-chain cleavage enzyme, and 3β-Hydroxysteroid dehydrogenase were involved in mitochondrial synthesis of cholesterol into placental progesterone (Strushkevich et al., 2011). We suggest that MT protects placental mitochondrial function to synthesize progesterone, thereby inhibiting myometrial contraction. All available evidence demonstrated that MT had positive effects on LPS-induced PTB, which was strongly associated with its antioxidative and anti-inflammatory activities, thus improving placental function and maintaining pregnancy.
PTB remains a leading cause of perinatal brain damage and subsequent neurological dysfunction. Epidemiological and experimental data suggest that placental dysfunction affects fetal neurodevelopment through the placenta–brain axis in the short and the long term (Zeltser and Leibel, 2011). We have shown that PTB is associated with placental dysfunction, including placental inflammation and placental oxidative stress. Placental inflammatory mediators, including cytokines and ROS, pass through the blood–brain barrier to affect the regulatory ability of vascular endothelial growth and damage cerebrovascular function, as well as increasing the probability of cerebral white matter damage (Leviton and Gressens, 2007). Moreover, these mediators directly inhibit the maturation and regeneration of nerve cells and exacerbate the degree of brain injury (Kwon et al., 2022; Shiow et al., 2017). In addition, fetal brain is susceptible to oxidative stress. Excess ROS promotes the production of inflammatory mediators and activates multiple neuroinflammatory pathways in microglia and astrocytes, thus leading to a vicious circle. Both microglia and astrocytes are innate immune cells of the central nervous system, which are existing in the fetal brain during embryonic development period and participate in neuroinflammation (Billiards et al., 2006; Gowrisankar and Clark, 2016). Previous studies have reported that activated microglia and astrocytes may be the therapeutic target cells for fetal brain injury induced by intrauterine inflammation (Cloarec et al., 2018). Treatment of microglia or astrocyte-mediated neuroinflammation may delay brain damage progression and prolong the time window for neonatal postnatal treatment, thereby allowing the nervous system to develop normally. Since neuroinflammation is mostly related to mitochondrial dysfunction, MT is used to improve mitochondrial function and inhibit neuroinflammation in recent studies (Zhelev et al., 2013). Previously, we have proved that MT is effective in preterm labor treatment, and we further valued the potential application of MT as a neurotherapeutic drug in preterm brain injury. We suggested that MT could protect fetal brain damage by reducing delivery of placental proinflammatory factors to the fetus. In this study, we found that LPS not only caused acute brain damage in fetus by activating microglia and astrocytes and increasing neuronal apoptosis but also adversely affected fetal brain development with long-term consequences in adulthood, as the fetal brain was damaged during the immature stage, whereas MT suppressed PTB and protected offspring from deleterious effects caused by LPS. Specifically, prenatal prophylactic use of MT could decrease expression of neuroinflammatory protein and inactivation of glial cells in fetal brain, thereby repairing neural damage and protecting the structural integrity of fetal brain. Studies have shown that maternal inflammation may cause long-term epigenetic changes in offspring brain and increase the risk of neurodevelopmental disorders (Han et al., 2021). We observed the behavioral changes of offspring mice at 35 and 56 days after birth and found that MT improved spatial exploration ability and cognitive function of the offspring exposed to maternal inflammation. The protective effect of MT on offspring brain function is a long-term process that remodels nerve cells to be more regenerative and able to cope with various injuries caused by inflammation.
Based on the positive results in vivo, we used LPS-stimulated trophoblast cell lines to further investigate the mechanism of the protective effects of MT against oxidative stress and inflammation. Consistent with in vivo study, in trophoblast cells, MT treatment suppressed LPS-triggered productions of ROS and cytokines and inhibited NF-κB activity and NLRP3 pathway. Meanwhile, MT enhanced antioxidant gene expression such as NQO1, HO-1, GCLC, and GCLM, which are transcripted by Nrf2. Previous studies have revealed that MT can activate Nrf2 pathway in different cell lines and tissues (Barzegari et al., 2020). We proposed that MT inhibited PTB dependent mainly on its capacities to stimulate Nrf2 activation. In this study, we revealed that Nrf2 knockdown strongly suppressed MT-induced antioxidant gene expressions, as well as reversed the inhibitory effects of MT on LPS-triggered inflammatory response in trophoblast cells. Consistently, animal experiments also showed that the therapeutic effects of MT on PTB were abolished when treated mice with Nrf2 inhibitor. All of these demonstrate the significance of Nrf2 activation in the beneficial effects of MT. However, the specific mechanism by which MT activates Nrf2 pathway remains unknown. Nrf2 activity is suppressed by binding with Keap1 in the cytoplasm where the Keap1/Nrf2 complex induces Nrf2 degradation by ubiquitin-proteasome pathways. Within stress conditions, Nrf2 is dissociated from Keap1 and translocates into the nucleus to bind to small proteins Maf, namely, MafF, MafG, and MafK. These proteins form a heterodimer to initiate the transcription of several downstream target genes at the sites of AREs (Taguchi et al., 2011). Thus, Nrf2 activity was partly dependent on the Keap1-Nrf2 interaction. In recent years, several molecules and proteins have been discovered to exert antioxidant, as well as anti-inflammatory effects, by inhibiting Keap1/Nrf2 binding (Lu et al., 2016). As such, we reasoned that MT might influence the Keap1-Nrf2 interaction. To confirm this hypothesis, we conducted molecular docking and proved that MT competitively bound to Keap1 and occupied the Nrf2 binding site, leading to the Nrf2 release and activation in trophoblast cells. Our study uncovered a novel mechanism whereby MT activated Nrf2 signaling pathway by suppressing the Keap1-Nrf2 interaction to treat PTB.
Our study had some limitations. First, the survival rate of offspring born in the LPS-induced PTB mouse model used in this study was extremely low or even stillborn, which may be due to immature development of fetus and its inability to survive leaving the intrauterine environment before gestational age of 19 days or because of inflammatory damage to fetus after exposure to high levels of inflammatory mediators in uterus; thus, our study lacked data on the growth and neurodevelopment of offspring born in the LPS-treated pregnant mice. Second, the LPS-induced PTB mouse model only simulated the pathological processes of inflammation-induced PTB; therefore, we could not study whether MT had protective effects on PTB caused by other stimuli.
Taken together, we demonstrated that MT could treat PTB and fetal brain damage through inhibiting oxidative stress and inflammation, improving mitochondrial function and maintaining placental function. The underlying mechanism is to promote Nrf2 activation via Keap1/Nrf2 interaction. Our findings provide evidence for MT as a potential drug for treatment of PTB via Keap1/Nrf2/ARE signaling pathway.
MT during pregnancy not only effectively prevented PTB and inhibited placental inflammation through activating Nrf2 antioxidant pathway but also prevented neuroinflammation in fetal brain and improved cognitive function of offspring in the long term, thus providing a new therapeutic strategy for PTB and fetal brain injury.
Materials and Methods
Human tissue collection
The Ethics Committee of Women’s Hospital, School of Medicine, Zhejiang University granted approval for this study (grant number: IRB-20200160-R). Before they were recruited, every single woman gave their written informed consent. We obtained placental tissues from women undergoing vaginal delivery at preterm (30 weeks–36 weeks and 6 days, PTL, n = 21) or term (37 weeks–39 weeks, TL, n = 16) with evident onset of labor and women having cesarean sections at term (37 weeks–39 weeks, TNL, n = 12) in the absence of labor. All of the placental tissues from PTL had pathological diagnosis of chorioamnionitis. Exclusion criteria included multiple pregnancies, serious maternal medical conditions, fetal aneuploidy, or lethal fetal abnormalities. Individual demographic and baseline features in age, parity, and body–mass index have not shown any statistically significant results between three groups. After delivery of placenta, placental tissues and villus lobules were immediately collected (2 cm from the placental margin, excluding placental infarcts) and rinsed in PBS to remove blood. Some samples were fixed in 4% paraformaldehyde for staining, some samples prepared for transmission electron microscopy by fixation in 2.5% glutaraldehyde, and some samples snap-frozen in liquid nitrogen and stored at −80°C until further examination.
IF
Placental villi were cut into 5 μm thick cryosections and preserved with primary antibodies against VEGF (1:100, 19003-1-AP, Proteintech), Glut1 (1:100, 21829-1-AP, Proteintech), Glut3 (1:200, 20403-1-AP, Proteintech), and β-hCG (1:100, 11615-1-AP, Proteintech) overnight at 4°C. For cellular IF, cells were cultured on slides that were positioned within the 12-well plates, following that, they were fixed for a period of 1 h in 4% paraformaldehyde, permeabilized utilizing 0.2% Triton X-100, and inhibited with 1% BSA for a period of 1 h. After that, cells were preserved with a primary antibody against Phospho-NF-κB p65 (1:200, #3033, CST) overnight. The sections were then hydrated with PBS and subsequently supplemented with the secondary antibodies, Alex Fluor®488-conjugated anti-rabbit antibody (1:250, SA00006-2, Proteintech) or CY3-conjugated anti-mouse antibody (1:200, SA00009-1, Proteintech) for 1 h in the dark at room temperature (RT). Nuclei were stained by treating slices with 5 μg/mL DAPI (Sigma) for 10 min. Ki67 staining and terminal deoxynucleotidyl TUNEL assay were conducted utilizing an anti-Ki67 primary antibody (Abcam) and a fluorescein-based cell death detection kit (Sigma). Using a confocal fluorescent microscope, fluorescent images were obtained, and the staining intensity was analyzed with the Image-Pro Plus 6.0 software. Each segment was measured across five distinct fields.
TEM
TEM was conducted as previously described (Chen et al., 2019). Placental tissues were fixed in 2.5% glutaraldehyde for over 4 h at 4°C, followed by 1 h of incubation at RT with 1% osmic acid. The specimens were dried with ethanol solution (50, 70, 90, and 100%, respectively) and 100% acetone solution, embedded for 4 h, and then cut into 70 nm ultrathin sections. Before analysis, the sections were mounted on copper grids and stained with uranyl acetate and lead citrate. They were detected with transmission electron microscope (FEI Tecnai G2 Spirit 120 kV) to examine mitochondrial distribution and morphology.
Targeted energy metabolomics
Placental samples were sent for targeted metabolomics analysis (LC-Bio Technology), and clinical statistics are shown in Supplementary Table S1. Briefly, we mixed 100 mg placental tissues with 1 mL cold methanol/acetonitrile/H2O solution. The mixture was sonicated for 30 min on ice, incubated at −20°C for 1 h to allow protein precipitation, and centrifuged for 20 min (14000 g, 4°C). The supernatants were extracted and dehydrated in a vacuum lyophilizer. The dehydrated specimens were dissolved in 100 μL acetonitrile/H2O solution and centrifuged for 15 min (14000 g, 4°C). The supernatants were extracted in order to perform the LC-MS/MS analysis. Using an UHPLC (1290 Infinity LC, Agilent Technologies) and a QTRAP (AB SCIEX 5500), analyses were performed.
Oxidative stress parameters
The oxidized and reduced forms of nicotinamide adenine dinucleotide (NAD+ and NADH) levels were identified utilizing commercial kits (Beyotime). The levels of 8-OHdG, MDA, GSH, SOD2, and T-AOC were measured by their commercial kits (Beyotime) according to the kits’ protocols. The placental levels of NAD+, NADH, 8-OHdG, MDA, GSH, SOD2, and T-AOC were adjusted to the placental total protein, which were determined utilizing BCA protein assay kit (Thermo). Placental villi were cleaved into 5 μm thick slices, which were subsequently subjected to an incubation with primary antibody against 4HNE (1:100, ab46545, Abcam) or TOMM20 (1:200, 11802-1-AP, Proteintech) for 1 h and secondary antibody (DAKO) at RT for a period of 30 min. DHE (Beyotime) assay kit was used to evaluate placental ROS by staining sections for 30 min. They were examined with a microscope (Leica LMD). For mtROS and cellular ROS, cells were treated with 10 μM DCFA (Beyotime) or 5 μM MitoSox Red dye (M36008, Invitrogen) for 30 min at 37°C and detected with a microscope (Leica LMD).
Reverse transcription and quantitative real-time polymerase chain reaction
Total RNA was collected from placental tissues and cells utilizing the RNAiso Plus reagent (Takara Bio) with the manufacturer’s guidelines. After that, the RNA was reverse transcribed to cDNA utilizing the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara Bio). Reverse transcription and quantitative real-time polymerase chain reaction (PCR) was conducted using the TB Green Premix Ex Taq™ Kit (Takara Bio) in a 7900HT Sequence Detection System (Applied Biosystems). The PCR program included 40 cycles of 95°C for 30 s, 95°C for 5 s, and 60°C for 30 s. We adjusted triplicate wells for every specimen to compute its mean cyclic threshold (Ct) value. The levels of target gene relative expression were investigated utilizing the 2−ΔΔCt method. Relative mtDNA copy number was computed through quantitating mtDNA copy number relative to nuclear DNA, as described previously (Miller et al., 2003). Primers for mitochondrial ND1 were used for determination of mtDNA content. All primers used for quantitative real-time PCR were developed by Tsingke Biotech (Beijing) and presented in Supplementary Table S2.
ELISA
Placental tissue homogenate and mouse serum were centrifuged at 2500 g at 4°C for 15 min to layer, and supernatants were preserved at −80°C for later ELISA. For human samples, IL-1β (EK101BHS), IL-6 (EK106HS), IL-18 (EK118), and TNF-α (EK182HS) ELISA kits (MultiSciences Biotech) were used to examine the cytokine concentrations in conformity to the instructions. For mouse samples, IL-1β (EK201BHS), IL-6 (EK206HS), and TNF-α (EK282HS) ELISA kits (MultiSciences Biotech) were utilized to examine the IL-6 and TNF-α concentrations according to the instructions. The progesterone levels were evaluated based on the manufacturer’s guidelines of the ELISA kit (E-EL-0154, Elabscience).
Animals
The Animal Ethics and Welfare Committee of Zhejiang Chinese Medical University approved all animal experiments (grant number: IACUC - 20200601-04). The ICR mice (8–10 weeks old; male mice: 30–35 g; female mice: 25–30 g) were preserved in a controlled environment (humidity: 50 ± 5%, temperature: 20°C–25°C) on a 12-h light/12-h dark cycle and fed with a standard laboratory diet. For coupling purposes, females were housed with males overnight at a 2:1 ratio at 9:00 P.M. Females underwent checking by 8:00 A.M. the following morning. A vaginal plug presence was marked as gestational day D0.
Experiment 1. To examine the functions of MT on LPS-stimulated PTB, all pregnant mice were separated into Control, LPS, MT, and LPS+MT groups, as depicted in Supplementary Figure S1. Pregnant mice were assigned randomly to either intraperitoneal (i.p.) injection with MT (Sigma, 2 mg/kg, dissolved in saline) or saline once daily from D13 to D17. The dose of MT used was selected similarly to the doses utilized in earlier investigations (Dikalova et al., 2017). All pregnant mice were i.p. injected with saline or LPS (Sigma, 250 μg/kg, dissolved in saline) on D15. The LPS dose was selected on basis of earlier studies (Cao et al., 2016) and preliminary experiments.
Experiment 2. To analyze the effects of Nrf2 activation on MT inhibiting LPS-stimulated PTB, some pregnant mice were separated into LPS+MT+ML385 group. They were i.p. injected with MT (2 mg/kg) from D13 to D17. On D15, they were i.p. injected with ML385 (Nrf2 inhibitor, from MCE, 10 mg/kg) 1 h before i.p. injection with LPS (250 μg/kg).
All pregnant mice were monitored carefully for checking the signs of preterm labor (like vaginal bleeding), and we defined PTB as delivery before D19. Some pregnant mice in every group were sacrificed 5 h after injection with saline or LPS on D15 to collect serum, amniotic fluid, placentas, fetal brains, and uterus tissues for further study.
Immunohistochemistry
The F4/80 molecule was considered as a unique marker of murine macrophages (Lin et al., 2005). We examined F4/80 expression in mice myometrium by immunohistochemistry. In brief, mouse myometrial tissues were sliced into 5 μm sections. After deparaffinization and dehydration, antigen retrieval was conducted by heating for a period of 10 min. The endogenous peroxidase activity was quenched by preserving with 3% hydrogen peroxide for 30 min after cooling to RT and rinsing in PBS. The samples were preserved with a primary antibody against F4/80 (1:100, 14–4801-81, Thermo) for a duration of 1 h and a secondary antibody for 30 min. Negative controls were conducted via replacing the primary antibody with the appropriate non-immune IgG isotypes. Samples were examined under a microscope (Leica LMD) and documented as positive if they were brown. The staining intensity of sections was semiquantitatively analyzed using Image-Pro Plus 6.0 software. The integrated optical density (IOD) of the brown part was divided by the total area to calculate the mean value of IOD, which was correlated to the corresponding protein expression. Each section was evaluated in five distinct fields.
Western blot analysis
The extraction of proteins was conducted via lysing tissues or cells for a period of 30 min at 4°C utilizing RIPA lysis buffer, followed by centrifugation (14,000 rpm, 15 min) at 4°C. Protein concentration was measured by BCA assay kit (Thermo). On a 12% SDS-PAGE, equal protein samples were separated, transferred onto a PVDF membrane (Bio-Rad), and inhibited with 5% BSA (Sigma) for a period of 1 h at RT. Incubation of the membrane was performed with primary antibodies overnight at 4°C, followed by incubation with HRP-conjugated secondary antibodies for 1 h at RT, and finally exposed to enhanced chemiluminescence detection reagent (Sigma). Band intensities were assessed by ImageJ software and normalized by GAPDH or ACTIN expression. The antibodies used were as follows: NLRP3 (1:1000, #15101, CST), p-p65 (1:1000, #3033, CST), COX2 (1:500, ab179800, Abcam), p-IκBα (1:1000, ab133462, Abcam), NRF2 (1:1000, ab137550, Abcam), GFAP (1:500, sc33673, Santa Cruz), iNOS (1:1000, 18985-1-AP, Proteintech), IBA1 (1:1000, 10904-1-AP, Proteintech), Caspase 1 (1:1000, 22915-1-AP, Proteintech), IL-1β (1:1000, 16806-1-AP, Proteintech), IL18 (1:1000, 10663-1-AP, Proteintech), Keap1 (1:1000, 10503-2-AP, Proteintech), NQO1 (1:1000, 11451-1-AP, Proteintech), HO-1 (1:1000, 10701-1-AP, Proteintech), GAPDH (1:2000, ab8245, Abcam), and β-Actin (1:2000, ab8226, Abcam).
Quantitative inflammation array
Amniotic fluid was extracted 5 h following LPS or saline injection on D15. The concentrations of 40 cytokines in amniotic fluid were measured using the Quantibody Mouse Inflammation Array Kit in accordance with the manufacturer’s guidelines (RayBiotech).
Open field test
Mice were exposed to the open field (45 × 45 × 45 cm square chamber) for 5 min free activity. A computer software (Smart 3.0-Panlab Harvard Apparatus) connected to a camera above the chamber recorded and analyzed the behaviors of mice. To measure the anxiety of the mice, the behavioral parameters were recorded during 5 min, including distance traveled, average speed, and time in the center.
Morris water maze test
The Morris water maze (MWM) test was conducted in a cylindrical tank (1.8 m in diameter, 0.5;m in height). The tank was full of water (23 ± 2°C). As external geographical clues for four distinct directions, four objects of various colors and shapes were affixed to the wall. In the center of the northwest quadrant, a transparent circular platform (10 cm in diameter) was loaded almost 1 cm beneath the water surface. Throughout the 5-day acquired training, mice were positioned in water, oriented toward the pool’s wall at four distinct sites on a daily basis, and the duration of the mice’s journey to the platform was documented. If the mouse did not locate the platform throughout 60 s, the latency was measured to be 60 s, and the mouse was directed to the platform to stay for 15 s. In order to perform the probe trial on the sixth day, the platform was eliminated, and every mouse was released from the southeast quadrant. The duration of time spent swimming in the target quadrant in 60 s interval was documented. The swimming trajectory and performance of the mice were recorded automatically and investigated with animal behavior analysis software (Smart 3.0-Panlab Harvard Apparatus).
Cell culture and treatments
The human trophoblast cell line HTR-8/SVneo cells (ATCC) were cultured in RPMI 1640 medium (BI) coupled with 10% fetal bovine serum (FBS, Gibco), and BeWo cells (ATCC) were cultured in Ham’s F-12K (Gibco), including 10% FBS. All cultures were preserved at 37°C in a humidified 5% CO2 atmosphere. Cells were pretreated with 100 μM MT for 4 h and then 1 μg/mL LPS for another 20 h. CHX concentrations were selected based on a publication (Li et al., 2021). CHX (50 μM) was added to incubate with cells for 0, 2, 4, 6, 8, and 10 h separately.
Cell proliferation assay
Cells were plated in 100 μL medium at a density of 5 × 103 per well in four 96-well culture plates and treated with different MT concentrations for 0, 24 h following the adherent cell development. Incubation of the cells were conducted with 10 μL/well Cell Counting Kit-8 (CK04-13, DOJINDO) solutions at 37°C for a period of 3 h, preventing from exposure to light. The optical density (OD) in the culture medium was assessed at 450 nm by a microplate reader.
Molecular docking
To gain molecular insights into the binding mode of MT at the Keap1 and Nrf2 binding sites, molecular docking approach was conducted by Maestro program in Schrödinger software (New York). The 3D structure of MT was converted from 2D version by LigPrep panel in Maestro. The crystal structures of Keap1 (PDB ID: 4L7B) and Nrf2 (PDB ID: 2LZ1) were acquired from Protein Data Bank (PDB), and the structures were processed and optimized by Protein Preparation panel in Maestro. The docking workflow was carried out based on Receptor Grid Generation panel, whereas receptor pocket was permitted to interchange based on ligand conformations, with their limited positions. Protein–ligand interactions were docked by Ligand Docking panel, and molecular graphics were done using Maestro program.
RNA interference by small interfering RNA
Scramble nontargeting control and Nrf2-specifc siRNA oligonucleotides were synthesized by GenePharma Corporation with the following target sequences:
5′-CGCUCAGUUACAACUAGAUTT-3′ (sense) and
5′-AUCUAGUUGUAACUGAGCGTT-3′ (antisense).
Statistical analysis
Data are presented as mean ± standard deviation of three independent experiments. All data analyses were conducted by the SPSS 12.0 statistical software (SPSS, Inc.). Variables in paired data were evaluated by parametric Student’s t-test. Multiple treatments were evaluated by one-way analysis of variance, and the means were separated by Tukey’s tests. p-Value of <0.05 is considered statistical significance. Electronic laboratory notebook was not used.
Footnotes
Acknowledgments
The authors thank all the pregnant women who participated in this study and thank all the clinicians and laboratory staff for their support and assistance in this study.
Authors’ Contributions
C.C. conducted the experiments, performed the experimental data analyses, and wrote the article. S.Z. and T.F. assisted with the experiments and collected human placental samples. L.B. and Y.C. edited the article and provided their valuable input for modification of the experimental design. D.C. supervised the project and revised the article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work was supported by the
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Data S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.