
Abstract
Significance:
Nitric oxide (NO) plays several distinct roles in endothelial homeostasis. Except for activating the guanylyl cyclase enzyme-dependent cyclic guanosine monophosphate signaling pathway, NO can bind reactive cysteine residues in target proteins, a process known as S-nitrosylation (SNO). SNO is proposed to explain the multiple biological functions of NO in the endothelium. Investigating the targets and mechanism of protein SNO in endothelial cells (ECs) can provide new strategies for treating endothelial dysfunction-related diseases.
Recent Advances:
In response to different environments, proteomics has identified multiple SNO targets in ECs. Functional studies confirm that SNO regulates NO bioavailability, inflammation, permeability, oxidative stress, mitochondrial function, and insulin sensitivity in ECs. It also influences EC proliferation, migration, apoptosis, and transdifferentiation.
Critical Issues:
Single-cell transcriptomic analysis of ECs isolated from different mouse tissues showed heterogeneous gene signatures. However, litter research focuses on the heterogeneous properties of SNO proteins in ECs derived from different tissues. Although metabolism reprogramming plays a vital role in endothelial functions, little is known about how protein SNO regulates metabolism reprogramming in ECs.
Future Directions:
Precisely deciphering the effects of protein SNO in ECs isolated from different tissues under different conditions is necessary to further characterize the relationship between protein SNO and endothelial dysfunction-related diseases. In addition, identifying SNO targets that can influence endothelial metabolic reprogramming and the underlying mechanism can offer new views on the crosstalk between metabolism and post-translational protein modification. Antioxid. Redox Signal. 40, 186–205.
Introduction
Nitric oxide (NO) was first identified as the endothelium-derived relaxing factor (Ignarro, 1989; Vanhoutte and Shepherd, 1989). Its role in vasodilation through smooth muscle cell soluble guanylyl cyclase (sGC) to produce cyclic GMP (cGMP) has been fully described (Arnold et al., 1977; Denninger and Marletta, 1999; Hussain et al., 1999). In mammals, NO is predominantly produced by three nitric oxide synthases (NOSs), including neuronal NOS (nNOS; NOS1) (Bredt et al., 1990; Gross et al., 1991), inducible NOS (iNOS; NOS2) (Gross et al., 1991), and endothelial NOS (eNOS; NOS3) (Gross et al., 1991).
nNOS is constitutively expressed in neurons and is also found in the peripheral circulation system (Boulanger et al., 1998; Xu et al., 1999). iNOS substantially increases in many cell types in response to inflammatory stimuli; it catalyzes the production of large amounts of NO to mediate the pathology of the inflammatory response (Kroncke et al., 1998). eNOS is primarily expressed in endothelial cells (ECs), and found in neurons and other tissues (Zhao et al., 2015b). It binds to plasma membranes and is typically associated with caveolin. eNOS is activated by calcium entry through membrane-bound receptors and phosphorylation at The, Ser, and Tyr (Forstermann and Sessa, 2012).
NOS3 regulates the dilation of blood vessels, prevents platelet aggregation and coagulation, and inhibits inflammatory response (Zhao et al., 2015b). Recent work shows that NO exerts multiple biological and physiological effects through the post-translational thiol modification of proteins by S-nitrosylation (SNO). There are many reviews about the multiple functions of protein SNO (Foster et al., 2003; Hess et al., 2005; Lima et al., 2010; Marshall et al., 2004; Martinez-Ruiz and Lamas, 2004b; Nakamura and Lipton, 2007; Nakamura et al., 2013; Plenchette et al., 2015; Sun and Murphy, 2010; Zhao et al., 2015a; Zhou et al., 2022). SNO can regulate cellular function by influencing enzyme activity, protein–protein interaction, protein stability, subcellular localization, ion channel conductivity, and transcriptional activity (Stomberski et al., 2019b).
The transfer of the NO group between donor and acceptor Cys thiols via trans-SNO is believed to be the primary mechanism for protein SNO (Williams, 1999). On the contrary, denitrosylases remove NO groups from protein S-nitrosothiols and reduce the level of protein SNO (Stomberski et al., 2019b).
The thioredoxin (Trx) and thioredoxin reductase (TrxR) system are the major protein denitrosylases (Sengupta and Holmgren, 2013). In addition to the Trx/TrxR system, there are three kinds of low-molecular-weight thiol-cofactor–dependent denitrosylases: S-nitrosoglutathione reductase (GSNOR), SNO-CoA reductase (SNO-CoAR), and aldo-keto reductase family 1 member A1 (AKR1A1), thioredoxin-related denitrosylases (Stomberski et al., 2019a; Stomberski et al., 2019b).
The vascular endothelium is a constitutive part of the vasculature. It is formed by a monolayer of ECs, and is the interface between the circulating blood and other vascular components. The endothelium controls vascular relaxation and constriction in response to multiple intra- and intercellular stimuli, including hormones, metabolites, macromolecules, mechanical stretch, and blood flow (Kruger-Genge et al., 2019). In addition to barrier function, resting ECs play essential roles in regulating regional blood flow, antiplatelet aggregation and coagulation, anti-inflammation, and angiogenesis (Kruger-Genge et al., 2019; Pober and Sessa, 2007; Sang et al., 2021).
In physiological conditions, NO in ECs is produced mainly by eNOS, a homeostatic regulator of endothelial function. However, in the presence of high glucose, oxidized low-density lipoprotein (oxLDL), and angiotensin II, iNOS is increased in ECs, produces high levels of NO, and induces endothelial dysfunction (Chao et al., 2021; Gunnett et al., 2005; Pan et al., 2020; Zhao et al., 2022b).
Evidence demonstrates that protein SNO is involved in endothelial homeostasis and dysfunction, leading to multiple diseases, including atherosclerosis, hypertension, aortic dissection, and diabetic vascular complications (Chao et al., 2021; Pan et al., 2020; Zhao et al., 2022b). This review summarizes the current findings on the roles of protein SNO in regulating intracellular signaling pathways in ECs.
Proteomics of S-Nitrosylated Proteins in ECs
A growing number of SNO proteins have been identified over the past decades in ECs. In response to different environments, the SNO targets differ in their number and molecular functions, indicating the distinct role of protein SNO in endothelial homeostasis and dysfunction (Fig. 1). For example, steady laminar shear flow has been reported to protect ECs through activating eNOS via the PI3K/Akt pathway, leading to an increase in nitric oxide NO production (Dimmeler et al., 1999).
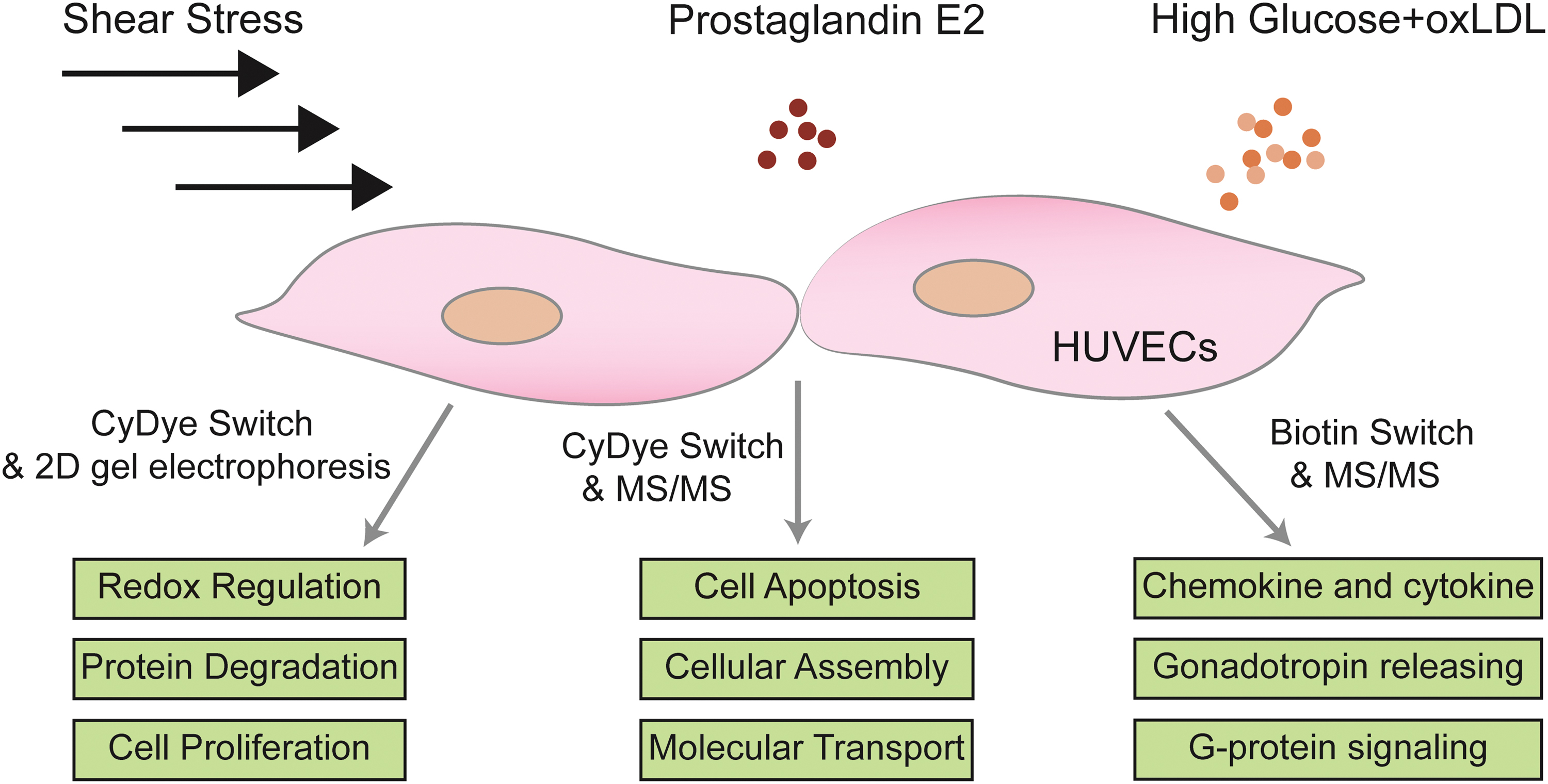
Using the CyDye switch and two-dimensional gel electrophoresis method, Huang et al. (2009a) found 12 proteins in human umbilical vein endothelial cells (HUVECs) subjected to shear flow with increased SNO. These SNO proteins show functions including redox regulation, protein degradation, cell proliferation, and cell defense (Huang et al., 2009a). This observation indicates that under physiological conditions, eNOS-derived NO preserves endothelial homeostasis at least partly through increasing protein SNO.
Estradiol-17beta (E2) is one of the most important estrogenic hormones with potential vascular dilation activity. In ECs, long-term E2 incubation transcriptionally enhances NOS3 gene expression through classical nuclear hormone receptors of estrogens, while short-term incubation with E2 stimulates eNOS-derived NO and relaxes blood vascular without changing eNOS expression levels.
A recent study found that the vascular relaxation activity E2 is related with protein SNO. In HUVECs stimulated with 10 nM E2 for 30 min, CyDye switch coupled with 2D-DIGE, MALDI-TOF, and MS/MS identified 58 SNO proteins. Among them, 28 are upregulated and 13 are downregulated. Pathway analysis identified that these different regulated SNO proteins are involved in pathways, including cell viability/apoptosis, cellular assembly and organization, cellular function and maintenance, and molecular transport (Zhang et al., 2010). This observation also apparently indicates that protein SNO is an important mechanism for cells to respond to extracellular stimuli and might mediate the vasodilation activity of E2.
In HUVECs stimulated with high glucose and oxLDL, Chao et al. (2021) observed enhanced endothelial inflammation with increased expression of chemokines and adhesion factors such as CXCL4, CXCL8, ICAM1, and VCAM1. Interestingly, they found an increased level of total protein SNO, and inhibition of protein SNO by iNOS inhibitor 1400W could alleviate endothelial inflammation.
LC-MS/MS identified 342 targets, and KEGG pathway analysis found that these SNO proteins are mainly involved in inflammation mediated by chemokine and cytokine signaling pathway (Fig. 1, not published). These observations reveal a relationship between protein SNO and endothelial inflammation in pathological condition.
Collectively, above literatures suggest the distinct role of protein SNO in endothelial homeostasis and dysfunction. Literatures also report the proteomics of SNO targets in ECs subjected to other stimuli, including high glucose, VEGFA, NO donors, and hypoxia (Chen et al., 2008; Huang et al., 2012; Huang et al., 2009b; Martinez-Ruiz and Lamas, 2004a; Satohisa et al., 2014; Wadham et al., 2007; Xu et al., 2019; Zhang et al., 2016; Zhang et al., 2012).
Roles of Protein SNO in NO Bioavailability
Reduced expression and dysregulation of eNOS lead to decreased NO bioavailability and endothelial dysfunction. eNOS is synthesized as monomers, and the formation of dimers is necessary for eNOS activation and NO production with L-arginine as substrate. Otherwise, the monomers generate O2 to induce oxidative stress (Forstermann and Munzel, 2006; Rafikov et al., 2011).
The binding of cofactor tetrahydrobiopterin (BH4) to the N-terminal oxygenase domain, nicotinamide-adenine-dinucleotide phosphate (NADPH), flavin adenine dinucleotide, and flavin mononucleotide to the C-terminal reductase domain is necessary for the catalyzing activity of eNOS (Yuyun et al., 2018). eNOS contains a calmodulin (CaM)-binding domain, which can respond to increases in the intracellular concentration of calcium and promote eNOS-dependent production of NO (Zhao et al., 2015b).
Phosphorylation of eNOS can also regulate the enzyme's activity independently of the calcium concentration, in which Thr495 is an inhibitory site. At the same time, Ser635 and Ser1179 are activation sites (Zhao et al., 2015b). The SNO of proteins and their effects in regulating NO bioavailability are summarized in Figure 2.
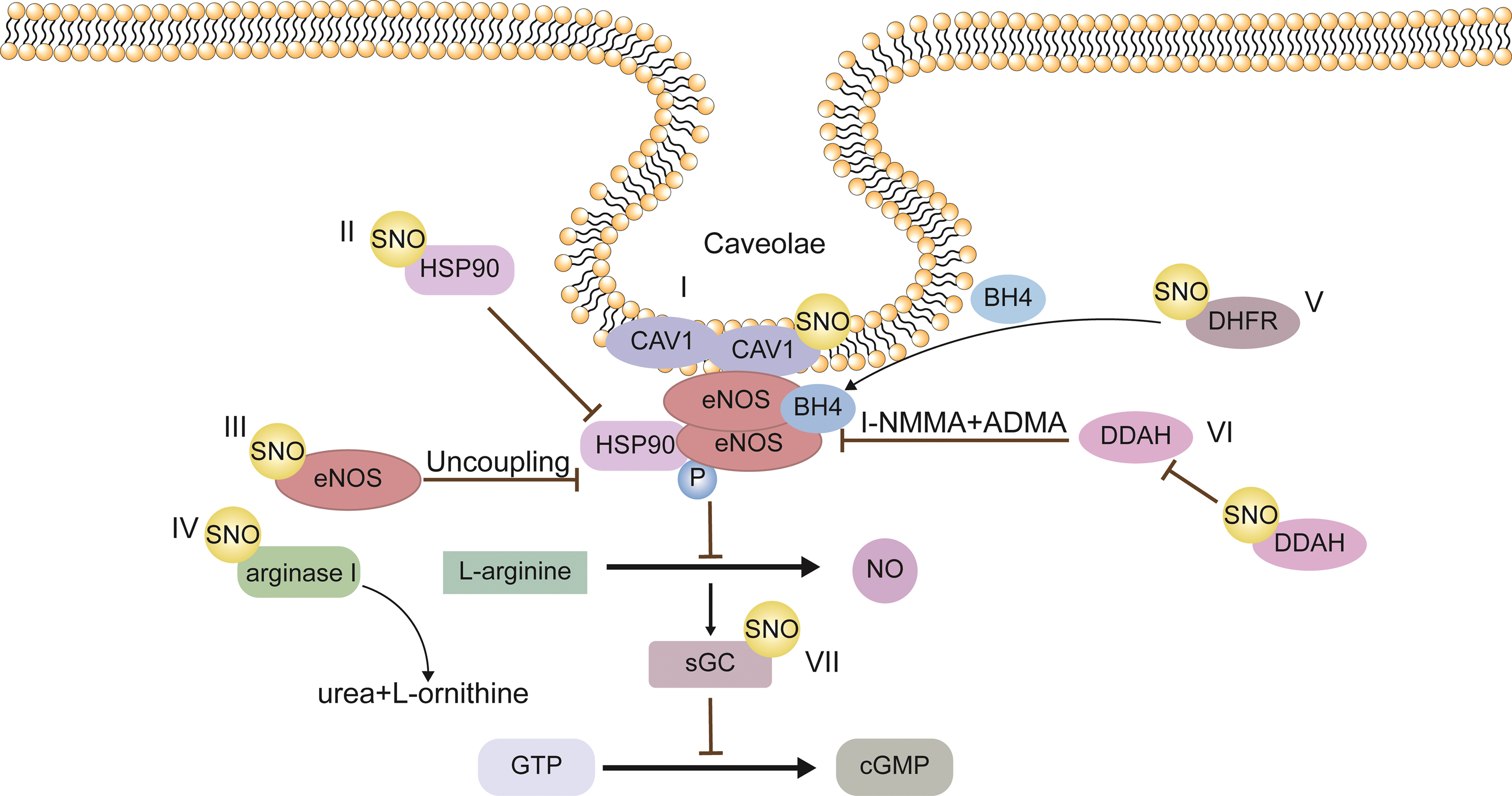
SNO of eNOS
Erwin et al. (2005) found that the activity of eNOS is inhibited by SNO. In this study, vascular endothelial growth factor (VEGF), a robust eNOS activator in bovine aortic endothelial cells (BAECs), was applied to investigate the regulation of eNOS activity by SNO. VEGF denitrosylated eNOS within 5 min, and interestingly, eNOS phosphorylation at Ser1179 was increased at this time point. The inverse relationship between eNOS SNO and phosphorylation at Ser1179 suggests that eNOS SNO inhibits eNOS activity. The cysteines of the zinc-tetrathiolate that comprise the eNOS dimer interface (Cys96 and Cys101 of each subunit) are the targets of SNO (Erwin et al., 2005).
Mechanism study found that eNOS SNO inhibits enzyme activity by perturbing the dimerization of eNOS (required for enzyme activity) (Erwin et al., 2005). Similarly, Ravi et al. (2004) found that in BAECs, exogenous NO donor treatment increased SNO of Cys99 in eNOS, which could reduce eNOS activity by interfering with its homodimerization. The presence of the Trx/TrxR system protected the eNOS dimer from disruption and restored eNOS activity (Ravi et al., 2004).
In another work, Wang et al. (2019) found that oxLDL induced the SNO of eNOS at Cys94 and Cys99, enhanced the interaction of eNOS with β-catenin, resulting in increased transcriptional activity of β-catenin on cell migration and adhesion genes in HUVECs, and eventually mediated atherosclerosis.
Protein SNO Regulates L-Arginine–Related Metabolites
NOSs produce NO by converting L-arginine to L-citrulline in the presence of molecular oxygen and cofactors. Arginase is a crucial element of the urea cycle, converting L-arginine to urea and L-ornithine. L-arginine homeostasis is characterized by the competition between NOS and arginase for the available intracellular L-arginine (Berkowitz et al., 2003; Hey et al., 1997). Santhanam et al. (2007) observed an increased level of arginase1 SNO in aged endothelium at C168 and C303. SNO of C303 stabilizes the arginase1 trimer and increases its enzymatic activity. This allows competition with NOS for L-arginine, impairing NO bioavailability and inducing endothelial dysfunction (Santhanam et al., 2007).
Dimethylarginine dimethylaminohydrolase (DDAH) consumes endogenous methylated arginine such as NG-monomethyl-L-arginine (L-NMMA) and asymmetric dimethylarginine (ADMA) to produce citrulline and either mono- or dimethylamine, respectively. There are two isoforms of DDAH, with DDAH1 mainly present in neuron and DDAH2 in vascular endothelium. Because ADMA and L-NMMA are endogenous inhibitors of NOSs, the decrease in DDAH expression and activity is believed to mediate eNOS dysfunction in ECs (Ogawa et al., 1989).
In sEnd.1 cell lines that stably overexpress DDAH2, lipopolysaccharide, IFN-γ, and tumor necrosis factor-α promote iNOS expression and NO production, excessive NO inhibits the activity of DDAH through enhancing Cys-249 SNO in DDAH2 and results in accumulation of L-NMMA and ADMA, which in turn inhibits activity of constitutively expressed eNOS in ECs (Leiper et al., 2002). The elevation of ADMA is observed in the development of endothelial dysfunction in many cardiovascular and metabolic diseases (Arrigoni et al., 2010; Pope et al., 2009), whether SNO of DDAH is involved in the progression of these diseases requires further investigation.
Protein SNO Regulates BH4 Concentration
BH4 is essential for the enzymatic activity of eNOS, and a lack of BH4 may lead to eNOS uncoupling. Two key enzymes regulate the concentration of BH4: GTP cyclohydrolase I (GTPCH) controls the de novo biosynthesis of BH4, and dihydrofolate reductase (DHFR) is the key enzyme rescuing BH4 from BH2 (Crabtree et al., 2009). Recent studies indicate that in streptozotocin-induced diabetes mellitus, high glucose increases both O2–· and ONOO− in HUVECs.
Acute high glucose down regulates GTPCH expression, decreases BH4 levels, and increases eNOS uncoupling in ECs through oxidative stress (Xu et al., 2007). In physiological condition, however, eNOS-derived NO was found to increase DHFR expression through inhibiting DHFR ubiquitination and proteasome degradation.
Mechanistically, eNOS-derived NO induces SNO of DHFR at Cys7, prevents DHFR degradation, and increases DHFR-dependent BH4 regeneration, thus increasing eNOS activity. This reciprocal regulation between eNOS activity and DHFR protein level via DHFR SNO constitutes a novel regulatory circuit with the potential for maintaining endothelial homeostasis (Cai et al., 2015).
Protein SNO of eNOS Interactors
Caveolin-1 (Cav1) is a membrane protein that forms large homo- and hetero-oligomeric complexes that promote the self-assembly of caveolae; ECs are one of the cell types with the highest expression of Cav1 (Sargiacomo et al., 1995). In quiescent ECs, the binding of eNOS with Cav1 in caveolae quenches eNOS activity by preventing the interaction of CaM with eNOS (Ju et al., 1997).
Upon addition of the Ca2+ ionophore A23187 to activate eNOS, NO production increases to induce SNO of Cav1 at Cys156, SNO-Cav1 dissociates the high-molecular-weight Cav1 oligomers and relieves the inhibition of eNOS activity. In addition to increasing eNOS activity, SNO-Cav1 enhances caveola-mediated albumin and insulin endocytosis in ECs, which mediates crosstalk of ECs with extracellular components (Chen et al., 2018b).
Hsp90 is a molecular chaperone that promotes the maturation, structural maintenance, and appropriate regulation of specific client proteins. Post-translational Hsp90 modifications, including phosphorylation, acetylation, and SNO, are essential for regulating its activity and interactions with different client proteins (Zhao et al., 2022a). Martinez-Ruiz et al. (2005) demonstrated that in ECs, GSNO and activators of eNOS could increase the SNO of HSP90 at Cys596/597. These cysteine residues are identified as reactive cysteines in HSP90 and reside in the interaction region of Hsp90 with eNOS (Fontana et al., 2002).
Further study found that SNO reduced Hsp90 ATPase activity needed for its function as a chaperone protein and a coactivator of eNOS (Martinez-Ruiz et al., 2005). Importantly, the inherent ATPase activity is essential for Hsp90 to perform its correct function in the folding of a large number of client proteins, the elevation of HSP90 SNO, thus impairing protein homeostasis in endothelium and inducing endothelial dysfunction.
Protein SNO Regulates sGC Sensitivity
The classical NO sensor is the heme-containing heterodimer sGC, composed of α and β subunits (Oppermann et al., 2011). When NO binds to the heme domain in sGC, its catalytic activity increases by several orders of magnitude to produce the second messenger, cGMP (Oppermann et al., 2011). However, little is known about the mechanism that regulates the sensibility of sGC to NO. In HUVECs treated with VEGF, sGC could be S-nitrosylated at C243 in the α and C122 in the β subunits. SNO of sGC could lead to reduced responsiveness to NO, as evidenced by the loss of NO-stimulated sGC activity.
Further mechanism study revealed that C122 resides near the heme domain of sGC. Its SNO could dampen NO activation by affecting the positions of key residues interacting with the heme domain (Sayed et al., 2007). As a result, SNO may functionally regulate the activities of sGC and provide a negative feedback mechanism for limiting NO signaling.
Roles of Protein SNO in Endothelial Inflammation
ECs have long been known to modulate inflammation. Emerging evidence demonstrates that ECs exert immunomodulatory activities, including expressing inflammatory factors and chemokines (Amersfoort et al., 2022; Pober and Sessa, 2007; Sjoholm and Nystrom, 2005). Indeed, endothelial inflammation is believed to initiate atherosclerosis, hypertension, diabetic vascular complications, and heart failure (Daiber et al., 2017; Gimbrone and Garcia-Cardena, 2016; Jia et al., 2022; Paulus and Tschope, 2013; Watson et al., 2008).
On the contrary, NO is believed to inhibit the inflammatory activation of ECs in the resting condition and is an initiator of endothelial inflammation in pathological conditions. The complex roles of NO in endothelial inflammation at least partly rely on the SNO of target proteins (Daiber et al., 2017). The SNOs of proteins and their effects in regulating endothelial inflammation are summarized in Figure 3.

SNO of N-Ethylmaleimide-Sensitive Factor
In resting conditions, NO production in ECs negatively regulates endothelial inflammation. The Weibel-Palade body is the storage vesicle for the von Willebrand factor and P-selectin. Release of the Weibel-Palade body from ECs could induce vascular thrombosis and inflammation (Block et al., 1988). N-ethylmaleimide-sensitive factor (NSF) can alter the conformation of the stable soluble NSF attachment protein receptor (SNARE) complex on vesicles and mediate their exocytosis (Block et al., 1988).
Matsushita et al. (2003) found that in human aortic endothelial cells (HAECs) stimulated with thrombin, exocytosis of Weibel-Palade bodies was enhanced. Further mechanism study revealed NO nitrosylate cysteine residues 91 and 264 of NSF. In resting conditions, SNO of NSF prevents the exocytosis of Weibel-Palade bodies through inhibition of NSF-dependent SNARE disassembly (Matsushita et al., 2003).
NO donor 2-(N,N-diethylamino)-diazenolate-2-oxide increased SNO of NSF, reduced its activity, and prevented exocytosis of Weibel-Palade bodies (Matsushita et al., 2003). The above study indicates that SNO of NSF could be one possible mechanism for the anti-inflammatory effects of NO.
SNO of Proteins in NF-kB Signaling Pathway
Under some pathological conditions, increased production of NO induces expression of inflammatory factors, suggesting that SNO might positively regulate endothelial inflammation (Bessa et al., 2002; Joussen et al., 2002). Nuclear factor-κB (NF-κB) is a heterodimer classically formed with p50 and p65, a core inflammatory regulator maintained in latent form in the cytoplasm. Its activity is sequestrated by inhibiting κB (IκB) (Thanos and Maniatis, 1995).
In one study, Wadham et al. (2007) incubated ECs with 5.5 or 30 mM glucose for 3 days, and found that high glucose reduced the S-nitrosylated protein content in HUVECs. Using mass spectrometry (MS) analysis and western blotting, they identified 7 targeting proteins with reduced SNO levels, including diacylglycerol kinase-α, eNOS, ERK-1, GRP78, H-ras, NF-κB, vimentin, and vinculin.
Among them, inhibition of SNO-NF-κB was found to promote the activation of NF-κB signaling pathways and induce endothelial inflammation. Interestingly, high-glucose–induced reduction of global protein SNO was restored to basal level with the superoxide scavenger TEMPOL or mitochondrial ROS scavengers, suggesting a key role for ROS in mediating high-glucose–induced reduction of protein SNO (Wadham et al., 2007). However, how ROS inhibits protein SNO in HUVECs stimulated with glucose, and why SNO of NF-κB enhances its activity are uninvestigated in this work.
A more deliberate-design work examined time-dependent change of eNOS, iNOS, and protein SNO in HUVECs stimulated with hyperglycemia combined with hyperlipidemia (palmitic acid, PA) (HG + PA), an in vitro model mimicking diabetic endothelial complications. The total protein SNO decreased at 2–3 days of exposure to HG+PA and subsequently reversed on days 4–5, while the activity of eNOS reduced at 2–3 days and the level of iNOS increased on days 4–5; this observation clearly suggests that SNO was mediated by NO, as reflected from the corresponding biphasic regulation of eNOS/iNOS activity or expression (Chen et al., 2021).
In this study, HG + PA impaired cell migration and tube-forming capacity of HUVECs, which were alleviated by bFGF pretreatment. Mechanism study further found that HG and PA reduced SNO of κB kinase subunit β (IKKβ) at Cys179 and P65 at Cys38, promoted nuclear translocation of p65, and increased NF-kB DNA-binding and transcriptional activity, thus promoting endothelial inflammation responses. On the contrary, bFGF could restore the SNO of IKKβ and p65 to counteract endothelial inflammation (Chen et al., 2021).
Similarly, in the process of experimental allergic encephalomyelitis, Prasad et al. (2007) found that GSNO could inhibit the adhesion of mouse monocyte cells to activated bEND.3 mouse brain ECs by increasing SNO of p65. This consequently inhibited the nuclear translocation of NF-κB and reduced the transcription of EC adhesion molecules (Prasad et al., 2007). On the contrary, C-reactive protein reduces p65 SNO and increases NF-κB transcription in HUVECs, which may contribute to the endothelial inflammatory injury (Wang et al., 2013b).
Zhao et al. (2022b) found another mechanism of SNO-mediated activation of the NF-κB signaling pathway during atherosclerosis. In oxLDL-treated HUVECs, the SNO level of HSP90 at Cys521 increased significantly, and SNO-Hsp90 at Cys521 promoted the association between Hsp90 and cell division cycle 37 (Zhao et al., 2022b). CDC37 is a key factor for assembling the Hsp90/CDC37/IKK complex to further mediate NF-κB signaling activation (Chen et al., 2002; Hinz et al., 2007).
SNO-Hsp90 increased the interaction between Hsp90 and CDC37, and activated NF-κB signaling during endothelial inflammation and oxidative stress. Transfection of SNO-resistant Cys521A mutant of HSP90 could relieve endothelial inflammation, thus alleviating atherosclerosis (Zhao et al., 2022b).
SNO of Guanine Nucleotide-Binding Protein G(i) Subunit Alpha-2
Interestingly, Chao et al. (2021) found increased protein SNO in coronary artery of diabetic patients with coronary heart disease. Using high glucose and oxLDL to induce endothelial injury, they found a significant increase in total protein SNO (Chao et al., 2021). Further proteomics and KEGG pathway analysis found that many SNO proteins belong to the NF-kB, TNF-α, chemokine, and chemokine receptor signaling pathways, confirming the importance of protein SNO in regulating endothelial inflammation (Chao et al., 2021).
The Hippo-YAP pathway, including MST1/2-LATS1/2-YAP as core components, has been well documented in inflammation (Zheng et al., 2022). The inhibition of the Hippo-YAP pathway, along with reduced phosphorylation of MST1/2 and LATS1/2, and nuclear translocation of YAP, promotes endothelial inflammation and monocyte attachment in unidirectional shear flow-induced atherosclerosis (Wang et al., 2016). In mouse liver injury, the Hippo-YAP pathway also controls the activation of NLRP3 and governs mesenchymal stem cell immunoregulation (Li et al., 2019).
Chao et al. (2021) found that inducible SNO of guanine nucleotide-binding protein G(i) subunit alpha-2 (GNAI2) at Cys66 increased the coupling of GNAI2 with CXCR5, reduced cAMP level, and dephosphorylated LATS1/2, resulting in enhanced nuclear translocation of YAP and increased expression of adhesion factors and chemokines.
Furthermore, in streptozotocin-treated Ldlr−/− mice fed with a high-fat diet, transfection of AAV vector containing SNO-insensitive GNAI2-C66S mutant (mutation of Cys66 to Ser66) alleviated the development of atherosclerosis. They also observed a reduction of melatonin concentration in plasma of diabetes-accelerated atherosclerosis mouse, and supplementation of melatonin inhibited SNO GNAI2, delayed plaque formation, and improved plaque stability (Chao et al., 2021).
SNO of PKCζ
Leukocyte recruitment is an essential cellular process in endothelial inflammation. Recruitment of leukocytes to ECs is coordinately regulated by recognition between adhesion proteins in the EC surface and specific integrins in leukocytes (Kolaczkowska and Kubes, 2013; Mitroulis et al., 2015; Muller, 2009).
Aguilar et al. (2021) found that, at the early stages of TNF-α–induced inflammation in EAhy926 cells, the eNOS-derived elevation of NO increased the SNO of PKCζ at Cys 503. They further found that SNO-PKCζ increased the interaction of PKCζ with ICAM1-1, promoting ICAM-1 clustering and phosphorylation, a fundamental process involved in early leukocyte adhesion to the endothelium (Aguilar et al., 2021). These observations confirm SNO of PKCζ an important regulatory step in early leukocyte adhesion in inflammation.
Estrogen induces NO in the endothelium, and appears to protect against inflammation and atherosclerosis; 17beta-estradiol (E2β) is the primary form of estrogen in the body (Prossnitz and Barton, 2011). Chakrabarti et al. (2010) found that a 24 h treatment of HUVECs with E2β increased protein SNO levels. Further study found that E2β activated ER (including Erα and ERβ), increased eNOS expression, and eNOS phosphorylation/activation (Chakrabarti et al., 2010).
Pretreatment with E2β completely abrogated the increase in ICAM-1 in HUVECs stimulated with Ang II. Also, DTT treatment reversed the increase in protein SNO and further enhanced the effects of Ang II on ICAM-1 expression (Chakrabarti et al., 2010).
Roles of Protein SNO in Oxidative Stress in ECs
Oxidative stress involves the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS molecules include superoxide (O2 −), hydrogen peroxide (H2O2), hydroxyl radical (•HO), and others (Abot et al., 2022). RNS includes radical compounds, such as nitric oxide (•NO) and its derivatives, including peroxynitrite (ONOO−) and dinitrogen trioxide (N2O3) (Lushchak and Lushchak, 2021). ROS and RNS act as messengers at cellular concentrations, coordinating the signaling transduction in ECs. However, uncontrolled ROS or RNA production reacts rapidly with membrane lipids, proteins, and nucleic acids to induce cell injury (Forman and Zhang, 2021).
NADPH oxidases (NOX) are a family of transmembrane oxidoreductases that produce O2− and other ROS (Qian et al., 2012). There are seven enzymes in the NOX family: Nox1-5 and double oxidase 1-2 (DUOX1-2). NOX enzymes in the human body play important roles in various biological functions and are differentially expressed in different tissues (Taylor and Tse, 2021). Endothelial NOXs are a major source of O2− in the vascular and are associated with the oxidative stress that induces vascular diseases (Selemidis et al., 2007).
Activation of NOX2 requires p22phox, p47phox, p67phox, p40phox, and Rac. Among them, p47phox is thought to be the organizing subunit of NOX2, and its phosphorylation is required for NOX2 activation (Bedard and Krause, 2007). Selemidis et al. (2007) found that in human-cultured microvascular endothelial cells, NO inhibits NOX2-dependent O2− production by inhibiting NOX2 activity.
Mechanism study revealed that NO could increase SNO of p47phox, which might account for the reduction of O2− production (Selemidis et al., 2007). Similarly, Qian et al. (2012) revealed that NO reduces NOX5 activity through reversible SNO. Both exogenous and endogenous NO reduced NOX5 activity in a dose-dependent manner in HAECs, as evidenced by reduced O2− production (Qian et al., 2012). NOX5 was S-nitrosylated at four sites (C107, C246, C519, and C694); among them, C107, 519, and 694 are highly conserved among different species and located within the functional cytoplasmic regions of NOX5.
Further study found that mutation of these sites all had inhibitory effect on NOX5 catalytic activity, while only C694, located at NADPH-binding domain of NOX5, was regulated by iNOS (Qian et al., 2012). In addition, NO-dependent inhibition of NOX5 activity was reversible by the denitrosylation enzymes GSNOR and Trx (Qian et al., 2012).
Trx is a 12 kDa disulfide oxidoreductase, which serves as a protector against oxidative stress. Trx contains two cysteines in an active CXXC motif; oxidized Trx is reduced by TrxR, which uses NADPH as a source of electrons (Haendeler et al., 2004). Under physiological conditions, NO induces SNO of Trx at cysteine 69, which is required for the redox-regulatory activity of Trx and its ability to reduce ROS in ECs (Haendeler et al., 2002).
Further research found that statins potentiated the antioxidant effect of Trx through inducing SNO Trx at C69 in HUVECs (Haendeler et al., 2004). In addition, the atorvastatin-induced increase in Trx SNO was entirely inhibited by NOS inhibitors L-NMMA, indicating that increased Trx SNO by atorvastatin requires NOS activation (Haendeler et al., 2004).
Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor (erythroid-derived 2)-like 2 (Nrf2) signal is a key regulator in the pathophysiological response of oxidative stress (Itoh et al., 1999; Shih et al., 2003). Under nitrosative stress, melatonin can restore the activity of the Nrf2/Keap1 antioxidant signal, maintain the integrity of ECs by inhibiting Keap1SNO, and alleviate ischemic injury (Tao et al., 2013).
AKT binds to the inositol 3’-OH group of plasma membrane phospholipids once phosphorylated and activated by PI3K. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a dual phosphatase with both protein and lipid phosphatase activities (Chen et al., 2018a).
PTEN has lipid phosphatase activity, which can antagonize the phosphorylation function of phosphoinositide 3-kinase (PI3K) through dephosphorylating phosphatidylinositol 3,4,5-trisphosphate (PIP3) to generate phosphatidylinositol 4,5-bisphosphate (PIP2), thus inhibiting the activation of AKT (Maehama and Dixon, 1998). It has been found that GSNO mediates the SNO of PTEN in pulmonary artery ECs in a time- and dose-dependent manner. SNO-PTEN reduces the dephosphatase activity of PTEN, leading to the translocation of AKT to the cell membrane and inducing the activation of downstream HIF-1 signaling under nonhypoxic conditions (Carver et al., 2007).
In addition, Gupta et al. (2017) found that in the core and penumbra regions of ischemic mouse brain, where NO concentrations were low, SNO-PTEN at Cys83 promoted its ubiquitin degradation, relieved the inhibition of PI3K activity, and activated AKT. This process was expected to contribute to Akt-dependent cell survival. On the contrary, in the ischemic core, SNO-Akt was increased, and the neuroprotective effects of Akt signaling were inhibited (Numajiri et al., 2011).
Moreover, PTEN SNO was observed in human cancer cells with increased PI3K/Akt signaling to enhance cell survival and proliferation under conditions of energy deprivation (Gupta et al., 2017). SNO of proteins and their effects in regulating endothelial oxidative stress are summarized in Figure 4.

Roles of Protein SNO in Endothelial Integrity
The vascular endothelium is a semiselective permeable barrier that regulates the material exchange between blood and surrounding tissue, and its permeability depends on its integrity (Lai et al., 2011; Yu et al., 2013). Maintaining endothelial barrier function and integrity is vital to normal vascular function and homeostasis (Mehta et al., 2014).
Many pathological stimuli, such as dyslipidemias, high blood glucose, Ang II, disturbed flow, and smoking, can increase endothelial permeability. The integrity of the endothelial barrier depends largely on adherens junctions, formed mainly with vascular endothelial-cadherin (VE-cadherin), β-catenin, p120-catenin, and α-catenin (Giannotta et al., 2013).
Roles of Protein SNO in Adherens Junctions
VEGF induces tyrosine phosphorylation of VE-cadherin and β-catenin, and increases endothelial permeability (Esser et al., 1998). Thibeault et al. (2010) found that VEGF could significantly increase the β-catenin SNO at Cys619 in BAECs, the S-nitrosylated site located within the interface between VE-cadherin and β-catenin. SNO of Cys619 promotes its dissociation from VE-cadherin, contributes to VEGF-stimulated disassembly of adherens junctions, and increases endothelial permeability (Thibeault et al., 2010).
Furthermore, inhibition of β-catenin SNO with NOS inhibitor NG-monomethyl-L-arginine acetate (L-NMMA) has been found to prevent eNOS-dependent dissociation of β-catenin from VE-cadherin, ultimately inhibiting the increase of VEGF-stimulated endothelial permeability (Thibeault et al., 2010).
Proinflammatory stimuli, such as VEGF and histamine, can stimulate the disassembly and internalization of adherens junction proteins, leading to increased endothelial permeability (Esser et al., 1998; Shasby et al., 2002). In addition, platelet-activating factor (PAF), an edematogenic mediator, increases endothelial permeability by inducing translocation of eNOS to cytosol and stimulation of eNOS-derived nitric oxide signaling cascade (Marin et al., 2012).
Marín et al. (2012) found that PAF could stimulate SNO of β-catenin and p120-catenin (p120) in EAhy926 EC line and bovine heart ECs. The levels of SNO-β-catenin and SNO-p120 were correlated with their diminished accumulation at the adherens junctions and increased endothelial permeability (Marin et al., 2012).
As expected, NOS inhibitors blocked PAF-induced SNO and endothelial hyperpermeability. TNF-α, rather than acetylcholine (not increase permeability), induces NO production, SNO of β-catenin and p120, and increases endothelial permeability. These results indicate that SNO is a mechanism shared among agents that cause endothelial permeability.
Likewise, PAF was found to induce SNO of VE-cadherin and its phosphorylation, which disrupted association of VE-cadherin with β-catenin and enhanced endothelial permeability. Inhibition of eNOS with NG-methyl-L-arginine, or indirect blockade of the VE-cadherin SNO with N-acetylcysteine prevented PAF-induced SNO and phosphorylation of VE-cadherin, and eventually prevented endothelial disruption (Guequen et al., 2016).
Per these findings, sphingosine-1-phosphate (S1P2) exerts a protective effect on vascular hyperpermeability through suppression of anaphylaxis-associated eNOS stimulation. Further study found that S1P2 inhibited PAF-induced disassembly of adherens junctions by inhibiting NO production and preventing SNO of β-catenin (Cui et al., 2013).
Roles of Protein SNO in Focal Junctions
Focal adhesions mediate the connections of ECs to the extracellular matrix. They are essential cellular structures that modulate endothelial barrier function and control microvascular permeability (Schlegel and Waschke, 2009; Wu, 2005). Reorganization of focal adhesions represents an important step in the process of increased vascular permeability.
One focal adhesion protein is the actin-binding, vasodilator-stimulated phosphoprotein (VASP), belonging to the Ena/VASP family of actin-regulatory proteins (Schlegel et al., 2008). Studies have shown that a VASP deficiency leads to impaired vascular structural integrity and increased endothelial permeability in the basal state (Furman et al., 2007; Schlegel and Waschke, 2009). Zamorano et al. (2017) demonstrated that SNO of VASP at Cys64 induced the onset of PAF-induced endothelial hyperpermeability.
They found that in different types of ECs, such as EAhy926 and coronary postcapillary venular ECs, proinflammatory agents, including PAF and TNF-α, could induce eNOS-dependent SNO of VASP (Zamorano et al., 2017). SNO of VASP inhibits its function in barrier integrity and contributes to the onset of PAF-stimulated endothelial monolayer hyperpermeability. SNO of proteins and their effects in regulating endothelial integrity is shown in Figure 5.

Roles of Protein SNO in Proliferation, Migration, and Apoptosis of ECs
The SNO modification of a protein by NO is an integral part of EC proliferation, migration, and apoptosis (Fig. 6), related to vascular endothelial dysfunction, and causes vascular diseases, such as atherosclerosis and hypertension (Rychter et al., 2016; Sun and Murphy, 2010).
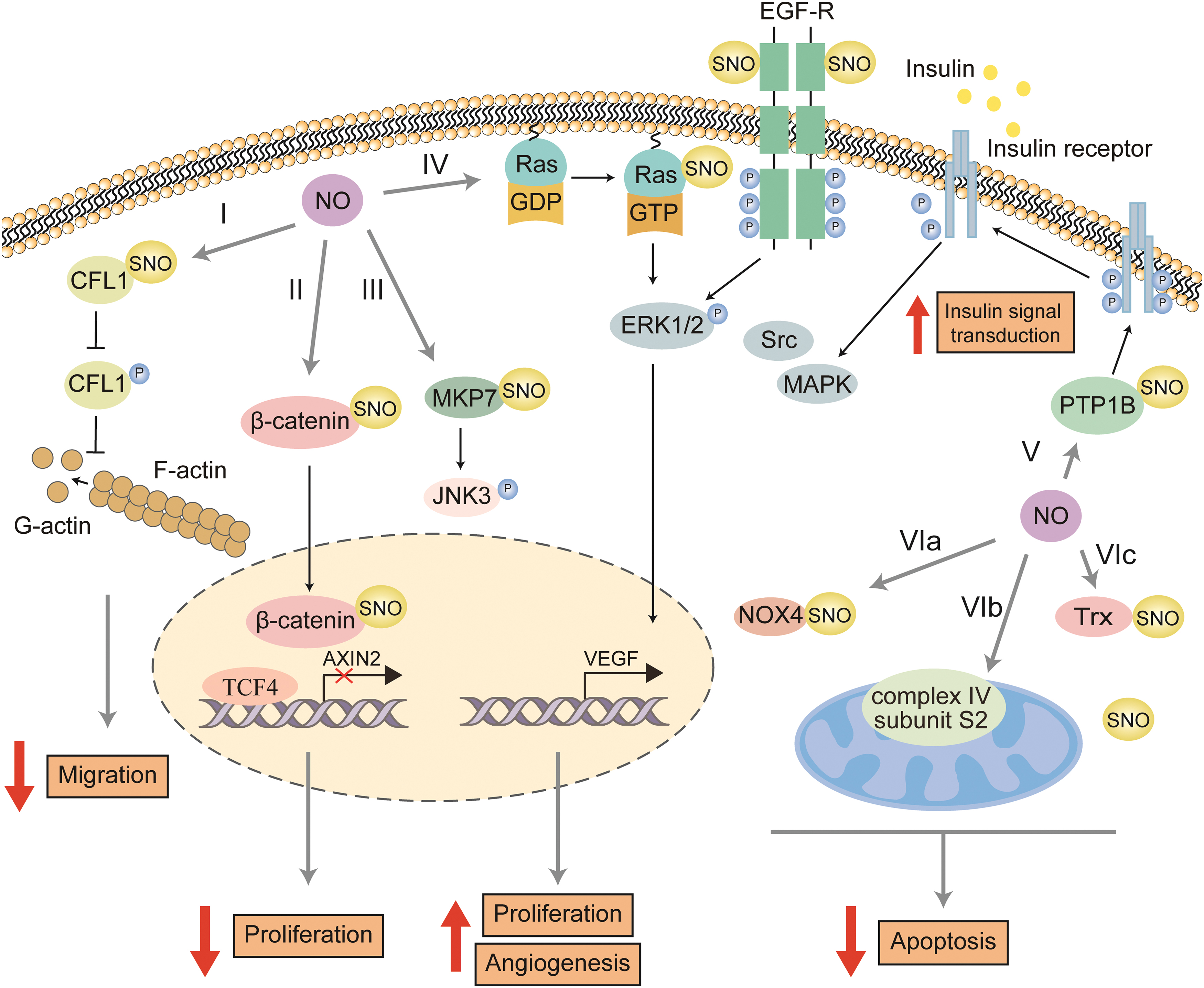
SNO in EC Proliferation
In ECs, β-catenin SNO was primarily found to disrupt endothelial intercellular connectivity, and increases EC permeability as described previously (Thibeault et al., 2010). Excepting for its role in the intercellular junction, the SNO modification of β-catenin on Cys466 by eNOS-derived NO leads to the dissociation of β-catenin from the transcription factor 4 (TCF4), which reduces the expression of the target genes and consequently inhibits EC proliferation (Zhang et al., 2017).
RAS is mainly located in the inner part of the cell membrane, and plays a vital role in cell growth and differentiation. However, several studies have shown that RAS is also distributed in the endosome, endoplasmic reticulum, Golgi apparatus, and mitochondria (Fehrenbacher et al., 2009). In Hela cells, GSNO directly induces SNO modification of RAS on the cell membrane.
In addition, GSNO induces the SNO modification of Src kinase, and then initiates the signaling cascade of PLC-c. This results in the translocation of RAS from the cell membrane to the Golgi apparatus, activating the ERK signaling pathway and promoting cell proliferation with SNO-RAS on the cell membrane (Batista et al., 2013).
In HUVECs, the activation of RAS induced by NO occurs only on the cell membrane (Batista et al., 2013). Evidence indicates that RAS can be S-nitrosylated at Cys118 in a NO-dependent manner, which triggers guanine nucleotide exchange activity and activates the ERK1/2-MAP kinase signaling pathway, promoting the cell cycle from the G2 phase to M phase.
Further mechanism study revealed that Cys118 locates in the highly conserved region of RAS superfamily, and is responsible for the direct interaction between GTP and GDP (Batista et al., 2013; Lander et al., 1997; Oliveira et al., 2008). Also, Moraes et al. (2014) found that in ECs, Bradykinin (BK)-mediated phosphorylation of eNOS increased the production of NO, and enhanced the SNO of PTPN6 (Protein Tyrosine Phosphatase Nonreceptor Type 6) and EGF-R (epidermal growth factor receptor), which could cooperate with SNO-RAS to activate the ERK1/2 signaling pathway. This could promote EC proliferation and VEGF expression to promote angiogenesis (Moraes et al., 2014).
SNO in EC Migration
The actin-binding protein Cofilin-1 (CFL1) regulates cytoskeletal remodeling by severing actin filaments. CFL1's ability to bind to actin is inhibited when phosphorylated at Ser3. After dephosphorylation, CFL1 is activated, and actin filaments (F-actin) are depolymerized to monomeric actin (G-actin) (Condeelis, 2001). Zhang et al. (2015) found two major SNO sites on CFL1, Cys80, and Cys139. SNO of CFL1 significantly inhibits the phosphorylation of CFL1 at Ser3, increases the actin filament depolymerization activity of CFL1, and reduces filopodium formation, leading to reduced migration ability of the ovine fetoplacental artery and uterine artery ECs.
Stromal cell-derived factor-1-α (SDF-1α) is one of the CXC chemokines that can promote angiogenesis (Mirshahi et al., 2000). Pi et al. (2009) explored the signaling pathway involved in the SDF-1α–induced migration of BAECs. They found that SDF-1α promotes endothelial progenitor cell migration by activating eNOS to produce NO. This subsequently modifies Cys244 of MKP7 (mitogen-activated protein kinase phosphatase 7), a JNK3 (c-Jun N-terminal kinase 3) phosphokinase, and activates JNK3 (Pi et al., 2009).
SNO in EC Apoptosis
Apoptosis is the active death of cells, manifesting as cell shrinkage, membrane blebbing, and chromatin condensation (Steller, 1995). ECs are more resistant to apoptosis than other cell types because of the upregulated expression of eNOS and NO synthesis by shear stress, which induces SNO of redox, senescence, and apoptosis-related factors (Nicotera et al., 1997; Uematsu et al., 1995).
ICE/CPP-32–like protease (interleukin-1β–converting enzyme-like and cysteine protease protein-32-like cysteine proteases) is a major path activated in the cell suicide program.
Dimmeler et al. (1997) found that the inflammatory cytokine TNF-α affected EC viability and induced apoptosis through activating ICE/CPP-32-like proteases (Robaye et al., 1991). Interestingly, they found that shear stress could completely inhibit TNF-α–induced EC apoptosis. Mechanism study revealed that shear stress-induced eNOS activation and NO production suppressed ICE/CPP-32-like protease activity by increasing CPP-32 SNO at Cys163 (Dimmeler et al., 1997), thus inhibiting TNF-α–induced endothelial apoptosis.
Kim and their coworker found that Korean red ginseng extract activates eNOS in an Akt-dependent manner and enhances NO synthesis. Furthermore, NO prevents apoptosis of ECs induced by serum deprivation through inhibiting caspase-3 activity via SNO (Kim et al., 2013).
The binding ability of extracellular ligands and membrane receptors is regulated by endocytosis. Large GTPase dynamin-2 plays an essential role in this progress by reducing the combination of apoptosis-associated factors and membrane receptors. Its function is regulated by multiple post-translational modifications (Ahn et al., 2002; Shajahan et al., 2004). In a TNF-α–induced endothelial apoptosis model, VEGF could promote ECs survival in an eNOS-dependent manner.
Further study revealed that NO-dependent SNO of Cys86 and 607 of dynamin-2 was required for the protective effects of VEGF. SNO of dynamin-2 promotes endocytosis and downstream survival signaling in BAECs. Structural analysis revealed that Cys86 resides in the GTPase domain, and Cys607 resides in the PH domain of dynamin2 (Kang-Decker et al., 2007).
Redox regulator thioredoxin 1 (Trx) is crucial for cell growth and the inhibition of apoptosis (Holmgren, 1989). Haendeler et al. (2002) show that SNO can modify Trx at Cys69 to enhance its redox enzyme activity, reduce ROS production, and inhibit apoptosis of ECs.
As previously mentioned, Nrf2 is one of the critical transcription factors that can bind to antioxidant response elements to initiate the transcription of the enzymes responsible for the elimination of superoxide and hydroperoxides, such as glutathione peroxidase and superoxide dismutase (Li and Kong, 2009). Evidence shows that Nrf2-deficient ECs exhibit a senescent phenotype with an increased level of SNO protein (Kopacz et al., 2020).
Kopacz et al. (2020) found that during premature senescence of ECs, Keap1 interacts with GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and NOS to form a complex, mediated SNO of NOX4. This inhibits the activity of NOX4, restrains oxidative stress damage and apoptosis of HAECs, thus ameliorating premature senescence of ECs (Kopacz et al., 2020).
During EC senescence, the expression of eNOS is significantly decreased, NO synthesis impaired, and the SNO protein level diminished, leading to the augmented sensitivity to apoptosis, which cannot be inhibited by shear stress (Hoffmann et al., 2001). This observation further confirms the protective role of protein SNO in premature senescence ECs.
Other Roles of Protein SNO in ECs
Protein SNO regulates mitochondrial function in ECs
In contrast to the bioenergetic role of mitochondria in high-energy–demanding tissue, such as the heart and liver, endothelial mitochondria play a prominent role in signaling transduction because glycolysis is the major source of ATP production in the endothelium (Kluge et al., 2013). The relationship between NO and mitochondrial function is evidenced by the existence of a functional NOS in rat liver mitochondria, whose activity is highly relevant to mitochondrial function (Ghafourifar and Richter, 1997).
A later study found that targeting eNOS to mitochondria decreased endothelial oxidative stress and facilitated vasodilation in fetal pulmonary circulation at birth (Konduri et al., 2015). Also, Clementi et al. (1998) found that in the physiological condition, NO-mediated protein SNO regulates complex IV activity in the mitochondrial respiration chain, and prolonged exposure to NO led to persistent inhibition of complex I activity in mitochondria, and induced cell pathology eventually.
Similarly, Zhang et al. (2005) also found that long-term exposure of porcine lung ECs to NO donor inhibited complex IV activity and reduced GSH/GSSG. Further research found that subunit II (S2) of complex IV has two NO-sensitive cysteine residues: Cys196 and Cys200 (Zhang et al., 2005). Mutation of these two sites significantly attenuated the NO-induced SNO of complex IV (Zhang et al., 2005).
Protein SNO regulates the communication between vascular ECs and smooth muscle cells
Vascular smooth muscle cells and ECs are connected through myoendothelial junctions (MEJs) for cell-to-cell communication, vascular contraction, and relaxation. Gap junctions at MEJs are intracellular signaling channels formed by hexamer hemichannel and the channel protein connexin (Cx) (Heberlein et al., 2009). A study found that eNOS/GSNOR regulates vascular contraction and relaxation through the SNO/denitrosylation of Cx43. SNO of Cx43 at Cys 271 also augments the permeability of gap junction channels, while denitrosylation attenuated channel permeability (Straub et al., 2011).
Protein SNO regulates insulin signaling in ECs
eNOS-dependent NO synthesis is essential for activating insulin signaling in islet ECs (Hill et al., 2021; Montagnani and Quon, 2000). Dephosphorylation of insulin receptors and their substrates by protein-tyrosine phosphatases (PTPs) reduces insulin action. It enhances insulin resistance in insulin-responsive cells (Asante-Appiah and Kennedy, 2003). The cysteine residues in the active site of PTPs are inactivated by SNO, indicating that NO can mediate the enhancement of insulin response by inhibiting the activity of PTPs (Hsu and Meng, 2010).
NO-mediated inactivation of PTP1B, a member of the PTPs superfamily, has been found to enhance insulin transport and signal transduction (Wang et al., 2013a). Hsu's team explored the mechanism using quantitative MS and X-ray crystallography, and confirmed that Cys215 in the active site of PTP1B is highly susceptible to SNO. In insulin-stimulated ECs, SNO PTP1B interacts with insulin receptors in the membrane, allowing its rapid phosphorylation to enhance insulin responsiveness; this observation indicates the vital role of SNO PTP1B in the NO-mediated insulin signaling pathway in ECs (Hsu et al., 2016).
Protein SNO regulates transdifferentiation of fibroblasts into ECs
Transdifferentiation is the process by which somatic cells differentiate into other types of somatic cells and plays a role in the physiological response to injury. For its application in cardiovascular regenerative medicine, several researchers have differentiated fibroblasts into ECs by overexpressing specific transcription factors (Margariti et al., 2012). However, the integration of exogenous DNA may induce genomic dysregulation.
Meng et al. (2016) developed a novel transdifferentiation strategy to activate innate immunity through small molecules and growth factors to promote the reprogramming of fibroblasts into ECs, in which iNOS plays an important role (Lee et al., 2012; Sayed et al., 2015). Knockdown of iNOS significantly reduces transdifferentiation efficiency. Detection of several important epigenetic modifiers reveals that SNO modified RING1A (ring finger protein 1a) during transdifferentiation.
Further investigation by MS shows that RING1A was heavily S-nitrosylated at Cys 398. RING1A is one of the key components of the polycomb repressive complex (PRC) 1 and is required for stabilizing H3K27 trimethylation (H3K27me3). SNO-RING1A may promote endothelial lineage gene expression by disrupting the PRC1complex and reducing H3K27me3 (Meng et al., 2016).
Protein SNO regulates the formation of thrombin
Protein disulfide isomerase (PDI), a multifunctional thiol isomerase, is essential for protein folding and forming a disulfide bond. Inhibition of PDI blocks platelet accumulation and fibrin formation in multiple vascular injury models (Reinhardt et al., 2008; Schulman et al., 2016). However, the molecular mechanism remains unknown. Studies show that endogenous NO modifies Cys397 and Cys400 on the catalytic motif of PDI oxidoreductase CGHC (Cys-Gly-His-Cys).
SNO-PDI inhibits the thiol isomerase reductase activity of PDI in ECs and reduces thrombin formation on the surface of ECs, thereby preventing platelet adhesion to the endothelium and ameliorating thrombosis (Bekendam et al., 2018). The S-Nitrosylated site (s) and cellular effects of different S-nitrosylation proteins in endothelial cells are listed in Table 1.
S-Nitrosylated Site (s) and Cellular Effects of S-Nitrosylation Proteins in Endothelial Cells
ADMA, asymmetric dimethylarginine; Cav-1, caveolin-1; CFL1, cofilin-1; CPP-32, cysteine protease protein-32; DDAH2, dimethylarginine dimethylaminohydrolase 2; DHFR, dihydrofolate reductase; eNOS, endothelial NO synthases; GNAI2, G(i) subunit alpha-2; ICE/CPP-32–like protease, interleukin-1β–converting enzyme-like, and cysteine protease protein-32–like cysteine proteases; MKP7, mitogen-activated protein kinase phosphatase 7; NSF, N-ethylmaleimide-sensitive factor; PAF, platelet-activating factor; PDI, protein disulfide isomerase; PRC1, polycomb repressive complex 1; PTEN, phosphatase and tensin homolog deleted on chromosome 10; PTP1B, protein-tyrosine phosphatase 1B; RING1A, ring finger protein 1a; sGC, soluble guanylyl cyclase; VASP, vasodilator-stimulated phosphoprotein.
Conclusions and Remarks
Protein SNO regulates multiple facets of endothelial function, including NO bioactivity, endothelial integrity, secretion of inflammation and adhesion factors, monocyte adhesion, response to oxidative stress, proliferation, migration, and apoptosis. Thus, SNO plays an essential role in both endothelial homeostasis and dysfunction. Single-cell transcriptomic analysis of ECs isolated from different mouse tissues shows heterogeneous gene signatures (Kalucka et al., 2020).
Besides, biotin-switch and MS analysis of S-nitrosylated proteins in HUVECs subjected to different stimuli found enrichment of proteins in different pathways. Above findings indicate that protein SNO is a process that exquisitely regulates the endothelial function and response to environmental change. Precisely deciphering the effects of protein SNO in different conditions is necessary to further characterize the relationship between protein SNO and endothelial functions.
New evidence indicates that metabolic reprogramming regulated by protein SNO is essential in kidney injury (Zhou et al., 2019). Quantitative proteomic analysis of VEGF-responsive protein SNO in ECs revealed that SNO proteins are enriched in the endothelial metabolism pathway (Zhang et al., 2016).
Moreover, glycolysis and mitochondrial function change are involved in endothelial proliferation, migration, and integrity. Therefore, protein SNO is vital in the metabolic reprogramming of ECs. Identifying these proteins and deciphering their underlying mechanisms may offer new interventional strategies for endothelial dysfunction-related diseases.
Authors' Contribution
Y.J., L.X., and S.L. conceptualized and designed the article; S.L., D.Y., Y.W., and X.W. wrote the article draft; S.L., D.Y., Y.W., and X.W. designed the figures; and X.L. revised the article. All coauthors have reviewed and approved the final version of the article.
Footnotes
Author Disclosure Statement
The authors claim that there is no conflict of interest related to this work.
Funding Information
This work is supported by the grants from National Key Research and Development Program of China (2019YFA0802704) and National Natural Science Foundation of China (81820108002, 82121001, 82030013, 82241211, 82270504).