
Abstract
Aims:
Endoplasmic reticulum (ER) degradation via autophagy is a process that maintains ER homeostasis when cells are in a state of stress and is associated with many diseases; however, the role of hypoxia inducible factor-1α (HIF-1α)-mediated ER degradation and the related regulatory pathway in acute kidney injury (AKI) still needs to be further established.
Results:
In the present study, an in vivo AKI model was induced in mice via the ischemia–reperfusion (IR) method. The results revealed that HIF-1α and BNIP3 were increased, and autophagy and ER degradation were activated in the kidneys of AKI mice, whereas HIF-1α knockout significantly inhibited BNIP3, autophagy and ER degradation, accompanied by aggravated kidney injury. Overexpression of HIF-1α in vitro significantly increased BNIP3, autophagy and ER degradation, whereas inhibition of BNIP3 significantly reversed the effects of HIF-1α. In addition, the in vitro inhibition of autophagy with chloroquine significantly reversed the effects of HIF-1α on cell apoptosis. Moreover, selectively overexpressing BNIP3 on the ER membrane significantly increased ER degradation via autophagy and decreased cell apoptosis in vitro.
Innovation and Conclusion:
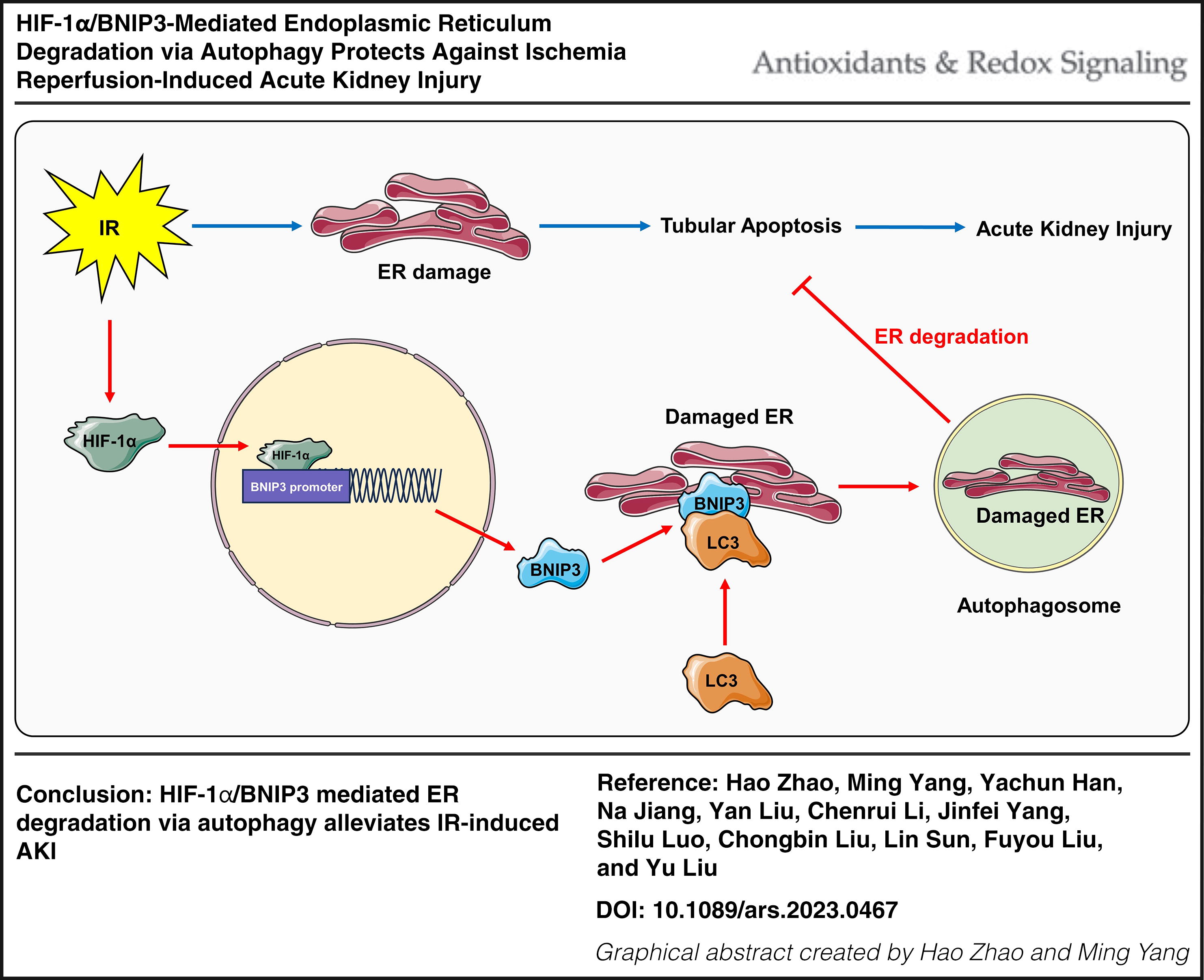
These data indicate that HIF-1α/BNIP3-mediated ER degradation via autophagy in tubular cells protects against IR-induced AKI. Antioxid. Redox Signal. 42, 212–227.
Introduction
Acute kidney injury (AKI) is characterized by a rapid decline in renal function and is caused mainly by ischemia–reperfusion (IR), nephrotoxins, and sepsis (Mehta et al., 2016). The main pathological change associated with AKI is renal tubular damage, which is characterized by tubular cell death and dysfunction (Sancho-Martínez et al., 2015). In clinical practice, the morbidity and mortality rates of AKI are still high (Bellomo et al., 2012). Many studies have reported the pathogenesis of AKI in recent years, involving different cell types, cellular processes, and molecular regulators, but the specific mechanism is still unclear (Bonventre and Yang, 2011; Sharfuddin and Molitoris, 2011). IR injury is defined as sudden temporary impairment of blood flow to particular organs and is one of the most common causes of human AKI (Malek and Nematbakhsh, 2015). During the process of IR-induced AKI, hypoxia-inducible factor 1α (HIF-1α) increases and plays protective roles (Bruzzese et al., 2021), but the underlying mechanism is still worthy of further study.
The endoplasmic reticulum (ER) is the largest organelle in cells and has fundamental functions such as protein folding, processing and transport, lipid and steroid synthesis, calcium storage, and detoxification (Schwarz and Blower, 2016). The half-life of the proteins and lipids of ER is as short as 3–5 days (Omura et al., 1967). To maintain the normal function and integrity of the ER, dynamic remodeling of the ER to maintain it in a homeostasis is important (Chino and Mizushima, 2020; Hubner and Dikic, 2020). Autophagy is an evolutionarily conserved process by which intracellular macromolecules are broken down into their constituent parts by lysosomes for further degradation or recycling. Autophagy comprises three subtypes: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy (Kim and Lee, 2014). ER degradation via autophagy (autophagy-induced ER degradation) is a type of macro-autophagy that selectively degrades the damaged ER when cells are under stress (Song et al., 2018) and plays a critical role in controlling ER homeostasis (Hubner and Dikic, 2020). During the process of autophagy-induced ER degradation, specific receptors in the ER or cytosol are recruited to the ER membrane, double-membrane vesicles combine with the receptors to package the damaged ER and form autophagosomes, and then, the autophagosomes fuse with lysosomes for subsequent ER degradation (Loi et al., 2018; Yang et al., 2021). Recently, autophagy-induced ER degradation has been shown to play important roles in many diseases, such as cancers, viral infections, and Alzheimer’s disease, but its role in AKI has not been reported until now.
Innovation
ER degradation via autophagy is a process that maintains ER homeostasis when cells are in a state of stress, but the role of HIF-1α-mediated ER degradation and the related regulation pathway in AKI remains unclear. Here, we showed that HIF-1α and BNIP3 were increased, and autophagy and ER degradation were activated in the kidneys of mice with AKI, while HIF-1α knockout significantly inhibited BNIP3, autophagy, and ER degradation, accompanied by aggravated kidney injury. Overexpression of HIF-1α in vitro significantly increased BNIP3, autophagy, and ER degradation, whereas inhibition of BNIP3 significantly reversed the effects of HIF-1α. In addition, inhibiting autophagy with chloroquine in vitro significantly reversed the effects of HIF-1α on cell apoptosis. Moreover, selectively overexpressing BNIP3 on the ER membrane significantly increased ER degradation via autophagy and decreased cell apoptosis in vitro. These data indicate that HIF-1α/BNIP3-mediated ER degradation via autophagy in tubular cells protects against IR-induced AKI.
Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) is an atypical BH3-only protein. BNIP3 is located on the outer mitochondrial membrane and functions as a receptor to induce mitochondrial degradation via autophagy, but BNIP3 is also located on the ER membrane and can induce ER degradation via autophagy (Hanna et al., 2012). The mitochondrial autophagy mediated by BNIP3 has been shown to protect against IR-induced AKI (Fu et al., 2020), but the role of BNIP3-mediated ER degradation via autophagy in AKI is still unclear. The induction of HIF-1α is a hallmark change in the kidney during AKI. Previous studies have shown that HIF-1α plays protective roles in AKI, but the underlying mechanism is still unclear. Interestingly, BNIP3 is a direct downstream target of HIF-1α (Erkan et al., 2005; Swiderek et al., 2013), and we assumed that HIF-1α may protect against IR-induced AKI through BNIP3 by mediating ER degradation via autophagy.
The results showed that AKI model mice were successfully induced by the IR method, HIF-1α, and BNIP3 were increased, and autophagy and ER degradation were activated in the kidneys of AKI mice. Furthermore, HIF-1α knockout significantly decreased BNIP3, autophagy, and ER degradation, accompanied by aggravated kidney injury. Overexpression of HIF-1α in vitro increased BNIP3, autophagy, and ER degradation, whereas inhibition of BNIP3 significantly reversed the effects of HIF-1α on autophagy and ER degradation. In addition, inhibiting autophagy with chloroquine in vitro obviously reversed the effects of HIF-1α on cell apoptosis. Moreover, selectively overexpressing BNIP3 on the ER membrane significantly increased autophagy-induced ER degradation and decreased cell apoptosis. Taken together, these data indicate that HIF-1α/BNIP3-mediated ER degradation via autophagy in tubular cells can protect against IR-induced AKI.
Results
Autophagy and ER degradation are activated in the kidney after IR injury
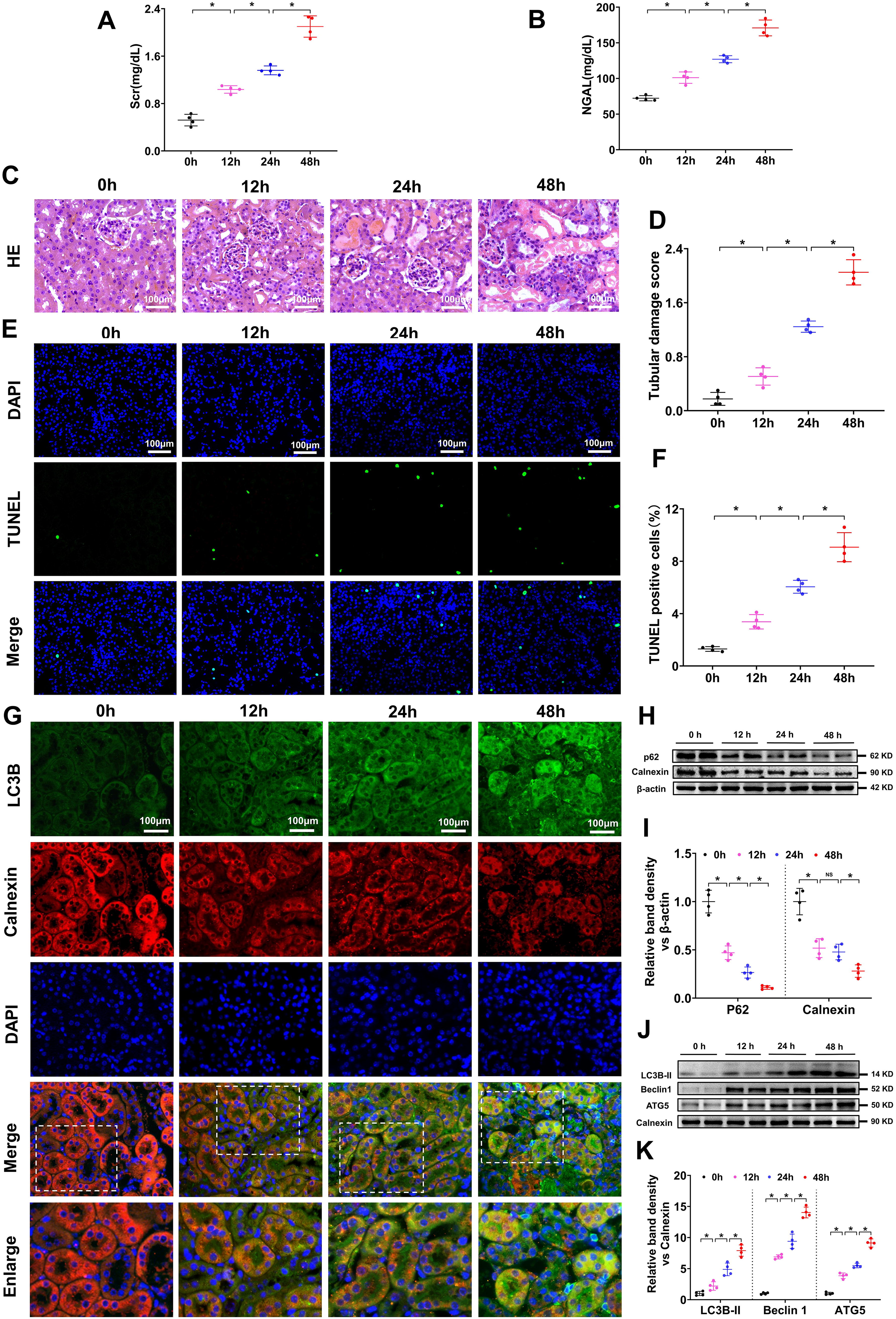
Mouse serum creatinine (Scr), neutrophil gelatinase-associated lipocalin (NGAL), and kidney hematoxylin and eosin (HE) staining and terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assays were used to valuate kidney injury after the IR operation. The Scr and NGAL levels in the mice gradually increased at 12 h, 24 h, and 48 h after the IR operation (Fig. 1A, B), as did tubular injury and tubular cell apoptosis, as shown by HE staining (Fig. 1C, D) and TUNEL assays (Fig. 1E, F). These results revealed that the mouse AKI model was successfully induced by the IR operation, and the degree of injury progressively increased after the operation at 12 h, 24 h, and 48 h. To observe autophagy and ER degradation in mouse kidneys after IR injury, we conducted immunofluorescence (IF) staining and Western blot (WB). IF analysis of LC3B (an autophagy marker) and calnexin (an ER marker) in mouse kidney sections revealed that LC3B gradually increased and calnexin decreased, and the yellow fluorescence of LC3B and calnexin overlay gradually increased after IR treatment (Fig. 1G). WB analysis of p62 (an autophagy adaptor that is digested during autophagy; thus, a decrease in p62 can indicate the activation of autophagy) and calnexin revealed that p62 and calnexin (an ER marker; a decrease in calnexin in total cell protein can indicate the activation of ER degradation) gradually decreased after IR treatment (Fig. 1H, I). Further WB analysis of representative autophagy markers (LC3B-II, Beclin-1, and ATG-5) in the ER protein extracted from the kidney cortex revealed that the LC3B-II, Beclin-1, and ATG-5 levels gradually increased after IR treatment (Fig. 1J, K). In the present study, when the WB analysis was conducted with the ER protein, calnexin was used as a loading control; when the WB analysis was conducted with total cell or tissue protein, calnexin was used as an ER marker to indicate the amount of ER in the cells, and β-actin was used as a loading control. These results showed that autophagy and ER degradation were gradually activated in the mouse kidney after IR injury and indicated that ER degradation might be induced by autophagy.
HIF-1α and BNIP3 levels are increased in the kidney after IR injury
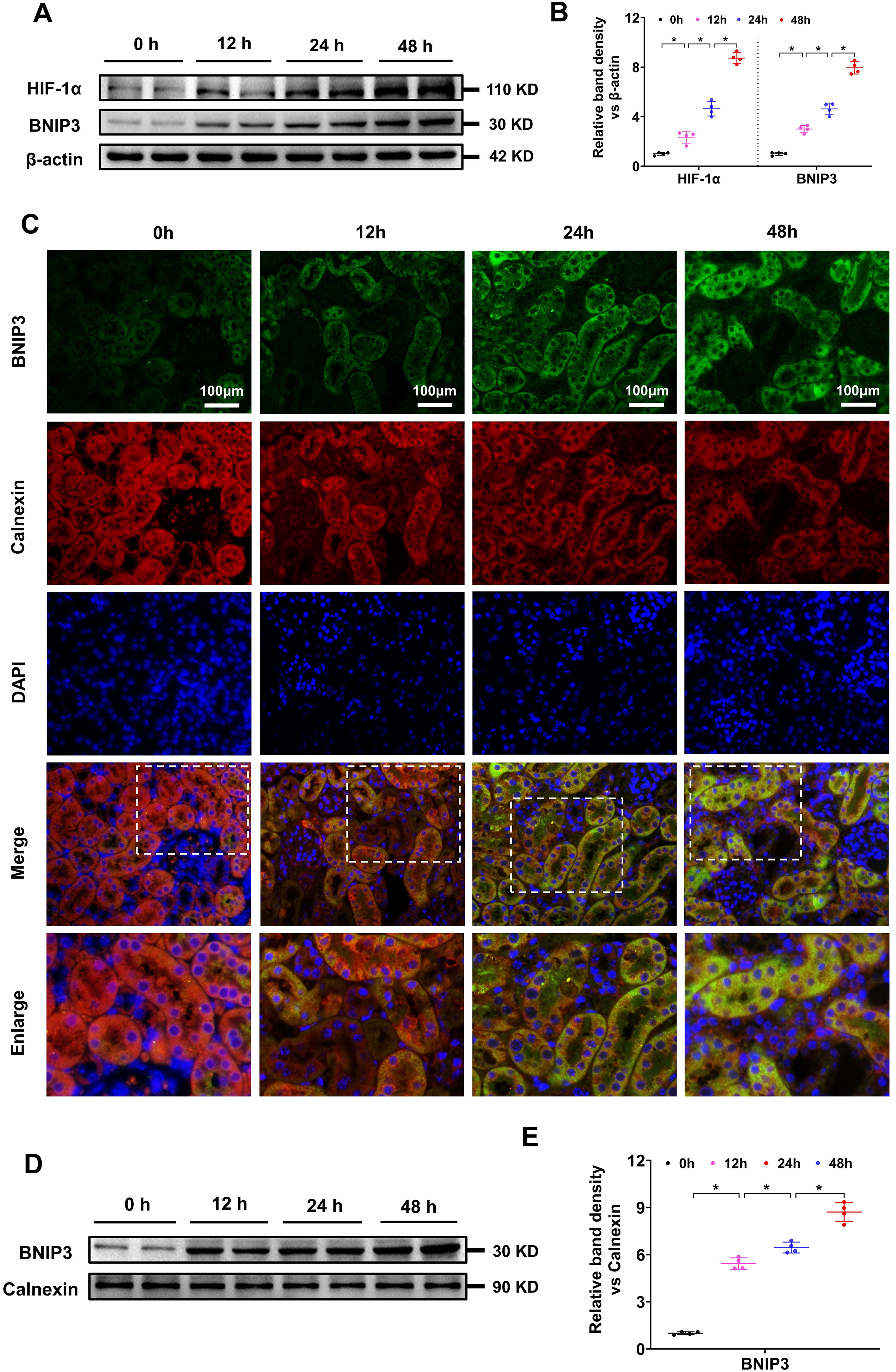
To observe HIF-1α and BNIP3 expression in the kidney after IR injury, WB and IF analyses were conducted. WB analysis revealed that HIF-1α and BNIP3 protein levels gradually increased in the kidney after IR injury (Fig. 2A, B), indicating that the HIF-1α/BNIP3 signaling pathway is activated in IR-induced AKI. IF colocalization of BNIP3 and calnexin was used to investigate the involvement of BNIP3 in ER degradation, and the results revealed that BNIP3 gradually increased, calnexin decreased, and the yellow fluorescence of BNIP3 and calnexin overlay increased in the kidney cortex after IR injury (Fig. 2C). We then extracted the ER protein from the kidney cortex and analyzed the BNIP3 protein level in the ER via WB analysis. The results revealed that the BNIP3 level in the ER gradually increased after IR treatment (Fig. 2D, E). These results suggested that the HIF-1α/BNIP3 signaling pathway was activated and might be involved in tubular cell ER degradation after IR injury.
HIF-1α gene knockout decreased BNIP3 and aggravated kidney injury in IR induced AKI
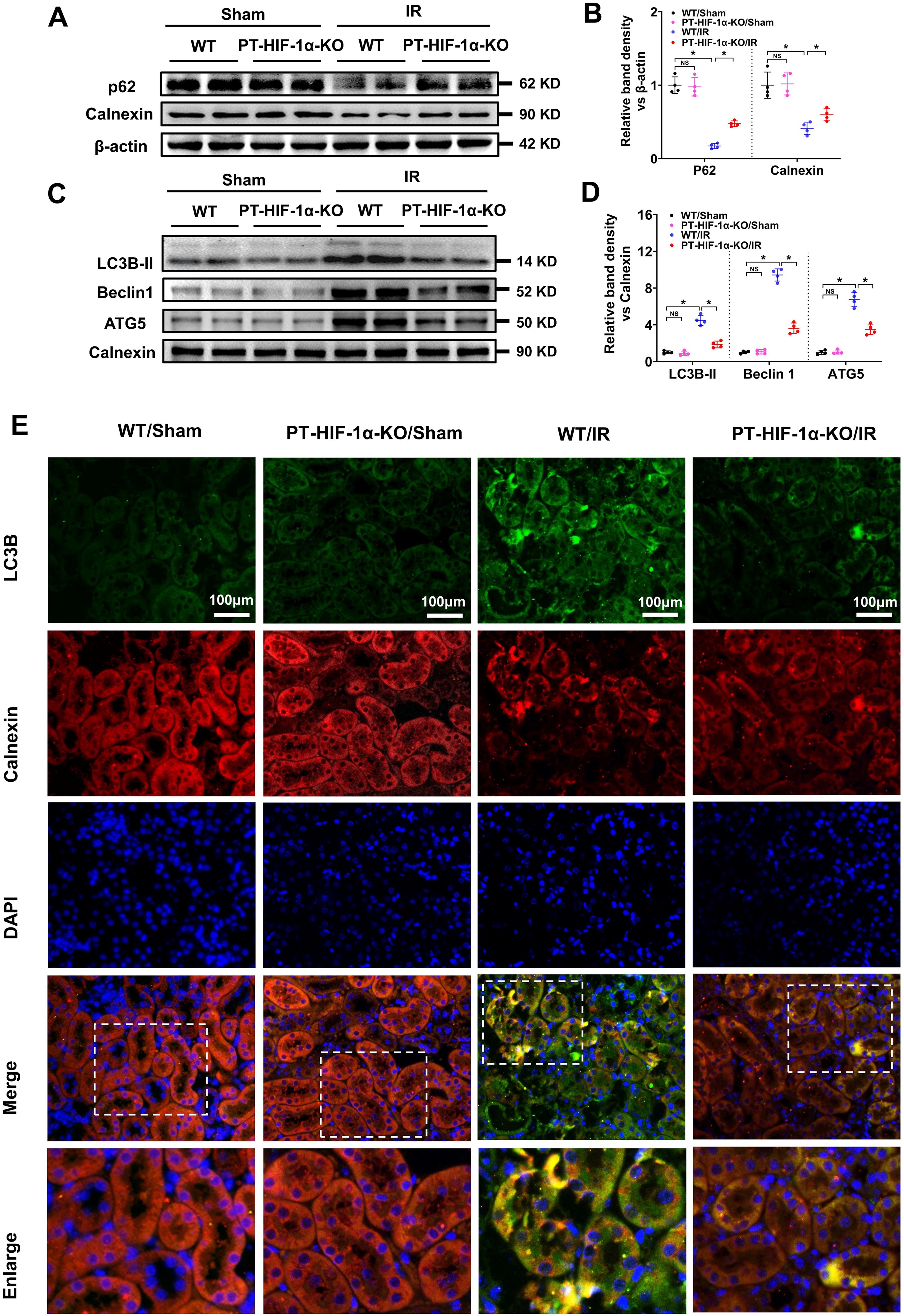
To further explore the role of HIF-1α and its relationship with BNIP3 in IR-induced AKI, we established PT-HIF-1α-KO (Knockout) mice, in which HIF-1α was conditionally knocked out in kidney proximal tubular cells (PTCs). After 48 h of IR, HE staining and TUNEL assays were conducted to analyze pathological changes in the kidney and tubular cell apoptosis, respectively. The results revealed that kidney injury and tubular cell apoptosis were mild and did not differ before the IR operation but were significantly increased in the WT/IR mice and further increased in the PT-HIF-1α-KO/IR group (Fig. 3A–D). WB results revealed that HIF-1α and BNIP3 protein levels were obviously increased in the IR-treated WT mice but decreased in the IR-treated PT-HIF-1α-KO mice (Fig. 3E, F). To further observe BNIP3 expression in the ER, we conducted BNIP3 and calnexin IF colocalization analyses. The yellow fluorescence of BNIP3 and calnexin overlay was low in both the (Wild Type) WT/Sham and PT-HIF-1α-KO/Sham groups but significantly increased in the WT/IR group compared with the PT-HIF-1α-KO/IR group (Fig. 3G). We further extracted ER protein from the kidney cortex and analyzed BNIP3 expression via WB. The results revealed that BNIP3 expression in the IR-treated WT mice was greater than that in the IR treated PT-HIF-1α-KO mice (Fig. 3H and 1). These data indicate that HIF-1α gene knockout in mouse PTCs can inhibit BNIP3 expression in the ER and aggravate kidney injury in IR-induced AKI.

HIF-1α gene knockout inhibited autophagy and ER degradation in IR-induced AKI
We further investigated the effects of HIF-1α gene knockout on autophagy and ER degradation in IR induced AKI. WB analysis revealed that both p62 and calnexin protein levels in the WT mice were significantly lower than those in the PT-HIF-1α-KO mice after IR injury (Fig. 4A, B), indicating that HIF-1α gene knockout in PTCs could inhibit autophagy and ER degradation in IR induced AKI. To further observe autophagy in the ER, we extracted ER protein from the mouse kidney cortex and analyzed the expression of autophagy indicators (LC3B-II, Beclin1, and ATG5) in the ER. The results revealed that the LC3B-II protein level in the extracted ER protein was low and not different between the WT/Sham and PT-HIF-1α-KO/Sham group and was significantly increased in the WT/IR group but was partly decreased in the PT-HIF-1α-KO/IR group, as were the levels of Beclin1 and ATG5 (Fig. 4C, D). IF colocalization analysis of LC3B and calnexin revealed that the yellow fluorescence of LC3B and calnexin overlay was significantly increased in the kidney cortex of the IR-treated WT mice but partly decreased in that of the IR-treated PT-HIF-1α-KO mice (Fig. 4E), indicating that autophagy in the ER was activated after IR injury and could be hindered by HIF-1α knockout. These results indicated that HIF-1α gene knockout in PTCs could inhibit autophagy and ER degradation in IR-induced AKI.
Inhibiting autophagy inhibited the effects of HIF-1α on ER degradation and cytoprotection
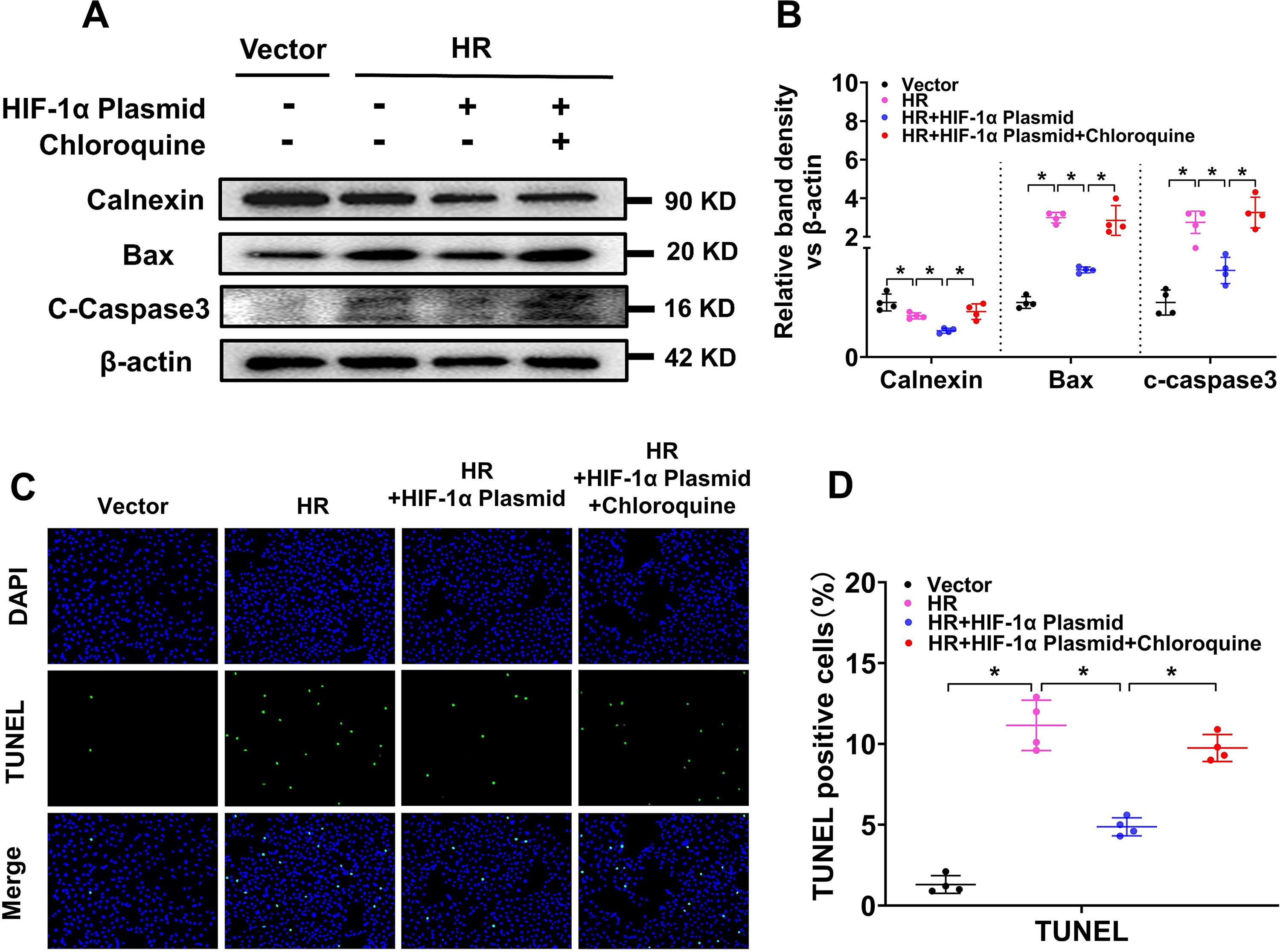
To analyze whether HIF-1α promotes ER degradation and cytoprotective effects via autophagy, we inhibited autophagy via chloroquine in hypoxia–reperfusion (HR)-treated HK-2 cells. WB results revealed that the level of calnexin was notably decreased in HR-treated HK-2 cells and further decreased by transfection with the HIF-1α plasmid; however, calnexin was partially increased by cotreatment with chloroquine, indicating that HIF-1α might promote ER degradation via autophagy. WB analysis of apoptosis markers (Bax and c-caspase3) revealed that both Bax and c-caspase3 were increased by HR treatment, and cotreatment with the HIF-1α plasmid decreased the Bax and c-caspase3 protein levels, which was partially reversed by chloroquine (Fig. 5A, B). TUNEL staining further revealed that cell apoptosis was increased by HR treatment and decreased by transfection with the HIF-1α plasmid but was reversed by cotreatment with chloroquine (Fig. 5C, D). These results indicate that HIF-1α might promote ER degradation and inhibit tubular cell apoptosis through autophagy in IR-induced AKI.
Inhibiting BNIP3 reversed the effects of HIF-1α on autophagy, ER degradation, and cytoprotection
To analyze the role of BNIP3 in HIF-1α in IR-induced AKI, we inhibited BNIP3 expression via BNIP3 siRNA in HR-treated HK-2 cells. WB results revealed that HIF-1α and BNIP3 protein levels were notably increased in HR-treated HK-2 cells and were further increased by transfection with the HIF-1α plasmid. When the cells were cotreated with BNIP3 siRNA, BNIP3 expression was significantly reduced, but no obvious effects were observed on HIF-1α expression. The protein levels of p62 and calnexin were significantly decreased by HR treatment and were further decreased by transfection with the HIF-1α plasmid but were partially increased by cotreatment with BNIP3 siRNA (Fig. 6A, B). Further WB analysis of BNIP3 and autophagy marker proteins (LC3B-II, Beclin1, and ATG5) in the ER extracted from HK-2 cells revealed that BNIP3 in the ER was notably increased by HR treatment and further increased by the HIF-1α plasmid but decreased by cotreatment with BNIP3 siRNA, as were LC3B-II, Beclin1, and ATG5 (Fig. 6C, D).
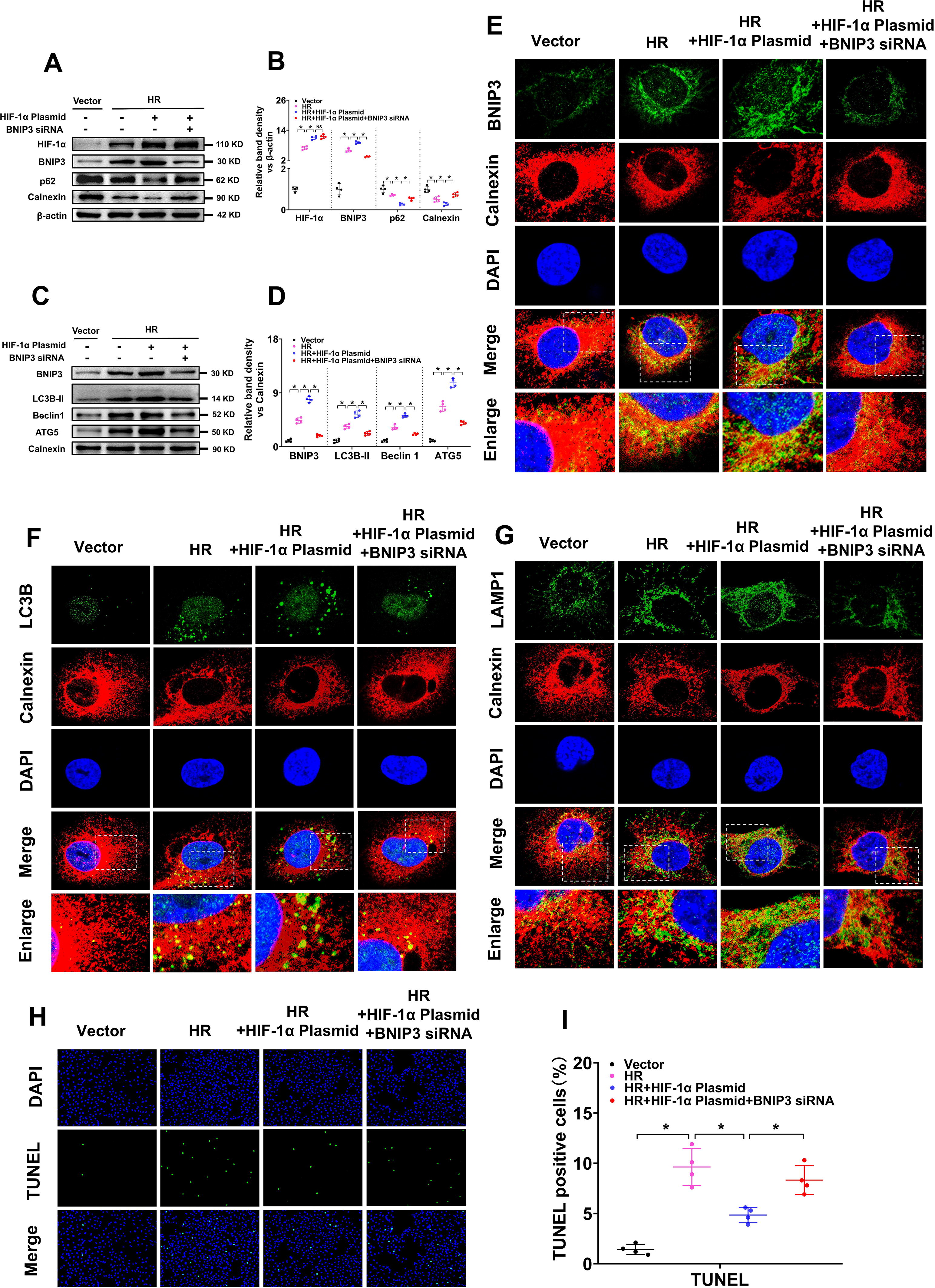
IF co-location analysis of BNIP3 and calnexin revealed that the yellow fluorescence of BNIP3 and calnexin overlay was significantly increased in HK-2 cells after HR treatment, and was further increased by HIF-1α plasmid, but was partially decreased by co-treatment with BNIP3 siRNA (Fig. 6E). So were the co-location analysis of LC3B and calnexin (Fig. 6F), LAMP1 (LAMP1 is a lysosome marker) and calnexin (Fig. 6G). The increase in the overlay of LC3B with calnexin, and LAMP1 with calnexin, indicates that ER degradation via autophagy is increased. The TUNEL assay revealed that HK-2 cell apoptosis increased after HR treatment and was decreased by the HIF-1α plasmid but was partially reversed by the cotreatment with BNIP3 siRNA (Fig. 6H, I). Taken together, these results indicate that inhibiting BNIP3 hindered the effects of HIF-1α on autophagy-induced ER degradation and cytoprotection in HR-treated HK-2 cells.
Specific overexpression of BNIP3 on the ER membrane promoted ER degradation via autophagy and protected against cell injury
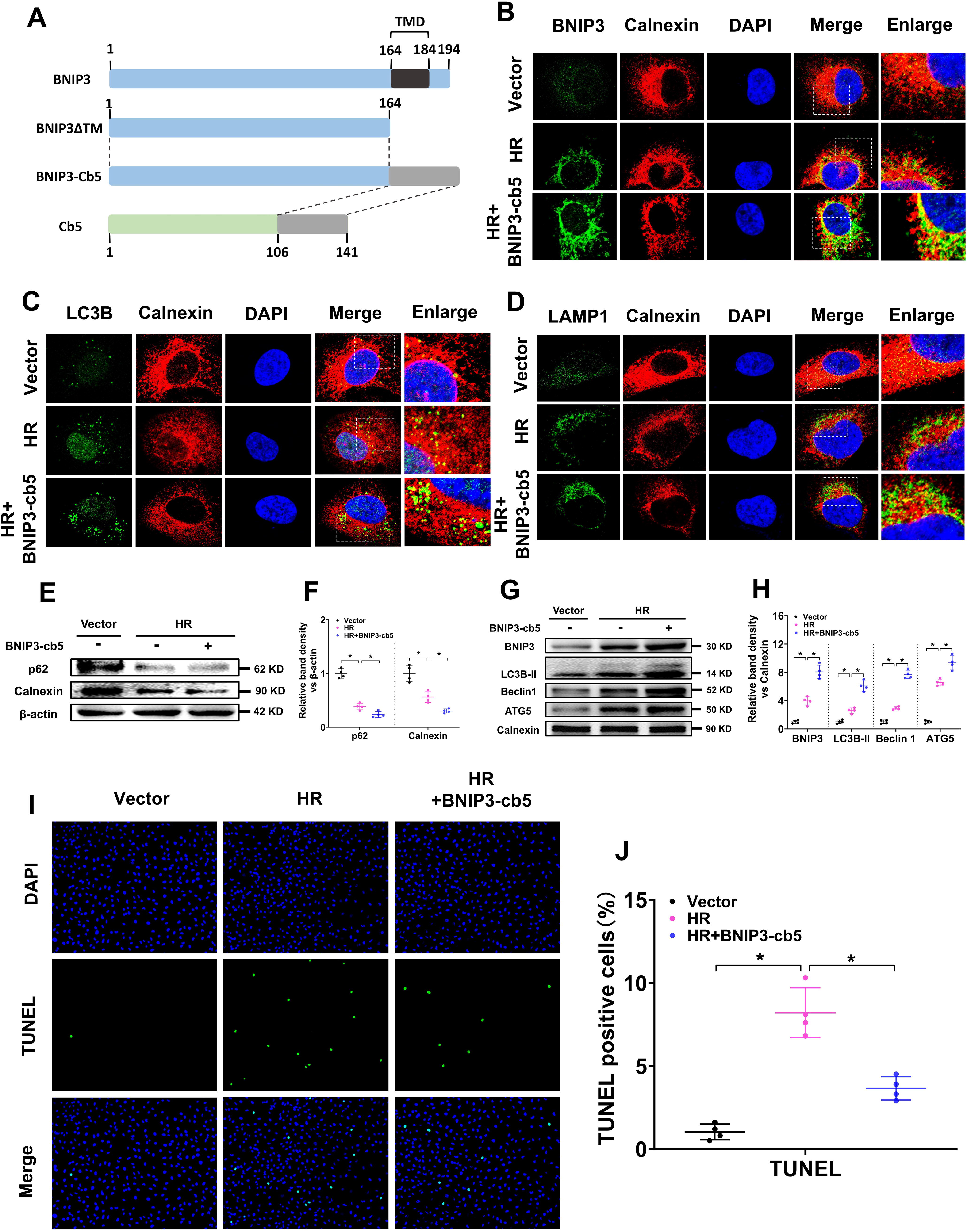
As an autophagy receptor, BNIP3 is involved in both autophagy-induced mitochondrial and ER degradation. To evaluate the role of BNIP3-mediated autophagy-induced ER degradation in IR-induced AKI, we constructed an ER-targeted BNIP3 overexpression plasmid (BNIP3-Cb5), in which the C-terminal 30-aa residue of wild-type BNIP3, including the putative transmembrane domain, was deleted (BNIP3ΔTM), and then the C-terminal 35-aa residue of human cytochrome b5 (Cb5) was ligated (Fig. 7A) without affecting the normal functions of BNIP3 as previously described (Hanna et al., 2012; Wang et al., 2001). Cb5 is an integral protein located on the ER membrane, and the C-terminal 35-aa residue of Cb5 can selectively target heterologous proteins to the ER membrane (Zhu et al., 1996). IF colocalization analysis of BNIP3 and calnexin revealed that the yellow fluorescence of BNIP3 and calnexin overlay was significantly increased in HK-2 cells after HR treatment and was further increased by transfection with the BNIP3-Cb5 overexpression plasmid (Fig. 7B), indicating that BNIP3-Cb5 successfully targeted BNIP3 to the ER membrane. Colocation analysis of LC3B and calnexin revealed that the yellow fluorescence of LC3B and calnexin overlay was significantly increased by BNIP3-Cb5 (Fig. 7C), as was the colocalization of LAMP1 and calnexin (Fig. 7D). WB results revealed that p62 and calnexin levels were significantly decreased in HK-2 cells after IR treatment and were further decreased by transfection with the BNIP3-Cb5 overexpression plasmid (Fig. 7E, F). Further WB analysis of BNIP3 and autophagy marker proteins (LC3B-II, Beclin1, and ATG5) in the ER of HK-2 cells revealed that the BNIP3, LC3B-II, Beclin1, and ATG5 protein levels in the ER of HK-2 cells were increased by HR treatment and further increased by transfection with the BNIP3-Cb5 overexpression plasmid (Fig. 7G, H). Both IF colocalization and WB analysis revealed that BNIP3 on the ER membrane could induce ER degradation via autophagy. The TUNEL assay revealed that HK-2 cell apoptosis was increased after HR treatment but was significantly decreased by transfection with the BNIP3-Cb5 overexpression plasmid (Fig. 7I, J), revealing that selective expression of BNIP3 on the ER membrane can inhibit cell apoptosis in HR-treated HK-2 cells. Taken together, these results indicate that BNIP3 overexpression on the ER can promote ER degradation via autophagy and protect against cell injury in HR-treated HK-2 cells.
Discussion
In the present study, our results revealed that HIF-1α and BNIP3 were increased and autophagy and ER degradation were activated in the kidneys of WT mice after IR injury but significantly reversed in PT-HIF-1α-KO mice, accompanied by severe kidney injury and tubular cell apoptosis. In vitro tests also revealed that HIF-1α and BNIP3 were increased and autophagy and ER degradation were activated in HR-treated HK-2 cells. Overexpression of HIF-1α further increased BNIP3 expression and autophagy-induced ER degradation, which was accompanied by decreased cell apoptosis. In addition, inhibiting autophagy with chloroquine in vitro significantly reversed the effects of HIF-1α on cell apoptosis. Moreover, inhibiting BNIP3 significantly reversed the effects of HIF-1α on autophagy, ER degradation, and cell apoptosis. Selectively overexpressing BNIP3 on the ER membrane significantly activated autophagy-induced ER degradation and decreased cell apoptosis. These results indicate that ER degradation via autophagy in renal tubular cells can be activated by the HIF-1α/BNIP3 signaling pathway, which plays a renoprotective role in IR-induced AKI.
Autophagy is an evolutionarily conserved cellular process, whereby intracellular macromolecules are broken down into their constituent parts within lysosomes (Kim and Lee, 2014). Autophagy has been demonstrated to be involved in various experimental models of AKI in recent years (Kaushal and Shah, 2016). The inhibition of autophagy leads to enhanced AKI, whereas the activation of autophagy has protective effects, suggesting a renoprotective role of autophagy in AKI (Jiang et al., 2010; Periyasamy-Thandavan et al., 2008). The cytoprotective role of autophagy in AKI is as follows: on the one hand, autophagy can degrade dysfunctional organelles that are potentially cytotoxic to cells; on the other hand, the autophagy process shares some important molecules responsible for cell death; as a result, the activation of autophagy may decrease cell death by regulating key molecules involved in the cell death process (Kang et al., 2011; Pattingre et al., 2005). However, it remains largely unclear as to what type of organelle autophagy plays a more important role in AKI.
ER degradation via autophagy is a type of macro-autophagy that can selectively degrade damaged ER when cells are under stress. The ER is one of the most important organelles involved in various cellular processes, such as protein synthesis, transport, modification, and intracellular calcium storage. Almost all secretory and membranous proteins must be folded and assembled in the ER. The homeostasis of the ER microenvironment is very important for its normal function (Oakes and Papa, 2015; Rashid et al., 2015). However, the ER is very sensitive to internal and external stimuli, such as oxidative stress and high glucose (Chen et al., 2018; Dandekar et al., 2015). When faced with such a stimulus, ER homeostasis is disrupted, resulting in the accumulation of unfolded proteins and subsequent ER stress and ER damage (Song et al., 2018). Autophagy-induced ER degradation is a newly described cellular process that counteracts excess ER stress and ER damage to restore the homeostasis and normal functions of the ER. The impairment of autophagy-induced ER degradation destroys the normal functions of the ER, eventually causing a number of diseases such as cancers, viral infections, and Alzheimer’s disease (Hubner and Dikic, 2020), but its role in kidney disease including AKI is still unknown. Degradation of the ER via autophagy must be mediated by specific receptors. Proteins that reside in the ER or cytosol, such as FAM134B, SEC62, CCPG1, TEX264, and ALT3, can be recruited on the ER membrane when needed (Chino et al., 2019;Fumagalli et al., 2016; Smith et al., 2018). In addition to these receptors, new receptors, such as BNIP3, are being discovered. BNIP3 was first found to be located on the outer mitochondrial membrane and is involved in autophagy-induced mitochondrial degradation (O’Sullivan et al., 2015; Zhang and Ney, 2009). BNIP3 directly binds to LC3 through its LC3-interacting region, thus facilitating the clearance of damaged mitochondria via autophagy (Ney, 2015; Tang et al., 2019). However, BNIP3 is also located on the ER membrane and is increased when cells are subjected to ER stress (Bozi et al., 2018; Zhang and Ney, 2009; Zhang et al., 2010). These findings suggest that BNIP3 may function as a receptor for ER degradation via autophagy.
HIF-1α is a key regulator under hypoxic conditions. There are accumulating data showing that HIF-1α protects against IR-induced AKI (Zou et al., 2017; Zhang et al., 2016). For example, preconditional activation of HIF-1α with the specific prolyl-hydroxylase inhibitors FG-4487 and FG-4497 attenuated IR-induced AKI (Bernhardt et al., 2006; Rosenberger et al., 2008). Further study revealed that downstream genes of HIF-1α such as vascular endothelial growth factor, erythropoietin, heme oxygenase 1, and glucose transporter type 1 could protect against IR-induced kidney injury (Matsumoto et al., 2003). Consistent with these findings, our present study demonstrated that HIF-1α plays a protective role in IR-induced AKI since HIF-1α knockout in PTCs significantly aggravated IR-induced kidney injury. Given that BNIP3 is a known direct downstream regulator of HIF-1α (Fu et al., 2020; Lin et al., 2021; Wu et al., 2019), we wondered whether the protective role of HIF-1α in AKI is associated with BNIP3-mediated autophagy. The results of the present study showed for the first time that HIF-1α/BNIP3-mediated ER degradation via autophagy could protect against IR-induced AKI.
BNIP3 was initially identified as a prodeath protein (Kale et al., 2018). For example, in IR-induced myocardial infarction animal models, BNIP3 increased in ventricular myocytes during the early cardiac ischemia period and persisted throughout reperfusion, accompanied by increased cardiomyocyte death (Chaanine et al., 2013). In addition, inhibition of BNIP3 reduces myocardial apoptosis and myocardial injury after cardiac IR injury (Hamacher-Brady et al., 2007; Diwan et al., 2007), suggesting a pathogenic role of BNIP3 in cardiac infarction. On contrary, emerging evidence has also revealed a cytoprotective role of BNIP3 in many diseases (Li et al., 2018; O’Sullivan et al., 2015; Zhang et al., 2016). Our present study revealed a renoprotective role of BNIP3 in IR-induced AKI. The discrepancy concerning the function of BNIP3 in different tissues may suggest that BNIP3 has cell type-specific functions.
In summary, the present study demonstrated the ER degradation via autophagy in tubular cells mediated by the HIF-1α/BNIP3 signaling pathway could protect against IR-induced AKI. These findings may provide some pharmacological targets for the prevention and treatment of AKI.
Materials and Methods
Inducing the AKI model
Male C57BL/6 mice (8–10 weeks, 20–25 g) were purchased from SJA Laboratory Animal Corporation (China). Mice with HIF-1α gene knockout in PTCs (PT-HIF-1α-KO) and their wild-type mice littermates were generated and identified as previously described (Zhao et al., 2020). Mouse AKI models were induced via the bilateral renal IR method as previously described (Tang et al., 2018). Briefly, the mice were anesthetized with pentobarbital (50 mg/kg) and then kept on a Homeothermic Blanket Control Unit (Harvard Apparatus Ltd., United Kingdom) to maintain body temperature at 36.5°C. The bilateral renal pedicles were exposed and clamped to induce ischemia for 30 min, after which the clamps were released for reperfusion. The color change of the kidney was observed as a visual symbol to monitor the success of IR. The sham group underwent the same operation but without clamping the renal pedicles. The mice were euthanized at 12, 24, or 48 h after the operation. All animal experiments were conducted according to the guidelines of the Animal Experimentation Ethics Committee of Second Xiangya Hospital of Central South University.
Cell culture and treatment
The human PTC line HK-2 (CRL-2190) was obtained from the American Type Culture Collection and cultured according to a standard protocol. HR treatment was performed as previously described (Fu et al., 2020). Briefly, the cells were first cultured in serum-free medium for 24 h and then transferred to a humidified hypoxic incubator (1% O2, 5% CO2, and 94% N2) and cultured for another 24 h. After hypoxia treatment, the cells were transferred to a normoxic incubator (21% O2, 5% CO2, and 74% N2) for another 24 h with fresh culture medium supplemented with 10% fetal bovine serum. The cells cultured with normal medium in a normoxic incubator were used as controls. For experiments with siRNA or plasmid interventions, cells were transfected with siRNA or plasmid via Lipofectamine 3000 (Invitrogen, USA) in accordance with the manufacturer’s instructions before HR intervention. HIF-1α and BNIP3-Cb5 overexpression plasmids were purchased from Sino Biological, China. BNIP3 siRNAs were purchased from RiboBio, China.
Physiological parameters analysis
Blood samples were collected from the mice and stored at −80°C. Scr levels were measured via a creatinine assay kit (BioAssay Systems, USA), and NGAL levels were measured via an enzyme-linked immunosorbent assay kit (R&D Systems, USA) according to the manufacturer’s instructions.
Morphological assessment
HE staining was performed as previously described to analyze the histopathological changes in the kidney (Yang et al., 2017). Briefly, mouse kidneys were collected, fixed with 4% buffered paraformaldehyde, and embedded in paraffin. Experiments were performed on 4-μm-thick kidney sections according to the instructions of the HE staining kit (Servicebio, China). The degree of tissue damage was scored according to the percentage of damaged tubules (0, no damage; 1, <25%; 2, 25%–50%; 3, 50%–75%; and 4, >75%) in HE-stained sections as previously described (Lin et al., 2005).
Analysis of apoptosis
TUNEL staining was used to detect the degree of tubular cell apoptosis according to the instructions of the cell apoptosis detection kit (Servicebio, China).
Tissue IF staining
Briefly, 4-μm-thick kidney sections were rehydrated and labeled with target antibodies, against LC3B (1:50, Proteintech, 18725-1-AP, USA), BNIP3 (1:100, Abcam, ab109362, USA), LAMP1 (1:100, Abcam, ab208943), and calnexin (1:400, Proteintech, 66903-1-lg). The slides were subsequently with Alexa Fluor 488-conjugated goat antirabbit or Alexa Fluor 594-conjugated goat anti-mouse antibodies. Nuclei were stained with Hoechst. The images were obtained with a confocal laser scanning microscope (Zeiss LSM 780).
Cell IF staining
Briefly, HK-2 cells treated as indicated were fixed with 4% paraformaldehyde for 5 min, permeabilized with methanol at −20°C for ∼10 min, and blocked with blocking buffer (phosphate-buffered saline [PBS] + 1% bovine serum albumin + 0.3% Triton X-100) for 1 h at 37°C. For LC3B-calnexin or BNIP3-calnexin costaining, the cells were incubated with primary antibodies against LC3B (1:50, Proteintech, 18725-1-AP) and calnexin (1:400, Proteintech, 66903-1-lg) or BNIP3 (1:100, Abcam, ab109362) and calnexin (1:400, Proteintech, 66903-1-lg) overnight at room temperature. Then, the cells were washed with PBS and incubated with Alexa Fluor 488-conjugated goat antirabbit antibodies and Alexa Fluor 594-conjugated goat anti-mouse antibodies for 1 h at 37°C. For LAMP1 and calnexin costaining, the cells were incubated with primary antibodies against LAMP1 (1:100, Abcam, ab25630) and calnexin (1:50, Proteintech, 10427-2-AP) overnight at room temperature, washed with PBS, incubated with Alexa Fluor 488-conjugated goat anti-mouse antibodies and Alexa Fluor 594-conjugated goat anti-rabbit antibodies for 1 h at 37°C, and then stained with Hoechst to delineate the nuclei. Images were obtained with a confocal laser scanning microscope (Zeiss LSM 780).
WB analysis
WB was performed in a conventional way. Briefly, equal amounts of protein extracted from the kidney cortex or HK-2 cells were electrophoresed and separated via sodium dodecyl sulfate polyacrylamide gels, electrotransferred onto nitrocellulose membranes, blocked in PBST (with 5% skim milk for 1 h, and incubated with primary antibodies against HIF-1α (1:1000, Abcam, ab17948), BNIP3 (1:1000, Abcam, ab109362), LC3B (1:1000, Proteintech, 18725-1-AP), Beclin1 (1:1000, Proteintech, 11306-1-AP), ATG5 (1:1000, Proteintech, 10181-2-AP), p62 (1:1000, CST, 5114S), calnexin (1:5000, Proteintech, 10427-2-AP), and β-actin (1:5000, Abcam, ab8226) at 4°C overnight. The membranes were subsequently washed with PBST solution, incubated with rabbit or mouse horseradish peroxidase-conjugated secondary antibody for antibodies 45 min, and visualized with an enhanced chemiluminescence reagent (Merck Millipore, USA).
Statistical analysis
The SPSS software, version 13.0 (SPSS Inc., Chicago, IL, USA) was used to perform the statistical analysis. The experimental data are presented as the means ± standard deviations. Differences among the groups were tested using one-way analysis of variance with Tukey’s post hoc analysis. A p value <0.05 indicated that the difference was statistically significant.
Footnotes
Acknowledgment
The authors are grateful to Dr. Tang (Second Xiangya Hospital, Central South University, Hunan, China) for kindly providing the PEPCK-Cre mice.
Author’s Contributions
H.Z. and M.Y. performed the experiments and wrote the article. Y.H., N.J., Y.L., C.L., J.Y., S.L., and C.L. provided technical support for this study. L.S. and F.L. participated in the discussion about this study. Y.L. designed the study and discussed the article. All the authors approved the final version to be published.
Data Availability
The raw data of this study are available from the first author and corresponding author upon reasonable request. Electronic laboratory notebook was not used.
Author Disclosure Statement
The authors declare they have no competing interests in the present study.
Funding Information
The research was supported by Natural Science Foundation of Chongqing (Grant No. CSTB2024NSCQ-MSX1166) and
Supplementary Material
Supplementary Data S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.