
Abstract
Significance:
Redox stress underlies numerous vascular disease mechanisms. Metabolic adaptability is essential for vascular cells to preserve energy and redox homeostasis.
Recent Advances:
Single-cell technologies and multiomic studies demonstrate significant metabolic heterogeneity among vascular cells in health and disease. Increasing evidence shows that reductive or oxidative stress can induce metabolic reprogramming of vascular cells. A recent example is intracellular L-2-hydroxyglutarate accumulation in response to hypoxic reductive stress, which attenuates the glucose flux through glycolysis and mitochondrial respiration in pulmonary vascular cells and provides protection against further reductive stress.
Critical Issues:
Regulation of cellular redox homeostasis is highly compartmentalized and complex. Vascular cells rely on multiple metabolic pathways, but the precise connectivity among these pathways and their regulatory mechanisms is only partially defined. There is also a critical need to understand better the cross-regulatory mechanisms between the redox system and metabolic pathways as perturbations in either systems or their cross talk can be detrimental.
Future Directions:
Future studies are needed to define further how multiple metabolic pathways are wired in vascular cells individually and as a network of closely intertwined processes given that a perturbation in one metabolic compartment often affects others. There also needs to be a comprehensive understanding of how different types of redox perturbations are sensed by and regulate different cellular metabolic pathways with specific attention to subcellular compartmentalization. Lastly, integration of dynamic changes occurring in multiple metabolic pathways and their cross talk with the redox system is an important goal in this multiomics era. Antioxid. Redox Signal. 41,793–817.
Introduction
Oxidation–reduction (redox) reactions are essential for all biological processes. Perturbed redox homeostasis underlies a broad spectrum of diseases, including the great majority of common cardiovascular diseases.
Increasing evidence has linked deranged vascular cellular metabolism with important vascular pathologies, including impaired angiogenesis (De Bock et al., 2013), pulmonary hypertension (Cao et al., 2019), and (nonlaminar) flow-mediated atherosclerosis (Yang et al., 2018). There has been growing attention to understanding how vascular cells utilize fuel sources for energy production and maintenance of redox homeostasis in the face of potentially adverse external perturbations. This crosstalk between redox stress and cellular metabolism in vascular cells is an important area of investigation and may open new therapeutic opportunities for vasculoprotection as well as prevention of cardiovascular diseases.
In this review, we first summarize how vascular cells utilize different energy sources via complementary metabolic pathways and maintain redox homeostasis in health and disease. We then highlight some of the recent advances in the field demonstrating the crosstalk between redox stress and vascular cellular metabolism.
Metabolism of Vascular Cells
Metabolism of endothelial cells
Endothelial cells (ECs) directly interface with numerous extracellular physicochemical stressors, including mechanical, biochemical, and inflammatory mediators, among other processes. ECs are critical regulators of vascular function, including hemostasis, vasomotor tone, angiogenesis, and protection against pathological thrombotic and inflammatory processes. Endothelial dysfunction is apparent in patients with cardiac risk factors (Celermajer et al., 1994, Hamburg et al., 2008), and signifies increased risk for atherothrombotic cardiovascular diseases and other vascular pathobiologies (Gimbrone and García-Cardeña, 2016).
Metabolic adaptation in response to extrinsic stimuli and stressors is critical in preserving vascular homeostasis and function (reviewed in Rohlenova et al., 2018). Increasing evidence suggests that inadequate metabolic adaptability or maladaptation resulting in metabolic perturbations in ECs can result in pathological phenotypic changes, such as impaired proliferation or migration (De Bock et al., 2013, Huang et al., 2017, Kim et al., 2017a, Schoors et al., 2015); increased pathological vascular permeability (Patella et al., 2015, Ziogas et al., 2021); endothelial-to-mesenchymal transition (Xiong et al., 2018); increased susceptibility to atherosclerosis associated with disturbed flow (Yang et al., 2018); and pulmonary hypertension (Cao et al., 2019).
Heterogeneity in endothelial functional and metabolic profiles and sex differences
With increasing availability of powerful single-cell technologies, there is growing appreciation of the considerable heterogeneity in endothelial metabolic signatures depending on the organ system and anatomical vascular bed, as well as specific physiological or pathological states (Becker et al., 2023, Dumas et al., 2020a, Geldhof et al., 2022, Jambusaria et al., 2020, Kalluri et al., 2019, Pasut et al., 2021, Rodor et al., 2022, Wang et al., 2022).
We now have the capacity to appreciate distinct and dynamic metabolic signatures expressed by heterogeneous EC types within and across a large number of distinct vascular beds in healthy and various disease states. For example, Kalucka and colleagues established a comprehensive single-cell atlas of the murine endothelium, including 32,567 ECs from 11 distinct organ systems (Kalucka et al., 2020). ECs demonstrated a strong heterogeneity in their organ-specific functional profiles. For example, ECs isolated from the heart and skeletal muscle demonstrated increased gene expression levels of membrane transport proteins and proteins regulating redox homeostasis, while ECs originating from the liver and spleen expressed higher levels of immunomodulatory genes. Within a given organ, vascular cell function and metabolic connectivity differ depending on their vessel calibers and vascular beds (e.g., artery, arteriole, capillaries, veins, lymphatics) (Becker et al., 2023), and their functional phenotypes (e.g., proliferative vs. quiescent; healthy vs. disease state) (Rohlenova et al., 2018). Within the same anatomical compartments, further heterogeneity exists between different endothelial subsets. Twenty-four distinct EC subsets have been identified within the kidney (Dumas et al., 2020b). Aerocytes and general capillary ECs with distinct functional profiles from the mouse alveolar microvasculature are additional examples (Kalucka et al., 2020). Such functional heterogeneity in ECs was accompanied by great heterogeneity with respect to their metabolic gene expression profiles. As expected, brain ECs expressed higher levels of the genes encoding transporters for robust nutrient uptake, while those involved in lipid metabolism were upregulated in skeletal muscle and the heart (Kalucka et al., 2020).
In addition, sex differences in endothelial biology are important determinants of the differential cardiovascular clinical outcomes in men and women (reviewed in Regitz-Zagrosek and Kararigas, 2017, Stanhewicz et al., 2018). Such differences are most pronounced in premenopausal women and often abrogated in ovariectomized animals, highlighting sex hormones as the key mediators of the difference. As a recent example, female mice with endothelial-specific deletion of the mitochondrial calcium uniporter proteins demonstrated impaired maximal vasodilation in response to acetylcholine, while no differences were seen in male transgenic animals, suggesting sex differences in endothelium mitochondrial calcium handling and its regulation of vasoreactivity (Damacena de Angelis et al., 2022). In the same study, cultured human aortic ECs treated with estradiol but not testosterone demonstrated increased mitochondrial calcium uptake capacity. This observation correlated with greater mitochondrial mass, higher membrane potential, and less mitochondrial reactive oxygen species (ROS) levels. It has been well established that estrogen provides vasculoprotective effects by promoting prostacyclin production, increasing nitric oxide (NO•) availability, and countering the renin–angiotensin system activity among additional mechanisms. Androgen has been found to counter many of these effects (reviewed in Stanhewicz et al., 2018). Estrogen is also known to be protective against ROS-dependent redox stress (reviewed in Kander et al., 2017, Miller et al., 2007, Regitz-Zagrosek and Kararigas, 2017). The widespread effects of sex hormones on vascular function and the redox system have been extensively studied. In addition, the intrinsic cellular mechanisms independent of sex hormones have been recognized (Hartman et al., 2020).
How vascular cells utilize fuel sources and control bioenergetics in a sex-specific manner is an important unanswered question. In the study comparing the metabolic profiles of human umbilical vein ECs (HUVECs) derived from male and female dizygotic twin pairs, the ECs originating from male donors were more susceptible to intracellular adenosine triphosphate (ATP) depletion upon serum starvation compared with those from female donors in vitro (Lorenz et al., 2019). In another study of male and female twin pairs, HUVECs originating from male donors were found to have their transcriptomic signature enriched in the pathways involved in oxidative phosphorylation and MYC targets (Hartman et al., 2020). Additional investigation is warranted to correlate these observations in gene expression signatures with the sex-specific metabolic activities of the ECs and their functional consequences in health and disease settings.
Endothelial metabolic pathways
With this endothelial heterogeneity in mind, we next summarize select prior studies that have helped define our current understanding of the central metabolic pathways in the ECs. This subject is growing rapidly and has also been reviewed by others in detail (Li et al., 2019b, Theodorou and Boon, 2018).
Glycolysis
Metabolic control of endothelial function has been best illustrated in the studies of angiogenesis (Cantelmo et al., 2015). ECs are largely quiescent in adults but can readily start proliferating upon proangiogenic stimulation, such as with hypoxia or growth factors.
Migrating and proliferating ECs, specifically migratory tip cells and proliferating stalk cells, prefer glucose as their main energy source via glycolysis (De Bock et al., 2013). During glycolysis, one molecule of glucose is ultimately converted to 2 molecules of pyruvate and a total of 2 molecules of ATP (Glucose + 2 NAD+ + 2 ADP + 2 Pi → 2 Pyruvate + 2 NADH + 2H+ + 2 ATP + 2 H2O). Proliferating ECs largely rely on glycolysis for up to 80% of ATP production, even when there is an adequate oxygen supply (Culic et al., 1997, De Bock et al., 2013, Krutzfeldt et al., 1990). This behavior is similar to that of many cancer cells (Warburg effect; reviewed in Vander Heiden et al., 2009). Compared with oxidative phosphorylation in the mitochondria, which produces 36 ATP molecules per one glucose consumed, glycolysis produces far less ATP. Although the net ATP produced per glucose molecule via glycolysis is markedly less, its production is considerably more rapid through glycolysis when compared with mitochondrial oxidative phosphorylation. Several additional advantages of endothelial dependence on aerobic glycolysis have been proposed (Li et al., 2019a). First, ROS production can be minimized, the bulk of which are then metabolically neutralized via a range of antioxidant enzymes, including those that utilize reduced glutathione (GSH) generated via the pentose phosphate pathway (PPP) as a source of reducing equivalents, as detailed in a later section. Second, by avoiding oxygen consumption in the endothelial mitochondria, delivery of oxygen to parenchymal tissues can be maximized. Third, glucose flux through glycolysis and the PPP is essential for biomass synthesis for proliferating ECs and redox homeostasis.
Hexokinase 2 (HK2), phosphofructokinase 1 (PFK1), and pyruvate kinase (PK) are the three rate-controlling enzymes of glycolysis. 6-Phosphofructo-2-kinase/fructose 2,6-bisphophatases are the bidirectional regulators of fructose-2,6-biphsphate, which is an important allosteric activator of PFK1 and, therefore, of glycolytic activity. De Bock et al. established PFKFB3 as a key regulator of endothelial glycolytic activity. PFKFB3 silencing inhibited endothelial glycolytic flux, which resulted in impaired vessel sprouting in cultured human ECs and inhibited ocular vascular development in mice in vivo (De Bock et al., 2013). Conversely, overexpression of PFKFB3 in the EC spheroid promoted a phenotype change toward vascular tips and increased vessel sprouting. Enriched glycolytic activity was noted in the lamellipodia of migrating tip cells where the key glycolytic enzymes colocalized with actin filaments to support angiogenesis. This seminal study illustrated that endothelial metabolic activity determines vascular function, and that glycolysis, in particular, is indispensable for angiogenesis (De Bock et al., 2013). Subsequently, a partial inhibitor of PFKFB3, 3-(3-pyridinyl)−1-(4-pyridinyl)−2-propen-1-one (3PO), helped reduce pathological retinal neovascularization in mice in vivo, providing an example of a therapeutic strategy specifically targeting endothelial metabolism for a vascular disease (Schoors et al., 2014).
In contrast to the migrating tip and proliferating stalk ECs, quiescent phalanx ECs demonstrate attenuated glycolytic activity. Wilhelm et al. showed that this endothelial metabolic modulation was mediated by transcriptional regulation by forkhead box protein 1 (FOXO1), whose antiglycolytic and antiangiogenic effects were mediated by its suppression of c-MYC activity (Wilhelm et al., 2016).
Shear stress by physiological and pathological flow is another important mediator of endothelial homeostasis and susceptibility to atherosclerosis (Dai et al., 2004, Garcia-Cardena and Gimbrone, 2006). The effect of mechanosensory transduction by shear stress on the endothelium has also been established as an integral aspect of endothelial metabolic regulation. Physiological laminar shear stress activates a transcription factor, Krüppel-like factor 2 (KLF2), in ECs, which results in gene expression changes promoting endothelial protection against an atherogenic phenotype, including downregulation of inflammatory genes (Parmar et al., 2006). KLF2 also mediates flow-induced metabolic regulation of ECs (Doddaballapur et al., 2015). ECs exposed to physiological laminar shear stress demonstrate decreased glycolytic activity as well as decreased mitochondrial respiration. Mechanistically, such flow-induced metabolic modulation was mediated by increased KLF2 activity, which partially inhibits PFKFB3 promoter activity. By contrast, disturbed flow with oscillatory stress activates endothelial glycolysis via hypoxia-inducible factor 1α (HIF1α) activation and limits mitochondrial respiratory capacity in human aortic ECs (Wu et al., 2017).
The PPP
In addition to the glycolytic pathway, a fraction of glucose 6 phosphate (G6P) can enter the PPP (Fig. 1). Glucose flux through the PPP is essential for redox homeostasis via reduced nicotinamide adenine dinucleotide phosphate (NADPH) production and biosynthesis of nucleic acids, lipids, and amino acids (Li et al., 2019a, Xiao and Loscalzo, 2020). The PPP consists of the oxidative and nonoxidative phases. The oxidative phase of the PPP (oxPPP) involves two irreversible, sequential, oxidative reactions mediated by glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD), which convert G6P ultimately to ribulose-5-phosphate while producing NADPH from each reaction. As discussed in detail in the next section, NADPH is needed by glutathione reductase (GR) to regenerate the reduced form of glutathione (GSH) needed to counter oxidative stress. In addition, NADPH modulates endothelial nitric oxide synthase (eNOS) activity and regulates NO• production in ECs, and is involved in fatty acid synthesis. Ribulose-5-phosphate subsequently enters the nonoxidative phase of the PPP and contributes to the synthesis of important precursors for nucleotide synthesis.

G6PD is the rate-determining enzyme for the oxPPP and has been extensively studied for its role in regulating endothelial redox homeostasis and vascular function. Complete G6PD deficiency is embryonically lethal, and its inhibition impairs cell survival (Vizán et al., 2009). High glucose resembling the diabetic milieu depleted NADPH in bovine aortic ECs by impairing G6PD activity and promoted cell death (Zhang et al., 2000). This effect was, at least in part, mediated by cyclic adenosine monophosphate-dependent protein kinase A. This study established the role of G6PD in protection against high-glucose-induced endothelial toxicity. Leopold et al. next demonstrated that pharmacologic inhibition of G6PD with dehydroepiandrosterone or by silencing gene expression increased ROS accumulation in bovine aortic ECs exposed to a pro-oxidant, hydrogen peroxide. Importantly, impaired G6PD activity was associated with decreased NO• bioavailability in these ECs (Leopold et al., 2001). This study highlighted increased ROS accumulation and decreased NO• bioavailability as the key mechanisms linking G6PD deficiency and endothelial dysfunction. Conversely, overexpression of G6PD in bovine aortic ECs resulted in decreased ROS accumulation in response to pro-oxidant stressors, such as hydrogen peroxide, tumor necrosis factor-alpha (TNFα), or xanthine oxidase, and maintained intracellular glutathione balance as a consequence of increased GR activity (Leopold et al., 2003). G6PD overexpression in ECs also provided protection against high-glucose-induced growth inhibition and cell death by restoring redox balance (Zhang et al., 2012).
Fatty acid metabolism
Increasing evidence suggests that ECs have an active role in regulating fatty acid transport and lipoprotein metabolism and their implications in vascular pathology (reviewed in Goldberg and Bornfeldt, 2013, Kim and Arany, 2022). The mechanisms involving endothelial uptake of lipids and its regulation have been extensively studied and remain an important area of ongoing investigation (reviewed in Abumrad et al., 2021, Hagberg et al., 2013, Kim and Arany, 2022). Depending on the specific vascular bed, vessel caliber, and the organ systems involved, the cellular pathways enabling fatty acid uptake and metabolism reflect great heterogeneity. CD36 is a transporter protein of long-chain fatty acids highly expressed in the capillary vessels supplying the organs consuming high levels of fatty acids, such as cardiomyocytes, skeletal muscle, and brown adipose tissue. Son and colleagues demonstrated that mice lacking CD36 in the endothelium but not in the parenchymal cells had impaired long-chain fatty acid uptake into these tissues, defining a major role of the endothelium in regulating lipid delivery and utilization in vivo (Son et al., 2018). In addition, lipoprotein lipase on the luminal aspect of capillary vessels hydrolyzes triglycerides and enables fatty acids to enter the ECs (Goldberg et al., 2021). Among the various mechanisms controlling endothelial fatty acid transport, vascular endothelial growth factor (VEGF) B has been shown to promote endothelial uptake of long-chain fatty acids from the circulation to local parenchymal tissues by transcriptional activation of fatty acid transport proteins via the endothelial VEGF receptor 1- and neuropilin-1-dependent mechanisms (Hagberg et al., 2010).
As a next step for fatty acid catabolism, fatty acids are activated on the outer mitochondrial membrane to form acyl-coenzyme A (Acyl-CoA) catalyzed by acyl CoA synthetase. Acyl-CoAs subsequently enters the mitochondrial matrix, where they undergo beta-oxidation (Houten et al., 2016). Transport of long-chain fatty acids from the cytosol to the mitochondria is mediated by carnitine palmitoyltransferase 1 (CPT1), which is located on the outer mitochondrial membrane, and CPT2, located on the inner mitochondrial membrane (Bonnefont et al., 2004). CPTs are the rate-determining enzymes for fatty acid oxidation (FAO). During subsequent beta-oxidation, flavin adenine dinucleotide (FADH2), NADH, and acetyl-CoA are formed, which contribute the reducing equivalents for the mitochondrial electron transport chain (ETC) and ATP production.
Loss of CPT1A impairs the proliferation and vascular sprouting of arterial ECs as well as HUVECs (Schoors et al., 2015). Similarly, loss of lymphatic endothelial-specific CPT1A impairs lymphatic vessel development (Wong et al., 2017). Endothelial FAO increases as cells assemble into a vascular network in a Matrigel system in vitro in a study combining proteomics and metabolic modeling (Patella et al., 2015). In this study, CPT1A inhibition induced endothelial hyperpermeability in association with deranged intracellular calcium signaling. Inhibition or silencing of CPT1A gene expression can also lead to a senescent phenotype in cultured HUVECs (Lin et al., 2022). This phenotypic change was ameliorated by overexpression of CPT1A in ECs or supplementation of short-chain fatty acids, acetate, and propionate. Acetate treatment in vivo improved senescence phenotype in the aorta of angiotensin II-infused mice and helped lower blood pressure.
While these studies established the role of endothelial FAO in regulating various key vascular functions, the precise regulatory mechanisms linking endothelial FAO and redox metabolism warrant further clarification. The relationship between perturbed redox homeostasis and altered endothelial fatty acid metabolism has been reported largely in the context of altered extracellular fuel availability. For example, endothelial exposure to excess exogenous free fatty acids has been demonstrated to increase ROS, exacerbate inflammation, inhibit eNOS and prostacyclin activation, and induce endothelial dysfunction (Du et al., 2006, Tripathy et al., 2003, Wang et al., 2006). In addition, ECs are known to activate FAO when they are cultured in glucose-depleted medium via activation of AMP-activated protein kinase (AMPK) (Dagher et al., 2001). Hypoglycemia-induced increase in mitochondrial ROS in cultured bovine aortic ECs was ameliorated by pharmacologic inhibition of FAO with etomoxir, implicating endothelial FAO in increased oxidative stress (Kajihara et al., 2017). The precise molecular and biochemical mechanism(s) underlying this observation, however, needs further elucidation.
Kalucka and colleagues discovered that quiescent ECs when compared with proliferating ECs were noted to have threefold greater basal FAO activity (Kalucka et al., 2018). These cells had higher gene expression levels of FAO enzymes, including CPT1A, and demonstrated relatively lower glycolytic activity in comparison. This metabolic reprogramming was mediated by increased Notch signaling. Interestingly, despite their higher FAO activities in comparison with proliferating ECs, these quiescent ECs had less basal ROS levels together with a lower proportion of oxidized glutathione and higher basal NADPH levels. These findings correlated with the observations that they had increased gene expression of the enzymes involved in NADP+ to NADPH conversion and the NAD(P) synthesis pathway. It remains to be clarified if this increased basal reductive drive seen in quiescent ECs is the result of their FAO activation or an alternative or additional mechanism independent from EC fatty acid metabolism. In the same study, inhibition of endothelial FAO via CPT1A deletion or etomoxir exacerbated oxidative stress with greater intracellular ROS levels in vitro and in vivo. Endothelial-specific CPT1A deletion in an experimental model of colitis resulted in a more severe disease form with increased mortality. Taken together, it was proposed that endothelial basal FAO is important in protection against oxidative stress. Of note, this understanding involving basal endothelial fatty acid metabolism is distinct from other systems in which excess FAO activation has been demonstrated to exacerbate oxidative stress (Batista-Gonzalez et al., 2019, Du et al., 2006).
Amino acid metabolism
Glutamine is abundant in circulation and is consumed in large quantities by ECs (Li et al., 2019a). Glutamine can provide anaplerotic substrates to the endothelial TCA cycle as established in cancer cells (DeBerardinis et al., 2008). Glutamine provides a large portion of the carbon sources for the TCA cycle in ECs (Kim et al., 2017a, Schoors et al., 2015). Glutaminase (GLS) is expressed by ECs and converts glutamine to glutamate and ammonia (Fig. 1). As detailed in Section 2, glutamine is an important source of GSH synthesis. Glutamine depletion, therefore, can perturb redox homeostasis. Deletion of endothelial-specific GLS1 impairs angiogenesis in mice in vivo, while extracellular glutamine depletion or GLS inhibition in vitro impairs endothelial proliferation (Huang et al., 2017, Kim et al., 2017a). Upon glutamine starvation, asparagine administration restores endothelial proliferative capacity (Huang et al., 2017). Asparagine synthetase converts nitrogen derived from glutamine and aspartate to asparagine. When the expression of this enzyme was silenced, endothelial proliferation became impaired. Impaired glutamine metabolism in cell culture prevents endothelial proliferation in association with loss of TCA intermediates (Kim et al., 2017a).
Of note, senescent ECs demonstrate a sixfold increase in glutamine consumption together with increased GLS expression (Unterluggauer et al., 2008). Despite this increased glutamine consumption, the intracellular ATP content is markedly depleted in the ECs with senescent phenotype. Interestingly, inhibition of GLS impairesproliferation and induces premature senescence, as well.
Endothelial metabolic changes in disease states
In addition to impaired angiogenesis, endothelial metabolic derangements drive various other vascular pathologies. In pulmonary arterial hypertension (PAH), a metabolic shift away from glucose oxidation toward increased glycolysis in pulmonary artery ECs and vascular smooth muscle cells (VSMCs) has been identified as a central pathological mechanism (reviewed in Xu et al., 2021). Inhibition of glycolytic activation ameliorated disease severity in experimental models of PAH (Cao et al., 2019). In addition, Bertero and colleagues found that pulmonary vascular extracellular matrix stiffening in pulmonary hypertension activates glutaminolysis and glycolysis in diseased vessels and supports the metabolic demands of hyperproliferating vascular cells (Bertero et al., 2016).
Altered endothelial metabolism is also gaining increasing attention in diseases associated with vascular inflammation (reviewed in Li et al., 2019b, Theodorou and Boon, 2018). The key glycolytic enzyme, PFKFB3, has been found to regulate nuclear factor-kappa B (NF-κB)-mediated vascular inflammation in ECs associated with tumor cells (Cantelmo et al., 2016) or those treated with proinflammatory cytokines, such as TNFα (Xiao et al., 2021, Zhang et al., 2019). Furthermore, Xiao et al. demonstrated increased glucose flux through the PPP and TCA cycle in these ECs in addition to increased glycolytic activity. Mechanistically, suppression of the FOXO1-PDK4 (pyruvate dehydrogenase kinase 4) axis mediated activation of oxidative phosphorylation. In the same study, inhibition of G6PD, the rate-determining enzyme of the PPP, exacerbated endothelial inflammation in vitro, while increasing mitochondrial respiration by pharmacological blockade of PDKs ameliorated vascular inflammation in LPS-treated mice in vivo (Xiao et al., 2021) (Fig. 2).

Yang and colleagues reported increased expression of the key metabolic regulator AMPK in the mouse aortic areas that were exposed to disturbed flow, together with increased glycolysis. Deletion of endothelial AMPK predisposed the animals to develop more severe atherosclerosis, and overexpression of glucose transporter 1 (GLUT1) in ECs helped attenuate accelerated atheroma progression, highlighting the importance of endothelial glucose metabolism in defense against flow-mediated vascular susceptibility to atherosclerosis (Yang et al., 2018).
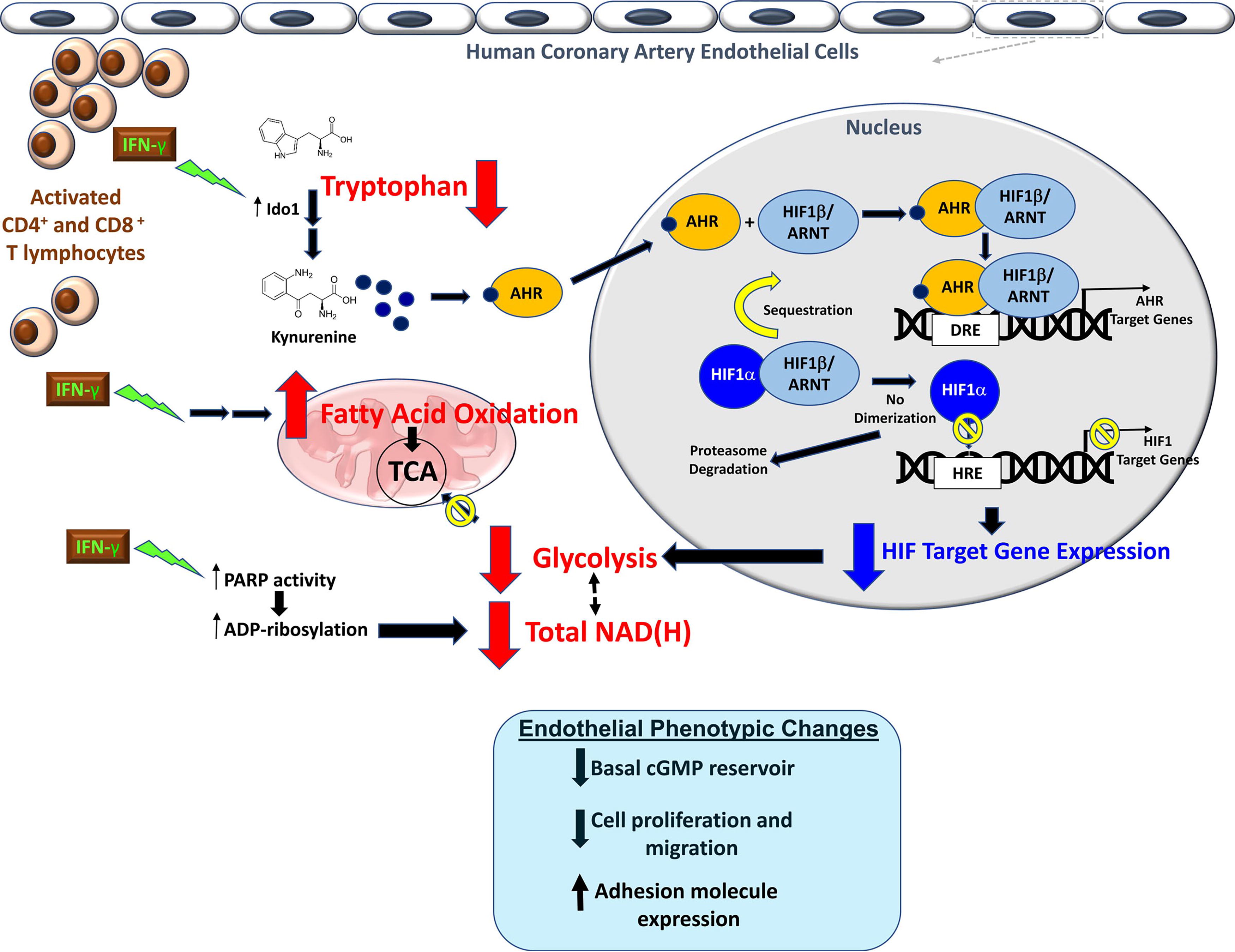
Lee et al. recently demonstrated that nonproliferating human coronary artery ECs undergo metabolic reprogramming from impaired glycolysis toward FAO upon exposure to a proatherogenic cytokine, interferon-gamma (Lee et al., 2021). Mechanistically, IFN-γ activates the catabolism of the essential amino acid tryptophan to kynurenine, leading to activated aryl hydrocarbon receptor’s outcompeting HIF1α for their shared binding protein HIF1β. Decreased HIF1 availability resulted in global transcriptional suppression of endothelial glycolytic enzymes. IFN-γ also depletes intracellular NAD(H). Compensatory FAO activation was necessary to maintain intracellular energy balance. Such widespread metabolic derangements are associated with pathological endothelial phenotype switches that are hallmarks of endothelial dysfunction and increased susceptibility to atherosclerosis (Lee et al., 2021) (Fig. 3). This study highlights the closely intertwined nature of multiple metabolic pathways, their cross talk in vascular metabolism, and consequences for endothelial phenotype.
Metabolism of VSMCs
Similar to ECs, great heterogeneity and plasticity in VSMC phenotypes have been appreciated in different vessel beds and organ systems (as reviewed in Lacolley et al., 2017). In adults under physiological condition, VSMCs in general are fully differentiated with high expression of proteins important in vascular contractile function (“contractile phenotype”) and minimal proliferation [as reviewed in (Owens et al., 2004)]. Various pathological stimuli can trigger a switch to a more “synthetic” phenotype, where VSMCs demonstrate greater proliferative and migratory capacities (Rensen et al., 2007, Rzucidlo et al., 2007). Impaired contractile function and increased proliferative activity of VSMCs underlie many important vascular pathologies, especially atherosclerosis and pulmonary hypertension [as reviewed in (Allahverdian et al., 2018, Chiong et al., 2014, Xu et al., 2021)].
Early studies established that VSMCs at baseline exhibit high rates of glucose utilization (Paul, 1983). Similar to ECs, aerobic glycolysis is preferred despite adequate oxygen tensions. In one study, lactate production reflecting glycolytic activity was noted to be of similar magnitude as the rate of oxygen consumption even when there is abundant oxygen (Paul, 1983). In addition, VSMC overexpression of a glutamine transporter, solute carrier family 1 member 5, promotes mammalian target of rapamycin complex 1 activation and VSMC proliferation (Osman et al., 2019).
Pathological proliferation and migration of VSMCs from the media to the subintimal space is a key mechanism in atherosclerosis (Bennett et al., 2016). Metabolic reprogramming in VSMCs has been recognized as one of the major drivers of pathological VSMC phenotypic changes and vascular pathogenesis (as reviewed in Salabei and Hill, 2013, Shi et al., 2020). Bernal-Mizrachi and colleagues demonstrated that impaired mitochondrial metabolism in the blood vessel wall results in hypertension and accelerates atherosclerotic lesion development by overexpressing uncoupling protein-1 in aortic VSMCs. This change was accompanied by increased superoxide production and decreased NO• availability (Bernal-Mizrachi et al., 2005). VSMCs undergoing a platelet-derived growth factor-mediated switch to synthetic phenotype demonstrated mitochondrial fragmentation and decreased mitofusin 2 with a 20% decrease in glucose oxidation (Salabei and Hill, 2013).
Synthetic VSMCs exhibit higher glycolytic activities (Werle et al., 2005), and HIF1 activation has been identified as one of the key regulatory mechanisms (Lambert et al., 2010) for this enhanced glycolysis. Inhibition or deletion of glycolytic enzymes, including HK2, pyruvate kinase M2 (PKM2), and lactate dehydrogenase B, has been shown to attenuate VSMC migration and proliferation (Kim et al., 2017b, Lambert et al., 2010, Zhou et al., 2019).
VSMC glucose metabolism also intersects with and promotes vascular inflammation. Wall and colleagues demonstrated that TNFα promotes GLUT1 expression in VSMCs, which increases further chemokine expression. Higher levels of GLUT1 expression by VSMCs, but not myeloid cells, accelerated atherosclerotic lesion growth in an animal model of metabolic syndrome (Wall et al., 2018).
Enhanced vascular cell proliferation and decreased apoptosis are key pathogenic features of PAH (as reviewed in Colon Hidalgo et al., 2022). Increased glycolytic enzyme expression via an HIF-mediated process is noted in pulmonary artery VSMCs of PAH patients (Dabral et al., 2019). Impaired mitochondrial ETC complex I-III activities together with increased ROS production have been observed in animal models of PAH (Rafikov et al., 2015). Impaired VSMC mitochondrial metabolism and overactivation of glycolysis have been proposed as therapeutic targets for PAH (Diebold et al., 2015, Marsboom et al., 2012). Taken together, these studies highlight the significance of altered fuel utilization of ECs and VSMCs in various vascular pathobiologies and the functional importance of preserving metabolic homeostasis in vascular cells.
Redox Homeostasis and Redox Stress in Vascular Cells
Energy metabolism in vascular cells is interconnected with cellular redox homeostasis. Cellular metabolism provides essential electron donors and acceptors required for redox reactions, while the cellular redox niche governs energetics through redox modifications of metabolic enzymes and signaling pathways (Xiao et al., 2018, Xiao and Loscalzo, 2020). The cellular redox environment is dynamically maintained via fine-tuning of cellular pro-oxidant and antioxidant levels. Such balance maintains cellular pro-oxidants at physiological levels so that they act as (normal) signaling molecules to regulate vascular cell biological functions. By contrast, disruption of this fine-tuned balance results in redox stress, including oxidative stress and reductive stress, both of which are known to be detrimental to the physiological functions of the vasculature leading to frank vascular dysfunction and consequent vascular diseases.
Redox network and its compartmentalization in vascular cells
The cellular redox network is formed by pro-oxidants [ROS and reactive nitrogen species (RNS)] and antioxidants (enzymatic and nonenzymatic antioxidants); taken together, these species dictate cellular redox homeostasis (Xiao and Loscalzo, 2020). Vascular cells produce ROS and/or RNS in mitochondria predominantly by the ETC and in extramitochondrial compartments mainly enzymatically, such as via NADPH oxidases (NOXs), NOS1-3, cytochrome P450 enzymes, xanthine oxidase, and lipoxygenases (Fukai and Ushio-Fukai, 2020, Handy and Loscalzo, 2016, Xiao and Loscalzo, 2020). Vascular cells also have dedicated antioxidant systems to counteract these oxidants. Superoxide dismutases (SOD1-3) metabolize superoxide anion (O2 •−) to hydrogen peroxide (H2O2); the latter and other organic peroxides (ROOH) can be further reduced to water by catalase, glutathione peroxidases (GPX1-8), peroxiredoxins (PRX1-6), and thioredoxins (TRX1-2) (Xiao and Loscalzo, 2020).
Of note, GPX, PRX, and TRX all require the redox couples GSH/GSSG or NADP+/NADPH as electron donors for their peroxidase activities (Schafer and Buettner, 2001, Xiao et al., 2018, Xiao and Loscalzo, 2020). GSH provides two electrons to GPX to reduce H2O2 forming water and GSSG; the latter is then recycled back to GSH by GR using electrons from NADPH (Xiao and Loscalzo, 2020). In addition to GR, thioredoxin reductases also use electrons from NADPH to reduce oxidized TRX (TRX-S2) to its active dithiol form TRX-(SH)2, which then donates electrons to PRXs in the reductions of H2O2, and ROOH to water and (organic) alcohols, respectively (Xiao and Loscalzo, 2020). Given their centrality, the GSH/GSSG and NADP+/NADPH redox couples are key regulators of cellular redox homeostasis.
One distinct feature of the cellular redox network is compartmentalization: pro-oxidant-producing sources, antioxidant enzymes, and redox couples NADP+/NADPH and GSH/GSSG all are highly compartmentalized (Xiao et al., 2018, Xiao and Loscalzo, 2020). In mitochondria, oxygen molecules gain electrons leaked primarily from respiratory complexes I, II, and III via forward and reverse electron transfer actions producing O2 •− in vascular cells (Read et al., 2021). In complex I, O2 •− production occurs at its iron–sulfur (Fe-S) cluster, the flavin mononucleotide site, and the quinone binding site, and is exclusively released into the mitochondrial matrix; likewise, complex II also generates O2 •− exclusively in the matrix at its flavin site (Read et al., 2021, Xiao et al., 2016) (Fig. 4). In contrast, O2 •− produced by the quinone cycle of complex III can enter the mitochondrial matrix and intermembrane space (IMS) (Muller et al., 2004, Read et al., 2021). Since O2 •− anion is negatively charged, is impermeable to mitochondrial membranes, and has a very short half-life (milliseconds), it can only function as a signaling molecule or oxidize cellular macromolecules locally. Normally, O2 •− in the matrix and IMS is often subsequently metabolized to H2O2 by mitochondrial matrix-localized SOD2 (manganese SOD; MnSOD) and IMS-localized SOD1 (copper zinc SOD; CuZnSOD), respectively. The resulting H2O2 is then reduced to water by mitochondrial peroxidases (catalase, GPX4, and the PRX3/5-TRX2 system) using mitochondrial pools of GSH/GSSG and NADP+/NADPH as electron donors (Xiao and Loscalzo, 2020) (Fig. 4). Interestingly, compared with O2 •−, mitochondrial H2O2 is less reactive, has a greater capability to traverse mitochondrial membranes into the cytosol, and has a relatively longer half-life; therefore, it can serve as a signaling molecule and oxidize its molecular targets both locally (i.e., near the source of synthesis) and remotely (Handy and Loscalzo, 2016).

In extramitochondrial compartments, NOXs are the primary ROS-producing enzymes in vascular cells. Vascular ECs and VSMCs express NOX1, NOX2, NOX4, and NOX5, among which NOX4 is the most abundant isoform (Fukai and Ushio-Fukai, 2020, Lassègue et al., 2012). NOXs were mainly localized to the plasma member, (peri)nucleus, and endoplasmic reticulum (ER); unlike other NOX enzymes, NOX4 is also expressed in mitochondria (Lassègue et al., 2012). NOX isoenzymes catalyze one-electron reduction of oxygen into O2 •− using NAD(P)H as an electron donor; the resulting O2 •− is subsequently released into the cytosol and/or extracellular space depending on the subcellular locations of a specific NOX enzyme (Fig. 5). In the cytosol, SOD1 catalyzes the conversion of O2 •− into H2O2 followed by peroxidase (catalase, GPX1, and the PRX1/2/4/6-TRX1 system)-catalyzed reduction into water. In the extracellular space, the metabolism of O2 •− is specifically mediated by extracellular SOD (EcSOD or SOD3) and then by GPX3 and its cofactor GSH (Fig. 5).

In addition to NOXs, NOS enzymes are also major contributors of ROS/RNS in the cytoplasmic compartment of vascular cells. Neuronal NOS (nNOS or NOS1) is constitutively expressed in ECs and smooth muscle cells (SMCs); inducible NOS (iNOS or NOS2) expression in the vasculature is normally low but can be induced by external (inflammatory) stimuli, including cytokines; and endothelial NOS (eNOS or NOS3) is predominantly and constitutively expressed in ECs (Garcia and Sessa, 2019, Tejero et al., 2019). All three NOS isoforms are membrane bound due to fatty acylation at the N-terminus and catalyze the same reaction to produce NO•:
These enzymes function as homodimers and require FMN, FAD, tetrahydrobiopterin (BH4), ferrous heme (Fe2+), and calcium as cofactors (Gantner et al., 2020). Notably, dissociation of the homodimeric structure and deficiency in their cosubstrates’ and cofactors’ availability, especially L-arginine, oxygen, and BH4, result in a situation where NADPH consumption is not stoichiometrically correlated with NO• generation, collectively termed “eNOS uncoupling,” leading to a shift in the production of NO• to O2 •− (Gantner et al., 2020, Vásquez-Vivar et al., 1998) (Fig. 5). Indeed, S-glutathionylation of cysteine (Cys) 689 and Cys908 at its reductase domain uncoupled eNOS and decreased its activity leading to elevated O2 •− production and impaired endothelium-dependent vasorelaxation (Chen et al., 2010).
eNOS is the most significant source of NO• in the vasculature and has great impact on vascular function under (patho)physiological conditions (Tejero et al., 2019). NO• as an endothelium-derived relaxing factor executes pleiotropic vasoprotective effects in the blood lumen and the vascular wall ranging from inhibition of leukocyte activation/adhesion and platelet aggregation (anti-inflammatory and antithrombotic), to vasodilation and regulation of vascular tone, and to suppression of VSMC proliferation and migration (Förstermann and Sessa, 2012, Tejero et al., 2019). For example, in the vascular wall, NO• diffuses to adjacent VSMCs where it activates soluble guanylyl cyclase to convert GTP into cyclic GMP, which, in turn, activates protein kinase G and, as a consequence, triggers a cascade of phosphorylation reactions leading to reduction in intracellular calcium levels and, thereby, VSMC relaxation and vasodilation (Furchgott, 1999, Soundararajan et al., 2023) (Fig. 6).

Of particular interest, in the vascular wall, NO• can readily and rapidly react with O2 •− (estimated rate constant of 6.7–10 × 109 M−1 s−1) forming a stronger oxidant, the peroxynitrite anion (ONOO-), leading to direct loss of NO• bioactivity in conjunction with oxidative inactivation of eNOS and guanylyl cyclase; together, these redox reactions compromise the vasoprotective functions of NO• (Fattman et al., 2003, Fukai et al., 2002). One decisive factor that circumvents these reactions is the antioxidant protein EcSOD. Physiologically, EcSOD is highly expressed in the arterial wall and accounts for roughly 50–70% of the total SOD activity; VSMCs are the principal sources of this enzyme, and these cells can secret large amounts of EcSOD into the extracellular matrix (Fattman et al., 2003, Oury et al., 1996, Strålin et al., 1995). In the extracellular space, EcSOD can stably exist, bind to the heparan sulfates on ECs’ glycocalyx, and be internalized by these cells. VSMC-secreted EcSOD maintains the bioavailability of endothelium-derived NO• by catalyzing the conversion of O2 •− into H2O2 and, thereby, limiting the consumption of NO• by O2 •−. The rate constant for the EcSOD-catalyzed reaction is estimated to be approximately 2–3 × 109 M−1 s−1 (Fattman et al., 2003, Fukai et al., 2002), which is 3 to 5 times lower than that of the NO• and O2 •− reaction. Owing to the high concentration of EcSOD in the arterial wall (an ∼30–55 μg/g tissue with the estimated activity of ∼3560–6440 Units/g tissue) in the face of the measured physiological levels of NO• (revised from earlier higher estimates to 100–5000 pM) (Hall and Garthwaite, 2009, Strålin et al., 1995), and to the observation that NO• stimulates EcSOD expression via a feed-forward mechanism in the vasculature (Fukai et al., 2000, Fukai et al., 2002), EcSOD is sufficient to sustain NO• bioactivity in the vascular wall (Fig. 6).
Redox homeostasis and redox stress in vascular cells
Redox homeostasis is finely controlled by cellular pro-oxidants and antioxidant systems. Under physiological conditions, vascular cells are capable of maintaining cellular pro-oxidants at normal steady-state levels to support various biological processes, including cell proliferation and growth, cell signaling, cell differentiation, and many others (Handy and Loscalzo, 2012, Handy and Loscalzo, 2016). Physiological levels of pro-oxidants (intracellular H2O2 and O2 •− at low nM and pM range, respectively) are beneficial to cellular functions, denoted “oxidative eustress” (Sarsour et al., 2009, Sies, 2021, Sies et al., 2022). Loss of such a state impairs normal vascular cell function. For example, endothelial functions require NOX-mediated ROS signaling. Using EC-specific NOX4 knockout and NOX4 transgenic mice models, Craige and colleagues showed that NOX4-generated H2O2 activated eNOS signaling leading to the promotion of angiogenesis as evidenced by accelerated recovery from hind limb ischemia in vivo; enhanced aortic capillary sprouting ex vivo; and increased tube formation, cell proliferation, and migration in vitro (Craige et al., 2011). Subsequently, two separate studies showed that endothelial NOX4 was required for angiogenesis, likely dependent upon H2O2 production and activation of transforming growth factor β signaling (Chen et al., 2014, Peshavariya et al., 2014). Furthermore, a feed-forward mechanism among NOX4-produced H2O2, NOX2, and the mitochondria-generated ROS cascade was reported to activate vascular endothelial growth factor receptor 2 signaling and, thereby, enhance endothelial angiogenesis (Kim et al., 2017c). Likewise, physiological levels of ROS are also crucial for the biological functions of VSMCs. Cumulative evidence from in vivo and in vitro studies supports that ROS production is required for growth factor-induced arterial VSMC proliferation and migration, with suppression of such ROS flux inhibiting these biological effects (Hsieh et al., 2010, Lu et al., 2018, Sturrock et al., 2006, Wu et al., 2022, Zhang et al., 2018). Taken together, these lines of evidence highlight the importance of redox homeostasis in controlling normal vascular cell functions.
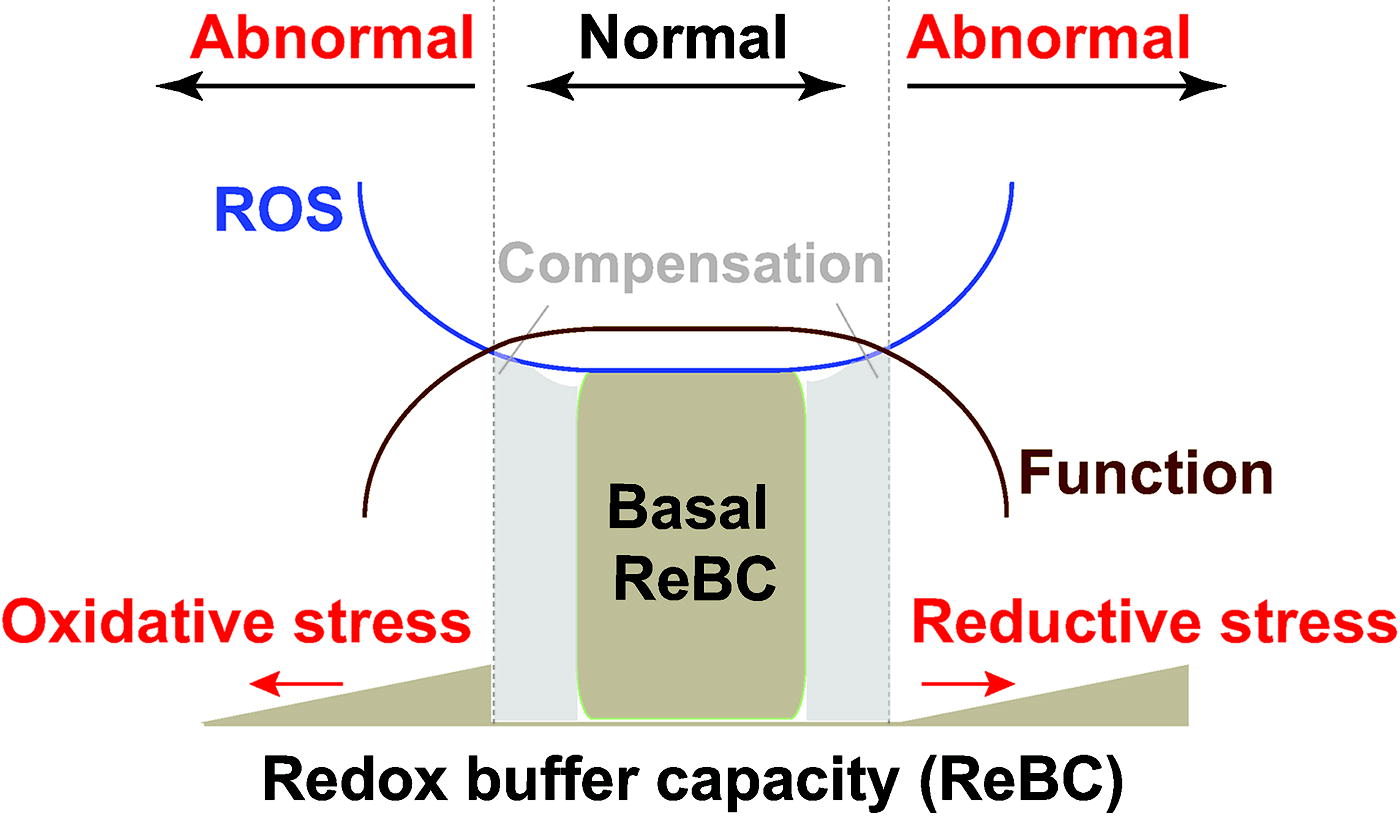
The delicate redox balance between pro-oxidants and antioxidants is often adversely perturbed under pathological conditions, which results in redox stress, a term used to denote oxidative stress and reductive stress (Fig. 7). The concept of “oxidative stress” was first introduced by Paniker and colleagues in 1970 in studies of the GSH/GSSG couple in H2O2-stimulated normal and GR-deficient human erythrocytes (Paniker et al., 1970). We define this term as the imbalance between cellular pro-oxidant levels and antioxidant capacity in favor of the former (Handy and Loscalzo, 2012, Handy and Loscalzo, 2016, Xiao and Loscalzo, 2020), implying that excess in oxidant levels (supraphysiological range) leads to oxidative stress (Fig. 7). By contrast, reductive stress has been underappreciated for a long period of time in the field but has been increasingly recognized of late. This term was first described by Gores and coworkers in a chemical-induced hypoxia model in 1989; the authors proposed that electron carriers became reduced due to limited oxygen availability (e.g., hypoxia) and were reoxidized during reoxygenation (e.g., reperfusion) leading to a burst of ROS generation, termed “reductive stress” (Gores et al., 1989). We recently defined reductive stress as an excess of reducing equivalents [largely NAD(P)H and GSH] that exceeds the capacity of endogenous oxidoreductases (Xiao and Loscalzo, 2020). It should be noted that, under reductive stress, cellular ROS levels can either decrease or paradoxically increase depending upon the mechanisms of action involved in their metabolism or functional consequences, as we reviewed recently (Xiao and Loscalzo, 2020) (Fig. 7).
It is now clear that both oxidative stress and reductive stress are deleterious to vascular cell functions. Oxidative stress has been widely linked to vascular dysfunction and disease as summarized in recent reviews (Chen et al., 2018, Forrester et al., 2018, Galley and Straub, 2017, Tejero et al., 2019, Ushio-Fukai et al., 2021). As with oxidative stress, reductive stress is deleterious to cells and has been implicated in many pathological conditions, including cardiovascular diseases and diabetes mellitus (Handy and Loscalzo, 2016, Lloret et al., 2016, Wu et al., 2016). Specifically, Ali et al (2014) showed that homozygous deletion of GPX1 induced reductive stress as evidenced by increased total GSH, the GSH/GSSG ratio, and dihydroethidium oxidation (indicative of O2 •− production), which resulted in hyperproliferation and increased migration in aortic VSMCs, and abnormal vascular remodeling and plaque formation in the atherosclerotic aorta of ApoE-/- mice. Mechanistically, accumulation of GSH in VSMCs led to S-glutathionylation and inactivation of proto-oncogene tyrosine protein kinase (ROS1) tyrosine phosphatase (SHP2), leading to increased phosphorylation and activity of ROS1. Pharmacological inhibition of ROS1 and deglutathionylation of SHP2 normalized the pathological phenotypes in GPX1 − / − ApoE − / − mice (Ali et al., 2014). A more general example in this regard is ER stress. A relatively oxidizing environment in this organelle is critical for normal structural disulfide formation of membrane-bound and secretory proteins; yet when reductive stress occurs, its oxidizing niche becomes more reduced, leading to inappropriate protein folding and disulfide bond formation, activation of the unfolded protein response, and, as a consequence, ER stress (Maity et al., 2016).
Pro-oxidants regulate cellular metabolism
O2 •− and H2O2 can target directly metabolic enzymes and, thus, regulate cellular metabolism. O2 •− can oxidize the Fe-S centers of metabolic enzymes and inhibit their catalytic activity (Fig. 8). Aconitase, which catalyzes the interconversion of citrate, cis-aconitate, and isocitrate in the TCA cycle, contains a [4Fe-4S]2+ cluster and is a known target of O2 •− (Gardner and Fridovich, 1991, Hausladen and Fridovich, 1994, Patel et al., 1996). O2 •− reacts rapidly with the [4Fe-4S]2+ cluster (estimated rate constant = 106-107 M−1 s−1) yielding an inactive [3Fe-4S]+-containing enzyme, an iron ion (Fe2+), and H2O2 (Castro et al., 2019). Hydrogen peroxide produced by this reaction and generated from other sources is also able to react with the [4Fe-4S]2+ cluster, despite doing so at a slower rate (approximately 102 M−1 s−1) (Castro et al., 2019). What is more important is that the released Fe2+ and H2O2 can readily form the additional oxidizing species hydroxyl radical (HO•) via the Fenton reaction (Xiao and Loscalzo, 2020), triggering a cascade of oxidation and inhibition of Fe-S cluster-containing enzymes. Furthermore, ONOO−, derived from the reaction of O2 •− with NO•, can also oxidize the [4Fe-4S]2+ cluster in aconitase with an estimated rate constant of 1.4 × 105 M−1 s−1 (Castro et al., 1994, Castro et al., 2019, Hausladen and Fridovich, 1994). The metabolic consequences of oxidant-mediated inhibition of aconitase are (1) reduced substrate influx into the TCA cycle; (2) decreased production of TCA cycle metabolites, especially NADH, in mitochondria; (3) consequent inhibited mitochondrial respiration; and, as a result, (4) efflux of accumulated citrate from mitochondria into the cytosol for fatty acid synthesis (Castro et al., 2019), leading to metabolic reprogramming.

Hydrogen peroxide controls metabolic enzymes predominantly via direct modifications of Cys residues (Fig. 8). In human aortic ECs, H2O2 stimulation triggered S-glutathionylation at Cys247 of the glycolytic enzyme glyceraldehyde-3-phosphate (G3P) dehydrogenase (GAPDH) (Mustafa Rizvi et al., 2021). As a result, GAPDH activity is inhibited and translocated into the nucleus where it forms a complex with histone deacetylase Sirtuin 1 and inhibits its deacetylase activity leading to activation of p53 signaling and apoptosis (Mustafa Rizvi et al., 2021). Using a purified GAPDH enzyme, Hyslop and Chaney recently showed that H2O2 oxidizes GAPDH at three Cys residues (Cys152, 156, and 247) forming sulfenic (SOH) and sulfinic (SO2H) derivatives and causing conformational change that leads to catalytic inactivation (Hyslop and Chaney, 2022). Cys residues of the glycolytic enzyme PKM2 (Cys358) and mitochondrial enzyme PDK2 (Cys45 and 392) were also targeted by H2O2 for oxidation leading to inhibition of their activities and adaptive metabolic switching (Anastasiou et al., 2011, Hurd et al., 2012). Furthermore, in rat pulmonary arterial SMCs cultured under hypoxia, Guo et al. (2016) showed that H2O2-mediated phosphorylation and inactivation of PKM2 promoted cell proliferation and increased the GSH/GSSG ratio (indicative of enhanced PPP activity).
Modulation of cellular metabolism can also be mediated by ROS-dependent signaling pathways, such as HIF1α, AMPK, and nuclear factor erythroid 2 related factor 2 (NRF2) (Fig. 8). For example, under disturbed flow, H2O2 produced by NOX4 activated HIF1α signaling and upregulated its downstream target genes GLUT1, HK2, and PDK1 leading to a metabolic switch from mitochondrial respiration to glycolysis and endothelial inflammation in human aortic ECs in vitro and in the atherosclerosis-prone aortic arch of pigs in vivo (Wu et al., 2017). Activation of HIF1α, protein kinase C, and phosphoinositide 3-kinase signaling pathways was found to mediate H2O2-stimulated glucose uptake and glycolytic activity in HUVECs under hypoxia (Paik et al., 2017). Furthermore, hypoxia-stimulated rat pulmonary arterial SMCs produced high H2O2 levels leading to activation of AMPK signaling, a primary energy sensor and a master regulator of metabolic homeostasis (Mungai et al., 2011). As expected, AMPK activation enhanced glycolysis as evidenced by HIF1α activation, upregulation of glycolytic genes (GLUT1, HK2, LDHA, and PFKFB3), and elevation of lactate secretion in oscillatory flow-primed HUVECs in vitro and mouse aorta arch in vivo (Yang et al., 2018). These metabolic changes were abrogated when the AMPK catalytic subunit α1 PRKAA1 was silenced in vitro and depleted in mice in vivo, supporting the key roles of AMPK signaling in regulating the glycolytic phenotype of ECs (Yang et al., 2018).
In addition, ROS-mediated activation of NRF2 signaling also regulates cellular metabolism in vascular cells (Fig. 8). Upon oxidative insults, NRF2 signaling is activated by oxidation of Cys residues at Kelch-like ECH-associated protein 1 (KEAP1), with consequent loss of sequestration of the NRF2 protein as a result of dissociation of the NRF2-KEAP1 complex. Uncomplexed NRF2 then translocates into the nucleus where it binds to antioxidant response elements in its transcriptional target gene promoter region and, thus, activates a vast array of cytoprotective genes, including antioxidant proteins and metabolic enzymes (Cheng et al., 2011, Muri and Kopf, 2021). NRF2 activation enhanced cystine uptake and GSH biosynthesis by upregulation of cystine-glutamate transporter xCT and γ-glutamylcysteine lyase (GCL) catalytic and modifier subunits in human arterial ECs and SMCs (Cheng et al., 2011). In response to oxidized phospholipids, NRF2 activation upregulated the key glycolytic enzyme PFKFB3 expression to enhance cellular glycolysis and cell proliferation in HUVECs and did so through direct and indirect regulation at the transcriptional and epigenetic levels (Kuosmanen et al., 2018). Taken together, these data demonstrate that pro-oxidants regulate metabolic enzyme activity and cellular metabolism directly by oxidative modifications and indirectly by modulation of other signaling molecules.
Redox couples connect with cellular metabolism
As mentioned above and highlighted at length in our recent work (Xiao and Loscalzo, 2020, Xiao et al., 2018), cellular redox couples NAD(P)+/NAD(P)H and GSH/GSSG are responsible for the majority of electron transfers, and together with antioxidant enzymes are essential for the maintenance of cellular redox homeostasis. Of note, these redox couples are also critical connectors between cellular redox state maintenance and cellular metabolism. Specifically, NAD+ serves as an electron sink in support of glycolysis; NADH generated from glycolysis and the TCA cycle provides electrons for mitochondrial oxide phosphorylation (OXPHOS) and ATP production; NADP+ supports the PPP shunt to generate the NADPH that is indispensable for redox homeostasis and reductive biosynthesis of macromolecules (e.g., nucleotides, amino acids, and lipids); and GSH connects amino acid metabolism with cellular redox status (Xiao and Loscalzo, 2020, Xiao et al., 2018).
As a cofactor of many metabolic enzymes, NAD+ can be generated by the de novo pathway, the Preiss–Handler pathway, the salvage pathway, and the recycling of NADH (Xiao et al., 2018). The interconversion of NAD+ and NADH is mediated by glycolytic enzymes in the cytosol and by the TCA cycle enzymes in mitochondria (Fig. 9). In the cytosol, GAPDH and LDH catalyze the reversible conversion of NAD+ to NADH in glycolysis (Lunt and Vander Heiden, 2011, Xiao and Loscalzo, 2020, Xiao et al., 2018). Alcohol dehydrogenases and aldehyde dehydrogenases are two additional sources of cytosolic NADH (Cederbaum, 2012, Xiao and Loscalzo, 2020). Mitochondrial NAD(H) is generated by the pyruvate dehydrogenase (PDH) complex and the TCA cycle enzymes, isocitrate dehydrogenase-3 (IDH3), α-ketoglutarate dehydrogenase, and malate dehydrogenase 2 (MDH2) (Xiao and Loscalzo, 2020, Xiao et al., 2018). As NAD+-linked enzymes, malic enzyme and glutamate dehydrogenases also contribute to the mitochondrial NADH pool.
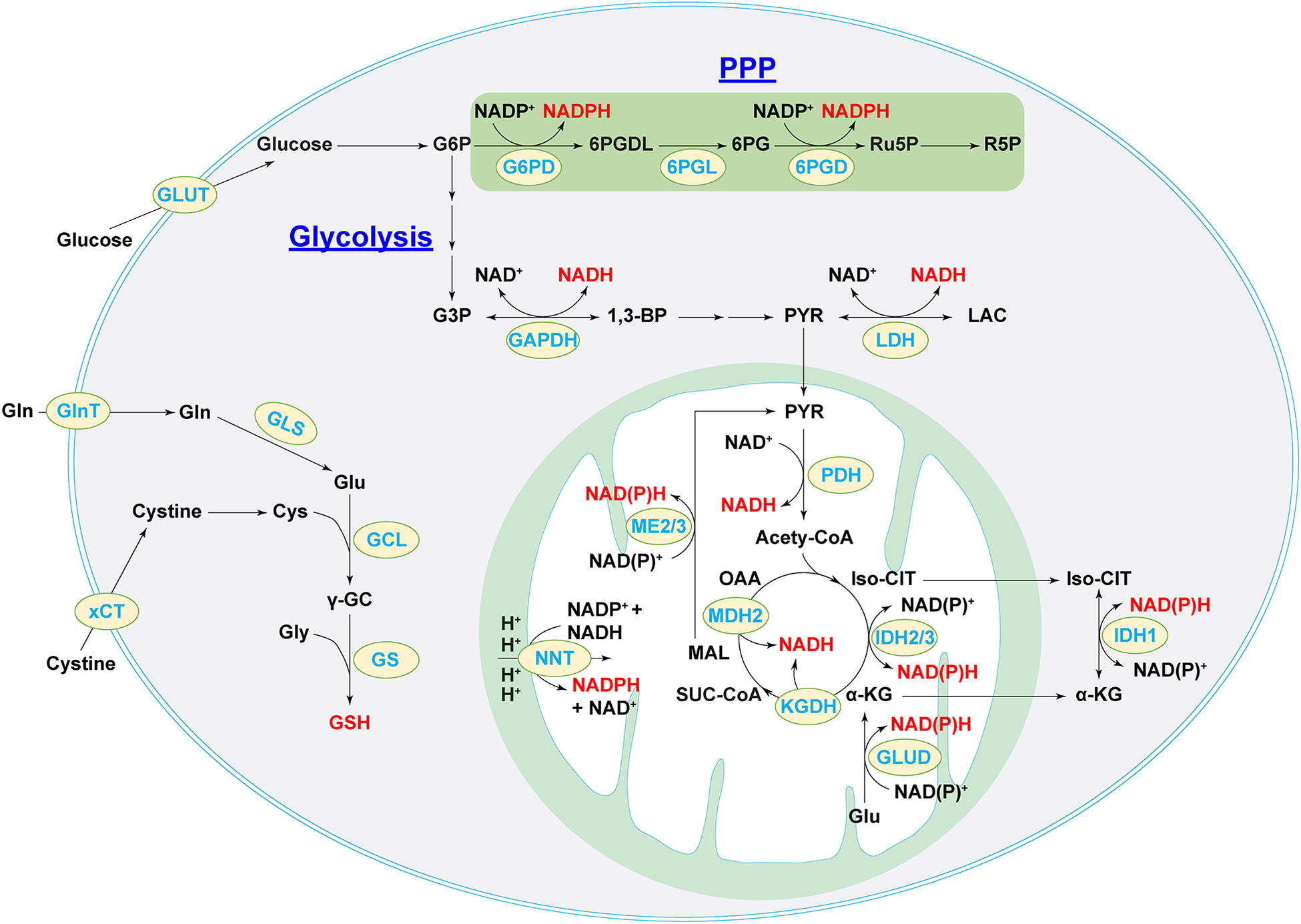
Formation of the phosphorylated product of NAD+, NADP+, is catalyzed by NAD+ kinases (Xiao and Loscalzo, 2020). Cytosolic and mitochondrial pools of NADP+ and NADPH are coordinated by many metabolic pathways and enzymes (Fig. 9). The PPP shunt enzymes G6PD and 6PGD are the primary contributors of cytosolic NADPH. Cytosolic and mitochondrial isozymes in glucose metabolism (IDH1/2 and ME1/3) and folate metabolism [methylene-tetrahydrofolate dehydrogenases and 10-formyl-tetrahydrofolate dehydrogenase] also significantly contribute to their corresponding pools of NADPH levels (Xiao and Loscalzo, 2020). Of particular interest, nicotinamide nucleotide transhydrogenase (NNT), a mitochondrial inner membrane enzyme, is another key source of mitochondrial NADPH; its function is driven by the proton gradient across the mitochondrial inner membrane to reduce NADP+ into NADPH via the reaction, NADH + NADP+ ←→ NAD+ + NADPH (Houtkooper et al., 2010, Rydstrom, 2006, Xiao and Loscalzo, 2020).
As with pro-oxidants and antioxidant enzymes, intracellular NAD(H) and NADP(H) pools are also highly compartmentalized, in part, due to specific localizations of their biosynthetic enzymes and membrane impermeability to these dinucleotides (Xiao et al., 2018, Xiao and Loscalzo, 2020). To circumvent this limitation, metabolic enzyme-mediated shuttling mechanisms between different compartments are used to coordinate NAD(P)H pools efficiently so that cells can maintain the overall cellular redox environment and redox-dependent functions. For example, two NADH shuttles, the malate–aspartate shuttle and the glycerol-3-phosphate shuttle, are responsible for the intercommunication between the cytosolic and mitochondrial pools of NADH (Houtkooper et al., 2010, McKenna et al., 2006, Xiao et al., 2018, Xiao and Loscalzo, 2020). The first shuttle requires cytosolic enzyme MDH1 and its mitochondrial isoform MDH2 as well as two transporters, α-ketoglutarate (α-KG)/malate antiporter (encoded by SLC25A11 gene) and aspartate-glutamate antiporter (encoded by SLC25A13 gene); by contrast, the second NADH shuttle is irreversible and requires only one enzyme, glycerol-3-phosphate dehydrogenase (Xiao et al., 2018, Xiao and Loscalzo, 2020). As with NADH, the exchange of cytosolic and mitochondrial NADPH pools is carried out by the isocitrate-α-KG shuttle through IDH1/2 isozymes and two carrier proteins, citrate carrier protein (encoded by SLC25A1 gene) and α-KG/malate antiporter, the same carriers as in the NADH malate–aspartate shuttle (Xiao et al., 2018, Xiao and Loscalzo, 2020,). These shuttle mechanisms closely tie cellular redox couples with energy metabolism to maintain homeostasis under physiological and pathological conditions.
GSH, the most abundant nonenzymatic antioxidant in vascular cells (at mM range), is synthesized from L-glutamate, L-Cys, and glycine via two ATP-dependent reactions catalyzed by the rate-limiting enzyme GCL and GSH synthetase (Lu, 2013, Schafer and Buettner, 2001, Xiao and Loscalzo, 2020) (Fig. 9). Thus, the availability of its constituent amino acids, especially Cys and Glu, and the enzymatic activity of GCL determine cellular GSH levels. Cellular Cys levels are controlled, in part, by the uptake of extracellular cystine via its transporter xCT (encoded by SLC7A11). Activation of NRF2 signaling by oxidative stress is known to increase cellular GSH synthesis as a counteractive response by upregulation of xCT and GCL expression and enhancement of cystine uptake in vascular cells (Cheng et al., 2011). In addition, the TCA cycle metabolite fumarate was also found to regulate GSH synthesis. Deletion of fumarate hydratase led to fumarate accumulation and formation of succinic GSH, a covalent adduct of fumarate and GSH, which triggered oxidative stress, a compensatory rise in GSH levels by stimulation of cystine transporter xCT expression and cystine uptake, and glutaminolysis-dependent GSH biosynthesis (Zheng et al., 2015). Collectively, redox couples NAD(H), NADP(H), and GSH are the principal links of cellular redox homeostasis and energy metabolism.
Metabolic Responses to Redox Stress in Vascular Cells
Metabolic response to reductive stress
As discussed in the next section, redox couples NAD(P)H and GSH link cellular redox homeostasis with cellular metabolism. Excessive accumulation of these molecules leads to reductive stress, which triggers cellular metabolic pathways to respond, adapt, and eventually function normally or with impairment.
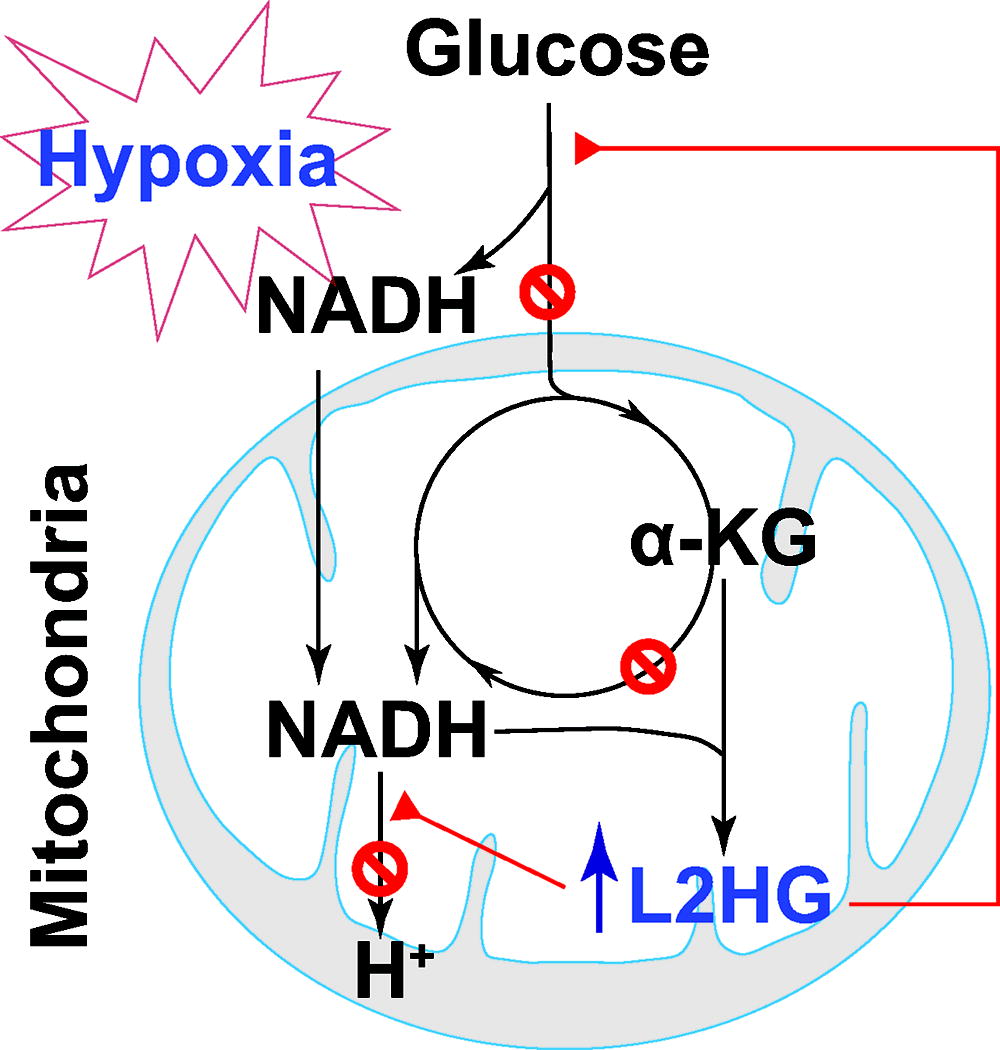
Produced during glycolysis in the cytosol, NADH is oxidized at mitochondrial complex I (NADH dehydrogenase). Under hypoxic conditions, glycolytic activity is enhanced in conjunction with limited mitochondrial respiration, which together lead to an increase in NADH and the consequent reductive stress. Accumulation of NADH by hypoxia provides more electrons for one-electron reduction of oxygen to O2 •− (Clanton, 2007). Indeed, elevated levels in the cytosolic NADH/NAD+ ratio and mitochondrial NAD(P)H were accompanied by increased mitochondrial O2 •− production likely by complex I in hypoxia-challenged bovine coronary artery SMCs (Gao and Wolin, 2008). Similarly, in primary human vascular cells, we demonstrated that hypoxia increased the cellular NADH/NAD+ ratio and mitochondrial O2 •− generation, indicative of reductive stress (Oldham et al., 2015). Mechanistically, we and others showed that L-2-hydroxyglutarate (L2HG), but not its enantiomer D2HG, a reduced metabolite of α-KG, selectively accumulated as a fundamental mechanism in response to hypoxia in various normal and tumor cells, including human pulmonary arterial ECs and SMCs (Intlekofer et al., 2015, Oldham et al., 2015). Hypoxia-induced L2HG accumulation was dependent on MDH1 and MDH2 activity since knockdown of either one or both enzyme(s) significantly abrogated such change under hypoxia (Oldham et al., 2015). Furthermore, increasing cellular L2HG levels by silencing L2HG dehydrogenase (L2HGDH; the only known enzyme that oxidizes L2HG back to α-KG) potentiated hypoxia-induced increase in the NADH/NAD+ ratio, but reduced mitochondrial oxygen consumption (indicative of mitochondrial respiration) and lactate production (indicative of glycolytic activity), all of which were recapitulated by exogenous addition of cell-permeable L2HG derivative. Therefore, these results support that L2HG accumulation inhibits both mitochondrial respiration and glycolysis to blunt their production of NADH and to ameliorate its attendant reductive stress (Oldham et al., 2015) (Fig. 10).
To study the in vivo and disease relevance of these in vitro observations, we recently used a L2HGDH-depleted mouse model to investigate how L2HG accumulation regulates reductive stress and energy metabolism in cardiac ischemia–reperfusion (IR) injury (Fig. 11). We first confirmed that L2HG levels are higher in the hearts of L2HGDH heterozygous (L2HGDH +/-) and homozygous (L2HGDH -/-) mice compared with wild-type littermates (L2HGDH +/+), supporting the view that genetic deletion of L2HGDH can sufficiently induce L2HG accumulation (He et al., 2022). Consistent with our in vitro findings, ischemia significantly increased myocardial L2HG levels in L2HGDH +/+ animals, and as expected, such increases were markedly exaggerated in L2HGDH +/- and L2HGDH -/- lines in a gene-dose-dependent manner (He et al., 2022). Interestingly, compared with wild-type mice under basal conditions, hearts from L2HGDH +/- and L2HGDH -/- mice had higher levels of NADH, NADPH, and GSH; comparable or lower levels of NAD+, NADP+, and GSSG; consequently higher ratios of each redox couple; and lower levels of H2O2, lipid peroxidation, and protein oxidation, indicative of reductive stress (He et al., 2022). When challenged by IR, severe oxidative injury was found in wild-type hearts as evidenced by increased levels of H2O2, lipid and protein oxidation, NAD+, NADP+, and GSSG; and decreased levels of NADH, NADPH, and GSH as well as the ratios, NADH/NAD+, NADPH/NADP+, and GSH/GSSG. All of these redox changes were gene-dose dependently attenuated in the hearts of L2HGDH +/− and L2HGDH −/− mice, and as a result, myocardial injury in these animals was less severe and energy states (ATP, inorganic phosphate, and free phosphocreatine) improved, supporting the view that L2HGDH deletion-induced L2HG accumulation protects the myocardium from IR injury (He et al., 2022). Mechanistically, L2HGDH depletion induced a glucose metabolic switch in myocardial tissue by decreasing flux into glycolysis and increasing flux into the PPP, leading to the accumulation of reducing equivalents NAD(P)H and GSH and protection from myocardial injury (He et al., 2022). Thus, L2HG accumulation by L2HGDH deletion shifts glucose metabolism to enhance PPP activity and reduce equivalent production, which together preserve and protect the hearts from IR injury (Fig. 11).
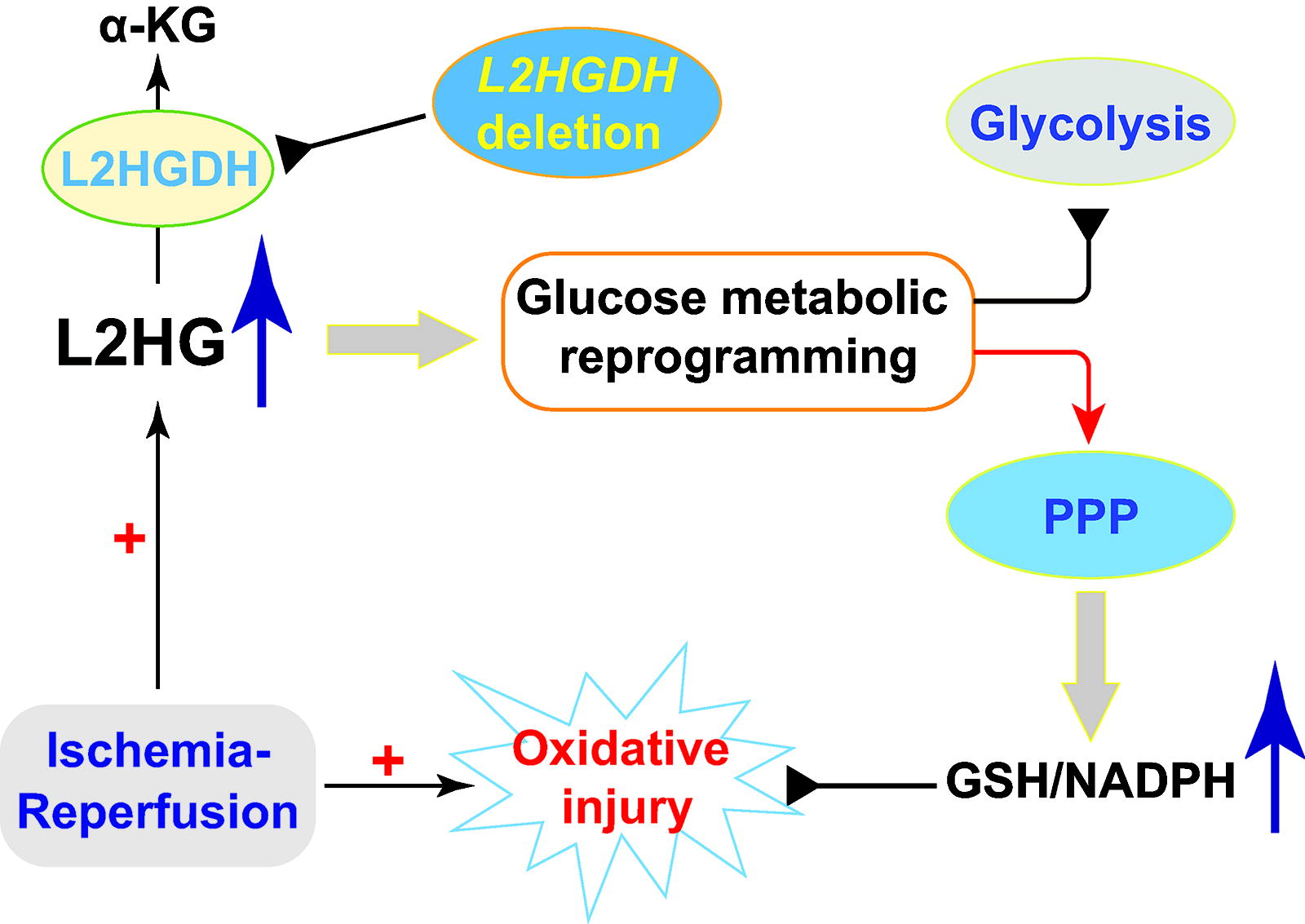
As mentioned above, NNT couples mitochondrial proton influx with the reduction of NADP+ to NADPH at the mitochondrial inner membrane, and, thus, its dysfunction could also lead to reductive stress and metabolic alterations. Using a spontaneous NNT loss-of-function mutant C57BL/6J mouse model (Toye et al., 2005), Nickel and coworkers showed that these mice were resistant to transverse aortic constriction-induced heart failure compared with NNT wild-type C57BL/6N mice (Nickel et al., 2015). This outcome was believed to occur owing to NNT’s operating through its reverse enzymatic mode in wild-type animals, which promoted NADH production at the expense of NADPH and its antioxidant capacity under pathological pressure overload (Nickel et al., 2015). As a result, the increased NADH production fueled mitochondrial OXPHOS, which, combined with reduced NADPH production, resulted in increased mitochondrial H2O2 production, oxidative injury, and cardiac dysfunction (Nickel et al., 2015). By contrast, under angiotensin II stimulation, aortic ECs isolated from C57BL/6J mice exhibited a lower oxygen consumption rate and GPX activity and higher O2 •− production than ECs of wild-type mice (Leskov et al., 2017). Thus, loss of NNT function impaired mitochondrial respiration in ECs in vitro, which correlated with resistance to endothelium-dependent vasodilation ex vivo and exacerbation of angiotensin II-induced hypertension in vivo (Leskov et al., 2017). Similarly, in human aortic ECs, NNT silencing exacerbated angiotensin II-induced decreases in NADPH/NADP+, eNOS activity, and NO• production, and increases in NAD+/NADH and ROS levels (O2 •− and H2O2), which were accompanied with reduced mitochondrial oxygen consumption rates and ATP production, and depolarization of the mitochondrial membrane, all indicating that mitochondrial function and activity are compromised by NNT silencing (Rao et al., 2020). Furthermore, increasing evidence strongly supports the view that reductive stress observed in C57BL/6J mice correlated with systemic metabolic syndromes, such as glucose intolerance, reduced insulin secretion and energy expenditure, and increased susceptibility to high-fat-diet-induced obesity (Fisher-Wellman et al., 2015, Freeman et al., 2006a, Freeman et al., 2006b, Nicholson et al., 2010, Roat et al., 2014). Taken together, these observations indicate that NNT is a key enzyme bridging cellular metabolism and reductive stress.
The NADH malate–aspartate shuttle is a primary mechanism for maintaining high cytosolic NAD+ levels and high mitochondrial NADH levels. Disruption of this exchange mechanism perturbs NADH/NAD+ in both compartments and, thus, alters cellular redox homeostasis and energy metabolism. In support of this concept, pharmacological inhibition of this shuttle with aminooxyacetic acid (a potent inhibitor of aspartate aminotransferase) augmented cytosolic NADH/NAD+ (indicated by elevated lactate/pyruvate) resulting in a more reducing environment in the cytosolic compartment in primary porcine aortic SMCs (Barron et al., 1998, Barron et al., 2000). Notably, such treatment also attenuated glucose oxidation and oxygen consumption in conjunction with increased glycolytic activity and lactate production, indicating a metabolic shift toward glycolysis (Barron et al., 1998, Barron et al., 2000). These results suggest that blockade of the malate–aspartate shuttle causes cytosolic retention of NADH and reductive stress, which decrease NADH availability in mitochondria and possibly increase NAD+ levels in the cytosol, thereby leading to inhibition of NADH-dependent mitochondrial respiration and enhancement of NAD+-dependent glycolysis.
Similar effects of this shuttle were also observed in vivo. Cardiac-specific deletion of the mitochondrial complex I subunit NDUFS4 increased NADH levels and NADH/NAD+ in the myocardium, which were associated with decreased H2O2 and O2 •− levels in cardiomyocytes and accelerated development of heart failure phenotypes (Karamanlidis et al., 2013, Lee et al., 2016), supporting the view that complex I dysfunction leads to reductive stress in the failing heart. Mechanistically, NADH accumulation in mitochondria repressed NAD+-dependent Sirtuin 3 deacetylase activity leading to hyperacetylation and inactivation of the malate–aspartate shuttle proteins MDH2, α-KG/malate carrier, and aspartate aminotransferase 2 (Karamanlidis et al., 2013, Lee et al., 2016). As a result, cardiac mitochondrial respiration and energy production were impaired. Therefore, these lines of evidence support the views that the malate–aspartate shuttle is essential for the coordination of cytosolic and mitochondrial NADH pools and that disruption of this shuttling mechanism leads to reductive stress and metabolic dysfunction.
In the PPP, G6PD is a predominant source of cytosolic NADPH, which serves as a cofactor for peroxide neutralization and is also a substrate for NOX-mediated O2 •− and H2O2 production. High G6PD activity produces high levels of NADPH, which could promote ROS production and reductive stress. Thus, loss of G6PD activity is expected to be beneficial in certain conditions. Indeed, male hemizygote G6PD mutant mice, despite loss of G6PD activity (10–20% of wild-type mice) and reduced NADPH levels in the aorta, showed resistance to angiotensin II-induced O2 •− production, thickening of the aortic medial layer, and an increase in blood pressure, indicating that G6PD deficiency protects against angiotensin-induced hypertension and aortic SMC hypertrophy (Matsui et al., 2005). In a later study, the same group reported that G6PD deficiency also decreased vascular O2 •− levels, serum cholesterol levels, and, importantly, atherosclerotic lesion abundance in high-fat-diet-fed ApoE −/− mice (Matsui et al., 2006). Similarly, pharmacological blockade of G6PD activity and NADPH-dependent NOX2 activity significantly decreased cellular O2 •− levels and increased acetylcholine-induced vasorelaxation in the aortae of diabetic rats (Serpillon et al., 2009).
Inhibition of G6PD activity was also found to abrogate interleukin 1β- and high glucose (22 mM)-induced activation of the PPP shunt, production of H2O2 by NOXs, inflammation, and/or vasoconstriction in human aortic SMCs and rat mesenteric vessels (Peiró et al., 2016). Furthermore, G6PD activity and reductive stress were shown to contribute to the development of cardiomyopathy. Mice with cardiac-specific overexpression of a mutated human αB-crystallin (R120G mutant) developed protein aggregation cardiomyopathy and exhibited higher G6PD activity and reductive stress in the heart (Rajasekaran et al., 2007). Notably, these pathological alterations were significantly normalized by replacing the wild-type G6PD with a hypomorphic G6PD mutant in the R120G mice (Rajasekaran et al., 2007). Mechanistically, the reductive stress in these mutants was induced by activation of the NRF2 signaling pathway, which transcriptionally upregulated the expression of several key antioxidant enzymes and GSH biosynthetic enzymes (e.g., GCL catalytic and modifier subunits) leading to enhanced antioxidant capacity and elevated GSH levels. Recently, Varghese et al (2021) demonstrated that G6PD-deficient mice developed spontaneous pulmonary hypertension characterized by elevated right ventricular systolic pressure, right heart hypertrophy, increased pulmonary vascular remodeling, and pulmonary arterial SMC hyperproliferation. Interestingly, profound and systematic metabolic reprogramming occurred in these animals to support such hemodynamic and pathological alterations as well as the hyperproliferative demands. Specifically, despite the expected inhibition of the oxidative phase activity of the PPP shunt and the concurrent increase in H2O2 production by G6PD deficiency, glucose uptake, and glycolytic activity were elevated in pulmonary arterial SMCs and lung tissues (Varghese et al., 2021). Because glycolytic enzyme phosphoglycerate mutase 1 activity, which catalyzes the conversion of 3-phophoglycerate (3PG) to 2-phosphoglycerate, was inhibited in G6PD-depleted lungs and VSMCs, glycolytic metabolites before this step were significantly increased (Varghese et al., 2021). As a consequence, G3P fed the nonoxidative branch of the PPP, and G6P was routed into the inositol pathway due to myo-inositol oxygenase upregulation as an alternate to bypass the defective PPP shunt. Furthermore, 3PG accumulation supported the increased biosynthesis of fatty acids and amino acids (Cys and serine); and glutaminolysis was also enhanced to replenish α-KG in the TCA cycle in pulmonary arterial VSMCs of diseased mice (Varghese et al., 2021). These metabolic alterations together provide sufficient nucleotides, nutrients, and energy sources for the hyperproliferative demands of vascular cells in this disease (Varghese et al., 2021). In short, G6PD deficiency and the resulting NADPH depletion perturb cellular redox homeostasis leading to redox stress and cellular metabolic reprogramming.
Inflammation and vascular redox metabolism
Vascular ECs play a critical role in regulating local and systemic inflammation through modulation of adhesion molecule expression on their cell surface and secretion of chemokines, thereby regulating the interaction between the blood vessel and circulating inflammatory cells (McEver et al., 1995). NF-κB regulates the expression of a large number of proinflammatory genes, including the key adhesion molecules, ICAM1 and VCAM1 (Collins et al., 1995).
Oxidative stress has been noted to modulate the major proinflammatory transcriptional factors in ECs, including NF-κB and AT-1 (Sen and Packer, 1996). ROS have been found to play key roles in regulating adhesion molecule expression by ECs (Takano et al., 2002). In an animal model of LPS-induced lung inflammation, endothelial NOX2 activation mediated increased adhesion molecule expression on ECs and was sufficient to induce proinflammatory endothelial phenotypic changes (Orndorff et al., 2014). Antioxidant SOD overexpression in ECs was able to counter TNFα-mediated upregulation of adhesion molecules (Chen et al., 2003).
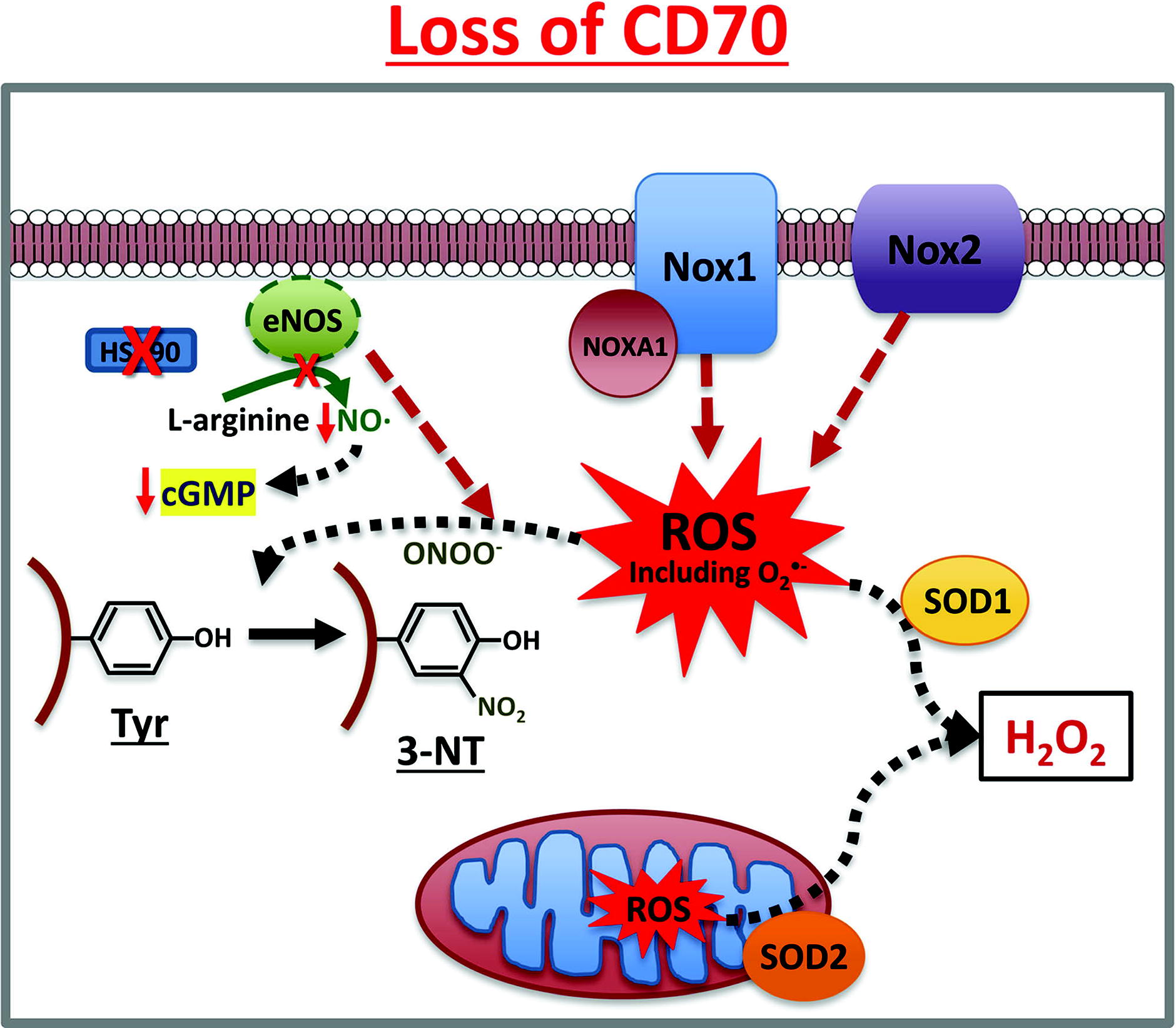
Cytokines, especially TNFα, have been shown to stimulate NOX1, 2, and 4 in vascular cells (Anilkumar et al., 2008 (Basuroy et al., 2009). We also observed that the inflammatory stimuli, TNFα and endotoxin (LPS), increased NOX4-protein expression, which correlated with metabolic and inflammatory stresses in human arterial ECs (Xiao et al., 2021). Loss of endothelial CD70, a TNFα superfamily member, increased NOX1-mediated H2O2 and O2 •− production, and decreased eNOS activity and NO• levels, leading to inhibition of cell migration and wound closure in normal human arterial ECs (Pandey et al., 2022) (Fig. 12). Beyond the ECs, NOXs in fibroblasts and other cell types in the adventitial layer promote inflammation and its consequences for various vascular pathologies, as highlighted in the review by Meijles and Pagano (Meijles and Pagano, 2016).
Concluding Remarks and Future Directions
Cellular redox state influences the phenotypes and biological functions of vascular cells. Physiological levels of ROS/RNS are essential for energy metabolism, angiogenesis, vasoconstriction and vasodilation, cell proliferation/growth, migration, and differentiation. By contrast, redox stress caused by imbalanced cellular pro-oxidants and antioxidant systems encompasses oxidative stress and reductive stress, both of which are deleterious to the vasculature and contribute to the development of vascular diseases. Therefore, targeting redox stress to reestablish a homeostatic redox state is a promising global therapeutic strategy for the treatment of vascular disorders. However, despite animal studies having unambiguously demonstrated the efficacy of such treatment strategies, clinical trials have not yet reached conclusive and favorable outcomes (Forrester et al., 2018). Thus, a better understanding of the complexity of the cellular redox niche landscape and redox stress, as well as their influences on cell functions particularly energy metabolism, offers the opportunity to develop effective antioxidative and antireductive interventional approaches to maintain/reestablish cellular redox balance.
ROS and RNS are broad generic terms commonly used and misused. Each molecule in ROS/RNS has distinct sources, metabolic fates, organelle/compartmental locations, biochemical properties, reactivity, stability, redox potentials, membrane permeability, biological targets, etc. For example, O2 •− often targets the Fe-S cluster-containing molecules proximate to the source, while H2O2 has longer diffusion distances, permitting oxidation of Cys residues in proteins distant from the source. Hydrogen peroxide is a normal signaling molecule under certain circumstances, while HO• is very toxic. Furthermore, ROS and RNS have dynamic metabolic fates, including initiation, propagation, and termination (Loscalzo, 2016). O2 •− is first converted into H2O2 by SODs, which is then either reduced to water by peroxidases or generates more oxidizing HO• via the Fenton reaction. For lipid oxidation, a cascade of oxidation steps occurs, including the initial generation of lipid radicals (L• and LO•), then lipid peroxyl radical, and finally end-product lipid oxidants and lipid dimers. Thus, we need to avoid using generic terms such as ROS/RNS, but, rather, specify the precise reactive species whenever possible. We also need to keep in mind that oxidation reflects a dynamic cascade of reactions and not a single snapshot event.
The distribution of pro-oxidants and antioxidants has compartmental heterogeneity in a cell. A relatively oxidizing environment in the ER is essential for appropriate protein folding and maturation, both structurally and functionally; by contrast, a reducing niche is required for the nucleus to minimize oxidative DNA damage. One determinant of such subcellular heterogeneity is the delicate interaction between pro-oxidants and antioxidants within each spatial location. Pro-oxidants from specific generating sources are countered by unique antioxidant enzymes in conjunction with their corresponding cofactors. For example, in mitochondria, respiratory chain-derived O2 •− is metabolized by specific mitochondrial antioxidant enzymes SOD2, GPX4, PRX3/5, TRX2, and TR2 using mitochondrial pools of GSH and NAD(P)H. To coordinate compartmental pools of redox couples, shuttle mechanisms are in place.
In addition to their role in the redox network, cellular redox systems also widely connect with many other biological processes, particularly cellular metabolism. The regulation of O2 •− and H2O2 on cellular metabolic activity could be executed directly by posttranslational modifications of metabolic enzymes and indirectly by regulation of other signaling molecules. In turn, cellular metabolism provides essential electron donors and acceptors for redox reactions. Such interactions are primarily mediated by three redox couples NAD(H), NADP(H), and GSH/GSSG. A delicate balance between the reduced and oxidized forms within each redox couple is a prerequisite for support of cellular redox homeostasis and energy metabolism. When that balance is lost, for example, an excess in cellular NAD(P)H and GSH levels leads to reductive stress, metabolic stress, and vascular dysfunctions.
As an emerging concept, reductive stress complicates, but expands, our knowledge of the cellular redox state. Traditionally, a reducing environment is thought to be beneficial for cell function and biological processes since an oxidizing niche can lead to oxidative damage to macromolecules. Reductive stress highlights the fact that excess reducing of equivalents [NAD(P)H and GSH] is also detrimental to cells, and exerts adverse effects through many mechanisms, including disruption of normal signaling roles of pro-oxidants, promotion of (adverse) ROS generation, perturbation of cell metabolism, and inhibition of protein disulfide formation. Thus, vascular cells must maintain a balanced redox environment to avoid both the excesses of oxidative and reductive damage.
To minimize the adverse effects of redox stress, vascular cells respond by reprogramming their metabolic fates to reestablish cellular redox homeostasis. Current knowledge about the underlying mechanisms of reductive stress and its biological consequences as well as how cellular metabolism responds to reductive stress remains limited, particularly in vascular cells. For example, although it is clear that NADH accumulation under hypoxia induces reductive stress, it is uncertain as to (1) how NADH accumulates under hypoxia beside inhibition of its oxidation by mitochondrial respiration; (2) whether or not NADH accumulation is compartment-specific; or (3) whether and how NADH accumulation affects the metabolic fates of glucose, lipids, and glutamine. The answers to these questions remain largely open and require future experimental efforts.
In addition, we and others have shown that accumulation of L2HG is a fundamental response to hypoxia-induced reductive stress; and that L2HG accumulation reprogrammed glucose metabolism: inhibition of glycolysis and mitochondrial respiration, and enhancement of the PPP. However, it remains unclear as to how L2HG mediates such metabolic switching in vascular cells. Previous reports and our unpublished data demonstrate that L2HG can inhibit prolyl hydroxylase domain-containing protein 2 and, thereby, activate HIF1α signaling (Burr et al., 2016), which is known to reprogram cellular metabolism under hypoxia. Whether HIF1α signaling mediates the L2HG-induced metabolic changes in vascular cells we observed is still unknown. Furthermore, in addition to PHD family enzymes, studies in cancer cells showed that L2HG can also inhibit the activity and functions of other α-KG-dependent dioxygenases, such as Jumonji histone lysine demethylases and ten-11 translocation enzymes (Intlekofer et al., 2015, Shim et al., 2014). Whether or not this holds true in normal vascular cells under hypoxia is an open question.
Little is known as to how GSH-induced reductive stress affects vascular cell metabolism. We herein summarized limited information on how reductive stress promotes GSH biosynthesis. GSH is known to modulate cellular metabolism through protein S-glutathionylation under oxidative stress, where GSH is oxidized to GSSG and the latter then reacts with cysteinyl residues (-SH) on proteins to form protein mixed disulfides (Mailloux et al., 2014). For example, glycolytic enzymes (e.g., enolase, aldolase, phosphoglycerate kinase, and triose phosphate isomerase), TCA cycle enzymes (i.e., aconitase, IDH3, and succinyl-CoA transferase), and ATP synthase all have been shown to be S-glutathionylated, which results in activation or inactivation of their enzymatic activities, thus modulating cellular energetics (Fratelli et al., 2002, Garcia et al., 2010, Han et al., 2005, Kil and Park, 2005). Whether S-glutathionylation of metabolic enzymes can occur under reductive stress (high GSH/GSSG) and how this modification influences metabolic activities in vascular cells deserve future investigation.
Footnotes
Acknowledgment
The authors thank Stephanie C. Tribuna for her expert technical assistance.
Authors’ Contribution
W.X.: writing—original draft (lead) and writing—review and editing (equal); L.L.: writing—original draft (equal) and writing—review and editing (equal); J.L.: conceptualization (lead); funding acquisition (lead); supervision (lead); writing—original draft (equal); and writing—review and editing (equal).
Author Disclosure Statement
The authors have nothing to disclose, and there are no conflicts of interest for this article.
Funding Information
This work was supported, in part, by National Institutes of Health (NIH) grants R01 HL155107, R01 HL155096, R01 HL166137, U01 HG007691; by AHA grants 957729, and AHA24Merit 11854447; and by an EU Horizon Health 2021 grant 101057619 to JL; and by an Alan Lerner Research Award and NIH grant K08 HL165047 to LYL.