
Abstract
Background:
Resistance to standard therapeutic methods, including chemotherapy, immunotherapy, and targeted therapy, remains a critical challenge in effective cancer treatment. Redox homeostasis modification has emerged as a promising approach to address medication resistance.
Objective:
This review aims to explore the mechanisms of redox alterations and signaling pathways contributing to treatment resistance in cancer.
Methods:
In this study, a comprehensive review of the molecular mechanisms underlying drug resistance governed by redox signaling was conducted. Emphasis was placed on understanding how tumor cells manage increased reactive oxygen species (ROS) levels through upregulated antioxidant systems, enabling resistance across multiple therapeutic pathways.
Results:
Key mechanisms identified include alterations in drug efflux, target modifications, metabolic changes, enhanced DNA damage repair, stemness preservation, and tumor microenvironment remodeling. These pathways collectively facilitate tumor cells' adaptive response and resistance to various cancer treatments.
Conclusion:
Developing a detailed understanding of the interrelationships between these redox-regulated mechanisms and therapeutic resistance holds potential to improve treatment effectiveness, offering valuable insights for both fundamental and clinical cancer research. Antioxid. Redox Signal. 00, 000–000.
Introduction—Overview of Therapy Resistance in Cancer
The issue of therapy resistance in cancer is a substantial obstacle in the management and treatment of this disease. Tumor recurrence or relapse arises when cancer cells acquire adaptive mechanisms to withstand and circumvent the therapeutic impact of diverse anticancer medications (Wang et al., 2019). Resistance can manifest in the following two forms: intrinsic resistance, which occurs before medication treatment, and acquired resistance, which emerges throughout the course of treatment (Emran et al., 2022). This phenomenon may occur as a result of various reasons, including as tumor cell heterogeneity, drug inactivation, modification of drug targets, DNA damage repair, drug efflux, suppression of cell death, and the epithelial–mesenchymal transition (EMT) (Housman et al., 2014; Mansoori et al., 2017). The issue of drug resistance in cancer is a multidimensional and intricate problem that necessitates a thorough comprehension of the underlying mechanisms and the formulation of innovative approaches to surmount it. Researchers and clinicians can strive for enhanced patient outcomes and more efficacious treatments by directing their efforts toward early detection, adaptive monitoring, innovative medication discovery, and the exploitation of cancer cell dependencies (Vasan et al., 2019). Recent research has indicated that the stress response elicited during tumor therapy, particularly oxidative stress, is a significant factor contributing to the evolution of drug-resistant tumor cells (Qin et al., 2022).
Currently, the primary modalities for cancer treatment encompass a range of conventional and innovative approaches, such as surgical intervention, immunotherapy, radiation, chemotherapy, and targeted therapy (Ward et al., 2021). Manipulating redox homeostasis has emerged as a promising therapeutic strategy to combat drug resistance in a variety of tumor types, as evidenced by recent studies (Cui et al., 2018; Hayes et al., 2020). The maintenance of cellular life and proper functioning relies on the regulation of redox homeostasis, a process that involves the careful management of reactive oxygen species (ROS) (generation and removal (Lennicke and Cochemé, 2021). ROS within cells primarily originate from the following three sources: NADPH oxidase (NOX), endoplasmic reticulum (ER) oxidase, and the mitochondrial electron transport chain (ETC) (Jakubczyk et al., 2020). The removal of ROS is achieved through the utilization of redox proteins, antioxidant enzymes, antioxidants, antioxidant transcription factors such as nuclear factor erythroid 2-related factor 2 (NRF2), and that are present in high quantities (Harris and DeNicola, 2020). The NRF2 transcription factor is implicated in the maintenance of cellular homeostasis and holds significant importance in the regulation of drug metabolism, proteasome degradation, energy metabolism, redox homeostasis, and various other physiological processes (Huang et al., 2015). In Lu and colleagues’ study (2016), it was observed that NRF2 exhibits a propensity to regulate genes that are closely linked to cellular defense mechanisms, such as antioxidant response elements (Lu et al., 2016). Nevertheless, within cells experiencing oxidative stress, the kelch-like ECH-associated protein 1 (KEAP1)-Cul3-RbX1 complex undergoes oxidation, leading to the destruction of crucial cysteine residues in Keap1. This oxidation process disrupts the ubiquitin degradation mechanism of NRF2, ultimately resulting in the heightened transcriptional activation of various genes such as heme oxygenase-1 (HO-1), glutathione (GSH), quinone oxidoreductase 1 (NQO1), and NADP(H) (Itoh et al., 1997; Ulasov et al., 2022) (Figs. 1 and 2). Consequently, this enhanced transcriptional activation contributes to the development of chemotherapy resistance (Wang et al., 2008). The ROS possess the potential to yield both positive and negative outcomes. The activation of ROS signaling in healthy cells has been shown to decrease the likelihood of cancerous lesion formation. However, in precancerous cells characterized by elevated ROS levels, the abnormal activation of ROS signaling may confer a survival advantage to cancer cells (Gañán-Gómez et al., 2013). When there is a balanced state of synthesis and removal of intracellular ROS, cells have the potential to maintain their survival and achieve redox homeostasis. In the past, ROS were regarded as intracellular by-products resulting from aerobic metabolism. However, recent research has revealed their role as second messengers, responsible for regulating many signaling pathways (Forman, 2016). The disulfide bond is a type of chemical connection that occurs when sulfhydryl (SH) groups from two distinct cysteine residues in a peptide chain interact with each other. This bond plays a crucial role in stabilizing the spatial conformation of a wide range of proteins, including enzymes, receptors, and other proteins (Hohl et al., 2012). According to Liu and coworkers (2016), the maintenance of redox homeostasis in cells significantly impacts the production of reversible disulfide bonds and is essential for the preservation of spatial structure and functional integrity (Liu et al., 2016). The phenomenon of tumor treatment resistance generated by oxidative stress is mediated by the combined effects of alterations in redox changes and redox-mediated pathways.
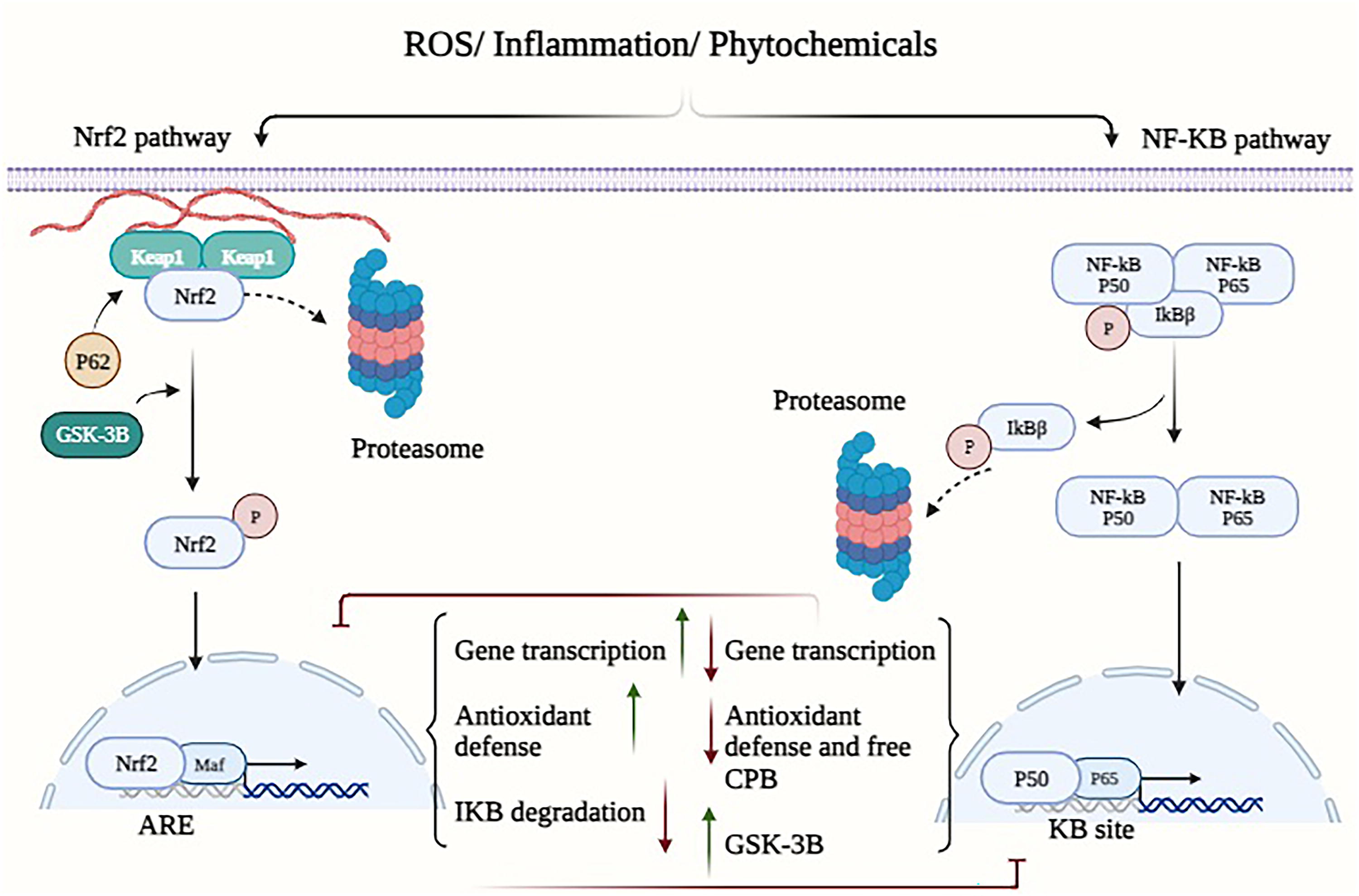
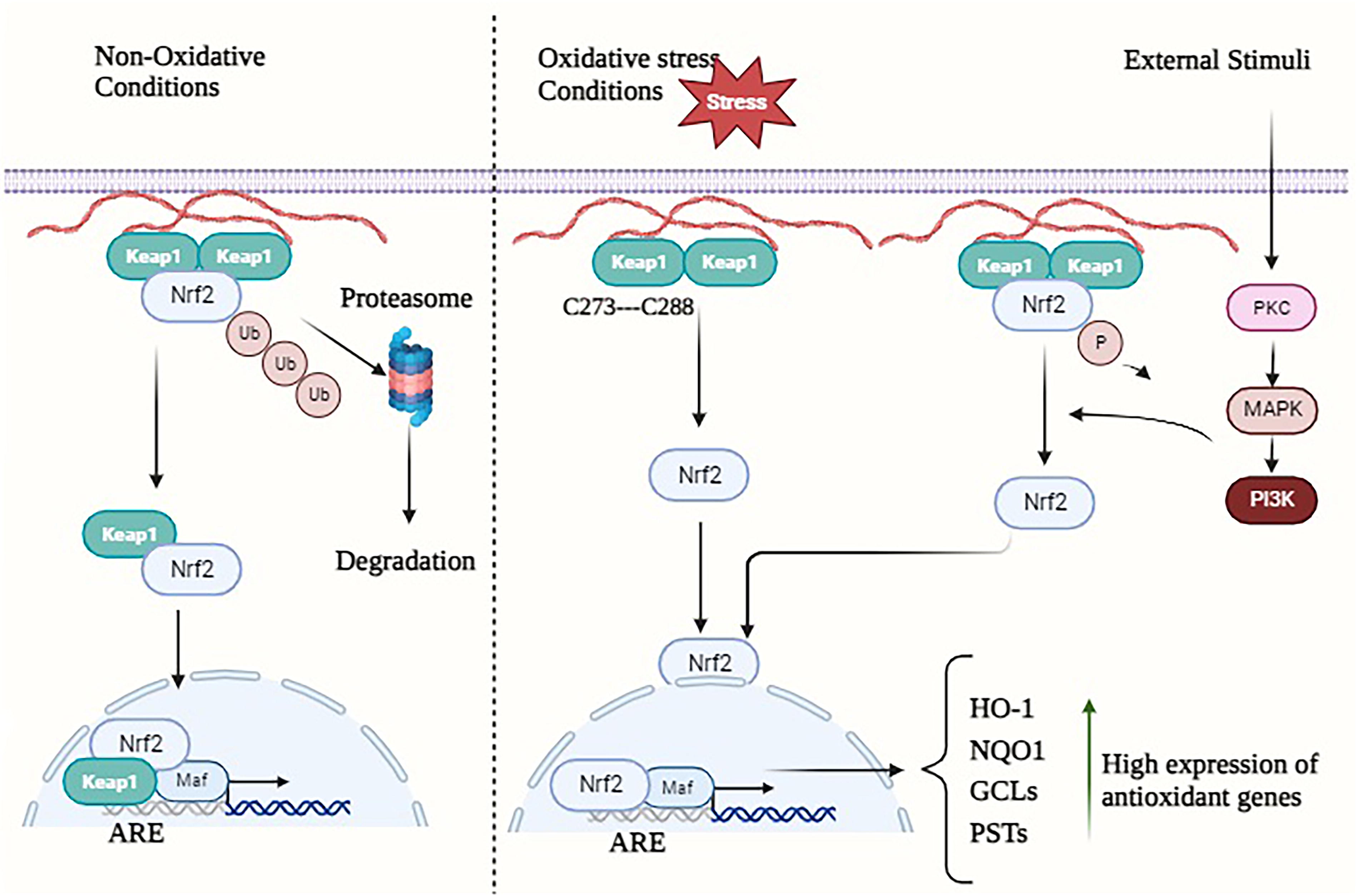
In a state of typical physiological conditions, the human body maintains redox homeostasis, which refers to the equilibrium between oxidants and antioxidants. In contrast to normal cells, cancerous cells have an elevated response to ROS during different therapies by promoting their antioxidant system (Jena et al., 2023). This phenomenon facilitates the development of resistance to tumor therapy through multiple pathways, including but not limited to changes in drug efflux, metabolism, and targeting, enhanced DNA damage repair, maintenance of stem cell-like properties, and alterations in the tumor microenvironment (TME) (Jomova et al., 2023). Enhancing treatment effectiveness can be facilitated by attaining a profound comprehension of the interconnections between various factors, as viewed through the lens of both fundamental and clinical research. The objective of this study is to provide a comprehensive overview of the molecular mechanisms behind redox-regulated resistance to cancer drugs, as well as to examine prospective clinical approaches for overcoming such drug resistance.
Redox Regulation in Cancer
Understanding the redox balance in cells
In multiple cancer cell variants, ROS serve as a pivotal factor in bolstering their viability, growth, and metastasis, by activating cellular signaling pathways that are conducive to tumor growth. The hypoxia-inducible factor (HIF)-1α, mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) are well-known signaling pathways in tumor progression and redox regulation. ROS have an important role in all these signaling pathways through inactivating and oxidizing negative regulators such as phosphatase and tensin homolog (PTEN), MAPK phosphatase, and prolyl hydroxylase (PHD)−2) (Guzy and Schumacker, 2006; Jomova et al., 2023; Seth and Rudolph, 2006). ROS have been found to facilitate the growth of cancer cells by activating the nuclear factor κ-light chain enhancer of activated B cells (NF-κB). A recent study indicated that mitochondrial ROS (mROS) are involved in the activation of NF-κB, and protein kinase D (PKD)−1 resulting in the upregulation of epidermal growth factor receptor signaling. This activation subsequently leads to the creation of preneoplastic lesions in the pancreas. The formation of anomalous pancreatic structures in living organisms was prevented by the administration of a mitochondria-targeted antioxidant known as mitoQ (Liou et al., 2016). Furthermore, ROS have a significant role in the promotion of metastasis through the activation of the protein-tyrosine kinase SRC/focal adhesion kinase PYK2 pathway. The upregulation of SRC/PYK2 signaling is facilitated by mROS, hence promoting the migration and invasion of many cancer cell phenotypes. Significantly, the injection of an mROS scavenger known as mitoTEMPO has been found to effectively inhibit the metastatic spread of breast cancer xenografts (Porporato et al., 2014). Hence, it is inferred that ROS play a significant role in the initiation and advancement of carcinogenesis (Fig. 3).
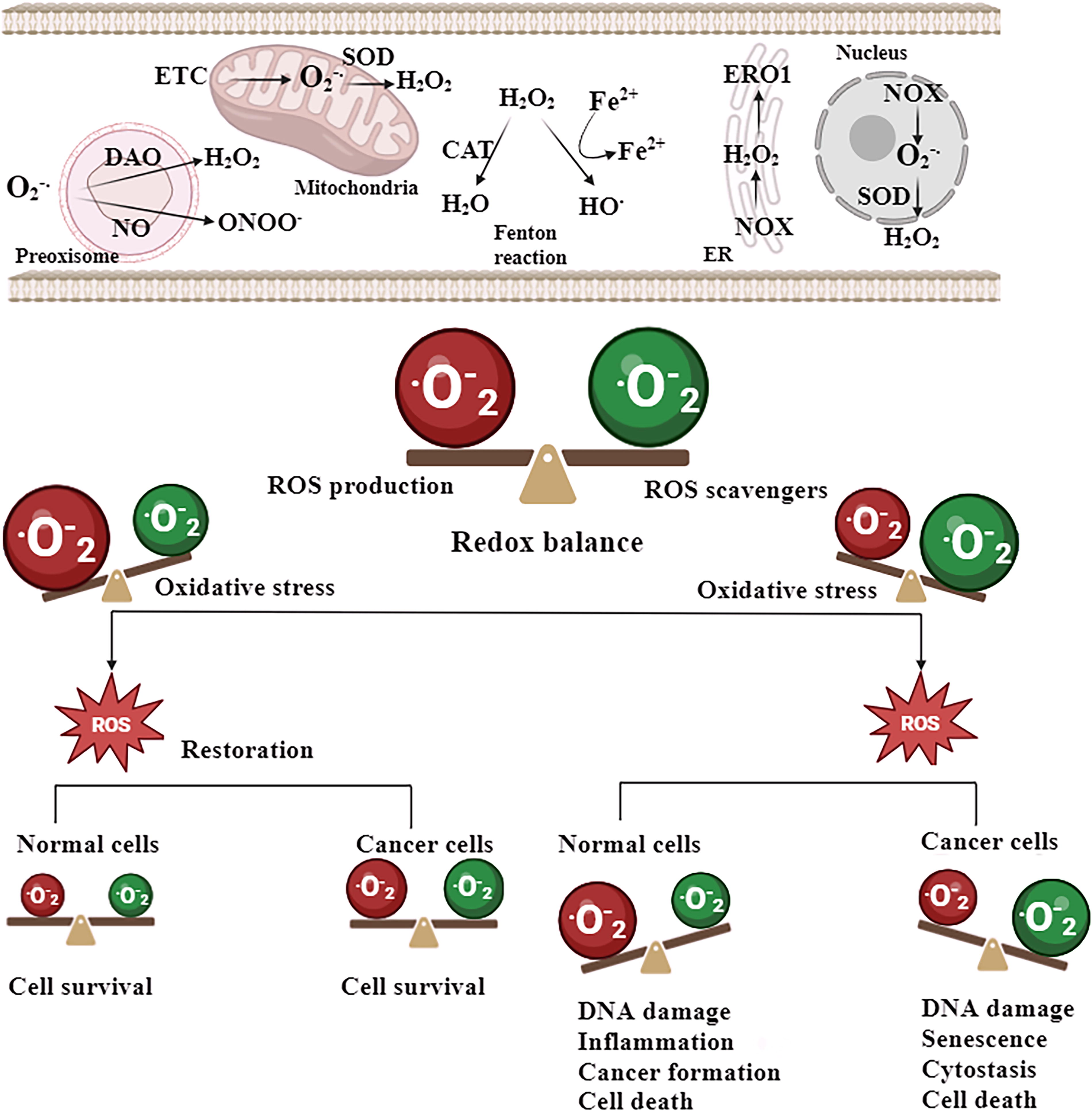
Superoxide dismutase in normal and cancerous cells—focusing on manganese superoxide dismutase
Mitochondria serve as the main organelles in cells for oxygen metabolism due to their involvement in oxidative phosphorylation. Therefore, mitochondria are the primary origin of ROS, which are by-products of oxygen metabolism, within cells (Fridovich, 1978). Superoxide radicals are the primary ROS generated in mitochondria (Frantz and Wipf, 2010). They play a role in the formation of other radical and nonradical species, including hydroxyl radical (Nelson et al., 2003) and peroxynitrite, which is a reactive nitrogen species (RNS) (Guzik et al., 2002). ROS disrupt cellular balance by modifying the function of various proteins, such as transcription factors HIF-1, p53 (Fojta et al., 1999; Galanis et al., 2008), NF-κB, and activator protein 1 (AP-1) (Kabe et al., 2005), as well as MAPKs, serine/threonine kinases (Schäfer et al., 2003), and tyrosine and serine/threonine phosphatases (Wright et al., 2009). Although first thought to exclusively cause harm to cells by damaging lipids, proteins, and DNA, meticulous research has uncovered significant roles of ROS in regular biological processes, including cell proliferation, differentiation, cell adhesion, death, and the immune response. ROS also serve as crucial second messengers in cellular communication (Sies, 2014). There is a delicate equilibrium between the creation and elimination of ROS. When this balance is disrupted, it can lead to abnormal accumulation of ROS, which can contribute to the development of various diseases, including neurological disorders and cancer. Basal levels of ROS can change through many causes, including as the presence of external agents that generate ROS, an increase in ROS generation from internal sources, a decrease in antioxidant capacity, or a combination of these factors. Due to the harmful and communicative impacts of ROS, the cell possesses a variety of enzyme and nonenzyme systems to eliminate ROS (Koehler et al., 2006). Superoxide radicals pose a significant risk since they actively contribute to the generation of other harmful ROS. Superoxide dismutases (SODs) are the primary enzymes responsible for eliminating ROS in cells (Fridovich, 1978). Among them, manganese SOD (MnSOD) is particularly crucial due to its placement in the mitochondria, which is the main site of ROS generation. Alterations in the expression or operation of MnSOD can have substantial effects on mitochondrial function and cellular balance due to oxidative harm to various proteins localized in mitochondria, leading to the development of diseases (Griess et al., 2017).
Cells possess many enzyme systems that neutralize ROS produced within the cell (Andreyev et al., 2005). SODs are the main enzymes responsible for neutralizing ROS in cells (Fridovich, 1978). These enzymes facilitate the conversion of superoxide into hydrogen peroxide (H2O2) and molecular oxygen through a process called dismutation (Fujii et al., 2022). Catalase facilitates the dismutation (disproportionation) of H2O2 by reducing it by two electrons to form water, H2O, and oxidizing it to produce O2. The peroxiredoxins (PRDXs) and glutathione peroxidases (GPXs) catalyze the conversion of H2O2 into water. Cells contain three distinct forms of SOD, each of which is encoded by its own set of genes (Zelko et al., 2002). Copper- and zinc-containing SOD (CuZnSOD, SOD1) is a homodimer primarily located in the cytoplasm (Slot et al., 1986). However, trace amounts have also been detected in the intermembrane space of mitochondria. Extracellular SOD (ECSOD, SOD3) shares 40%–60% similarity in amino acid composition with CuZnSOD. It contains copper and zinc in its active site. However, unlike CuZnSOD, ECSOD is located in the extracellular portion of the cell (Miriyala et al., 2011). MnSOD (SOD2) is a homotetramer that is specifically located in the matrix of mitochondria. MnSOD expression is remarkably conserved, since it has been found in prokaryotes, as well as in lower and higher order eukaryotes (Candas and Li, 2014).
MnSOD exhibits a multifaceted dual function in the initiation and advancement of cancer. Multiple studies have demonstrated that elevated MnSOD expression can augment the ability of cancer cells to survive, proliferate, spread to other parts of the body, and defy programmed cell death (Fu et al., 2016). Elevated MnSOD levels are linked to more advanced cancer stages and the spread of cancer to other parts of the body in several forms of cancer. Possible methods involve inhibiting the release of cytochrome c, enhancing the production of matrix metalloproteinase, and facilitating neuroendocrine differentiation (Robbins and Zhao, 2014). Nevertheless, additional research indicates that MnSOD has a role in suppressing tumor growth. Increased expression of MnSOD suppresses traits such as invasiveness, proliferation without attachment, and the ability to form tumors in different cancer models (Miriyala et al., 2012). The mechanisms involve making cells more sensitive to substances that generate ROS, decreasing ROS caused by carcinogens, promoting cell differentiation, and blocking the function of AP-1 (Milkovic et al., 2019). The divergent functions indicate a subtle impact of MnSOD, which is controlled by its regulation of H2O2 concentrations. A deficiency in MnSOD may result in an excessive production of ROS and the formation of tumors, whereas moderate increases in MnSOD levels could prevent the early development of cancer by reducing oxidative stress (Liou and Storz, 2010). However, in well-developed malignancies with elevated metabolic levels of ROS, the increase in MnSOD expression could potentially facilitate the spread of cancer cells to other parts of the body by boosting the creation of H2O2. In mouse skin carcinogenesis models, it has been observed that MnSOD levels decline in the early stages but increase in the later aggressive phases (Muchtaridi et al., 2024). To summarize, MnSOD has contrasting effects on tumor growth, either suppressing or stimulating it, depending on the specific circumstances. These effects are closely related to the impact of MnSOD on the amounts of H2O2 within cells, which varies depending on the stage of cancer. Given the crucial importance of MnSOD in preserving the various functions of mitochondria, it is undoubtedly valuable to use strategies that aim to maintain or enhance MnSOD expression and enzyme activity. These strategies include increasing the expression of MnSOD naturally present in the body, supplementing cells with externally sourced MnSOD protein, or using pharmaceuticals that imitate the effects of MnSOD. Such approaches hold great potential for treating and preventing various diseases associated with ROS.
Redox signaling pathways in cancer
Tumor cells have heightened intracellular levels of ROS to facilitate the promotion of tumorigenic redox signaling. Enhancement of ROS generation is initiated by the acquisition of oncogenes, such as the constitutively active isoforms of RAS. When H-RASV12 is overexpressed, human fibroblasts exhibit a significant increase in the production of O2̇̄ through NOX (Ogrunc et al., 2014). Moreover, it has been observed that the absence of functioning p53 in mouse embryonic fibroblasts leads to an upregulation of mROS generation with the expression of H-RASV12 or K-RASV12 (Weinberg et al., 2010). The presence of oncogenic K-RAS has been identified as a crucial factor in the development of lung adenocarcinoma and pancreatic preneoplastic lesions, with the induction of mROS playing a significant role in this process (Liou et al., 2016; Weinberg et al., 2010). Tumor hypoxia has been identified as a contributing factor to the elevation of ROS generation (Chandel et al., 1998). Furthermore, cancer cells have the ability to enhance the creation of ROS by reducing the activity of antioxidant systems at the specific locations where ROS are generated. The deacetylation and activation of SOD2 by mitochondrial sirtuin (SIRT)−3 are responsible for the regulation of mitochondrial O2̇̄ levels (Newman et al., 2012). Therefore, the reported depletion of SIRT3 in numerous cases of breast cancer leads to the buildup of mROS and stabilization of hypoxia-inducible factor 1 alpha (HIF-1α) (Finley et al., 2011).
The presence of increased levels of ROS within cancer cells is dependent on the inhibition of overall antioxidant mechanisms. Several tumor suppressors have been found to possess antioxidant properties in nontransformed cells. The enzyme responsible for sensing DNA damage, known as the ataxia-telangiectasia mutated (ATM) kinase, is regulated by redox reactions and has the ability to suppress the generation of ROS (Arfin et al., 2021). The breast cancer type 1 susceptibility protein, which is regulated by the ATM gene, has a role in inhibiting the ubiquitination process of NRF2 mediated by KEAP1. This interaction leads to the stabilization and activation of NRF2, which serves as a crucial regulator of antioxidant responses (Smolková et al., 2020). The p53 protein is a possible activator of NRF2 as a tumor suppressor, leading to an upregulation of antioxidant enzymes such as SOD2 and GPX1 (Fig. 1). In addition, p53 promotes the synthesis of NADPH, which serves as the required reducing equivalent for the reactivation of antioxidant systems (Shah and Rogoff, 2021). It is noteworthy that p53 has the ability to decrease the expression of solute carrier family 7 member 11 (SLC7A11), a transporter responsible for cysteine uptake in the synthesis of GSH. Consequently, p53 can exhibit a pro-oxidant function under some physiological circumstances (Wu et al., 2020). Nevertheless, it has been discovered that the antioxidative role of p53 is imperative for its capacity to inhibit the development of cancer (Budanov, 2014). Hence, the promotion of carcinogenesis is facilitated by the depletion of tumor suppressors, leads to high levels of intracellular ROS.
Interestingly, cancer cells simultaneously increase their antioxidant capacity to mitigate the potentially harmful effects of ROS reaching hazardous levels. An excessive quantity of ROS can have deleterious effects on the survival and proliferation rate of tumor cells. The presence of susceptibility can present a significant barrier in the process of metastasis, since cancer cells that become separated from the extracellular matrix encounter increased amounts of ROS (Gibellini et al., 2010). Notably, blood and viscera create oxidizing conditions that are unfavorable for the viability and growth of circulating cancer cells (Piskounova et al., 2015). Consequently, an abundance of ROS serves to restrict the progression of metastasis and cancer. As a result, upregulation of NRF2 is reported in several types of tumors, such as those affecting the skin, lungs, pancreas, breasts, ovaries, and prostate. The dysregulation of NRF2 can be modulated by loss- and gain-function mutations in KEAP1 and NRF2, respectively, which enable the continuous stabilization of NRF2 (Van Loenhout et al., 2020). Also, the initiation of the NRF2 antioxidant program occurs through the acquisition of some oncogenes, including MYC, BRAF, and K-RAS. Significantly, the presence of NRF2 is essential for the development of pancreatic and lung tumors in living organisms, as indicated by previous studies. This has given rise to studies on targeting NRF2-dependent malignancies. A recent work used chemical proteomic techniques to successfully discover a druggable target, namely, nuclear receptor subfamily O group B member 1, controlled by NRF2 in KEAP1-null non-small-cell lung tumors (Bar-Peled et al., 2017).
GSH is a significant antioxidant molecule within the context of tumor cells. Upregulation of GSH has been documented in the tissues affected by head and neck, breast, lung, and ovarian cancers (Bansal and Simon, 2018). The elevated presence of GSH in tumor tissues can be attributed to the heightened cellular accessibility of its biosynthetic precursors, namely, cysteine, glycine, and glutamate. The SLC7A11 antiporter, responsible for the transport of cystine and glutamate, exhibits significant expression levels in human malignancies (Jyotsana et al., 2022). In addition, it has been observed that the upregulation of the glutamate-cysteine ligase modifier subunit, a crucial component in the synthesis of GSH, occurs in several forms of human cancer (Di Giacomo et al., 2023). In addition to de novo production, the cellular level of GSH is influenced by another mechanism, namely, its regeneration. The process of reducing oxidized glutathione disulfide (GSSG) back to declined GSH is facilitated by the enzymes glutathione reductase (GR) and the reducing agent NADPH. To expedite this biological mechanism, cancer cells upregulate NADPH. The maintenance and raising of cellular GSH levels play a crucial role in the development and propagation of tumors (Lian et al., 2018). Furthermore, it has been discovered that a specific isoform of GPX, known as GPX4, is essential for the viability of cancer cells that are resistant to conventional therapies. This finding further emphasizes the significance of antioxidant pathways controlled by GSH in the advancement of cancer (Viswanathan et al., 2017).
Cancer cells exhibit an upregulation of metabolic pathways that are essential for the production of NADPH, a reducing potential. The oxidative pentose phosphate pathway (PPP), which is a significant contributor to the production of NADPH, diverges from the glycolytic pathway. Several regulatory enzymes involved in glycolysis, such as TP53-induced glycolysis regulatory phosphatase (TIGAR), pyruvate kinase isoform M2 (PKM2), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) have the ability to redirect glycolytic intermediates toward the oxidative PPP to produce reducing potential (Cho et al., 2018). It is noteworthy that the increased levels of PPP by GAPDH and PKM2 are contingent upon the oxidation of certain cysteine residues (Hayes et al., 2020). The overexpression of activators of the PPP has been evidenced in various cancer cell types. In addition, it is crucial to note that the NADPH-generating/antioxidant capacity of TIGAR and PKM2 has a vital role in the intestinal and lung cancer growth, respectively (Jin and Zhou, 2019). It is noteworthy that PKM2 has the ability to redirect glycolytic precursors onto an alternative pathway, known as one-carbon metabolism, which also generates NADPH (Hay, 2016). The process of one-carbon metabolism results in the production of NADPH through the catalytic activity of phosphoglycerate dehydrogenase (PHGDH), which is responsible for serine biosynthesis. Following this, serine hydroxy methyltransferase (SHMT) inserts a carbon unit from serine into the folate cycle. Within the context of the folate cycle, the enzymatic process of methylene-THF dehydrogenase facilitates the oxidation of 5,10-methenyl-THF, leading to the production of NADPH (Li and Ye, 2020). When cells that have been changed by MYC are subjected to a state of reduced oxygen availability, the transcription factors HIF-1α and MYC work together to enhance the production of the mitochondrial variant of SHMT, known as SHMT2. This process enables the generation of mitochondrial NADPH, which plays a role in mitigating the increased levels of mROS that occur during hypoxia. The survival of hypoxia transformed cells was diminished upon targeting SHMT2, but this effect was counteracted by the administration of the antioxidant N-acetylcysteine (Infantino et al., 2021). This finding provides evidence that hypoxic cancer cells increase the activity of one-carbon metabolism and elevate the level of mitochondrial NADPH to escape from cell death conducted by ROS. The production of mitochondrial NADPH is also of significant importance in metastasis. In the context of tumor spheroids that are not reliant on anchoring, the presence of cytosolic isocitrate dehydrogenase, mitochondrial isocitrate dehydrogenase, and mitochondrial citrate transporter facilitates the process of reductive carboxylation within the mitochondria, leading to the production of NADPH. The aforementioned procedure facilitates cellular maintenance of mitochondrial redox equilibrium and evasion of oxidative damage resulting from dissociation from the extracellular matrix (Martínez-Reyes and Chandel, 2014). In addition, folate cycle suppression reduces distant metastasis of melanoma cells (Piskounova et al., 2015), and targeting PHGDH eliminates the metastatic ability of breast cancer cells (Samanta et al., 2016).
In summary, cancer cells expand their capacity for both pro-oxidant and antioxidant processes with various strategies. For instance, APC-deficient intestinal cells can use Wnt signaling to increase both ROS production and antioxidant defense. Removing either pro-oxidant or antioxidant components slows down cell proliferation, but removing both at the same time has a more severe effect. This suggests that ROS generated by NOX promote proliferation, while TIGAR limits damaging ROS. It is proposed that localized ROS production activates protumorigenic signaling pathways, while antioxidants prevent the accumulation of harmful ROS, ultimately influencing cancer cell behavior.
Therapy Resistance and Redox Dysregulation
A glimpse into the mechanisms of treatment resistance in cancer
The development of resistance to therapeutic interventions by cancer cells might present significant challenges in the management of this disease, as it can occur through diverse methods. The following are several significant mechanisms involved in drug resistance: drug inactivation, multidrug resistance (MDR), regulating various cell death pathways, modification of drug metabolism, epigenetic changes, alteration in drug targets, promoted DNA repair, upregulated gene amplification, adaptive immune resistance, tumor heterogeneity, alterations in the TME, and cancer stem cells (CSCs). Comprehending these pathways is crucial for formulating strategies to surmount medication resistance in the context of cancer therapy (Ramos et al., 2021) (Fig. 4). This encompasses the advancement of novel pharmaceuticals capable of selectively targeting these resistance mechanisms, the implementation of combination therapy that can concurrently address several pathways, and the adoption of personalized medicine strategies that consider the distinct genetic and molecular attributes of individual patient’s tumors. In the subsequent sections, we provide a concise overview of the molecular mechanisms behind redox-regulated resistance to cancer drugs. In addition, we also review various clinical approaches aimed at circumventing drug resistance.
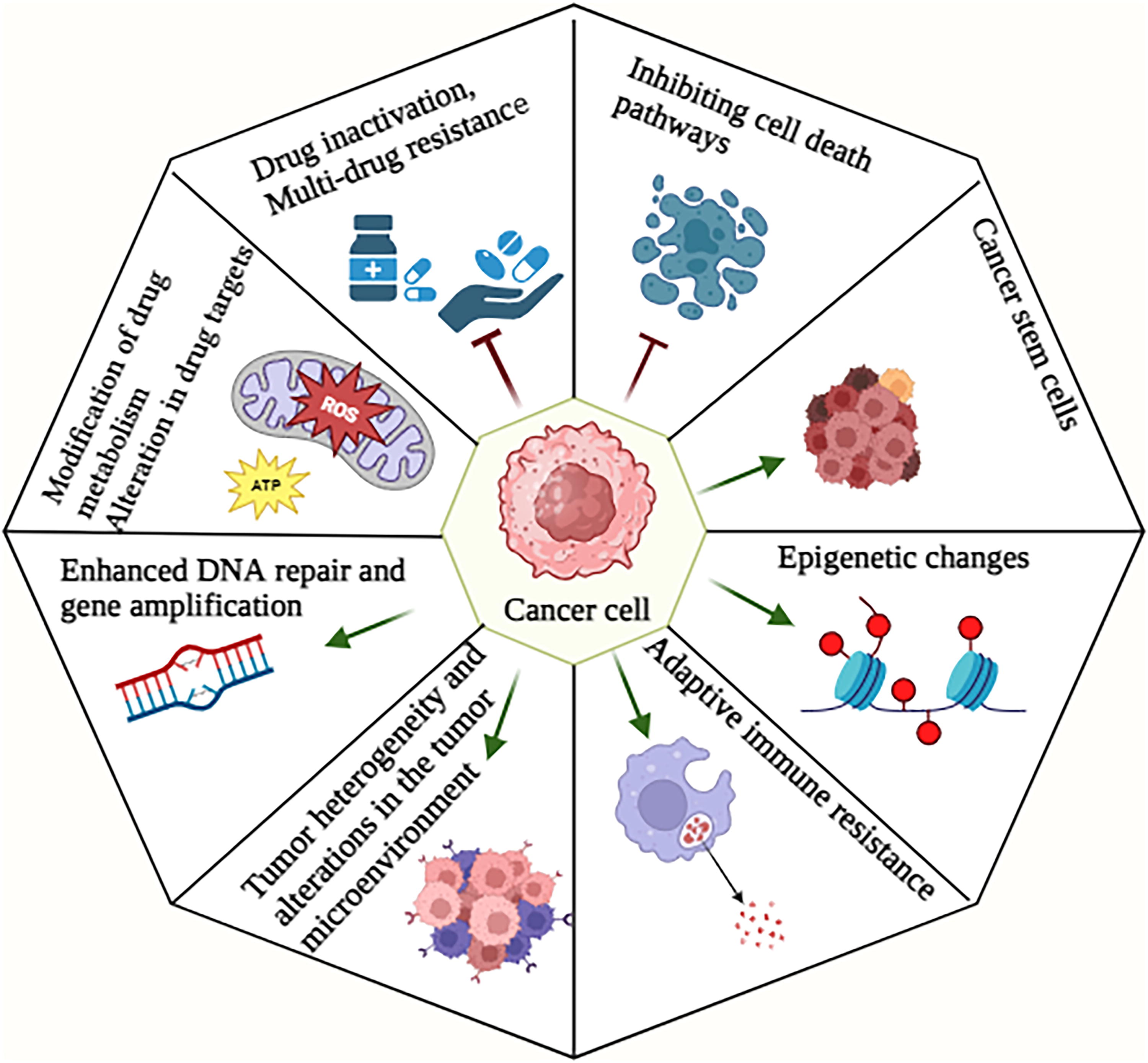
Implications of redox dysregulation in therapy resistance
Drug inactivation and drug efflux
The processes of drug inflow and efflux play a vital role in the regulation of intracellular drug concentration, a critical factor in achieving optimal efficacy in tumor cell eradication. ATP-binding cassette (ABC) transporters are of significant importance in the process of chemotherapy efflux and influx, since they facilitate the removal of key cancer chemotherapeutic drugs such as antimetabolites, taxanes, and topoisomerase inhibitors. ABC transporters, including MDR protein 1 (MDR1) and breast cancer resistance protein (BCRP), are pivotal in facilitating MDR by enhancing the efflux of anticancer medications, hence diminishing intracellular drug levels. ROS have the capacity to control ABC transporters by two distinct mechanisms (Duan et al., 2023). First, a moderate level of oxidative stress has been seen to stimulate the expression and activity of ABC transporters in cancerous cells. Conversely, an excessive level of oxidative stress has been found to impede the functionality of the transporter system (Yuan et al., 2022).
Multidomain peptides can be formed by combining ABC transporters, which are composed of two cytoplasmic nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs), by different methods. Modifications in these arrangements promote the transportation of drugs, resulting in a decrease in intracellular drug concentrations to levels that are below the threshold of lethality (Yuan et al., 2022). The transport function of MDR1 is associated with the oxidative-reductive (REDOX) condition of cysteine residues Cys431 and Cys1074 located in NBD1 and NBD2, respectively, which exhibit comparable structural characteristics. The MRP1 possesses a distinct topological arrangement and includes a TMD known as MSD0. This particular domain functions as a regulatory mechanism, controlling the transportation of drugs across the cellular membrane (Maiti, 2012). The creation of disulfide bonds through cysteine residues in MSD0 is responsible for the dimerization of MRP1. Structural alterations in these cysteine residues have the potential to impede dimer formation and disrupt the process of drug transport. ROS can be generated subsequent to the administration of some anticancer medications, hence facilitating the enhancement of the antioxidant system within malignant cells (van Norren et al., 2009).
The human BCRP, which falls within the ABCG2 classification, functions as a semimolecular transporter that plays a crucial role in the transportation of several medicines, such as mitoxantrone and SN-38. The homodimer drug transporter is of significant importance in the development of chemotherapeutic resistance. The cysteine residues Cys592 and Cys608 of ABCG2 are involved in the formation of intramolecular disulfide bonds, which play a role in regulating the substrate specificity of the protein. In addition, the cysteine residues Cys603 and Cys608 are responsible for the creation of intermolecular disulfide bonds, which are crucial in defining the localization of ABCG2 in the plasma membrane (Henriksen et al., 2005). The presence of both intermolecular and intramolecular disulfide bridges plays a crucial role in the functioning of ABCG2. The modulation of ABC transporters by ROS represents a novel approach for circumventing cellular drug resistance. According to research findings, it has been indicated that ROS can render transporters inactive by impacting the disulfide bonds within the dimeric structure of the transporter protein. This observation underscores the capacity of ROS to modify transporters, leading to a decrease in the efflux of drugs and an enhancement in their therapeutic effectiveness (Kage et al., 2005).
The available literatures suggest that drug efflux transporters can be regulated by redox processes, which can be mediated by several channels, including transcriptional mechanisms that are impacted by environmental variables. The induction of ER stress has been observed to result in the overexpression of MDR1 in hepatoma cells. Similarly, in Caco-2 human colon cancer cells, exposure to high concentrations of H2O2 has been found to enhance the expression of MDR1. The process of oxidative stress induces the oxidation of the SH group, resulting in the upregulation of NRF2 (Jakubczyk et al., 2020). This upregulation subsequently leads to an increase in the quantity and functionality of p-glycoprotein transporters, thereby restricting the permeability of the cell membrane. The activation of forkhead box O (FOXO), a transcription factor, has been observed to potentially have a role in the development of drug efflux transporters. This activation occurs through disulfide interactions with the transporters (Šereš et al., 2020).
Following the administration of chemotherapeutic medicines, the intracellular redox balance is disturbed, resulting in the buildup of ROS. This accumulation subsequently triggers FOXO activation, and the production of transporter proteins. One such instance involves the compound paclitaxel (PTX), which has been documented to elicit drug resistance in ovarian malignant tumor cells through the involvement of thioredoxin 1(Trx1) and the transcription component FOXO (Gomes et al., 2013). Oxidative stress has the ability to induce the transcription of many genes encoding transporter proteins, such as MDR, multidrug resistance-associated proteins (MRPs), and BCRP, through the facilitation of apurinic/apyrimidinic endodeoxyribonuclease 1 or redox factor I translocation. The phenomenon of drug resistance can be influenced by epigenetic mechanisms, which involve modifications to the structure of drug efflux pumps. For instance, hypermethylation in the region of the MDR1 promoter can lead to alterations in its configuration, hence affecting drug resistance. Gaining insight into the molecular underpinnings of oxidative stress has the potential to offer valuable insights into the management of clinical drug resistance (Peng and Zhong, 2015).
Cell death regulation
Apoptosis is a pivotal pathway during cellular development and maintenance of internal equilibrium governed by the death receptor and mitochondrial pathways. The contribution of ROS in the cell death pathways is of considerable importance as even minimal quantities can promote the formation of cancer cells. Significant quantities of a substance induce oxidative stress resulting in the initiation of apoptosis (Redza-Dutordoir and Averill-Bates 2016). Within the context of tumors the cells experience a loss of their capacity to perform the typical process of apoptotic induction resulting in unregulated cellular proliferation. Research findings indicate that VB1 supplementation induces an elevation in ROS levels inside melanoma cells treated with anti-BRAFi agents (Zhou et al. 2014). This rise in ROS then triggers DNA cytotoxicity and initiates the process of programmed cell death known as apoptosis. The regulation of apoptosis is significantly influenced by Bcl-2 family proteins. The expression and transportation of these proteins to the mitochondria are tightly controlled by ROS (Qian et al. 2022). The activation of p53 can be facilitated by the c-Jun NH2-terminal kinase (JNK) signaling pathway as a result of accumulated ROS hence boosting apoptosis. The protein c-FLIP which is found in the cytoplasm is known to block apoptosis mediated by death receptors. In certain types of cancer c-FLIP is increased leading to enhanced antiapoptotic capabilities (Redza-Dutordoir and Averill-Bates 2016).
Autophagy is a cellular catabolic process that occurs within the confines of the cell. In instances of food deprivation or stress-induced stimulation, the cellular mechanism involves the degradation of misfolded proteins and damaged organelles, resulting in the provision of energy to the cell. The involvement of autophagy in malignancies is characterized by a dualistic and intricate nature (Yun et al., 2020). Autophagy generally exerts a suppressive effect on tumors during the initial stages of tumorigenesis, whereas it selectively safeguards and promotes the survival of tumor cells. Research has demonstrated a strong correlation between REDOX signal transduction and alteration and the process of autophagy (Chavez-Dominguez et al., 2020). One illustrative instance involves the ability of ROS to modulate the mechanistic target of rapamycin activity via many pathways, including the PI3K/Akt, HMGB1, and LKB1/AMPK pathways, so eliciting the process of autophagy (You et al., 2017). The activation of autophagy in human breast cancer cells can also be facilitated by ROS through the phosphorylation of JNK. Furthermore, a variety of autophagy-associated proteins, such as ATG, P62L, and C3-II proteins, exhibited activation in H9c2 cells upon treatment with H2O2 (Sinenko et al., 2021). In the context of cancer laser therapy, the accumulation of ROS results in beclin-1 upregulation, consequently facilitating tumor autophagy and augmenting its resistance to therapeutic interventions. Nevertheless, numerous investigations have provided evidence indicating that oxidative alterations of autophagy regulatory components exert a suppressive influence on the process of autophagy (Poillet-Perez et al., 2015). The activity of tubulin 1A/1B LC3, which is involved in autophagy, is hindered by the oxidation of the autophagic proteins ATG4B at cysteine residues Cys292 and Cys361. Consequently, this oxidation results in a decrease in autophagic flux. Furthermore, the process of LC3 lipidation is hindered by the oxidative alteration of Cys263 and Cys572 (Zheng et al., 2020).
Furthermore, autophagy has the potential to establish a negative feedback mechanism aimed at mitigating oxidative stress, in addition to the two aforementioned elements of the impact of oxidative stress on autophagy (Chang et al., 2022; Zheng et al., 2020). The activation of MCOLN1 by elevated ROS triggers the release of Ca2+, resulting in the translocation of TFEB to the nucleus, so initiating the process of autophagy. The promotion of enhanced autophagy facilitates the removal of excessive ROS (Zhang et al., 2016b). Insufficient autophagy in the context of excessive ROS accumulation can potentially promote the demise of cancer cells. The introduction of novel targets has the potential to expedite the advancement of drug-resistant tumor therapy. However, it is imperative to conduct additional investigations to ascertain the precise impact of oxidative stress on autophagy (Zhao et al., 2023).
Necrosis is another form of programmed cell death that is controlled by death receptors and intracellular signaling molecules. Necrosis, which is characterized by cellular swelling and rupture, entails the deliberate killing of cells to circumvent the inhibition of cell apoptosis. The primary process of necroptosis signaling involves the activation of RIP3 by an upstream protein containing the RIP homotypic interaction motif (RHIM) domain (Galluzzi et al., 2017). Ligands such as tumor necrosis factor (TNF) and Fas ligand have the ability to initiate necroptosis. This process triggers the secretion of damage-associated molecular patterns (DAMPs), which subsequently activate inflammatory responses (Kim et al., 2019). Necroptosis is associated with the suppression of tumor growth in certain cancer types, whereas in esophageal, pancreatic, and colon cancer, it facilitates the advancement of tumors and their metastatic spread. Multiple studies have indicated a significant association and positive correlation between ROS and necroptosis, both of which are implicated in the development and progression of various pathophysiological conditions, such as malignancies (Gong et al., 2019). A study demonstrated that the alteration of specific cysteine residues (257, 268, and 586) by ROS might result in the creation of an intramolecular disulfide bond on RIP1 (Zhang et al., 2017). This disulfide bond facilitates the autophosphorylation of RIP1 at serine 161, which in turn enhances the recruitment of RIP3. The findings of this study indicate that ROS have a role in the initiation of necroptosis. The study revealed that with exposure to H2O2, RIP3 was observed to translocate to the ER, leading to an imbalance in calcium ions. This observation indicates that oxidative stress has the potential to trigger programmed necrosis in cardiomyocytes by upregulating RIP3 (Zhang et al., 2016a). Furthermore, the elimination of ROS using butylated hydroxyanisole demonstrated a substantial decrease in TNF-induced necrosis in a cellular model of mouse fibrosarcoma (L929) (Vanlangenakker et al., 2011). Further work is required to explore viable therapeutic approaches that can restrict the progression of cancer by concurrently controlling necrosis and ROS.
Cell reprogramming
Glucose undergoes metabolism in somatic cells through the tricarboxylic acid (TCA) cycle, subsequently being phosphorylated to generate energy. In situations where there is an inadequate supply of oxygen, anaerobic glycolysis leads to the production of a significant quantity of pyruvate, which is then transformed into lactic acid as a means of sustaining energy provision. Despite the presence of enough oxygen, tumor cells exhibit a preference for glycolysis as their primary metabolic pathway to fulfill the energy and food requirements necessary for their rapid multiplication. This phenomenon, commonly known as the Warburg effect, has been extensively studied (Liberti and Locasale, 2016). The alteration in metabolism is commonly recognized as a characteristic feature of cancer (Potter et al., 2016). An increasing amount of scholarly literature indicates a correlation between aerobic glycolysis and the proliferation of tumors, as well as the development of chemotherapy resistance (Sattler et al., 2010). Furthermore, it is worth noting that this observed phenomenon may not be attributed just to impaired mitochondrial respiration, but rather to the increased expression of glycolytic enzymes and glucose transporters (Wangpaichitr et al., 2021). The formation of ROS within cells and the maintenance of redox equilibrium are mostly influenced by cellular metabolism. However, it is worth noting that ROS can also impact tumor energy metabolism through the regulation of crucial metabolic enzymes (Quijano et al., 2016). Several glycolytic enzymes possess conserved cysteine residues that are prone to redox alteration in response to ROS (Lennicke and Cochemé, 2021). According to Heneberg (2019), hexokinase (HK) serves as the initial regulatory step in facilitating glucose metabolism, facilitating the prompt utilization of ATP (Heneberg, 2019). The utilization of 2-deoxy-D-glucose (2-DG) as an HK2 inhibitor to amplify the responsiveness of cancer cells has been shown to enhance the effectiveness of the cytotoxic medications doxorubicin and PTX, as demonstrated by Maschek et al. in 2004 (Maschek et al., 2004). Another study revealed that the covalent binding of dehydroascorbic acid to the active cysteine of HK1 results in the irreversible loss of its enzymatic activity (Fiorani et al., 2000). The enzyme PKM2 exhibits reduced pyruvate kinase activity, which effectively hinders the entry of glycolytic metabolites into the TCA cycle. Consequently, this mechanism supports the viability and persistence of cancerous cells (Christofk et al., 2008). According to Shi et al. (2010), the inhibition of PKM2 expression can elevate the accumulation of docetaxel within A549 lung cancer cells, hence augmenting its anticancer efficacy (Shi et al., 2010). Moreover, the introduction of H2O2 in lung cancer cells resulted in a gradual elevation of ROS levels. In addition, this led to the suppression of PKM2 function through the oxidation of cysteine Cys358 (Anastasiou et al., 2011). The isomerization of dihydroxyacetone phosphate to glyceraldehyde-3-phosphate is facilitated by the catalytic activity of triose phosphate isomerase (TPI) (Pekel and Ari, 2020). According to Dumont et al. (2016), research has indicated that ROS can induce oxidation of TPI by impacting the integrity of intramolecular disulfide bonds. Consequently, the oxidized TPI undergoes degradation, resulting in a shift from glycolysis to the PPP. The maintenance of tumor glycolytic characteristics is strongly influenced by the presence of elevated levels of GAPDH. According to Reisz et al. (2016), in conditions of oxidative stress, the amino acid Cys152 within the enzyme GAPDH can undergo oxidation, resulting in the formation of intermolecular disulfide bonds (Reisz et al., 2016). This process ultimately leads to the aggregation of GAPDH and subsequent cell death. Furthermore, the GAPDH activity is inhibited and metabolic flow is redirected from glycolysis to the PPP through redox alteration of cysteine (Ralser et al., 2007). According to Kuehne et al. (2015), tumor cells indicate higher ROS levels in contrast to healthy cells. As a result, alterations in glycolysis toward the PPP contribute to an increased reductive capacity of cancer cells, although to a certain degree (Kuehne et al., 2015). In this particular scenario, it is shown that ROS function as signaling molecules, effectively modulating tumor metabolism in accordance with alterations in the cellular milieu. In addition, this observation implies that the integration of metabolic recombination and ROS regulators in cancerous cells presents a novel concept for cancer therapy. Peroxynitrite is a highly reactive substance that is produced when nitric oxide (NO
It is crucial to maintain cellular oxidative homeostasis by carefully regulating the levels of ROS, RNS, and RSS. Imbalances in this equilibrium can result in oxidative stress and harm to cells, which contributes to the development of cancer. Hence, it is crucial to take into account not just ROS but also RNS and RSS while investigating the impact of oxidative stress on cancer cells. In summary, it can be concluded that the regulation of metabolic enzymes by RS-mediated redox alteration has the ability to control tumor metabolic reprogramming, therefore exerting a substantial influence on the process of carcinogenesis and development.
Enhancing DNA repair and cell survival
DNA damage can arise from exposure to a range of substances, resulting in alterations of a physical or chemical nature. Oxidative stress, which acts as a source of DNA damage in the context of carcinogenesis, has the potential to elevate rates of cellular mutation and facilitate the process of malignant transformation. ROS have the ability to induce DNA lesions by several mechanisms, including base modification, nucleotide removal, disruption of DNA structure, and crosslinking between DNA and proteins (Katerji and Duerksen-Hughes, 2021). The administration of ROS has the potential to cause DNA breakage in both the nuclear and mitochondrial genomes of sperm, leading to a decrease in transcription levels. Guanine exhibits a higher susceptibility to oxidation compared with other DNA bases, with 8-hydroxy-2′-deoxyguanosine being the prevailing form of oxidized guanine (Chianese and Pierantoni, 2021). The induction of lipid peroxidation by ROS can lead to the indirect occurrence of further cyclic DNA damage. Oxidative stress is also implicated in the occurrence of base lesions and degradation in mitochondrial DNA, crucially participating in the development of mitochondrial gene mutations (Barrera, 2012).
The cellular response to DNA damage is a multifaceted process that encompasses the activation or inactivation of many signaling networks and proteins, which exhibit variable regulation in distinct cancer types. Tumorigenic cells elicit the appropriate DNA damage response (DDR) to counteract DNA damage and sustain tumor viability. In typical cellular environments, the equilibrium between spontaneous DNA damage and subsequent repair mechanisms is often maintained (Soutoglou and Misteli, 2008). However, in the context of tumor cells, this equilibrium may be disrupted as a result of insufficient repair capabilities. ROS exert their inhibitory effects on the activation of the DNA repair enzyme 8-oxoguanine glycosylase by the oxidation of crucial cysteine residues. In addition, ROS impede the timely identification of damaged regions by influencing sensor kinases (Vlahopoulos et al., 2019). According to Gruosso and coworkers (2016), the process of continuous oxidative stress leads to an increased rate of degradation of H2AX, which is facilitated by the E3 ubiquitin ligase RNF168. This degradation process ultimately results in enhanced DNA damage within cancer cells. Given that ROS have the ability to impede the DDR of tumors to a certain degree, it is plausible that augmenting ROS levels through treatment could potentially facilitate the apoptosis of cancer cells (Gruosso et al., 2016).
The involvement of moderate levels of ROS in tumor treatment resistance is of significant importance, as evidenced by their influence on many pathways, including NF-κB, MAPK, and PI3K/AKT (Noorolyai et al., 2019). The activation of PI3K is associated with cellular processes such as growth, cell division, and survival, which are intricately linked to cancer development and tumor progression. The phenomenon of oxidative stress has the capability to cause the inactivation of PTEN, hence leading to the activation of the PI3K and AKT pathways, which are crucial for promoting cell survival (Fruman et al., 2017).
The activation of transforming growth factor, TGF-β1, through ROS is limited to the oxidation of Met253 on the latency-associated peptide β (Richter et al., 2015). Multiple studies have demonstrated that ROS induce an upregulation and activation of the PI3K/Akt signaling pathway via TGF-β1, resulting in the development of malignant alterations in renal cells. Furthermore, the PI3K/Akt pathway is known to enhance the production of GSH, hence increasing the resistance of breast cancers to oxidative stress (Lu et al., 2019). The NF-κB transcription factor is crucial for cell survival and proliferation, and its activation has been reported in the development of drug resistance in many cancer types during therapeutic interventions. The NF-κB upregulation triggered by oxidative stress is a complex mechanism that warrants further investigation. ROS have the capability to exert both promotive and inhibitory effects on NF-κB activity. In the majority of instances, NF-κB activity tends to be heightened in tumor cells (Drain et al., 2021). The MAPK signaling pathway has four distinct subfamilies, namely, ERK5, JNK, P38, and ERK. The dimerization of mitogen-activated protein kinase 3, a crucial upstream activator of p38, is influenced by the oxidative alteration of Cys119 and Cys162 residues inside p38 (Bassi et al., 2017). The EGF receptor activation by ROS subsequently activates the ERK signaling cascade. Recent research findings have indicated that the activation of the ROS/JNK/p53 pathway by ginsenoside Rh4 can induce autophagy and death in colorectal carcinoma cells (Li et al., 2023a).
Epigenetic modifications
Epigenetics pertains to the inheritance of phenotypic modifications through generations, without modifying the underlying DNA sequence. Epigenetic mechanisms encompass many processes such as DNA methylation/demethylation, histone changes, and control mediated by microRNAs. The DNA methylation process entails the enzymatic attachment of methyl groups to cytosine residues located within DNA molecules, catalyzed by enzymes known as DNA methyltransferases (Al Aboud et al., 2023). This results in the formation of a modified cytosine called 5-methylcytosine (5-mC). The aforementioned procedure inhibits the process of transcription. The process of DNA demethylation involves the utilization of enzymes known as ten-eleven translocation enzymes (TETs) to reverse the methylation of DNA. This is achieved by converting 5-mC into three distinct forms as follows: 5-carboxylcytosine (5-caC), 5-formylcytosine (5-fC), and 5-hmC. Afterward, these modified forms of cytosine are removed from the DNA by the action of glycosylases (Kumar et al., 2018). The process of histone methylation has the ability to either inhibit or facilitate transcription, contingent upon several parameters such as the methylation of arginine and lysine residues on histone H3 and H4, and the presence of mono-, di-, or trimethylated lysine residues. Histone demethylases, such as lysine-specific demethylase 1 and Jumonji domain-containing histone demethylases, participate in the process of demethylating lysins that have been methylated (Jambhekar et al., 2019).
The investigation of Keap1’s epigenetic regulation has been conducted in diverse cancer types. The initial findings by Wang et al. indicated that Keap1 exhibits high expression levels in BEAS-2B human normal bronchial epithelial cells. However, it was observed that the expression of Keap1 is declined in various lung cancer cell lines and tissues. The decrease in Keap1 expression was found to be associated with the hypermethylation of CpG sites in its promoter region (Guo et al., 2015). Furthermore, the downregulation of Keap1 was successfully reversed through treatment with 5-aza (Hanada et al., 2012). Comparable findings have been achieved in diverse cancer types, encompassing malignant gliomas as well as head and neck, thyroid, breast, colorectal, and prostate cancer cell lines. The association between tumor growth and resistance to treatments is linked to the activation of Nrf2 signaling through hypermethylation of the Keap1 promoter (Smolková et al., 2020). The study conducted by Li et al. demonstrated a substantial correlation between reduced expression of EZH2 and increased expression of HO-1, NQO1, and Nrf2 in both lung cancer cell lines and tissues. This correlation was mostly ascribed to a decrease in H3K27Me3 levels in the promoter region of Nrf2 (Li et al., 2014b). Recently, Kang and coworkers demonstrated the causal association between the expression of Nrf2 and epigenetic modifications, specifically DNA methylation at cytosine residues, in the context of oxidative stress generated by 5-fluorouracil (FU) treatment (Kang et al., 2014). It has been discovered that the induction of ROS by 5-FU leads to the activation of DNA demethylases, specifically TETs. This activation subsequently causes a decrease in methylation levels of the Nrf2 promoter, leading to the activation of Nrf2 production. Consequently, the upregulation of HO-1, which is a target of the transcription factor Nrf2, leads to the development of resistance against 5-FU. Furthermore, it was observed that the administration of 5-FU to mice implanted with shNrf2-transfected 5-FU-resistant SNUC5 (SNUC5/5-FUR) colon cancer cells can significantly reduce the tumor volume, size, and weight. In addition, this treatment increased the proportion of apoptotic cells compared with mice implanted with shControl-transfected SNUC5/5-FUR cells. Therefore, it can be inferred that oxidative stress-induced DNA demethylation, leading to the upregulation of Nrf2 expression, could potentially enhance resistance to 5-FU (Kang et al., 2014). The results of Kang and colleagues’ study provided evidence that, alongside DNA methylation, the state of histone methylation under oxidative stress generated by 5-FU plays a role in regulating the Nrf2 expression and its associated antioxidant enzymes. This ultimately can develop resistance to 5-FU (Kang et al., 2014).
The mixed lineage leukemia protein is a constituent of the Set1-like complex, which is part of the larger complex of proteins known as the complex of proteins associated with Set1 (COMPASS). Host cell factor 1 (HCF1), an essential constituent of the MLL complex, has a pivotal role in preserving the structural integrity of the MLL/COMPASS-like complex. According to Capotosti et al., it has been proposed that HCF1 undergoes GlcNAcylation and proteolytic maturation within the active region of O-GlcNAc transferase (OGT). The provided OGT connection is connected to the triggering HCF1’s regulatory roles (Capotosti et al., 2011). Furthermore, a prior investigation has proposed that HCF1 serves as a transcriptional regulator for the activity of MLL (Yokoyama et al., 2004). The application of small interfering RNA (siRNA) targeting HCF1 in SNUC5/5-FUR cells leads to a decrease in the abundance of MLL mRNA and protein. Consequently, this reduction in MLL expression results in a diminished quantity of Nrf2 protein (Kang et al., 2016). The findings of this study indicate that the MLL and HCF1 constituents of the MLL/COMPASS-like complex are key regulator of Nrf2 expression. The expression levels of MLL and its associated alteration, H3K4Me3, were found to be elevated in SNUC5/5-FUR cells compared with SNUC5 cells. Furthermore, the use of siRNA to knock down MLL in these cells resulted in a significant downregulation of Nrf2 and HO-1, as reported in a previous study (Kang et al., 2016). Moreover, Kang and coworkers provided evidence indicating that the TETs and MLL proteins establish an interaction with OGT and HCF1 to modulate the production of Nrf2 (Kang et al., 2016). Multiple studies have demonstrated that the interaction between TETs and OGT enhances the DNA demethylation activity of TETs and facilitates the recruitment of a histone methyltransferase complex that is essential for establishing a chromatin environment enriched with H3K4Me3. Consequently, this leads to the activation of transcription (Ardehali et al., 2011).
Recently, it is demonstrated that OGT can stabilize the MLL protein. Specifically, when OGT was depleted in cells, it led to a reduction in MLL levels through a process involving ubiquitin/proteasome-dependent proteolytic destruction. Conversely, the introduction of OGT protein by ectopic expression resulted in the suppression of MLL ubiquitylation (Ding et al., 2015). The mRNA and protein levels of MLL were lowered in SNUC5/5-FUR cells through the use of siRNA to knock down OGT, leading to a decrease in the expression of HCF1 and Nrf2 proteins (Kang et al., 2016). Furthermore, the confirmation of Nrf2 upregulation by the TET1-OGT-MLL complex was achieved through the use of a chromatin immunoprecipitation experiment. The results of this assay demonstrated a large increase in the H3K4Me3 level on the promoter of Nrf2 in SNUC5/5-FUR cells. Conversely, the level of H3K4Me3 on the β-actin promoter, which is not associated with 5-FU resistance, remained stable (Kang et al., 2016). Therefore, it can be observed that TET1, upon activation by oxidative stress, is responsible for the demethylation of DNA in the Nrf2 promoter. Furthermore, it has been observed that TET1 actively recruits OGT and the MLL/COMPASS-like complex to the promoter region. The complex facilitates the methylation of H3K4, which then induces the transcriptional activation of Nrf2, finally culminating in the development of chemoresistance.
Cancer stem cells
Initiating tumor cells or CSCs demonstrates precise control over levels of ROS, resistance to therapy, and the recurrence of tumors (Phi et al., 2018). The maintenance of stemness requires low levels of ROS, a condition facilitated by the action of NRF2 in preserving stem cells in an undifferentiated and stem cell-like state. The reduction of NRF2 leads to a decrease in the expression of stem cell markers in distinct cancer cell types and induces cellular differentiation in models of glioma (Kahroba et al., 2019). Breast CSCs exhibit elevated expression levels of the glutamate-cysteine ligase catalytic subunit (GCLC), an NRF2 target gene, hence contributing to the enhanced antioxidative capabilities of CSCs (Kim et al., 2020). The phenomenon of prolonged and low-level exposure to arsenic stress has been observed to result in the transformation of human bronchial epithelial cells into cancer stem-like cells. This transformation is characterized by the presence of characteristics associated with EMT and an increased resistance to chemotherapy. The expression levels of miR3065/223, which inhibit N-methyltransferase, are low in CSCs of non-small-cell lung cancer (NSCLC) (Kuo et al., 2022). This low expression leads to overexpression and high activity of NRF2, hence contributing to heightened chemoresistance to cisplatin. Emerging research indicates that the expression of histamine N-methyltransferase (HNMT), an enzyme subject to regulation by ROS, is concurrent with that of the human epidermal growth factor receptor (HER2) (Kuo et al., 2022). The levels of both HER2 and HNMT are elevated in breast and lung cancer, and HNMT has a direct role in modulating NRF2 DNA-binding activity and the regulation of target genes via interacting with HER2. The establishment of a CSC-like phenotype in NSCLC is influenced by the CD133/NRF2 axis (Park et al., 2021). The overexpression of the antioxidant gene peroxiredoxin-2 (PRDX2) has been linked to CD133+CD44+ cells and is correlated with the stemness of colon CSCs. The considerable suppression of stemness maintenance, migration, invasion, and metastasis is observed with depletion of PRDX2. The downregulation of PRDX2 results in an increase in ROS levels specifically in the CD133+CD44+ cell population, rendering CSCs more sensitive to oxidative stress and chemotherapy (Park et al., 2021). Multiple studies have identified a correlation between CSC markers and Nrf2. The population of CSCs characterized by upregulated CD44 and decreased CD24 expression demonstrated elevated levels of CSC markers and Nrf2. This upregulation was observed to be augmented by the introduction of hyaluronic acid; a ligand known to interact with CD44. Elevated CD133 and CD44 expression is positively correlated with heightened NRF2 activity and the upregulation of its downstream efflux transporters, including ATP-binding cassette subfamily C member 1, in ovarian CSCs (Goto et al., 2020). The relationship between the redox signaling pathway and CSCs is indicated in Figure 5.
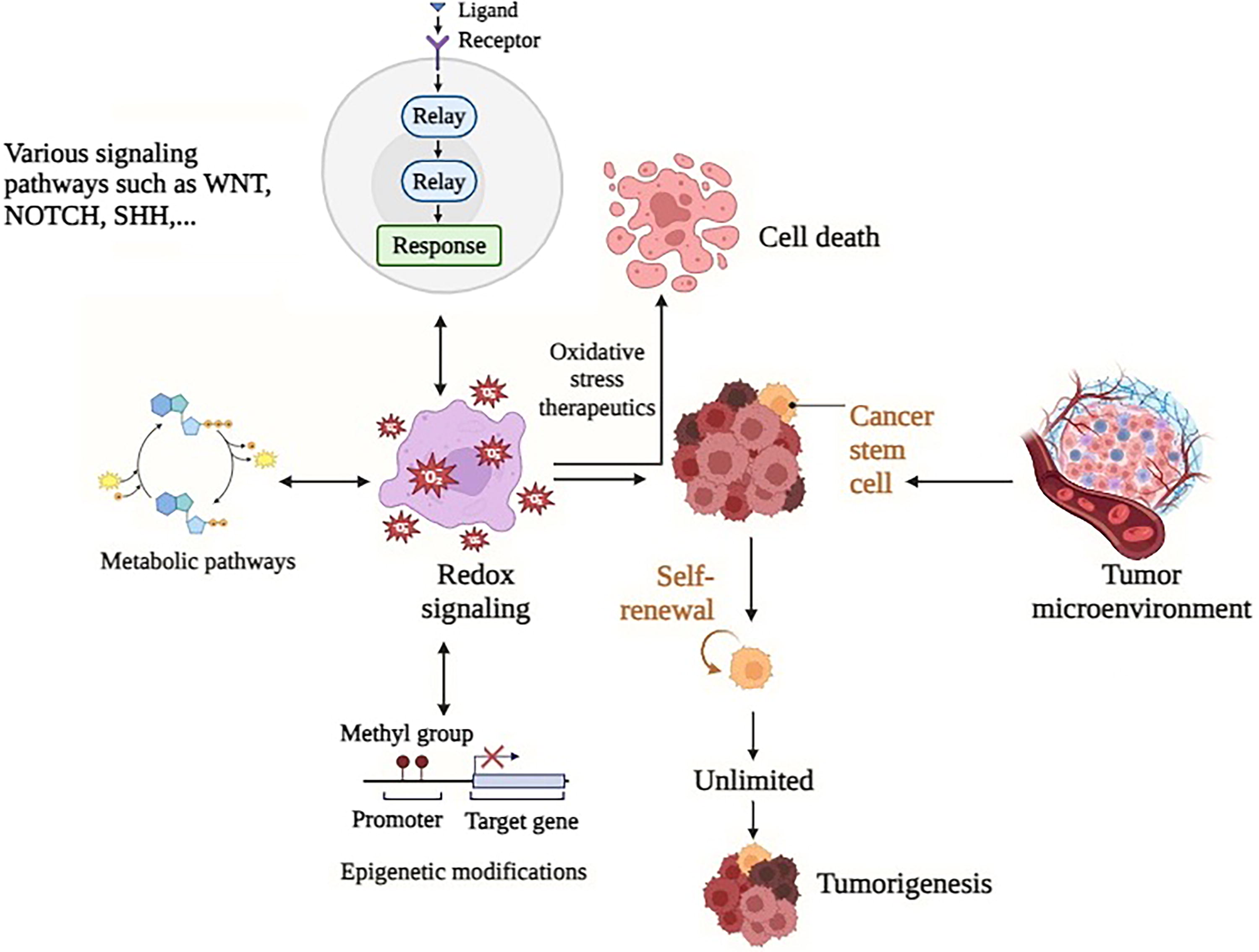
Alterations in the TME
The TME encompasses several components such as secretory products, stromal cells, tumor cells, immune cells, and noncellular constituents within the extracellular matrix. This complex milieu plays a critical role in regulating tumor growth and facilitating the process of metastasis. Hypoxia is a significant characteristic of the TME, facilitating cellular metabolic reprogramming, angiogenesis, proliferation, and conferring radio- and chemoresistance (Boveris, 1977; Li et al., 2021). The decrease in oxygen availability results in a decrease in electron transport through the mitochondrial complex, which subsequently causes an excessive generation of ROS. The process of angiogenesis plays a crucial role in facilitating the proliferation and spread of cancer cells. In the presence of hypoxia, tumor cells initiate the expression of angiogenic factors such as vascular endothelial growth factor (VEGF) through the activation of HIF proteins (Feng et al., 2018). The suppression of angiogenesis and inhibition of tumor growth in head and neck squamous cell carcinoma cells can be achieved by successfully lowering ROS, which in turn inhibits the transcription of oncogenic factors and angiogenic markers. ROS have the ability to stimulate tumor cells to release matrix metalloproteinases, hence facilitating the development of vascular structures within the TME (Engblom et al., 2016).
ROS play a pivotal part in the process of T cell activation and exert significant influence on the subsequent cellular responses, including the regulation of interleukin-2 (IL-2) and interleukin-4 (IL-4) production. The limitation of SENP7 cytosolic translocation and inhibition of CD8+ T cell metabolism in human colorectal cancer cells can be achieved by reducing T cell-intrinsic ROS (Wu et al., 2022). Nevertheless, the excessive presence of ROS caused by impaired mitochondrial function leads to the depletion of T cells. The reduction of tumor hypoxia has the potential to restrict T cell exhaustion. The redox state of T cells can additionally exert an impact on the receptors present on the membranes of these cells, thereby modulating signal transduction processes and potentially leading to alterations in immunological responses (Scharping et al., 2021). Immunosuppressive cells, such as myeloid-derived suppressor cells, have been observed to enhance the generation of ROS inside the TME, hence exerting a detrimental impact on immune responses (Wei et al., 2015). The dysfunctionality of T cells can be attributed to the presence of an immunosuppressive microenvironment. This observation implies that the modulation of ROS or the targeting of related signaling pathways could potentially increase the efficacy of cancer immunotherapy. Cancer-associated fibroblasts (CAFs) play a crucial role in the elevation of ROS levels within malignancies (Chan et al., 2017).
In addition, the involvement of NRF2 in macrophage polarization is significant, hence establishing macrophages as crucial contributors to the advancement of cancer (Jaganjac et al., 2020; Wu et al., 2019). The existence of tumor-associated macrophages (TAMs), which infiltrate both the tumor and its surrounding areas, has been frequently documented. This infiltration has been linked to a negative prognosis in solid tumors (Gentles et al., 2015). There is substantial evidence to support the notion that M2 macrophages play a crucial role in facilitating angiogenesis and neovascularization, as well as in the remodeling of stroma and the TME, ultimately leading to the dispersion of tumor cells. All of these processes have a role in the advancement of tumors. Solid tumors that exhibit rapid growth sometimes contain regions with low oxygen levels, known as hypoxic zones, as a result of inadequate oxygen supply. These regions are frequently situated within the central region of the tumor or in areas adjacent to the necrotic tissue. In a mechanistic manner, it has been demonstrated that the production of IL-4, reliant on ROS, plays a role in the recruitment and polarization of macrophages toward the M2 phenotype under hypoxic settings. TAMs have a preferential localization within hypoxic regions of a tumor, as indicated by previous research (Boveris, 1977). Tumor cells generate lactate through the process of anaerobic glycolysis, which in turn promotes the formation of ROS by macrophages and triggers the activation of Nrf2 (Feng et al., 2018). In addition, the activation of NRF2 in adjacent tumor cells is facilitated by M2-derived VEGF, hence promoting the process of EMT (Feng et al., 2018). The presence of TAMs lowers the efficiency of tumor therapeutic protocols, which applies to both radiotherapy and chemotherapy along with angiogenesis inhibition (Engblom et al., 2016). The NRF2 activation in tumor cells, achieved by the increase of target gene expression, can facilitate the growth of cancer, metastasis, and resistance to radiotherapy or chemotherapy (Rojo de la Vega et al., 2018).
Moreover, oxidative stress, specifically ROS, has the ability to alter the metabolic capabilities of regular fibroblasts, causing them to adopt the metabolic CAF phenotype within the TME. This phenomenon facilitates the process of glycolysis, hence promoting the metastasis of breast cancer cells (Zhou et al., 2017). Inflammation constitutes a significant characteristic of the TME, exhibiting a strong association with both the progression of cancer and the effectiveness of therapeutic interventions targeting cancer. The overabundance of ROS has the potential to stimulate the production of proinflammatory cytokines through the activation of NF-κB and AP-1 (Zuo et al., 2019). These transcription factors are responsive to alterations in the redox state and may have a part in inducing the NLRP3 inflammasome. The efficacy of aloin in mitigating Lipopolysaccharide (LPS)-induced inflammatory reactions has been demonstrated through its inhibition of JAK1-STAT1/3 signaling pathway activity triggered by ROS (Swanson et al., 2019). The formation of ROS also establishes a regenerative feedback loop by means of autocrine TNF-α-mediated production of inflammatory cytokines/chemokines, hence playing a role in the advancement of tumors and their therapy resistance (Lu et al., 2022).
Redox Regulation as a Therapeutic Target
Rationale for targeting redox regulation
Aforementioned, the modulation of redox processes is of significant importance in the initiation, progression, and management of malignancies, rendering it a very potential target for therapeutic interventions in the context of cancer therapy (Xing et al., 2022). The regulation of the cellular redox state is accomplished by many systems that adjust the cellular redox status by counteracting the detrimental effects of free radicals and ROS, or by reversing the creation of disulfides (Montero and Jassem, 2011). Cancer cells frequently exhibit a condition of redox imbalance, characterized by a disruption in the equilibrium between oxidants and antioxidants, leading to an elevated presence of oxidants within the cellular environment (Acharya et al., 2010). The susceptibility of cancer cells to oxidative signals is a promising therapeutic objective for the deliberate development of novel anticancer agents (Montero and Jassem, 2011). In addition, the maintenance of redox homeostasis is not just essential for physiological functions in healthy cells, but is also a significant objective for cancer cells. The antioxidant system serves a crucial function in the removal of excessive ROS in both healthy and malignant cells, with the antioxidant response being particularly enhanced in cancerous cells (Xing et al., 2022).
Redox-sensitive molecules in cancer cells
Numerous redox-sensitive compounds are known to exert a substantial influence on the behavior of cancer cells. As an illustration, H2O2 serves as a facilitative molecule in the advancement of cancer and is also a subject of interest in cancer therapy (Chaiswing et al., 2018). The upregulation of glutaredoxin (GRX), a molecule sensitive to redox reactions, has been shown to provide cellular protection against apoptosis triggered by H2O2 by modulating the cellular redox state (Montero and Jassem, 2011). The tumor protein p53 (TP53) tumor suppressor gene, responsible for encoding a DNA-binding transcription factor involved in the regulation of several cellular processes, serves as an additional illustration of a redox-sensitive protein that holds potential as a therapeutic target in the context of cancer (Eriksson et al., 2019).
As mentioned, H2O2 plays a crucial role in the advancement of cancer and has the potential to be targeted for treatment. H2O2 has the ability to regulate many signaling pathways that are involved in the processes of cell proliferation, survival, and metastasis. It does this by oxidizing cysteine residues in proteins that are sensitive to changes in the redox state (Chu et al., 2023; Li et al., 2023b). For example, H2O2 can deactivate protein-tyrosine phosphatases by oxidizing their catalytic cysteine residues, resulting in continuous activation of prosurvival pathways such as as PI3K/AKT and MAPK (Rahman et al., 2023; Xing et al., 2022). In addition to GRX and TP53, numerous additional redox-sensitive proteins and enzymes have significant impacts on the behavior of cancer cells and their resistance to therapy. For instance, the TRX system is a significant antioxidant system that controls the balance of cellular redox reactions. The overexpression of TRX and TRXR has been detected in several types of malignancies, leading to enhanced resistance against oxidative stress and chemotherapy drugs (Xing et al., 2022). In addition, PRDXs are enzymes that play a role in reducing ROS in cells. PRDXs are a group of antioxidant enzymes that facilitate the reduction of H2O2 and other peroxides. PRDX overexpression has been associated with the advancement of cancer, spread to other parts of the body, and resistance to therapy. This is achieved by safeguarding cancer cells from oxidative stress (Zhang et al., 2023a). Moreover, the Keap1-Nrf2 pathway is a biological mechanism that regulates the activity of the Nrf2 protein through the interaction with the Keap1 protein. The Keap1-Nrf2 pathway serves as a central controller of cellular antioxidant reactions. During oxidative stress, Nrf2 is liberated from its inhibitor Keap1 and moves to the nucleus, stimulating the production of different antioxidant and detoxifying enzymes. The activation of Nrf2, which is constantly present, has been reported in numerous malignancies, leading to resistance to therapy (Brzozowa-Zasada et al., 2024).
Furthermore, multiple kinases, such as ASK1, PKC, and PTEN, along with transcription factors such as NF-κB, HIF-1α, and AP-1, are sensitive to redox conditions and have significant impacts on the survival, growth, and spread of cancer cells. Their actions are regulated by oxidative alterations of cysteine residues, resulting in changes to signaling pathways and gene expression profiles (Butturini et al., 2021). The redox-sensitive molecules and pathways mentioned here have the potential to be targeted therapeutically to overcome resistance to cancer therapy. Current research in cancer is actively investigating strategies to regulate cellular redox homeostasis. These strategies include blocking antioxidant systems, causing oxidative stress, and targeting specific redox-sensitive proteins (Liu et al., 2023; Shi et al., 2023). Table 1 summarizes the redox-sensor factors and their correlation with cancer drug resistance.
Redox-Sensitive Factors as Potential Therapeutic Targets for Drug-Resistant Cancer Cells
Potential benefits and challenges of redox-based therapies
Redox-based therapeutics have demonstrated promising advantages in the context of cancer treatment. As an example, the elevation of the redox state to induce oxidative stress has been proposed as a potentially effective adjunctive treatment for aggressive forms of cancer (Chaiswing et al., 2018). Furthermore, recent preclinical investigations and clinical trials using pro-oxidant chemicals have yielded significant findings, demonstrating that these compounds elicit an activation of antioxidant defense response in healthy cells, while simultaneously triggering apoptosis in different types of cancer cells (Chaiswing et al., 2018). Nevertheless, redox-based medicines are also accompanied by several obstacles. One significant obstacle arises from the considerable variability in the redox responses of tumors across different individuals, rendering population-based treatment techniques impractical and highlighting the need for personalized therapy approaches suited to each patient (Benfeitas et al., 2017).
One further obstacle pertains to the possible adverse consequences of oxidative stress on regular cells, leading to redox imbalances and so facilitating cancer initiation and tumor progression (Xing et al., 2022). Hence, it is imperative to provide sensitive and pragmatic techniques for the identification of cellular redox status to effectively implement pro-oxidant therapy and enable the integration of redox therapy into the realm of precision medicine (Chaiswing et al., 2018). In summary, it can be inferred that redox regulation holds significant potential as a therapeutic target in the context of cancer treatment. However, it is imperative to conduct additional research to address the obstacles associated with redox-based therapies and effectively exploit their advantageous properties.
Emerging Therapeutic Strategies
Antioxidant-based approaches
The utilization of antioxidant-based strategies in the therapy of cancer is increasingly being recognized as significant, owing to the involvement of oxidative stress in the development and advancement of cancer (Luo et al., 2022b). Oxidative stress arises due to a dysregulation between ROS and antioxidants, hence causing molecular harm and perturbation of redox signaling (Gào and Schöttker, 2017). Antioxidants have the ability to mitigate the effects of damaging free radicals by their neutralization, consequently serving as a preventive or therapeutic measure against cancer (Debela et al., 2021). A number of antioxidant pharmaceuticals are presently undergoing preclinical and clinical trials, exhibiting potential in the realm of cancer therapy (Luo et al., 2022b). The aforementioned substances encompass nonenzymatic antioxidants, including vitamins, NRF2 activators, GSH esters, and N-acetylcysteine, in addition to enzymatic antioxidants, such as SOD mimics and NOX suppressors (Luo et al., 2022b).
Nevertheless, the utilization of antioxidant supplements in cancer treatment remains a subject of debate due to their potential to impede therapies that eliminate cancer cells through the production of free radicals (Singh et al., 2018). Hence, additional investigation is necessary to comprehend the function of antioxidants in the context of cancer treatment and to enhance their use.
Redox-modulating agents
Pharmacological chemicals known as redox-modulating agents have the ability to influence cellular redox homeostasis, either through direct or indirect means, by altering levels of ROS (Wondrak, 2009). The aforementioned medicines possess the ability to specifically target the unique vulnerabilities exhibited by malignant cells, hence presenting a highly promising avenue for the treatment of cancer (Wondrak, 2009). A number of small-molecule redox-active drugs, including vitamin C vitamin E, melatonin, and selenium, have demonstrated promise in the field of cancer therapy (Petronek et al., 2021). These compounds have the ability to function as pro-oxidants within cancer cells, so triggering oxidative stress and potentially resulting in cell death. However, they also maintain antioxidant properties in normal cells (Petronek et al., 2021). Nonetheless, the effectiveness of redox intervention can be compromised due to the intricate and diverse nature of redox dysregulation in malignancies. This dysregulation is influenced by factors such as the kind of tumor, stage of advancement, location, and previous exposure to chemotherapy (Wondrak, 2009). Hence, the implementation of customized medicine strategies may be imperative to effectively harness the capabilities of redox-modulating drugs for the purpose of cancer therapy.
Targeting redox-sensitive signaling pathways
The utilization of redox-sensitive signaling pathways represents a burgeoning therapeutic approach in the field of cancer treatment. The ability of ROS to engage with proteins and influence many signaling pathways implicated in the regulation of cellular growth and death has been documented (Perillo et al., 2020). The involvement of signaling pathways in cancer formation and progression is of utmost importance, and the strategic targeting of these pathways is being recognized as a viable strategy in cancer therapy. The aforementioned pathways play a role in a multitude of cellular functions, encompassing cell survival, proliferation, invasion, angiogenesis, and resistance to chemotherapy and radiotherapy (Luo et al., 2022b).
As previously stated, Nrf2 is a crucial redox-sensitive transcription factor with a significant role in activating the antioxidant response within cancer cells (Perillo et al., 2020). Under physiological circumstances, the degradation of Nrf2 occurs via its interaction with Keap1. However, when exposed to oxidative stress, Keap1 undergoes oxidation, resulting in the nucleus translocation of Nrf2. Therefore, Nrf2 induces the expression of several genes in the nucleus (Perillo et al., 2020).
Additional redox-sensitive signaling pathways encompass those that are governed by kinases and phosphatases, which oversee the phosphorylation state of transcription factors such as β-catenin, STAT, NF-κB, APE1/Ref-1, AP-1, FOXO, HIF-1α, and p53 (Gào and Schöttker, 2017). The possible prevention of tumor formation or the sensitization of cancerous cells to radiation can be achieved by targeting redox-sensitive signaling pathways (Perillo et al., 2020). However, additional study is necessary to achieve a thorough comprehension of these pathways and to formulate efficacious therapy approaches that specifically address them.
SOD mimetics
SOD mimetics are a notable breakthrough in the field of oncology, namely, as a treatment approach based on redox reactions. These chemicals are created to imitate the function of the natural enzyme SOD. This enzyme is important for controlling oxidative stress in cells by causing the breakdown of superoxide radicals into oxygen and H2O2.
Peptides have been used as ligands to produce metal complexes that imitate various SOD enzymes. Short linear sequences (Pep4-17) formed compounds with Cu(II) (Csire et al., 2017) or Ni(II) (Lihi et al., 2020) cations and exhibited encouraging inherent catalytic abilities. To overcome the significant flexibility of peptides, which reduces the likelihood of coordination, researchers have utilized a cyclic peptide (Pep3) or preorganized peptide scaffolds known as de novo miniproteins. A method that involves creating a collection of peptides and testing them with metal ions using an activity-based test has resulted in the identification of a copper complex that imitates SOD (Pep1). This complex has proven to be effective, even in cellular models of oxidative stress (Ben Hadj Hammouda et al., 2022). The application of this approach has effectively been expanded to the identification of the first peptidyl di-copper(II) complex (Pep2) that imitates the catalase enzyme (Ge et al., 2022). Notably, the selection of complexes with the intended activity depended on the specific activity assay utilized during screening. The desired activity refers to either a monoelectronic SOD activity with a peptide complex containing a single metal, or a bi-electronic catalase activity with a complex containing two copper atoms (Ge et al., 2022).
The clinical application of the native SOD enzyme is constrained by several limitations, such as its expensive manufacturing process, limited ability to penetrate cells, potential for causing immune reactions, and a short duration of circulation in the body. Similar to other drug enzymes, SODs have a high charge density and are quickly eliminated by the kidneys, which negatively impacts the enzyme’s pharmacodynamics and pharmacokinetics. In addition, the use of antioxidant therapy with SOD has not been widely adopted in clinical medicine due to its effects being characterized by a bell-shaped curve (McCord, 2008). The latter implies that SODs exhibit significant efficacy until a specific threshold, beyond which their protective effects drop sharply and may even exacerbate a condition (Batinic-Haberle et al., 2012). Nevertheless, the phenomenon of a bell-shaped curve has also been documented with SOD mimetics such mangafodipir (MnDPDP), as explained by Kurz et al. (2012). The successful creation of SOD mimetics proves the feasibility of producing small molecules that can perform the same functions as bigger enzyme systems. Consequently, a variety of metal complexes with therapeutic uses have been developed (El-Lateef et al., 2023). SOD mimetics, in contrast to SODs, possess lower molecular weights and have extended half-lives in the bloodstream. These attributes would enhance cellular accessibility, offer a more cost-effective alternative to SOD, and additionally mitigate immunogenic reactions (Riley, 1999).
The primary categories of manganese-based SOD mimetics presently consist of porphyrin, cyclic polyamines, nitroxides, salen, and PLED60 mimetics (Rosenthal et al., 2009). In vivo models have unequivocally demonstrated the advantageous impact of SOD mimetics in safeguarding against radiation injuries, enhancing the therapeutic index of anticancer drugs, mitigating ischemia-reperfusion injury, shielding against chemical stress, countering endotoxic shock, alleviating neuronal oxidative stress, managing diabetes, and combating inflammation. Riley et al. synthesized manganese cyclic polyamines, which are composed of seven-coordinate MnSOD mimic complexes utilizing the macrocycle 1,4,7,10,13-pentazazcyclopentadecane as the template ligand (Riley, 1999). The M40403 complex is the archetypal complex in its family. An MnSOD mimic functions as a catalyst to eliminate the superoxide radical. M40403 is a stable, low-molecular-weight molecule that contains manganese. It has a function and rate of catalysis that are similar to native SOD enzymes. However, M40403 is substantially smaller in size compared with native SODs. These synthetic enzymes have the benefit of not exhibiting a bell-shaped curve, unlike native SOD enzymes (Muscoli et al., 2003).
Masini et al. demonstrated the therapeutic efficacy of SOD mimetics, specifically the M40403 or M40401 complexes, in various experimental models of ischemia, intestinal reperfusion, and acute inflammation (Cuzzocrea et al., 2001). Oral mucositis, a painful condition, is a prevalent side effect experienced by certain people after radiation treatment and chemotherapy. Murphy et al. conducted a study to examine the effectiveness of M40403 in a hamster model of acute, radiation-induced oral mucositis. The results demonstrated that animals treated with M40403, as opposed to the placebo, experienced significantly reduced severity and duration of oral mucositis across various treatment schedules (Murphy et al., 2008). Filograna et al. recently conducted a study to explore the medicinal possibilities of the M40403 chemical, utilizing Drosophila melanogaster as the in vivo model. The results showed that M40403 can effectively counteract the absence of either Sod or Sod2 in Drosophila, which are equivalent to human SOD1 and SOD2 enzymes. In addition, M40403 offered defense against oxidative damage caused by paraquat treatment. This discovery is groundbreaking because it identifies specific SOD-mimetic chemicals as prospective therapeutic agents that could potentially decelerate the advancement of Parkinson’s disease (Filograna et al., 2016).
Porphyrins are linked to proteins involved in several physiological activities such as molecular oxygen transportation, cell respiration, cell apoptosis, and defense against oxidative stress (Battersby et al., 1980). An omnipresent metalloporphyrin in various biological processes is iron protoporphyrin IX, commonly referred to as a heme group. Nevertheless, metalloporphyrins have been artificially created to imitate the role of heme proteins, specifically cytochrome P-450 and SODs. Therefore, porphyrins function as both medicinal and catalytic agents. Modifying the central metal, substituents at the peripheral or meso locations, or the microenvironment, can change the properties of porphyrins. Distinct microenvironments have a role in the diverse functions of heme groups in heme proteins, such as oxygen and electron transportation, peroxide cleavage, and hydroxylation. By manipulating the structure and microenvironments of synthetic porphyrins, one can achieve varied functionality. The versatile nature of porphyrins is demonstrated by the replacement of the central metal in TMPyP [5,10,15,20-tetrakis(N-methyl pyridiniumL)porphyrin]. MnTMPyP functions as an antioxidant and demonstrates both SOD and GPX activity (Ferrer-Sueta et al., 2003). Manganese porphyrin (MnP) SOD mimetics exhibit therapeutic potential in various forms of cancer, such as head and neck, breast, brain, skin, and lymphoma. These specific SOD mimetics have demonstrated favorable attributes that could be utilized for the treatment of radiation injury, asthma, inflammation, bleomycin-induced pulmonary fibrosis, and amyotrophic lateral sclerosis (Batinic-Haberle et al., 2012). Mn porphyrins play a significant role in redox signaling in living organisms by modifying protein cysteines. This process is essential for controlling many cellular activities and maintaining a balance in the oxidation-reduction reactions within the cell. Protein cysteines have the ability to undergo different oxidative modifications, including S-glutathionylation, S-nitrosylation, and disulfide bond formation. These alterations have the potential to change protein function, activity, and relationships. MnPs assist these alterations by regulating the redox environment within cells. MnPs have the ability to engage with thiol groups in proteins, resulting in the creation of mixed disulfides or other oxidative alterations. This connection serves to safeguard proteins from irreversible oxidation and sustain their functional integrity amid circumstances of oxidative stress (Bonetta, 2018). MnPs can stimulate different antioxidant pathways by changing the redox status of cysteine residues. For example, the activation of the NRF2 pathway, which controls the expression of genes that protect against oxidative stress and cell damage, is affected by the redox state of cysteine residues in the KEAP1 protein, which inhibits NRF2. MnPs have the ability to facilitate the separation of NRF2 from KEAP1, resulting in its activation and subsequent transcription of genes that produce antioxidants (Sharma et al., 2023). MnP–s can potentially hinder proinflammatory pathways, such as the NF-κB pathway, by altering cysteine residues in crucial signaling proteins. This inhibition has the potential to decrease inflammation and shield tissues from oxidative harm (Sharma et al., 2023). MnPs have been shown to regulate redox signaling and provide protection against oxidative damage in many disease models, such as cardiovascular illnesses, cancer, and neurological disorders. MnTnBuOE-2-PyP has demonstrated its ability to safeguard cardiomyocytes from harm caused by hypoxia/reoxygenation by diminishing oxidative stress and regulating redox signaling pathways (Sharma et al., 2023; Soares et al., 2023). Mn porphyrin, specifically MnTE-2-PyP, has been shown to have an impact on many proteins and enzymes, including Keap1, NF-kB, and p38(MAPK). Although Mn porphyrins have the ability to scavenge superoxide when they come across it in living organisms, this action alone does not make them promising therapeutic agents in vivo. However, by activating the Nrf2 pathway through the oxidation of cysteine in Keap1, they increase the expression of MnSOD and other natural antioxidants. As a result, they indirectly reduce the amounts of superoxide and H2O2, thereby suppressing oxidative stress and preventing damage to normal tissue. MnPs play a role in redox signaling, which affects important metabolic processes that facilitate the repair of healthy tissue and promote apoptosis in tumors (Batinic-Haberle et al., 2018; Batinic-Haberle and Tome, 2019).
MnTnHex-2-PyP5+ is a lipophilic Mn porphyrin that has been extensively researched. The latter is particularly efficacious when used in models of oxidative stress injuries, especially in disorders pertaining to the central nervous system (Soares et al., 2022). Nevertheless, the compound MnTnHex-2-PyP5+ exhibits toxicity when administered either as a single large dose or in successive doses. The toxicity of the substance can be attributed to its micellar characteristics, which are characterized by the presence of hydrophobic alkyl chains and polar cationic nitrogens. An effective method for eliminating this toxicity is to substitute CH2 groups with oxygen atoms that are deeply embedded inside the lengthy heptyl chains. This prevents their excessive solvation, while maintaining the lipophilicity of the molecule. The inclusion of these oxygen atoms, in addition to the high lipophilicity, bestows exceptional chemical and physical characteristics upon Mn3+meso-tetrakis(N-n-butoxyethylpyridinium-2-yl)porphyrin, also known as MnTnBuOE-2-PyP5+ (MnBuOE, BMX-001) (Rajic et al., 2012). Ashcraft et al. conducted experiments to evaluate the radioprotective and radiosensitizing capabilities of MnBuOE in both normal and tumor tissues. MnBuOE effectively mitigated the adverse effects of radiation, including mucositis, xerostomia, and fibrosis, thereby safeguarding healthy tissue. Tumors treated with MnBuOE showed a significant increase in the presence of M1 TAMs, as assessed using immunohistochemistry. The latter provides a mechanistic understanding of the radiosensitizing effects in tumors. MnTnBuOE-2-PyP5+ was discovered to enhance the therapeutic range in a preclinical mouse model of head and neck cancer. This is achieved by reducing the radiation dosage needed to control the tumor, while simultaneously enhancing the resistance of normal tissues to damage caused by radiotherapy (Ashcraft et al., 2015). Table 2 summarizes the characteristics of various types of SOD mimetics and their relation to redox signaling pathways, as well as their potential in cancer therapy.
The Summary of the Different Types of Superoxide Dismutase Mimics
SOD, superoxide dismutase.
The Present Analysis Focuses on Preclinical and Clinical Investigations
Recent preclinical investigations and growing interest in exploring the potential of redox-based therapies
Recent preclinical investigations have yielded encouraging outcomes in the realm of redox-based interventions for the management of cancer. For example, a scholarly investigation emphasized the potential of enhancing the redox state to counteract oxidative stress as an effective supplementary treatment for very aggressive forms of cancer (Chaiswing et al., 2018). One potential strategy for addressing platinum-resistant tumors that are influenced by Nrf2 genes is the application of GSH inhibitors. Elevated concentrations of GSH within neoplastic cells have been observed to be correlated with the development of resistance to platinum-based chemotherapeutic agents. The process of GSH-platinum conjugation, facilitated by the enzyme glutathione S-transferase P1 (GSP1), plays a significant role in the deactivation of drugs. Moreover, it has been observed that certain platinum-resistant cell lines have elevated amounts of GSH synthesis enzymes, including GCLM and GCLC, as well as regeneration enzymes, including glucose-6-phosphate dehydrogenase and GR (Galluzzi et al., 2012; Meijer et al., 1992). The genes involved in the metabolism of GSH were under the regulation of Nrf2. The utilization of Nrf2 genes as a potential indicator of redox status in platinum-resistant malignancies is a plausible avenue of investigation. Therefore, the upregulation of these genes can be regarded as a potential prognostic biomarker for platinum-resistant malignancies, enabling the identification of individuals who would get therapeutic benefits from GSH inhibition as an adjunctive therapy. Both buthionine sulfoximine (BSO) and phenyl ethyl isothiocyanate have demonstrated potential in selectively inducing cell death in aggressive prostate cancer cells and in a preclinical animal model of ovarian cancer. BSO is a drug that inhibits the synthesis of GSH, while phenyl ethyl isothiocyanate directly binds with GSH, leading to their respective anticancer effects (Rocha et al., 2015). In support of this concept, the efficacy of NOV-002, a formulation of disodium GSSG mimetic is a liposomal compound that modulates the intracellular GSH/GSSG ratio, has been evaluated in a clinical trial including patients diagnosed with HER2-negative breast tumors, characterized by elevated GSH expression. The coadministration of NOV-002 alongside adjuvant chemotherapy, specifically doxorubicin-cyclophosphamide followed by docetaxel, yielded a favorable response rate and shown the ability to alleviate adverse effects compared with the use of adjuvant chemotherapy alone (Kirkpatrick and Powis, 2017; Sadhu et al., 2017). Several GSH inhibitors, such as Telista, Disulfiram, and sulforaphane, have undergone evaluation in clinical trials (Kirkpatrick and Powis, 2017). Due to these rationales, the utilization of GSH inhibitors in conjunction with radiation or chemotherapeutic agents that elicit cell death via oxidative stress has promise in the selective targeting of cancerous cells. Dual-function chemicals, such as MnP, possess the ability to create ROS and conjugate GSH, making them a favorable option for malignancies that exhibit resistance mediated by GSH.
A separate scholarly investigation examined the potential of redox cycling chemicals as a radical approach to selectively target cancer cells (Chaiswing et al., 2018). An illustration of this phenomenon can be observed in malignancies that have developed resistance to cisplatin, which demonstrate an elevated concentration of GSH. The formation of cisplatin-GSH complexes is associated with reduced efficacy in inducing DNA damage, decreased buildup of endogenous ROS, and altered redox state within cells. These phenotypic characteristics are commonly observed in drug-resistant malignancies. To address this disturbance, the concurrent administration of a mitochondrial SOD mimetic (MnP, which regulates mROS) alongside platinum-based medicines would promptly elicit both DNA damage and mitochondrial stress. The ability of cancer cells to effectively react to oxidative stress is compromised when numerous subcellular compartments are implicated. Consequently, various pathways leading to apoptosis-induced cell death are simultaneously triggered. Moreover, it has been observed that MnP, specifically MnTE-2-PyP, has the ability to catalyze oxidation of proteins into protein disulfides, which then react with GSH (Jaramillo et al., 2015; Jaramillo et al., 2012). This suggests that MnP may have the potential to hinder the formation of cisplatin-GSH conjugates by competitively binding to GSH. The potential exists for utilizing MnP as a therapeutic approach for the treatment of cisplatin-resistant cancer by elevating intracellular ROS levels within subcellular compartments beyond the antioxidant capability of cancer cells.
In a separate investigation, Chaiswing et al. utilized MnTnBuOE-2-PyP5+, Mn(III) meso-tetrakis (N-n-butoxyethylpyridinium-2-yl) porphyrin), or BMX-001, to examine the oxidative status of healthy ovarian cells in comparison with two types of ovarian cancer cells: OV90 human serous ovarian cancer cells and chemotherapy-resistant OV90 cells (OVCD) (Chaiswing et al., 2022). The analysis revealed that OVCD cells are under oxidative stress as a result of elevated levels of H2O2 and reduced activity of GPX and Trx1. In addition, OVCD cells exhibit heightened glycolysis activity and mitochondrial respiration in comparison to hTER7 immortalized ovarian cells and OV90 parental cancer cells. Their objective was to investigate the relationship between ovarian cell proliferation and the redox state of the cell. To achieve this, they used MnP (BMX-001), a redox-active MnSOD mimic, as a molecular tool to modify the redox state of ovarian cancer cells. OVCD cells exhibit a higher affinity for MnP compared with OV90 cells, resulting in enhanced suppression of cell growth, glycolytic activity, OXPHOS, and ATP levels in OVCD cells. The benefits were augmented when MnP was synergistically coupled with carboplatin. The effects were analyzed in relation to the rise in H2O2 levels, heightened oxidative stress, and diminished Nrf2 levels and its subsequent targets when cells were subjected to either MnP or MnP/carboplatin. It is important to highlight that MnP provides protection to the normal ovarian cell line, hTER7, from the harmful effects of carboplatin. Their findings indicate that incorporating MnP-based redox-active medications could potentially enhance the effectiveness of chemotherapy in cancer patients with platinum drug resistance by significantly boosting oxidative stress in serous ovarian cancer cells (Chaiswing et al., 2022). Moreover, the study conducted by Soares et al. aimed to demonstrate the impact of MnBuOE on the A549 and H1975 cell lines of NSCLC (Soares et al., 2023). The harmful effects of MnBuOE, either alone or in combination with cisplatin, were assessed using crystal violet and/or 3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)−2H-tetrazolium reduction tests. Two fluorescent probes were used to evaluate the levels of intracellular ROS. In addition, the effects of MnBuOE, either alone or in conjunction with cisplatin, on collective cell migration, individual chemotactic migration, and chemoinvasion were evaluated using the wound-healing and Transwell assays. The quantification of gene expression associated with migration and invasion was evaluated using reverse rranscription quantitative polymerase chain reaction (RT-qPCR). Although MnBuOE alone exhibited a decrease in H1975 cell survival at high doses, its combination with cisplatin significantly reduced the viability of the more invasive H1975 cell line, while having no effect on the A549 cell line. Nevertheless, the presence of MnBuOE alone resulted in a substantial reduction in the migration of both cell lines. The migratory inhibition was significantly enhanced when MnBuOE was administered in conjunction with cisplatin. Ultimately, the utilization of MnBuOE, either independently or in conjunction with cisplatin, resulted in a substantial decrease in cell invasion. In both cell lines, the administration of MnBuOE, either alone or in combination with cisplatin, resulted in the downregulation of MMP2, MMP9, VIM, EGFR, and VEGFA, as well as the upregulation of CDH1 (Soares et al., 2023). In summary, their data clearly show that MnBuOE has the ability to prevent the spread of cancer in NSCLC patients.
Jaramillo et al. discovered that the MnP MnTE-2-PyP5+ enhanced the effectiveness of corticosteroid (dexamethasone) treatment in a trial on lymphoma by acting on the NF-κB pathways (Jaramillo et al., 2012). Jagetia et al. recently showed that MnTE-2-PyP5+ deactivates complexes I and III in the mitochondrial ETC, leading to a decrease in mitochondrial ATP synthesis (Jagetia and Rajanikant, 2012). MnTE-2-PyP5+ exhibits glycolysis suppression when used in conjunction with 2-deoxyglucose, a compound that inhibits glycolysis (Jaramillo et al., 2012). The presence of MnTE-2-PyP5+ significantly diminishes the energy generated by the cell (Jagetia and Rajanikant, 2012). The research conducted by Rabbani et al. shown that the administration of MnTE-2-PyP5+ at a high concentration exhibited intrinsic anticancer effects in the 4T1 breast cancer mouse model. In the latter case, the significant levels of HIF-1α inhibition result in the suppression of angiogenesis (Ratcliffe et al., 2017). Li et al. synthesized a new lipophilic mimic based on MnP namely, manganese(III) 5, 10, 15, 20-tetrakis [3-(2-(2-methoxy)-ethoxy) ethoxy] phenylporphyrin chloride (molecular formula: C64H68ClMnN4O12), named HSJ-0017. This compound is intended for the treatment of disorders associated with free radicals. During their initial investigations, they discovered that HSJ-0017 is rapidly delivered to tissues with a high blood supply and accumulates in mitochondria after being injected intravenously (Li et al., 2013). Pharmacological studies initially showed that HSJ-0017 enhanced the therapeutic benefits and decreased the harmful effects caused by radiation and cyclophosphamide in C57BL/6 animals with B16/F1 murine melanoma. Recent research indicates that HSJ-0017 functions as a potent SOD/CAT mimic, augmenting the therapeutic impact of chemotherapy and radiotherapy on tumors, while mitigating their adverse side effects. Li et al. discovered that when HSJ-0017 is given at the same time as radiotherapy, it can counteract the beneficial effects of radiotherapy on tumor growth (Li et al., 2014a).
The field of redox-responsive nanoparticles (NPs) for integrated cancer therapy has been experiencing a surge in research activity (Semlali et al., 2023). The NPs have been specifically engineered to react to the redox species present in the TME, thus offering an intelligent and focused strategy for the therapy of cancer (Yang and Sun, 2022). Biothiols are frequently utilized in the development of GSH-responsive NPs due to their ability to cleave disulfide bonds (Lu et al., 2020; Li et al., 2020b; Yi et al., 2021). Disulfide linkages are frequently used in the polymer chain for the purpose of linking prodrug molecules or serving as a carrier for agent delivery. Amphiphilic polymers have the ability to undergo self-assembly, resulting in the formation of nanoscale particles. These particles exhibit an enrichment at the tumor site due to the enhanced permeability and retention effect. Furthermore, the presence of GSH triggers the cleavage of disulfide bonds, leading to the release of the encapsulated payloads. As an example, Gan et al. developed an amphiphilic polymer characterized by a significant disulfide density, which facilitated its self-assembly with a Pt(iv) prodrug (Luo et al., 2020). The assemblies demonstrated a strong sensitivity to GSH and a high efficiency in encapsulating platinum (Pt). Significantly, compared with free cisplatin, the NPs exhibited notable synergistic effects, including the clearance of GSH and induction of mitochondrial damage. These actions effectively counteracted cisplatin resistance, while also facilitating the transportation and release of a greater quantity of medicines to cisplatin-resistant cells (Hela-CDDP). Furthermore, the Hela-CDDP cells exhibited increased sensitivity to X-ray radiation as a result of the elevated intracellular platinum content of the NPs, hence conferring advantages for cancer chemoradiotherapy. The findings from in vivo investigations demonstrated that NPs had a notable ability to concentrate within tumors. Consequently, this led to a decrease in the required dosage of cisplatin administered through a single injection. As a result, the growth of tumors in cisplatin-resistant xenograft models was effectively hindered, and the incidence of severe side effects was alleviated (Luo et al., 2020). The utilization of X-rays in conjunction with NPs has been found to augment the therapeutic impact of NPs in synergistic chemoradiotherapy. Luo et al. established a connection between the chemotherapeutic agent DOX and GSH-responsive disulfide bonds, while also incorporating disulfide bonds into the polymer backbone to facilitate the delivery of the photosensitizer chlorine6 within tumor cells. The polymeric prodrug, which was crosslinked by disulfide bonds, exhibited a dual release mechanism triggered by GSH, resulting in the simultaneous release of both the drug and photosensitizer. This system holds promise for the combined application of chemo-photodynamic therapy (Luo et al., 2022a). Utilizing of NPs in redox-based anticancer therapy is shown in Figure 6.
Furthermore, empirical research has demonstrated that the modification of RS rhythms can enhance the effectiveness of redox-based anticancer therapy. The foundation of this technique is rooted in the discovery that a greater reset of rhythm was correlated with an increased therapeutic effectiveness of the drug (Kizhuveetil et al., 2020).
Clinical trials investigating the manipulation of redox processes in cancer therapy
Clinical trials have been undertaken to assess the treatment efficacy of innovative redox medicines in individuals diagnosed with cancer. The aforementioned trials have provided evidence that redox chemotherapy has successfully progressed from experimental laboratory settings to clinical application. One example is auranofin, a potentially effective anticancer compound that has been investigated for its ability to induce fast cancer cell death through redox-based pathways (Hatem et al., 2022). The findings of this study demonstrate encouraging outcomes, indicating potential for further investigation and analysis. Ongoing research is being conducted to build upon these first results and expand our understanding in this field. The findings derived from preclinical investigations as well as clinical trials have demonstrated encouraging potential within the realm of redox-based cancer treatments. For example, the utilization of redox-active pharmaceuticals such as auranofin has demonstrated promise in facilitating fast apoptosis of cancerous cells (Hatem et al., 2022).
As aforementioned, Mn(III) ortho-N-alkyl- and N-alkoxyalkyl porphyrins (MnPs) were originally created as mimics of SOD. Subsequently, it was demonstrated that these compounds can interact with a variety of RS, including ONOO−, H2O2, H2S, CO3•-, ascorbate, and GSH. Furthermore, the capacity of MnPs to oxidatively alter the functions of several proteins has been identified as their primary mode of operation, both in healthy cells and in cancer cells. The proteins include transcription factors such as NF-κB and Nrf2, MAPKs, antiapoptotic bcl-2, and endogenous antioxidative defenses. The efficacy of MnPs, specifically MnTE-2-PyP5+ (BMX-010, AEOL10113), MnTnBuOE-2-PyP5+ (BMX-001), and MnTnHex-2-PyP5+, was evaluated in various normal tissue injuries as well as in cellular and animal cancer models. MnTnBuOE-2-PyP5+ (BMX-001) has been used in four Phase II clinical trials to treat glioma (combined with radiation and temozolomide, NCT02655601), head and neck cancer patients (combined with radiation and cisplatin, NCT02990468), anal squamous cell carcinoma patients (combined with 5-FU/mitomycin, NCT03386500), and to protect the normal brain in cancer patients with brain metastases (combined with radiation, NCT03608020). MnP, MnTnBuOE-2-PyP (BMX-001), has recently finished Phase II clinical testing for high-grade glioma at nine clinical sites in the United States. The results indicate that patients saw a significant improvement in overall survival, with an average increase of around 6.6 months. Out of the 160 patients involved in the trial, 15 are still alive. The FDA has awarded it Orphan, Fast Track, and Breakthrough designations.
Metaphore Pharmaceuticals Inc. created the M40403 chemical, which has since been subjected to clinical testing. The clinical trials involving M40403 have been either discontinued (trial: NCT00033956) or terminated (trial: NCT00101621) (Weekley et al., 2017). A Phase II clinical research yielded favorable outcomes by augmenting the analgesic benefits for alleviating postoperative pain resulting from dental procedures by the combination of M40403 and morphine. A subsequent experiment was conducted on a model of postoperative pain following a bunionectomy, using a combination method (Jones and Thornback, 2007). The cyclic polyamine GC4419 SOD mimic, formerly referred to as M40419, has successfully concluded Phase 1 clinical trials conducted by Galera Therapeutics. It is intended for application in chemotherapy and radiation therapy for squamous cell carcinoma. Currently, the compound is undergoing Phase 2 b clinical trials. The latter entails assessing the effectiveness, safety, and tolerability of GC4419 as a therapy to decrease the duration of oral mucositis in head and neck cancer patients undergoing chemoradiation therapy (clinical trial: NCT02508389). Presently, investigations are underway to examine the pharmacokinetics, safety, and tolerability of the derivatives GC4702 (trial: NCT03096756) and GC4711 (trial: NCT03099824). In addition, MnBuOE is presently in the process of Phase 1/2 clinical trials for its potential application as a radioprotector of healthy brain tissues in patients with glioma (trial: NCT02655601). In addition to assisting in the survival of patients with high-grade glioma, the inclusion of MnBuOE in radiation therapy and temozolomide would also serve to prevent cognitive decline and worsening in quality of life. Holley et al. further emphasized the significance of MnBuOE in inhibiting abnormal cell signaling, which could potentially result in the development of cancer (Holley et al., 2014).
The majority of metalloporphyrins are manganese complexes, while a small number of iron porphyrins have also been examined. One of these iron porphyrins, namely, Fe(III) ortho N-substituted pyridyl analog known as INO-4885, is currently undergoing clinical trials (NCT01921426). In addition, the polyamine-based SOD mimic, GC4419, is currently undergoing clinical trials (Li et al., 2020a).
Ascorbate, a cancer-fighting agent and a redox-active compound, has shown no toxicity in Phase I clinical trials, with marginal efficacy. Its cytotoxicity is attributed to its oxidation catalyzed by endogenous metalloproteins, which lack the appropriate thermodynamics and kinetics for ascorbate oxidation (Glorieux and Buc Calderon, 2021). MnPs, which readily undergo cycling with ascorbate, may be better catalysts for its oxidation. Cellular and mouse studies support the anticancer therapeutic potential of the MnP/ascorbate system, and combined therapy has shown benefits. The efficacy of MnPs in terms of SOD mimicking parallels its ability to catalyze ascorbate oxidation, both dependent on the E1/2 of MnIIIP/MnIIP redox couple. MnPs, which are the best catalysts for ascorbate oxidation, are the most cytotoxic in cellular studies. MnPs were found to be the most successful, and were forwarded into a 4T1 sc mouse flank tumor model (Kim et al., 2012; Leu et al., 2017). The 4T1 cell line is particularly useful in evaluating drugs due to its short duration and high tumor growth success. Pharmacological ascorbate, administered intravenously to achieve high concentrations of ascorbate in the blood plasma, is a possible addition to chemotherapy treatment (Praditi et al., 2023). P-AscH- exhibits specific toxicity toward NSCLC while being relatively harmless to nonmalignant cells (Furqan et al., 2022). The selective toxicity of P-AscH- in cancer cells is attributed to inherent disparities in oxidative metabolism, particularly in iron metabolism, between malignant and nonmalignant cells (Mapuskar et al., 2017; Schoenfeld et al., 2017). Malignant cells exhibit elevated quantities of ROS, such as superoxide (O2 •-) and H2O2. These ROS can interact with iron-containing proteins, such as iron–sulfur cluster proteins and ferritin, leading to the release of redox-active iron. When P-AscH- is present, ferric iron that can undergo redox reactions is converted to ferrous iron, resulting in the production of H2O2. H2O2can exert direct toxicity on organisms by oxidizing essential biomolecules or by interacting with Fe2+ to generate hydroxyl radicals (HO•) through Fenton chemistry. HO• have the potential to cause harm to proteins, lipids, and DNA, resulting in increased sensitivity to platinum-based chemotherapy. Noncancerous cells exhibit reduced amounts of redox-active iron, H2O2, and O2 •-, resulting in a lower production of H2O2 after P-AscH- therapy (Schoenfeld et al., 2017).
Preclinical investigations conducted in laboratory settings and animal models have demonstrated that P-AscH- enhances the sensitivity of different types of solid tumors to chemotherapy and radiation treatment (Schoenfeld et al., 2017). P-AscH- has been assessed in the first phase clinical trials in conjunction with temozolomide and radiotherapy for patients with glioblastoma, with gemcitabine for locally advanced-stage pancreatic cancer, and with carboplatin-PTX for ovarian malignancies. No instances of toxicities that reached the maximum tolerated dose were recorded in the trials (Allen et al., 2019; Ma et al., 2014; Welsh et al., 2013). The treatment-related adverse events were comparable with the anticipated toxicity profile linked to typical anticancer medicines in isolation. Furthermore, throughout these first stages, the results of single-arm studies showed better outcomes compared with historical controls. P-AscH- shows promise as an additional therapy to platinum chemotherapy in patients with advanced NSCLC due to its capacity to improve the cytotoxic effects of chemotherapy and its favorable safety profile. Ascorbate is now being tested in many clinical trials, including a Phase II experiment that investigates the effectiveness and safety of combining P-AscH- with carboplatin and PTX in chemotherapy-naive patients with advanced-stage NSCLC (NCT02420314). The trial included 38 patients and successfully achieved its main goal by demonstrating an objective response rate of 34.2% (p = 0.03). All of the responses were validated as partial responses (cPR). The disease control rate was 84.2%, which includes both stable disease and complete response. The median duration of progression-free survival (PFS) was 5.7 months, while the median duration of overall survival was 12.8 months. The treatment resulted in adverse effects, including one instance of grade 5 neutropenic fever and five instances of grade 4 cytopenias. The cytokine and chemokine data indicate that the combination triggers an immunological response. Peripheral blood mononuclear cells were analyzed by immunophenotyping, revealing a higher presence of effector CD8 T cells in patients who had a PFS of at least 6 months (Furqan et al., 2022). The inclusion of P-AscH- in platinum-based chemotherapy enhanced the tumor response in advanced-stage NSCLC. P-AscH- seems to modify the immunological response of the host and requires additional research as a possible enhancer of immunotherapy. Moreover, ascorbate is in other clinical trials with pancreatic (NCT02905578), and brain (NCT02344355) cancers. All redox-based cancer therapies that are ongoing clinical trials are listed in Table 3.
Clinical Trials Related to Redox Regulation in Various Cancers
AGE, advanced glycation end-products; AR, androgen receptor; HNSCC, head and neck squamous cell carcinoma; NSCLC, non-small-cell lung cancer; GSH, glutathione; ALDH, aldehyde dehydrogenase.
Promising results and ongoing research
In addition, the modulation of RS dynamics has been linked to the enhanced therapeutic effectiveness of redox-based anticancer therapy (Kizhuveetil et al., 2020). In the study of Kizhuveetil’s team, an examination was carried out on the patterns of RS in cancer cells, with a specific focus on superoxide and hydroxyl radicals. The resetting of these rhythms is achieved through the administration of chemotherapeutic chemicals or pharmaceutical agents. The research investigation revealed that the temporal alignment of drug administration with the phase of the circadian rhythm known as the rest–activity cycle has a substantial influence on the effectiveness of anticancer drugs in inducing cell death. Two pharmaceutical compounds, namely menadione and curcumin, were subjected to experimental evaluation using two distinct types of cancer cell lines (Kizhuveetil et al., 2020).
The study revealed that curcumin had greater efficacy, as evidenced by the IC50 values of 15 µM for SiHa cells and 6 µM for HCT116 cells. The intervention caused a resetting of the cycles of superoxide and hydroxyl radicals, resulting in an increase in their respective amounts. The compound menadione exhibited IC50 values of 20 µM for SiHa cells and 17 µM for HCT116 cells, specifically impacting the levels of superoxide (Kizhuveetil et al., 2020).
The administration of the medication at various intervals within the respiratory system rhythm yielded a noteworthy increase of up to 27% in cytotoxicity. The research investigation also revealed a lack of link between the intracellular pseudo-steady-state RS and levels of antioxidants. This finding implies that the practice of utilizing antioxidant enzyme levels as indicators of intracellular oxidative stress levels may require reassessment. The study’s findings suggest that targeting the RS rhythm may have significant implications for enhancing cancer therapy. While the findings are promising, significant challenges still need to be addressed. The efficiency of redox intervention may be diminished due to the complexity and variety of redox dysregulation in tumors, which is influenced by factors such as tumor type, stage of progression, localization, and past exposure to chemotherapy (Kizhuveetil et al., 2020).
Current studies are primarily dedicated to the advancement of medications that specifically target the redox environment in cancer, with the objective of impeding crucial components of the redox system (Narayanan et al., 2020). Furthermore, ongoing research is being carried out to gain a comprehensive understanding of the involvement of mitochondrial activity in the production of ROS specific to tumors. This knowledge will have significant implications for the advancement of innovative therapeutic approaches that target redox processes (Ippolito et al., 2020). In summary, although redox-based therapies have exhibited potential in cancer therapy, additional investigation is necessary to understand these mechanisms and formulate efficacious therapeutic approaches that specifically address them.
Combination Therapies
Synergistic effects of redox-based therapies with conventional treatments
The integration of redox-based treatments with conventional therapies has demonstrated promise in augmenting the efficacy of cancer treatment. The pleiotropic mechanism of action exhibited by numerous redox chemotherapeutics, characterized by the simultaneous regulation of various redox-sensitive targets, has the potential to overcome drug resistance in cancer cells (Wondrak, 2009). Experimental evidence indicates that the coadministration of antitumoral drugs with distinct natural substances, such as curcumin and resveratrol, has been shown to augment the efficacy of cancer therapy (Castañeda et al., 2022). In addition, there has been progress in the development of NPs that are responsive to redox conditions, which can be utilized for the purpose of integrated cancer treatment. The NPs exhibit a responsive behavior toward the redox species present in the TME, so offering an intelligent and focused strategy for the therapy of cancer (Yang and Sun, 2022). Nevertheless, notwithstanding the encouraging outcomes, the investigation pertaining to redox-responsive NPs remains in the preclinical phase. It is imperative to enhance their targeting efficacy and ensure their biosafety before they can be used with confidence in forthcoming clinical research (Yang and Sun, 2022b).
The utilization of combinatorial strategies to address and overcome resistance
The development of resistance to standard protocols of radiation, chemotherapy, and molecular targeted therapy poses a substantial obstacle in the field of cancer treatment. The utilization of combinatorial techniques may prove to be more efficacious in addressing this resistance phenomenon (Cerrato et al., 2022).
One illustrative instance is ferroptosis, a type of controlled cellular demise, which has been empirically established to have a correlation with the development of resistance to cancer treatment. Recent research findings indicate that ferroptosis has a significant impact on cancer treatment through its disruption of target molecules and involvement in the progression of cancer. Furthermore, there have been reports indicating that the utilization of ferroptosis can serve as an approach to overcome resistance encountered in targeted therapy (Zhang et al., 2022). Ferroptosis is a regulatory mechanism of nonapoptotic cell death that is facilitated by elevated levels of lipid ROS. This process is intricately linked to cysteine metabolism and oxidative stress (Dixon et al., 2012). The xCT system suppression, located on the cell membrane, results in a decrease in the production of intracellular GSH synthesis precursors. This then leads to the indirect inhibition of GPX4, finally causing the accumulation of harmful lipid peroxides and triggering the process of ferroptosis (Imai et al., 2017). The typical manifestation of ferroptosis is the deadly metabolic imbalance resulting from the depletion of GSH or the inactivation of GPX4, as described by Friedmann Angeli et al. in 2014. There is a growing body of evidence suggesting that the induction of ROS has a role in the process of ferroptosis, which in turn contributes to the suppression of tumor growth and enhances the responsiveness of cancer cells to chemotherapy. The activation of NRF2 has been found to have a role in the development of resistance in head and neck cancer cells against sorafenib. Conversely, inhibiting the NRF2-antioxidant response element pathway has been shown to trigger ferroptosis and effectively counteract the resistance to chemotherapeutic drugs in head and neck cancer (HNC) cells (Roh et al., 2017). In a study conducted by Sato et al., (2018), it was observed that ovarian cancer cells that had developed resistance to cisplatin exhibited heightened sensitivity to cisplatin when pretreated with erastin, a compound known to promote ferroptosis. These findings indicate a positive synergistic impact of the two medications (Sato et al., 2018). Furthermore, the induction of ferroptosis through the use of erastin has been observed to effectively reverse cisplatin resistance in cells of HNC. In addition, erastin has been discovered to greatly increase the anticancer effects of the primary chemotherapeutic drugs cytarabine and doxorubicin in cells of HL60, a cell line commonly used in research (Roh et al., 2016). Hence, it is plausible that ferroptosis could exert a significant influence on the process of carcinogenesis. Consequently, the utilization of medications or compounds that instigate ferroptosis has potential as an adjunctive chemotherapeutic approach for managing drug-resistant tumors in the realm of clinical application. However, gaining a comprehensive understanding of the significance of oxidative stress and ferroptosis in cancer development and tumor progression could yield important insights.
Furthermore, the investigation of poly(ADP-ribose) polymerase (PARP) inhibitors in conjunction with other therapeutic modalities has been undertaken as a potential strategy to surmount resistance to PARP inhibitors in cancer treatment (Bhamidipati et al., 2023). PARP inhibitors have undergone extensive evaluation in clinical trials, particularly in the context of breast cancer and ovarian cancer management, and have demonstrated remarkable efficacy. The primary role of PARP is to detect and mend breaks in DNA strands. Consequently, the therapeutic impact of inhibiting PARP is commonly related to the disruption of DNA repair processes (Bhamidipati et al., 2023). Hou et al. (2018) revealed that the inhibition of PARP leads to an elevation in oxidative stress levels, which subsequently contributes to the observed antitumor effect. The researchers demonstrated that PARP1 has significant expression levels in samples of high-grade serous ovarian carcinoma, and its functionality is essential for the unimpeded proliferation of ovarian cancer cells (Hou et al., 2018). The inhibition or depletion of PARP results in an augmentation of DNA damage and a concomitant rise in ROS levels. Significantly, the antioxidant N-acetylcysteine effectively mitigated the induction of DNA damage and the disruption of cellular proliferation caused by PARP inhibition or depletion. The researchers additionally demonstrated that the expression levels of NOX 1 and 4 were dramatically increased as a result of PARP inhibition, and these enzymes played a partial role in the initiation of oxidative stress. The growth inhibition of tumor xenografts lacking PARP1 was partially alleviated by the depletion of NOX1 and NOX4. Their results indicated that, apart from impeding the repair of DNA damage, the inhibition or depletion of PARP may have an additional anticancer impact by increasing oxidative stress in cells affected by ovarian cancer (Hou et al., 2018).
Conventional chemotherapy exerts its cytotoxic effects by selectively producing an elevated level of oxidative stress within cancer cells, well described in Li et al. (2023b). According to Young et al., anthracycline medicines, including doxorubicin, daunorubicin, and adriamycin, have demonstrated anticancer properties against both solid tumors and hematological malignancies. This is attributed to their ability to impede DNA synthesis and stimulate the generation of ROS (Young et al., 1981). Moreover, empirical studies have shown that synergistically combining chemotherapy with certain natural compounds can potentiate the efficacy of cancer treatment. An illustration of the combined effect of anethole and adriamycin on the induction of ROS-mediated apoptosis in cells of triple-negative breast cancer has been demonstrated by Arumugam et al. (2021). According to Pan and colleagues, (2021), the coadministration of doxorubicin and oxymatrine demonstrated enhanced synergistic effects on colorectal cancer cells both in laboratory settings and in living organisms, surpassing the individual efficacy of either doxorubicin or oxymatrine alone. Platinum-based chemotherapeutic agents, namely cisplatin and carboplatin, have been observed to effectively sustain elevated levels of ROS (Pan et al., 2021). This mechanism leads to the initiation of DNA damage and the subsequent apoptosis in cancer cells by inducing oxidative stress. Notably, these drugs primarily exert their effects by promoting excessive generation of mROS while concurrently diminishing the intracellular levels of antioxidants, such as GSH, in certain types of human malignancies (Mikuła-Pietrasik et al., 2019). Nevertheless, the administration of cisplatin can induce oxidative stress, leading to adverse effects in healthy cells. One such impact is ototoxicity, which arises from the overproduction of ROS in cochlear cells (Sheth et al., 2017). Furthermore, it has been discovered that PTX, a substance utilized in the treatment of tumors to impede cell division, can stimulate ROS production by augmenting the functionality of NOX (Alexandre et al., 2007). The efficacy of ROS-based therapies can significantly differ based on factors such as the specific tumor type, its location within the body, and the stage of cancer progression. The efficacy of therapies might vary across different tumor types, and the improper use of ROS modulation therapy may not only lack effectiveness, but could even facilitate the advancement of malignancy. The resolution of the contentious adverse effects of pro-oxidative anticancer therapy is anticipated to be achieved by the implementation of more sophisticated regulatory measures (Wang et al., 2021).
In addition, the therapeutic effects of MnPs have been demonstrated in numerous diseases with oxidative stress, including prostate cancer, head and neck cancer, and glioma. MnP-based SOD mimics have shown remarkable efficacy in radioprotection of normal tissue and radio-, chemo-, and ascorbate-driven sensitization of tumor (Mapuskar et al., 2019). These differential effects have enabled MnTnBuOE-2-PyP5+ to enter Phase I/II clinical trials. In prostate cancer, MnPs have demonstrated remarkable radioprotective effects, such as radioprotection of erectile function, protection of testes, and preservation of prostate architecture during prostate radiation. In a nontumor bearing mouse model, MnTnBuOE-2-PyP5+ showed efficacy as a radioprotector of those tissues. In a mouse sc xenograft model of head and neck FaDu cancer cell line, MnP radiosensitized tumor suppressing its growth (Ashcraft et al., 2015). Glioma has also shown promising results in MnPs, protecting brain white matter, neurocognition, and hippocampal neurogenesis but not tumor (Leu et al., 2017; Yulyana et al., 2016). Remarkable radioprotection of the rectum by MnTE-2-PyP5+ was demonstrated in a rat model, suppressing radiation-based damage at both acute and chronic levels (Kosmacek et al., 2016). Several cellular studies confirmed the therapeutic potential of MnPs in the treatment of breast cancer, with MnP able to induce apoptosis of triple-negative mammary 4T1 cell line, MCF-7, and inflammatory breast cancer lines SUM-149 and SUM-190 when combined with H2O2, such as ascorbate (Tovmasyan et al., 2017).
In summary, the integration of redox-based treatments with conventional medicines has demonstrated promise in augmenting the efficacy of cancer treatment and surmounting resistance. Nevertheless, additional investigation is required to profoundly understand these pathways and formulate efficacious therapy approaches that specifically address them.
Challenges and Future Directions
Limitations of redox-based therapies
Redox-based therapies have exhibited potential in the realm of cancer treatment; yet, they encounter many obstacles. One of the primary constraints lies in the variety observed in the redox responses exhibited by tumors across various patients. The presence of variability within a population necessitates the use of patient-specific therapy techniques, while population-based treatment strategies may prove ineffective (Benfeitas et al., 2017).
One additional obstacle pertains to the limited comprehension regarding the specific targets of redox-based pharmaceutical agents. The precise targets of numerous medications remain incompletely elucidated, and there exists uncertainty over whether these drugs effectively target the optimal redox effectors. There are lingering uncertainties pertaining to the selectivity of the targeted compounds in their ability to treat cancer cells while sparing healthy cells. In addition, there is a question regarding whether these compounds represent the optimal target within the given pathway. Furthermore, the identification of combined therapeutic treatments that could enhance the efficacy of the treatment remains an area of inquiry (Benfeitas et al., 2017).
Potential toxicities and safety concerns
Although redox-based medicines have demonstrated potential efficacy, they are also accompanied by possible toxicities and safety considerations. For example, hemoglobin-based oxygen carriers, utilized in redox-based therapeutics, have been linked to temporary hypertension, gastrointestinal complications, increase of pancreatic/liver enzymes, and cardiac/renal damage in human subjects (Alayash, 2019). Furthermore, chemotherapy, a frequently used clinical modality for the management of cancer, predominantly elicits oxidative stress to increase the amounts of ROS and perturb the balance of redox homeostasis within cancerous cells. Exceeding a specific threshold, elevated levels of ROS might have detrimental effects on tumor cells. Nevertheless, the process by which acquired chemoresistance occurs may seem contradictory, given that recent research has indicated that administering medicines with antioxidant properties to resistant cells might actually decrease the levels of ROS within the cells (Ren et al., 2023).
Future research directions and innovative strategies
Despite the inherent difficulties and potential adverse effects, redox-based medicines exhibit encouraging prospects for future investigation and offer novel approaches. Nanoceria, also known as cerium oxide NPs, have garnered significant attention as a new emerging strategy for cancer therapy. The effects of nanoceria on ROS levels in biological systems are influenced by surface features and the surrounding environment (Tang et al., 2023). These effects can either be antioxidative or pro-oxidative in nature.
In addition, the notion of synthetic lethality, which refers to the selective toxicity of drugs toward cancer cells exhibiting mutations in tumor suppressor genes or increased expression of oncogenes, is highly promising (Wondrak, 2009).
The influence of the oxidative milieu on tumor cells, immune cells, and their interplay is of considerable consequence, indicating that ROS not only contribute to established anticancer treatments but also to tumor immunotherapy. The immune system is currently directing its attention on T cells as a potential therapeutic avenue for cancer treatment, owing to their cytotoxic properties. Adoptive cell therapy (ACT) and immune checkpoint inhibitors are key constituents of immunotherapeutic approaches used in the treatment of malignancies (Waldman et al., 2020). Immune checkpoint proteins serve the purpose of inhibiting excessive activation of T cells, hence safeguarding healthy tissues from immune-mediated damage. Nevertheless, neoplastic cells have the ability to enhance the expression of immune checkpoint proteins, such as programmed cell death protein 1/programmed death ligand 1 (PD-L1), hence evading the surveillance and response of the immune system. The relationship between PD-L1 and ROS is highly correlated, and the increased presence of PD-L1 on the cell membrane of cancer cells is linked to heightened levels of HIF-1α expression. The occurrence of hypoxia induces a prompt and specific upregulation of PD-L1 within tumors, and this increase is reliant on the presence of HIF-1α, which is closely linked to ROS. Multiple research studies have examined the impact of different pharmacological regulators of ROS, such as inhibitors of thioredoxin reductase 1, modulators of Nrf2 signaling, and scavengers of free radicals, including ethaselen, edaravone, and A. formosanus extract, on the expression of PD-L1. In addition, these studies have investigated the relationship between NF-κB signaling pathways, HIF-1α, ROS, and the expression of PD-L1 (Bailly, 2020). Gaining insight into this correlation can facilitate the development of novel medication combinations involving anti-PD-L1 and ROS modulators (Shurin and Umansky, 2022). The utilization of cytotoxic lymphocytes in ACT has proven to be a highly efficacious kind of immunotherapy in the treatment of tumors. Nevertheless, a TME that is not conducive to optimal conditions, characterized by increased levels of ROS, has the potential to hinder the functionality of T cells. Extensive research indicated that ROS play a significant role in the activation, proliferation, and survival of T cells. This suggests that manipulating ROS levels within the immunological milieu may have the potential to augment the immune response against tumors. The application of antioxidants, such as GSH and NAC, before treatment, resulted in a notable enhancement in the longevity of adoptively transferred cells. Certain medications have been documented to function as potent activators of Nrf2, thereby enabling cells to develop a heightened resistance to ROS. These treatments have the potential to augment the effectiveness of ACT (Renken et al., 2022).
Nevertheless, additional investigation is required to thoroughly understand the significance of the redox state in aggressive malignancies and determine how to effectively harness the alteration in redox status to enhance cancer treatment. The redox state’s biological importance presents a novel opportunity for the advancement of precision medicine in cancer treatment (Chaiswing et al., 2018). In summary, although redox-based medicines have demonstrated potential in cancer therapy, they encounter several obstacles and potential adverse effects. Additional research is indispensable to achieve a thorough insight of these pathways and to devise efficacious therapeutic approaches that specifically address them, notwithstanding the promising nature of future research areas and creative tactics.
Conclusion
The management of redox processes is of paramount importance in the context of cancer growth and in the acquisition of resistance to therapeutic interventions. ROS, known for their involvement in redox control, have been found to interact with multiple facets of cancer, including tumorigenesis, proliferation, metastasis, and treatment resistance (Xing et al., 2022). Cancer cells frequently exhibit an elevated level of ROS in comparison with normal cells, hence contributing to their aggressive phenotype and resistance to therapeutic interventions (Chaiswing et al., 2018). Furthermore, cancer cells have the ability to enhance the synthesis of antioxidants, resulting in a heightened resistance toward chemotherapy, radiotherapy, and immunotherapy (Narayanan et al., 2020). Hence, the comprehension and manipulation of the redox state within cancer cells present a viable approach toward enhancing cancer treatment and surmounting drug resistance (Xing et al., 2022).
There are a growing number of therapeutic approaches that are being developed to specifically address the redox status within cancer cells. One potential strategy involves stimulating the generation of ROS within cancer cells, thereby elevating oxidative stress levels and ultimately leading to cellular demise (Chaiswing et al., 2018). An additional approach involves the inhibition of crucial participants within the redox environment to regulate the generation of ROS, hence impeding the formation of tumors or enhancing the susceptibility of cancer cells to radiotherapy (Narayanan et al., 2020). Moreover, the notion of synthetic lethality, which refers to the selective cytotoxicity of drugs toward cancer cells exhibiting either loss-of-function mutations in tumor suppressor genes or overexpression of oncogene, holds significant potential (Wondrak, 2009). Furthermore, the scientific community has successfully engineered redox-responsive NPs to enable a synergistic and localized strategy for combating cancer (Mollazadeh et al., 2021).
The advancement of cancer treatment hinges upon a deeper comprehension of the involvement of redox regulation in cancer and the formulation of efficacious therapeutic approaches that specifically address this mechanism. The redox state holds significant biological importance and has the potential to open up new possibilities for precision therapy in the field of cancer research (Chaiswing et al., 2018). Nevertheless, there exist various obstacles that must be addressed to advance in this field. These problems encompass the variability in redox reactions observed in tumors across different individuals, the limited comprehension about the specific targets of redox-based medications, as well as the potential risks and safety considerations associated with their use (Zhang et al., 2023b). Hence, it is imperative to do additional study to clarify these pathways and devise efficacious therapeutic approaches that specifically address them. Redox-responsive treatments have great promise for achieving more favorable therapeutic outcomes in the future, owing to their improved tumor selectivity, biosafety, and sensitivity.
SOD mimetics are synthetic substances created to imitate the function of the natural SOD enzyme. The SOD enzyme is responsible for controlling oxidative stress by facilitating the conversion of superoxide radicals into oxygen and H2O2 through a process called dismutation. Their therapeutic potential stems from their capacity to regulate the redox environment within cancer cells, thereby affecting multiple cellular processes that contribute to the advancement of cancer and its resistance to treatment. Nonetheless, the practical implementation of SOD mimetics encounters various obstacles, such as the inconsistency in oxidative stress reactions among distinct tumors and within different areas of the same tumor. Consequently, it becomes arduous to anticipate their effectiveness on a broader scale. Another obstacle is the possible detrimental impact of oxidative stress on regular cells. SOD mimetics strive to specifically attack cancer cells, but they could disturb the redox equilibrium in healthy cells. This might result in unexpected side effects and perhaps aid in the onset and advancement of cancer. The inherent SOD enzyme is constrained by factors such as a substantial charge density, swift clearance by the kidneys, and a brief duration in the body’s circulation, which adversely affect its pharmacokinetics and pharmacodynamics. Although there are challenges, SOD mimetics show great potential for the future of cancer treatment, especially in addressing resistance. Progress in design has resulted in chemicals that have reduced molecular weights and longer half-lives in the bloodstream. This improves their ability to reach and specifically target tumor cells. SOD mimetics can enhance the effectiveness of traditional treatments by modifying the redox environment, hence increasing the sensitivity of cancer cells and enhancing therapeutic outcomes. Customized medical strategies are required to adapt SOD mimetic therapy to the unique redox characteristics of an individual’s tumor, resulting in enhanced and focused treatment results. Continuing preclinical and clinical research is essential to better understand the processes by which SOD mimetics work and improve their therapeutic effectiveness.
Footnotes
Author Disclosure Statement
The authors declare that they have no known conflicts of interest/competing interest.
Funding Information
No funding was received to assist with the preparation of this article.