
Abstract
Aims:
Endothelial inflammation is crucial in the initiation and progression of atherosclerosis, whereas cholesterol crystals (CCs) play a key role in atherogenesis. However, the effects and origination of CCs on endothelial inflammation are not well understood.
Results:
In the present study, we found that CCs appeared in the subendothelial space of the partially ligated carotid artery only 1 week after Western diet feeding, which was before immune cell infiltration. In vitro, CCs were generated by human aortic endothelial cells (HAECs) upon low-density lipoprotein treatment. These endothelial cell (EC)-derived CCs increased the expression of intercellular cell adhesion molecule-1 (ICAM1) and vascular cell adhesion molecule-1 (VCAM1). Mechanistic studies demonstrated that CC-induced pyroptosis was critical for endothelial inflammation. Knockdown of gasdermin D (GSDMD) inhibited CC-induced endothelial inflammation, attenuated plaque progression, and decreased macrophage content. Accordingly, the inhibition of GSDMD reduced the CC-induced increase of VCAM1 and ICAM1 in HAECs. Furthermore, CC-mediated pyroptosis was found to be caspase-1 (CASP1)-dependent. Inhibition of CASP1 also reduced endothelial inflammation and attenuated plaque progression. The expression of GSDMD was increased in human atherosclerotic plaques. These findings identify that EC-derived CCs may be an important driving force in the pathogenesis of atherosclerosis.
Innovation:
This study uncovered a new understanding that CC-induced pyroptosis contributes to endothelial inflammation in early atherogenesis.
Conclusion:
Targeting endothelial GSDMD or CASP1 contributes to the repression of vascular inflammation and atherogenesis. Antioxid. Redox Signal. 41, 201–215.

Introduction
Atherosclerosis is the most frequent underlying cause of coronary artery disease (CAD) (Libby and Theroux, 2005). Endothelial dysfunction and vascular inflammation are crucial in the initiation and progression of atherosclerosis (Xu et al., 2021). Studies have demonstrated that cholesterol cells (CCs) may be an important driving force in the pathogenesis of atherosclerosis, as they have been described as an important determinant of plaque stability (Geng et al., 2003). Plaque formation is a highly dynamic process that begins with entry of cholesterol into the arterial wall via low-density lipoprotein (LDL) particles. Once LDL is oxidized, it is assimilated by macrophages to become foam cells. It is also widely accepted that CCs originate from macrophage-derived foam cells during the development of atherosclerosis (Kellner-Weibel et al., 1999; Janoudi et al., 2016). In addition, CCs can be formed by smooth muscle cells and endothelial cells (ECs) at different stages of atherosclerosis (Baumer et al., 2020a, 2020b; Baumer et al., 2017; Ho-Tin-Noé et al., 2017). Thus, CCs can be produced in various ways during different stages of atherosclerosis, and the origin of CCs in early atherogenesis is yet to be determined.
Innovation
CC deposition occurs in the subendothelial space of carotid arteries before signs of macrophage and neutrophil infiltration. ECs generated CCs in the early stage of atherogenesis. CC-induced pyroptosis is essential for endothelial inflammation in early atherogenesis.
It is reported that the presence of CCs in early pathogenesis promotes plaque vulnerability and rupture (Baumer et al., 2020a, 2020b; Baumer et al., 2017; Ho-Tin-Noé et al., 2017). In the early stage, the appearance of CCs has been associated with the induction of endothelial inflammation (Baumer et al., 2020a, 2020b). The NLR family pyrin domain containing 3 (NLRP3) inflammasome has been reported to be critically involved in plaque formation (Duewell et al., 2010). CCs induce complement-dependent inflammasome activation and cytokine release (Samstad et al., 2014). In contrast, CCs also elicit a response toward inflammasome-independent production of interleukin 1 alpha (IL-1α) (Freigang et al., 2013). Therefore, the mechanisms through which CC-mediated endothelial inflammation contributes to early vascular damage are still not well understood. Previous study shows that small crystals appeared as early as 2 weeks after the start of an atherogenic diet in atherosclerotic sinus lesions, indicating that CCs are able to appear in the area where plaque is prone to form (Duewell et al., 2010). The curvature and bifurcations along the arterial tree, where shear stress is low and disturbed, are commonly considered as atheroprone vascular regions. Thus, we used partial ligation of the left common carotid artery (LCCA) along with Western diet in apolipoprotein E (ApoE)-deficient (ApoE−/−) mice to study the formation and effect of CCs during atherogenesis. In this study, we found that CC deposition in the subendothelial space of carotid arteries occurred after only 1 week of a Western diet, which was before signs of macrophage and neutrophil infiltration appeared. Through an in vitro investigation, we further confirmed that CCs were generated from ECs under conditions of hyperlipidemia. Furthermore, we found that CCs induced endothelial inflammation through caspase-1 (CASP1)-dependent pyroptosis. Knockdown or inhibition of gasdermin D (GSDMD) suppressed CC-induced endothelial inflammation, attenuated plaque progression, and decreased macrophage content and IL-1β level in plaques. Additionally, the expression of GSDMD increased in human atherosclerotic plaques. Essentially, these data identify the importance of EC-derived CC-induced pyroptosis in the initiation and progression of atherosclerosis. Therefore, targeting GSDMD and CASP1 in the early stage of atherosclerosis could help alleviate endothelial inflammation and atherosclerosis.
Results
CCs appeared in the carotid artery in the early stage of atherogenesis
The appearance of CCs in the subendothelial space was noted after only 1 week of Western diet, and its content increased in accordance with lipid deposition (Fig. 1A,B). Macrophage infiltration began to occur about 2 weeks after Western diet, which was after the appearance of CCs (Fig. 1A). Consistent with this, we also observed the presence of neutrophils, CD45+ immune cells, and CCs, and we found that CCs appeared in the subendothelial space before the infiltration of neutrophils and CD45+ immune cells 1 week after Western diet feeding (Fig. 1C,D). To further verify the appearance of CCs, we used fluorescent dye (cholesteryl-BODIPY-C12), which is a cholesterol analog that can label CCs with high affinity (Vedre et al., 2009) to stain CCs. Our result showed that CCs were stained with BODIPY and found to be protruding through the intimal surface (Fig. 1E). These results suggest that CC deposition in the subendothelial layer of the carotid artery occurs before signs of immune cell infiltration in the early stage of atherogenesis, indicating that CCs may act as the initiator of plaque formation.
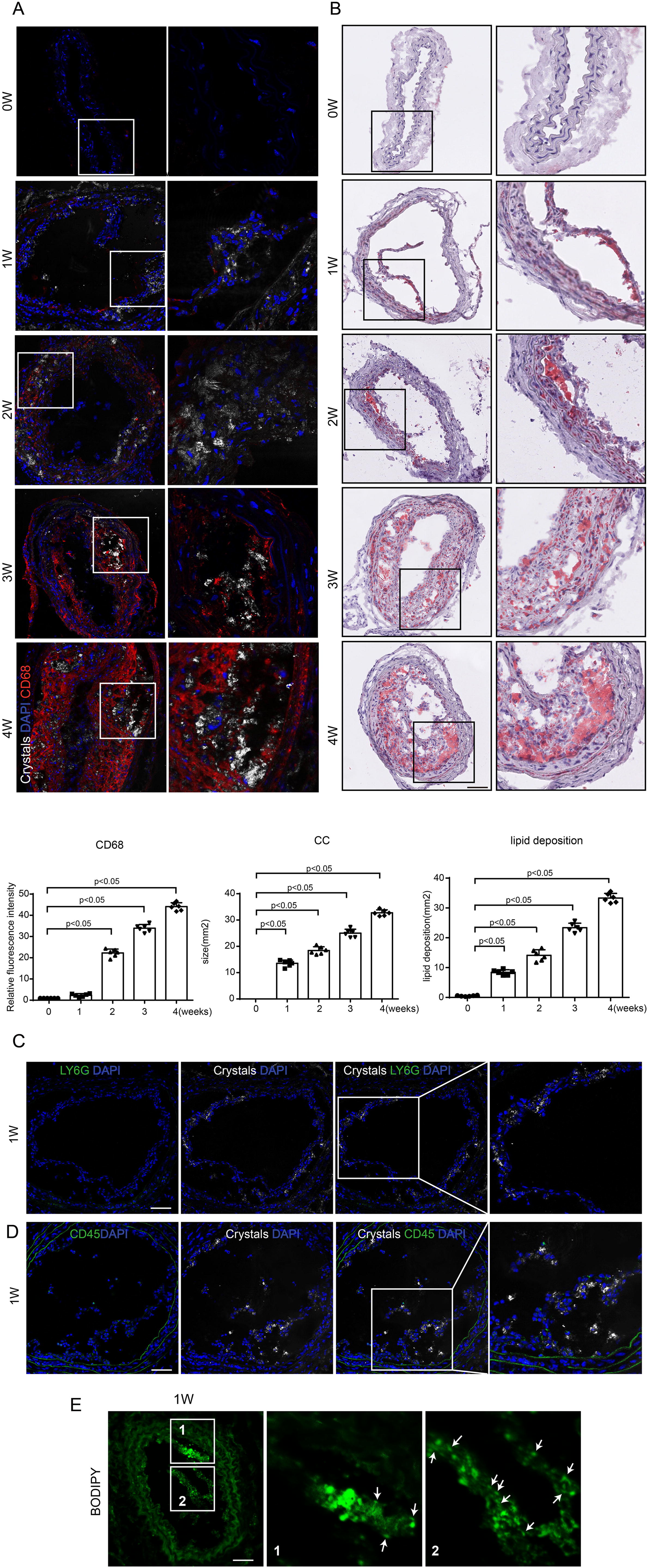
ECs generate CCs upon hyperlipidemic condition in vitro, which promotes endothelial inflammation
Plaque formation is a highly dynamic process that begins with entry of cholesterol into the arterial wall via LDL particles. In the context of atherosclerosis, LDL is transcytosed through ECs and deposited in the intima (Vasile et al., 1983). During this process, some of the lipoprotein particles become oxidatively modified, causing the activation of the overlying ECs and subsequent expression of adhesion molecules that attract monocytes to the area (Tabas et al., 2007). To determine if ECs can take up and metabolize LDL during a hypercholesterolemic state, HAECs were cultured and treated with Dil-labeled LDL or LDL followed by cholesteryl-BODIPY staining. We noted signs of robust LDL uptake with prominent lipid droplets visible within HAECs (Fig. 2A). Of particular interest, we observed needle-shaped CC formations that appeared 48 h after exposure to LDL (Fig. 2B). Then, we determined the colocalization of CCs, EC marker CD31, and CD68, as well as the colocalization of CCs, EC marker vWF, and CD45. It was seen that CCs accumulated close to the endothelium, and macrophages or CD45+ immune cells were barely seen (Fig. 2C,D).
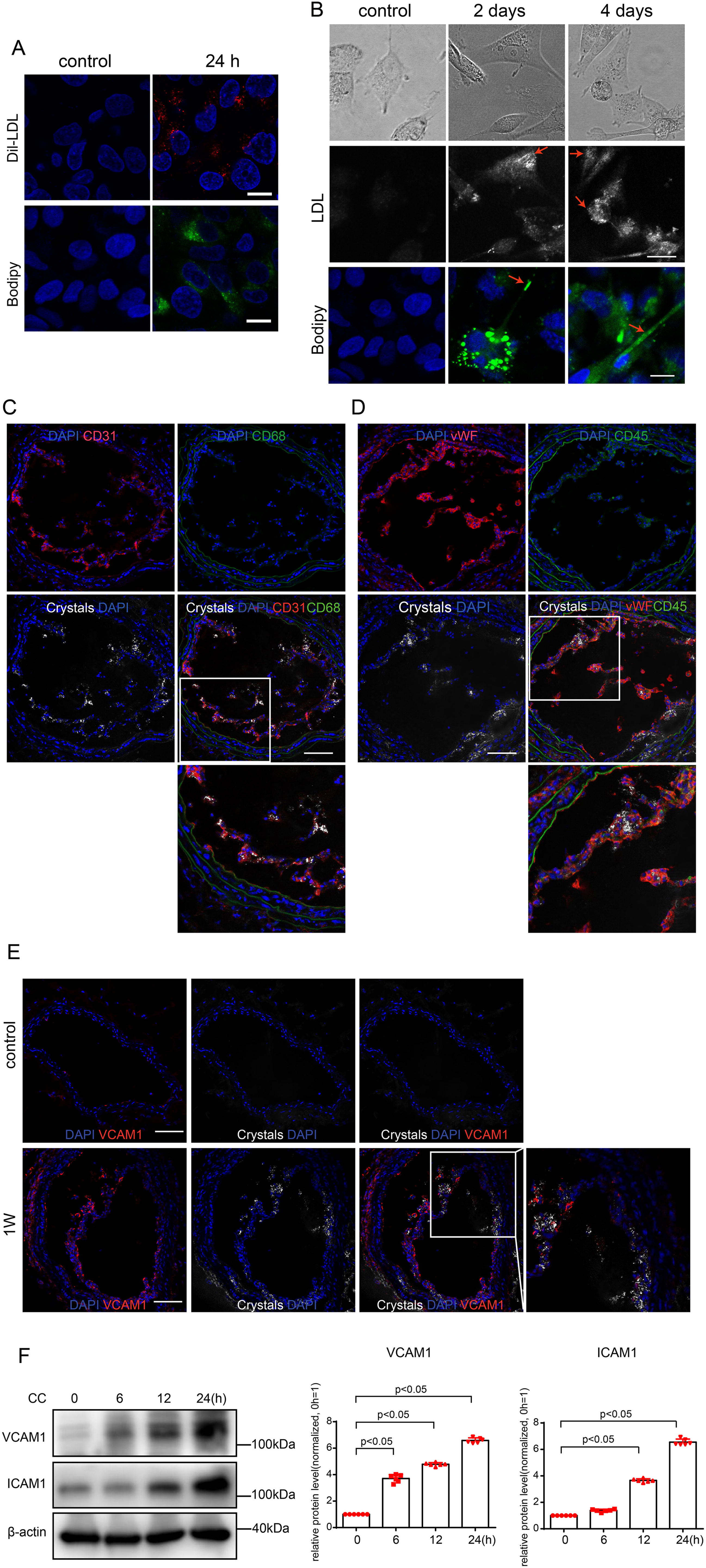
It was demonstrated that CCs promote the interaction of ECs and monocytes (Pichavaram et al., 2019). Next, we wondered whether such EC-derived CCs were sufficient to induce endothelial inflammation. Our data showed that the expression of vascular cell adhesion molecule-1 (VCAM1) increased, which was associated with CC accumulation (Fig. 2E). Accordingly, the expression of ICAM1 and VCAM1 in HAECs increased in a time-dependent manner after CC stimulation (Fig. 2F). These results suggest that EC-derived CCs promote endothelial inflammation and may therefore contribute to the development of atherosclerosis.
EC-derived CCs promote endothelial inflammation by inducing pyroptosis
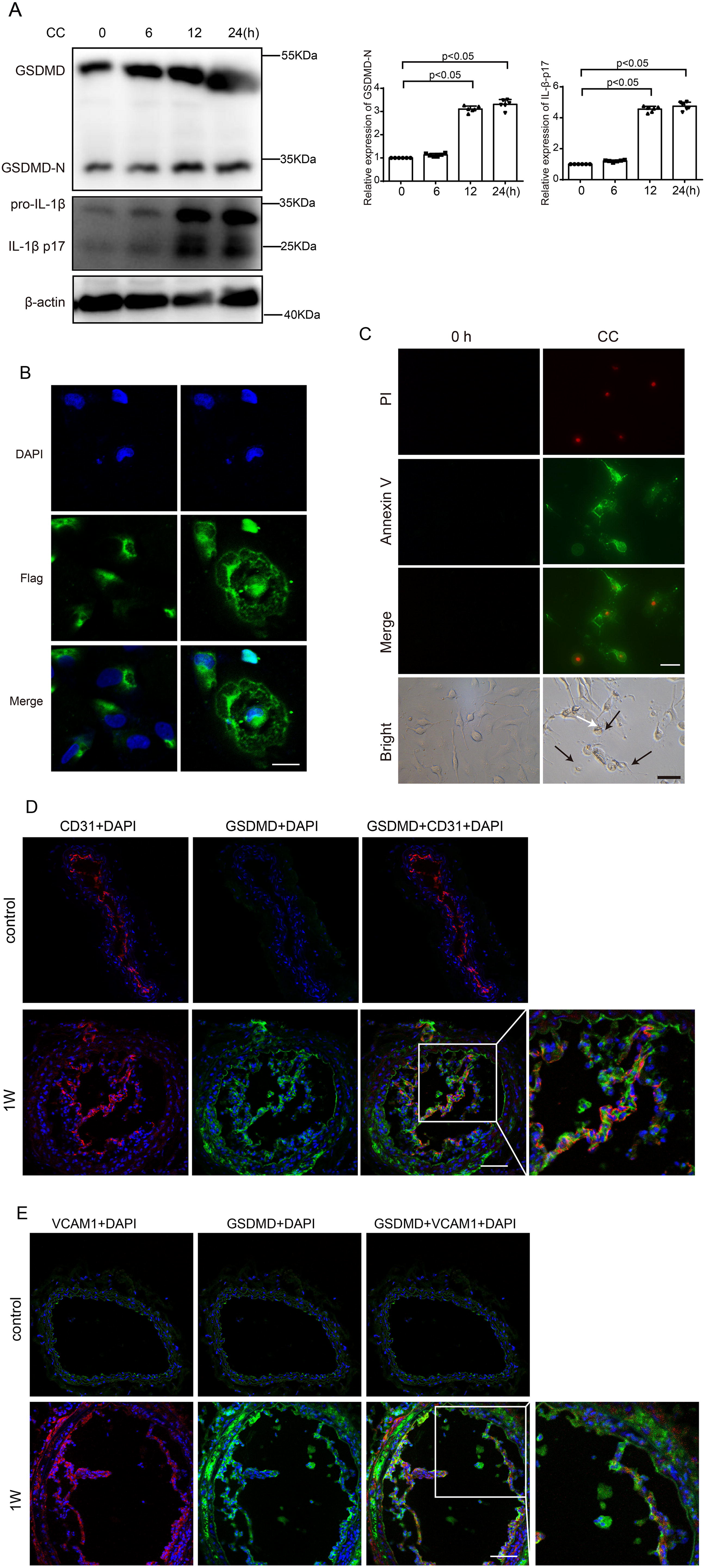
Pyroptosis is a critical immune-inflammatory response involved in atherosclerosis (Pichavaram et al., 2019). A previous study reported that CCs induced EC pyroptosis through AMPK/sirtuin 1 (SIRT1) pathway (Yang et al., 2020). To explore whether CCs promote endothelial inflammation through pyroptosis in atherogenesis, we detected the protein levels of IL-1β and GSDMD, a classic marker of pyroptosis. We found that the expression of GSDMD-N and IL-1β p17 in HAECs increased time-dependently (Fig. 3A). The translocation of GSDMD to the plasma membrane is a biological marker of pyroptosis. To investigate the subcellular location of GSDMD, a recombined plasmid harboring GSDMD-N-Flag was transfected in HAECs. Immunofluorescence results showed that the Flag was mainly distributed in the cytoplasm without CC stimulation. While cells were treated with CCs after GSDMD-N-Flag transfection, enriched Flag staining was observed on the plasma membrane, which indicated that GSDMD may translocate from the cytoplasm to the plasma membrane (Fig. 3B). In addition, cells stimulated with CCs exhibited both Annexin V and propidium iodide (PI) staining and swelled with characteristic bubbles (indicated by black arrows in Fig. 3C) on the plasma membrane. Accordingly, the level of IL-1β and IL-18 was increased in Lipopolysaccharide (LPS)-primed HAECs after CC stimulation (Supplementary Fig. S1). We also detected the expression of GSDMD in the endothelium and found a large proportion of GSDMD to be expressed in ECs within 1 week of Western diet intake (Fig. 3D). Furthermore, we observed the expression of VCAM1 and GSDMD. We found that the increased expression of VCAM1 was associated with GSDMD after 1 week of Western diet (Fig. 2E). These results suggest that CCs can induce endothelial pyroptosis in the early stage of atherosclerosis.
Knockdown of GSDMD inhibited CC-induced endothelial inflammation and attenuated plaque progression
To explore the role of CC-induced pyroptosis in endothelial inflammation, EC-specific GSDMD knockdown was achieved using EC-specific GSDMD adeno-associated virus type-ENT-GSDMD (AAV-EC-GSDMD) infection. We treated APOE−/− mice with AAV-EC-GSDMD and then performed partial carotid ligation surgery on the mice. After 1 week of Western diet, immunofluorescence staining was performed to verify GSDMD knockdown in ECs (Fig. 4B). The expression of VCAM1 decreased after 1 week of Western diet in AAV-EC-GSDMD group compared with the adeno-associated virus-negative control (AAV-NC) group (Fig. 4C). Moreover, after 4 weeks of Western diet, histological analysis demonstrated that EC-specific GSDMD knockdown reduced the size of atherosclerotic plaques Fig. 4D,E). Furthermore, the number of CD68+ macrophages within the lesions in carotid artery sections decreased, and the expression of IL-1β was reduced after GSDMD knockdown, compared with the AAV-NC group (Fig. 4F,G). To explore the role of endothelial GSDMD in atherosclerotic lesion formation in hypercholesterolemic mice, 12 weeks of Western diet was administered to induce hypercholesterolemia after AAV-EC-GSDMD or AAV-NC infection; knockdown of GSDMD reduced Oil Red O staining in en face aortas of mice, compared with AAV-NC group (Fig. 4H). Histological analysis of the aortic root demonstrated that knockdown of GSDMD reduced atherosclerotic plaque size (Fig. 4I).

To further investigate whether pyroptosis is involved in endothelial inflammation, we applied disulfiram (DSF; MedChem Express), which specifically inhibits GSDMD pore formation. We found that the expression of VCAM1 and ICAM1 decreased after DSF treatment (Fig. 4J). Thus, these results indicate that EC-specific GSDMD knockdown or inhibition alleviates endothelial inflammation and plaque progression.
EC-derived CC-induced pyroptosis was CASP1-dependent
Activated CASP1 can cleave GSDMD, precursors of IL-1β (pro-IL-1β) and IL-18 (pro-IL-18), to initiate pyroptosis (Rathinam and Fitzgerald, 2016). Thus, we evaluated whether CC-induced pyroptosis is CASP1-dependent. We found that the protein level of CASP1-p20 increased after CC application (Fig. 5A). Immunofluorescence data showed that CASP1-p20 was highly expressed in ECs after 1 week of Western diet (Fig. 5B,C). To explore whether inhibition of CASP1 ameliorates endothelial inflammation and plaque progression, mice were treated with the CASP1 inhibitor, VX765 (MedChem Express), by intraperitoneal administration every day for 1 or 4 weeks (Fig. 5D). Our result showed that inhibition of CASP1 ameliorated endothelial inflammation, reduced plaque size, and decreased macrophage content (Fig. 5E–G). Furthermore, VX765 suppressed the CC-induced increase of VCAM1, ICAM1, and GSDMD-N (Fig. 5H) in HAECs. Collectively, these observations indicate that CCs initiate atherosclerotic lesion formation via CASP1-dependent pyroptosis.

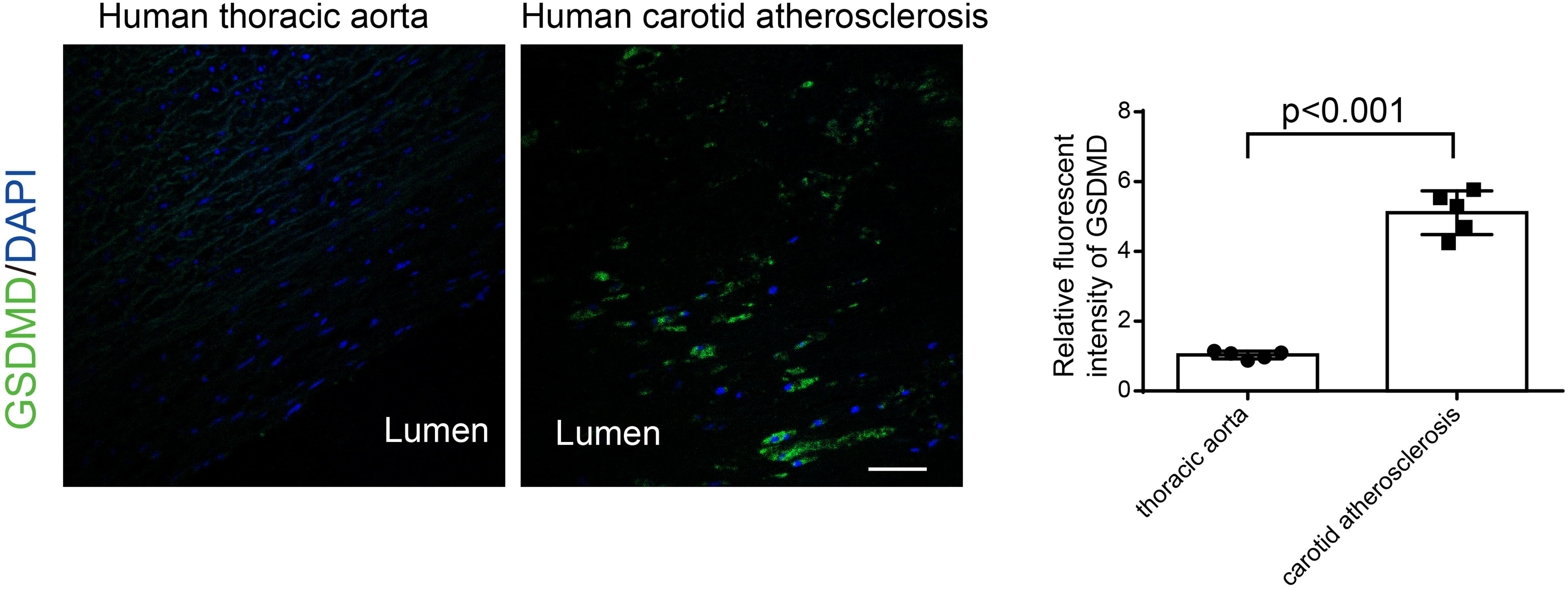
The expression of GSDMD increased in human atherosclerotic plaques
Immunostaining was used to analyze the expression and localization of GSDMD in human carotid lesions. Our data showed that GSDMD was highly expressed and localized in the intima of human carotid arteries with atherosclerotic plaques compared with healthy aortas (Fig. 6).
Discussion
CCs have been well-documented to be involved in the initial development of atherosclerosis. In fact, cholesterol-lowering treatment is protective in early atherosclerosis but ineffective at later stages (Björkegren et al., 2014). Thus, it is essential to figure out the specific mechanism through which CCs affect the pathogenesis of atherogenesis. In the present study, we revealed that EC-derived CC-induced pyroptosis may be a key initial factor in early atherogenesis. This conclusion is supported by several findings: (1) CCs appeared in the early stage of atherogenesis before the infiltration of immune cells. (2) ECs generated CCs upon hyperlipidemic condition. (3) CCs promoted endothelial inflammation by inducing pyroptosis. (4) Knockdown of GSDMD reduced endothelial inflammation and alleviated atherosclerotic lesion progression. (5) CC-induced pyroptosis was CASP1-dependent. (6) The expression of GSDMD increased in human atherosclerotic plaques. These findings revealed the novel notion that CC-induced pyroptosis of ECs is essential for endothelial inflammation in early atherogenesis.
Carotid atherosclerotic plaques with CCs were more likely to have concomitant macrophage and calcification accumulation (Shi et al., 2020). Extensive studies have found that CCs appeared in the initiation of atherosclerotic plaque and were associated with the early inflammatory response (Abela, 2010; Duewell et al., 2010). Our present study demonstrates that CCs are present in the subendothelial space during atherogenesis even when there is no immune cell infiltration, indicating that CCs may act as an initial inducement of atherogenesis.
It is generally believed that LDL taken up by ECs is transcytosed through the cell and deposited in the intima in the context of atherosclerosis (Vasile et al., 1983). ECs take up LDL or cholesterol through pathways that are either receptor-dependent or independent of any receptors (Simionescu, 2007; Frank et al., 2009). Thus, we wondered whether CCs are made by ECs in the early stage. In the next experiment, we treated HAECs with LDL for different time points and found signs of robust LDL uptake with prominent lipid droplets visible within HAECs. Furthermore, 48 h after LDL treatment, CCs began to appear. We also observed the localization of ECs, immune cells, and CCs in vivo and found CCs accumulated in the vascular intima where macrophages or CD45+ immune cells were barely seen. Based on these findings, we consider that EC-derived CCs may act as an initial factor in the early stage of atherogenesis. Our findings are in accordance with a previous study that found atherogenesis to be promoted by hyperlipidemia-induced CC production by ECs (Baumer et al., 2017).
Our present study proves that CCs can be generated by ECs in vitro, the formation of CCs in vivo is complex, and several physical conditions such as a rise in cholesterol saturation, a minor drop in local temperature (1°C–2°C), an alkaline pH, and hydration of the cholesterol molecule to form cholesterol monohydrate are recognized to enhance cholesterol crystallization (Baumer et al., 2017). It has also been shown that under certain circumstances, CCs can be formed by active intracellular processing of cholesterol in macrophages (Tangirala et al., 1994; Sheedy et al., 2013). The formation of CCs in EC can be inhibited by pharmacologically increasing endothelial cAMP (Baumer et al., 2017), indicating that endothelial cathelicidin antimicrobial peptide (cAMP) is involved in CC formation. In addition, interferon γ (IFNγ)/tumor necrosis factor α (TNFα) synergistically can increase LDL-induced CC formation in EC, which was accompanied by an increase in lysosomal pH and increased cellular free cholesterol (Baumer et al., 2020a, 2020b). Thus further work focused on the formation of CCs is needed.
Inflammation is critical in atheroma development, and EC activation is recognized as necessary for the recruitment of inflammatory cells to the plaque. However, it is not known whether CCs can induce inflammation and activate ECs. We found that the increase of VCAM1 was associated with the appearance of CCs 1 week after Western diet. In vitro, the expressions of VCAM1 and ICAM1 were also increased in HAECs after CC stimulation. Our results are in accordance with a previous study (Baumer et al., 2020a, 2020b).
Pyroptosis has been demonstrated to be involved in the initiation, progression, and complications of atherosclerosis (He et al., 2021; Qian et al., 2021). Furthermore, existing studies have indicated that ox-LDL, nicotine, and blood low-shear stress promote NLRP3-mediated EC pyroptosis (Zeng et al., 2019; Chen et al., 2021; Jiang et al., 2023). Thus, we wondered whether CCs can induce endothelial inflammation by pyroptosis. Previous evidence indicates that inflammasome-activated GSDMD causes pyroptosis by forming membrane pores (Liu et al., 2016). We examined pyroptosis markers, including GSDMD-N and IL-1β, in HAECs after CC stimulation, and we found that the protein levels of GSDMD-N and IL-1β p17 increased at 12 h and 24 h after CC stimulation. HAECs were treated with CCs after GSDMD-N-Flag transfection, and we observed the translocation of GSDMD from the cytoplasm to the plasma membrane. In addition, CC-stimulated cells exhibited both Annexin V and PI staining and swelled with characteristic large bubbles on the plasma membrane. To further investigate CC-induced EC pyroptosis, we detected the colocalization of GSDMD and ECs. We found that the expression of GSDMD increased in ECs 1 week after Western diet, which was in accordance with the expression of VCAM1. Therefore, these results indicate that CC-induced pyroptosis contributes to endothelial inflammation.
To illustrate the relationship between CC-induced pyroptosis and endothelial inflammation, we knocked down GSDMD in ECs using AAV-EC-GSDMD. Our results showed that the expression of VCAM1 was reduced 1 week after Western diet. Meanwhile, EC-specific GSDMD knockdown attenuated plaque progression and suppressed macrophage infiltration and IL-1β level. The GSDMD inhibitor, DSF, also suppressed CC-induced endothelial inflammation in vitro, suggesting that CC-induced pyroptosis may be the initial factor resulting in endothelial inflammation.
To investigate whether CC-induced pyroptosis was CASP1-dependent, we detected the expression of CASP1-p20 and found that CASP1-p20 was highly expressed in ECs after 1 week of Western diet. Furthermore, inhibition of CASP1 ameliorated endothelial inflammation, reduced plaque size, and decreased macrophage content. These results suggest that CC-induced CASP1-dependent pyroptosis contributes to endothelial inflammation and plaque progression.
It is generally accepted that oxidative stress and the associated cellular stress response are also underlying the pathogenesis of some chronic diseases (Calabrese et al., 2016). Modulation of endogenous cellular defense mechanisms represents an innovative approach to therapeutic intervention in diseases causing chronic tissue damage (Calabrese et al., 2010; Calabrese et al., 2007; Concetta Scuto et al., 2019). Thus intervention from the perspective of oxidative stress may be available for prevention and treatment of atherogenesis. Hydrogen sulfide (H2S) has been proposed to exert potentially significant effects on many physiological processes, especially in atherosclerosis (Mani et al., 2014; Zhang et al., 2020). Therefore, using H2S precursors can be considered to prevent atherogenesis. Further work will be done in this area.
Additionally, to investigate the relevance of our findings to atherogenesis in humans, we analyzed the expression and localization of GSDMD in human aortic lesions by immunostaining. As expected, GSDMD was highly expressed in the intima of human carotid arteries with atherosclerotic plaques. These findings suggest the human relevance of the CASP1-GSDMD pathway in regulating atherosclerosis progression.
Although we proved that CC-induced pyroptosis may be the initial cause of atherogenesis, we also showed that EC-specific GSDMD knockdown led to reduced endothelial inflammation. The mechanisms behind the deposition of CCs in ECs are yet to be fully elucidated. Further studies are still needed to explore the underlying mechanism. One additional aspect is that CCs tend to be formed at atheroprone vascular regions, such as the curvature and bifurcations along the arterial tree, where low shear stress occurs. In our present study, low shear stress combined with Western diet feeding was applied to induce CC formation. Although this approach can rapidly induce atherosclerosis, the rapid atherosclerosis development doesn’t truly reflect the pathological processes of atherosclerosis in humans. Therefore, a model of chronic atherosclerosis is also needed to investigate the role and mechanism of EC-derived CC in endothelial inflammation.
In conclusion, our study uncovered that EC-derived CCs may be an initial factor in early atherogenesis. CCs promote endothelial inflammation via pyroptosis, and endothelial-specific knockdown of GSDMD inhibits endothelial inflammation. These results identify that the GSDMD or its upstream regulators, including CASP1, in ECs might be novel promising therapeutic targets against atherosclerotic cardiovascular disease.
Materials and Methods
Animals
ApoE−/− mice were obtained from the Charles River Laboratory Animal Technology Co., Ltd. Seven-week-old male mice (18–20 g) were adapted to the environment (12/12-h light/dark cycle; 25°C ± 2°C) for 1 week before the study. Then, the mice received a Western diet (Research Diets) containing 21% fat, 50% carbohydrates, and 20% protein. To establish the carotid artery plaque mouse model, the LCCA of the mice was partially ligated as previously described (Yuan et al., 2021). Briefly, intraperitoneal injections of pentobarbital sodium (50 mg/kg) were used to anesthetize the mice, and their body temperatures were maintained at 37°C using heating pads. After blunt dissection to expose the distal branches of the LCCA, blood flow was reduced by ligating all branches of the LCCA except for the left thyroid artery. After that, the incision was closed with a suture. Mice were fed a Western diet immediately after surgery at different times. All animal experiments were approved by the Medical Ethics Committee of Shanghai Jiaotong University and were performed in strict accordance with approved guidelines.
Adeno-associated virus application
Concentrated stocks of EC-specific GSDMD adeno-associated virus type-ENT-GSDMD (AAV-EC-GSDMD) were obtained from Jikai Biotechnology Co. Ltd. AAV-EC-GSDMD is incorporated with the promoter of Tie2, which specifically targets ECs. The male ApoE−/− mice were administered with a single tail vein injection of AAV-EC-GSDMD or AAV-EC-NC with 1 × 1011 viral particles in a 100 μL volume of sterile PBS. The LCCA of the mice were partially ligated, and they were fed Western diet 4 weeks after infection.
Human sample collection
Endarterectomy plaques from human carotid stenosis were acquired from Shanghai Chest Hospital, Shanghai Jiaotong University School of Medicine. The tissues were placed in formalin, embedded in paraffin, and made into serial sections for histological analysis. For the control group, aortic tissues were obtained from age-matched organ donors undergoing heart transplant surgery without aortic dissection, aneurysm, coarctation, or previous aortic repair. This study was approved by the Ethics Committee of Shanghai Chest Hospital, Shanghai Jiaotong University School of Medicine. All procedures involving human samples were performed in compliance with the ethical guidelines of the Declaration of Helsinki.
Histology
The mice were perfused with ice-cold isotonic saline, after which their left carotid arteries or aortas were isolated. Tissue samples were fixed with 4% paraformaldehyde overnight and embedded at an optimal cutting temperature (OCT, Sakura Finetek). These OCT-embedded sections (5 μm thick) were cut at every 200 μm over a 2-mm length of carotid artery from the distal bifurcation of the carotid artery specimens or thoracic aortas from the proximal thoracic aorta specimens. The OCT- or paraffin-embedded carotid artery sections were stained with hematoxylin and eosin and Oil Red O staining as described previously (Yuan et al., 2021; Wang et al., 2023).
Immunofluorescence
Immunofluorescence was performed as previously described (Wang et al., 2023). The OCT-embedded sections were washed with PBS, fixed in 4% paraformaldehyde for 20 min, and permeabilized with 0.2% Triton X-100 for 10 min. The slices were washed with 0.01 M of PBS and blocked in 2% bovine serum albumin (BSA) for 1 h at room temperature. After blocking, the slices were incubated with the primary antibodies of anti-CD68 (BioLegend, 137002), anti-VCAM1 (ABclonal, A19131), anti-ICAM1 (Santa Cruz, sc-8439), anti-GSDMD (Santa Cruz, sc-393581), CD31 (Abcam, ab28364), anti-IL-1β (ABclonal, A11370), and CASP1-p20 (Santa Cruz, sc-398715) followed by the secondary antibody (Beyotime, A0516, A0568). The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Finally, light microscopy and confocal microscopy (Zeiss LSM 710) were used to detect the fluorescence.
Cholesteryl-BODIPY-C12 staining
Mice were perfused with ice-cold isotonic saline, after which their left carotid arteries were isolated. Tissue samples were embedded at an OCT without fixation. The OCT-embedded sections were washed with PBS and then stained with BODIPY (Thermo Scientific) for 5 min at 37°C. Confocal microscopy (Zeiss LSM 710) was used to detect the fluorescence.
Cell culture
The human aortic ECs (HAECs, ATCC® PCS-100-011™) were purchased from ATCC. The cells were cultured in Dulbecco’s modified Eagle medium (DMEM; 4.5 g/L glucose; Life Technologies) and supplemented with 10% fetal bovine serum and antibiotics (100 KU/L penicillin and 100 mg/L streptomycin) in an incubator at 37°C with 5% CO2. The cells were grown to a confluence of 70%–80% before the experiments were performed. Human LDL was obtained from Yeasen Biotechnology Co., Ltd.
Western blotting
Protein was extracted from the cultured cell lysates with Radio-Immunoprecipitation Assay (RIPA) lysis buffer using a phosphatase inhibitor (Roche). The protein concentration was determined by a BCA Protein Concentration Assay kit. Equal amounts (30–50 μg) of protein were subjected to 12% dodecyl sulfate, sodium salt (SDS)-Polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to the Polyvinylidene Fluoride (PVDF) membrane. The membrane was blocked with 5% fat-free milk at room temperature for 1 h and incubated by following primary antibodies: ICAM1 (Cell Signaling, 4915), VCAM1 (Cell Signaling, 13662), GSDMD (Cell Signaling, 69469), IL-1β (Abclonal, A16288), and CASP-1 (Cell Signaling, 3866) at 4°C overnight. On the second day, after 1-h incubation with horse radish peroxidase (HRP)-conjugated secondary antibodies, the labeled proteins were detected by enhanced chemiluminescence (ShareBio, SB-WB001). Image analysis was performed by Amersham Imager 680 scanning machine and intensity quantified with ImageJ. The optical density was normalized to that of β-actin (Beyotime, AF0003), which represented as relative optical density.
CC preparation
CCs were prepared as previously described (Samstad et al., 2014). Briefly, 25 g of anhydrous cholesterol (Sigma, cat.8667) was dissolved in 2 L of prewarmed (60°C) 95% ethanol. While warm, the mixture was filtered through a Whatman-1 filter paper to remove unwanted debris. The mixture was left overnight at room temperature, and the sedimented cholesterol crystals were filtered through the Whatman-1 filter paper and dried at room temperature. The same procedure was repeated twice to obtain pure monohydrated CCs. The crystals were then ground using a mortar and pestle, and they were stored in an amber-colored bottle at −20°C. The cholesterol crystals were used as needed.
Confocal laser reflection
Confocal microscopy was performed using Zeiss LSM 710. CCs were visualized by laser reflection microscopy as previously described (Duewell et al., 2010; Zimmer et al., 2016). In brief, the detector and the acousto-optical beam splitter were set to allow the detection of reflected laser light. Plaque CC content was quantified from three to four sections per mouse using Volocity Quantitation (PerkinElmer) and depicted as a ratio of total crystal reflection area to total plaque area. The researchers who performed the histological analyses were blinded to the treatment of the respective animals.
Data Analysis
All data are represented as mean ± SD. Statistical analysis between the two groups was performed by Student’s t-test, and one-way analysis of variance was used to compare the significance among three or more groups. In addition, the Bonferroni method was used to evaluate the significance conservatively. The calculations and data processing were performed using SigmaPlot 14.0 (Systat).
Footnotes
Acknowledgements
Authors’ Contributions
B.H. designed and supervised research. X.W. wrote the article. W.F., G.Z., and L.L. performed research; H.H. and X.S. assisted with data analysis. H.W., Y.W., X.H. and L.S. contributed to article revision.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This work was supported by National Natural Science Foundation of China (82130012, 81830010), Clinical Research Plan of SHDC (SHDC2020CR1039B), and SINO-German Mobility Program (M-0526).
Data Availability
All data are available in the main text. All data used in the current study are available from the corresponding author upon reasonable request.
Supplementary Material
Supplementary Figure S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.