
Abstract
Objective:
Lipotoxicity is a well-established contributor to cardiomyocyte death and heart damage, with ferroptosis being identified as a crucial death mode in cardiomyocyte disease. This study aims to explore the potential role and mechanism of ferroptosis in lipotoxicity-induced myocardial injury.
Methods:
Eight-week high-fat diet (HFD) Sprague-Dawley rat and H9c2 cardiomyocytes treated with palmitic acid (PA) were established for an in vivo and in vitro lipotoxic model. Ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1) were used to inhibit ferroptosis. Myocardial-specific stimulator of interferon genes (STING) knockdown rat (Sting myo-KD) with HFD was further introduced. Rat cardiac structure and function, cell viability, the level of lipid peroxidation, malondialdehyde (MDA), glutathione (GSH), mitochondrial function, ferroptosis-related proteins, and STING pathway-related proteins in H9c2 cells/myocardium were detected.
Results:
HFD rats with a ferroptosis inhibitor showed improved cardiac structure and function, reduced lipid peroxidation, and restored GSH, which was further confirmed in H9c2 cell. The time-dependent activation of the STING pathway following PA stimulation was observed. Knockdown of the expression of STING could reduce PA-induced cell death, lipid peroxidation, and MDA levels while restoring the GSH. In addition, both HFD Sting myo-KD rats and HFD rats with systematic inhibition by the STING inhibitor exhibited mitigating lipotoxicity-induced myocardial ferroptosis and reducing myocardial injury.
Innovation and Conclusion:
These findings suggest that lipotoxicity can induce ferroptosis in cardiomyocytes through the activation of the STING pathway, providing new targets and strategies for the treatment of lipotoxicity cardiomyopathy. Antioxid. Redox Signal. 42, 184–198.
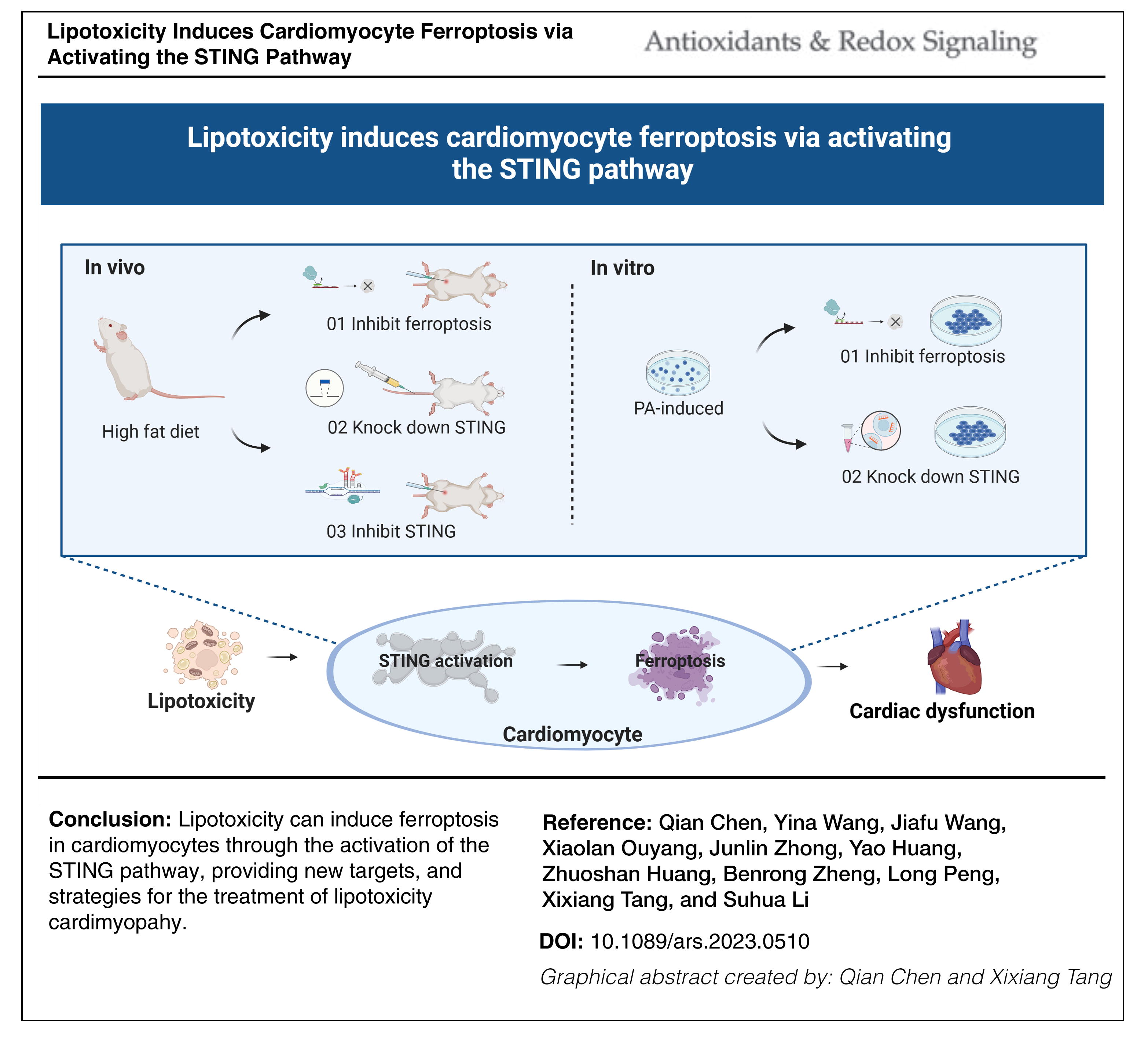
Innovation and Conclusion
Our study provides compelling evidence that ferroptosis plays a critical role in the pathogenesis and progression of lipotoxicity-induced myocardial injury. Particularly noteworthy is the novel finding that lipotoxicity can trigger ferroptosis in cardiomyocytes by activating the STING pathway, providing new targets and strategies for treating lipotoxicity cardiomyopathy.
Introduction
Cardiometabolic diseases are the leading cause of mortality in Western societies, and rapidly rising in low- and middle-income countries, seriously endangering human health, causing an increasing social and economic burden, and has become a major public health problem (GBD 2017 Risk Factor Collaborators, 2017; Miranda et al., 2019; Valenzuela et al., 2023). Cardiometabolic disease is a clinical condition characterized by cardiovascular damage resulting from metabolic disorders, including obesity, insulin resistance, diabetes, dyslipidemia, and hypertension. This condition can manifest as coronary artery disease, cardiomyopathy, arrhythmia, and other complications, greatly increasing the risk of heart failure (Hsu et al., 2023; Nakamura and Sadoshima, 2020). Metabolic cardiomyopathy is characterized by impaired myocardial metabolism, triglyceride (TG) buildup, and lipotoxic damage leading to changes in the cardiac structure and function (Chen et al., 2022).
Cardiac lipotoxic injury is a crucial mechanism in the occurrence and development of cardiometabolic diseases (Engin, 2017). Normally, the energy for adult cardiomyocytes is mainly derived through the oxidation of free fatty acids. However, under metabolic disorder conditions, toxic lipid intermediates such as diacylglycerol and ceramide are overproduced and accumulated around the myocardium, leading to an imbalance in lipid metabolism, damage to myocardial cells, and ultimately cell death (Goldberg et al., 2012). Clinical evidence supports the strong correlation between ventricular remodeling in patients with heart failure and the accumulation of toxic lipids (Lee et al., 2021; Peterson et al., 2020). The mechanisms behind cardiac lipotoxic injury include inflammation, insulin resistance, oxidative stress, the accumulation of ceramide and amyloid, endoplasmic reticulum stress, and programmed cell death (Hsu et al., 2023).
Ferroptosis, a form of regulated cell death, has recently emerged as a crucial player in various pathological conditions, including cardiovascular diseases (Hu et al. 2021). It is characterized by the accumulation of lipid peroxides, an iron-dependent oxidative burst, the depletion of glutathione (GSH), and decreased glutathione peroxidase 4 (GPX4) activity, leading to the disruption of cellular membrane integrity and subsequent cell death (Li et al, 2020). While the role of ferroptosis in cancer and neurodegenerative disorders has been extensively studied, its involvement in cardiac pathophysiology and, specifically, its relationship with lipotoxicity, remain poorly understood (Chen et al., 2021; Ou et al., 2022).
In this study, we sought to investigate the interplay between lipotoxicity and cardiomyocyte ferroptosis and elucidate the underlying molecular mechanisms. Specifically, we focused on the stimulator of interferon genes (STING) pathway, an essential component of the innate immune response responsible for sensing cytosolic DNA and initiating downstream signaling cascades (Garcia et al., 2023). Previous studies have shown that lipotoxicity could activate the STING pathway and damage target organs. Recent studies have also suggested the involvement of the STING pathway in various cardiovascular diseases; however, its involvement in lipotoxicity-induced cardiomyocyte ferroptosis has not been explored (Rech and Rainer, 2021; Xiong et al., 2021).
In the present study, we explored the link between lipotoxicity, the STING pathway, and cardiomyocyte ferroptosis in metabolic cardiomyopathy. Our results suggest that lipotoxicity could induce ferroptosis in cardiomyocytes through the activation of the STING pathway, providing new targets, and strategies for the treatment of lipotoxicity cardiomyopathy.
Results
Myocardial cell ferroptosis contributes to myocardial injury induced by lipotoxicity
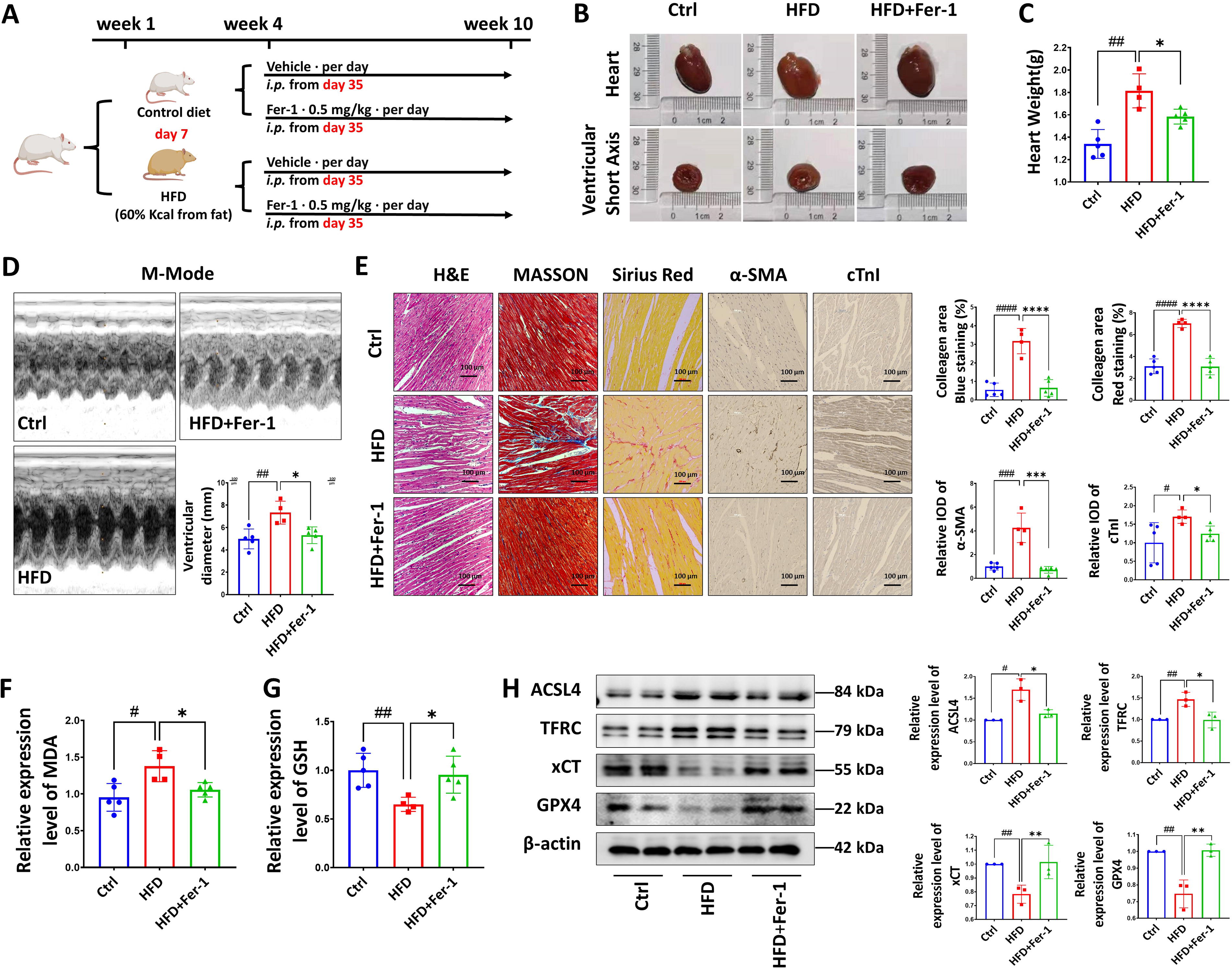
To investigate the involvement of cardiomyocyte ferroptosis in the development of myocardial injury induced by lipotoxicity, we developed a rat model of lipotoxicity through high-fat diet (HFD) feeding and subsequently treated them with a ferroptosis inhibitor (Ferrostatin-1) (Fig. 1A). Compared with rats fed a normal chow diet, HFD rats displayed an elevated level of body weight, cholesterol, low-density lipoprotein, and TG (Supplementary Figure S1), along with noteworthy cardiac hypertrophy and increased heart weight (Fig. 1B and C). Furthermore, cardiac color Doppler ultrasonography revealed an augmented inner ventricular diameter in HFD rats (Fig. 1D). Notably, the administration of a ferroptosis inhibitor mitigated the detrimental cardiac remodeling observed in HFD rats (Fig. 1B–D). While ultrasonography did not reveal any apparent lesions based on other indicators (Supplementary Figure S2B and C), the pathological analysis of myocardial sections in HFD rats indicated elevated troponin and aggravated fibrosis, along with the disrupted and disordered myocardial fibers, which could be alleviated by inhibiting ferroptosis (Fig. 1E). Cardiac tissue from HFD rat exhibited elevated levels of malondialdehyde (MDA) and decreased levels of the antioxidant substance GSH. However, treatment with a ferroptosis inhibitor successfully reversed these alterations in the HFD rat model (Fig. 1F and G). In addition, the administration of the ferroptosis inhibitor effectively mitigated the upregulation of ferroptosis-related proteins acyl-CoA synthetase long-chain family member 4 (ACSL4) and transferrin receptor (TFRC) and rescued the downregulation of system xc-(xCT) and GPX4 expression in the myocardium of HFD rats, as observed in our study (Fig. 1H). Together, these findings suggest that myocardial cell ferroptosis plays a pivotal role in causing lipotoxic injury within the heart.
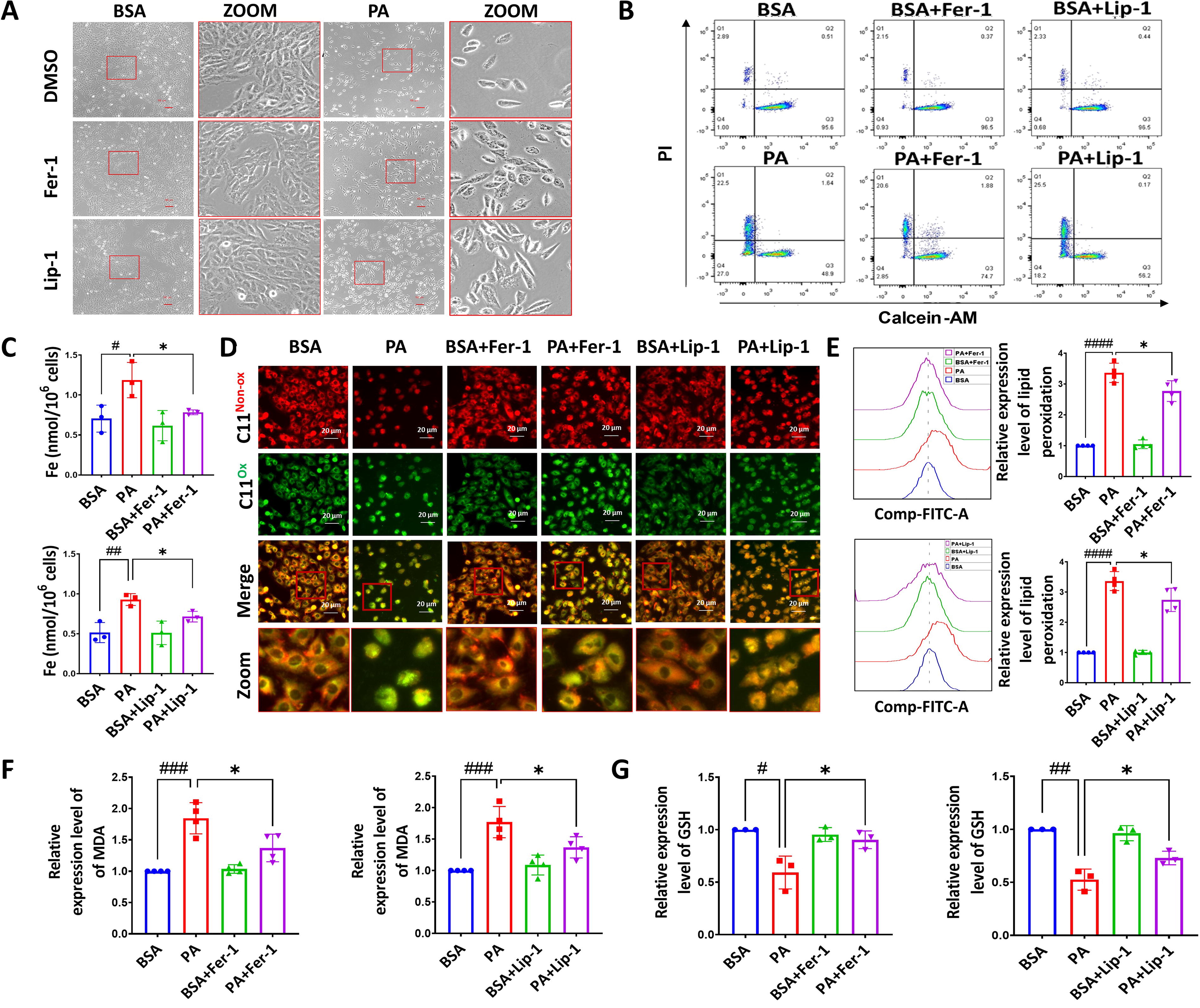
Palmitic acid induces ferroptosis and mitochondrial dysfunction in myocardial cells
We further verified our findings using in vitro experiments on rat H9c2 cardiomyocytes. Cells were treated with 0.5 mM palmitic acid (PA) for 24 h to establish a stable in vitro lipotoxic cell model (Supplementary Figure S3).
As illustrated in Figure 2A, it revealed that PA treatment induced notable shrinkage of H9c2 cardiomyocytes and a significant reduction in cell count. Furthermore, cells with the treatment of the ferroptosis inhibitors, Fer-1 and Lip-1, exhibited a remarkable attenuation of the cellular lipotoxic effects, as evidenced by the results obtained from the acetoxymethyl ester of calcein and propidium iodide (Calcein-AM/PI) kit (Fig. 2B). Cells challenged with PA showed an increased level of labile iron, which was decreased by ferroptosis inhibitors (Fig. 2C). In addition, the intervention with Fer-1 and Lip-1 yielded a substantial reduction in the levels of lipid peroxidation induced by PA (Fig. 2D–F), while the levels of the crucial antioxidant substance GSH were significantly restored by the administration of Fer-1 and Lip-1 (Fig. 2G). Collectively, these findings confirm the crucial involvement of ferroptosis in cell death induced by PA.
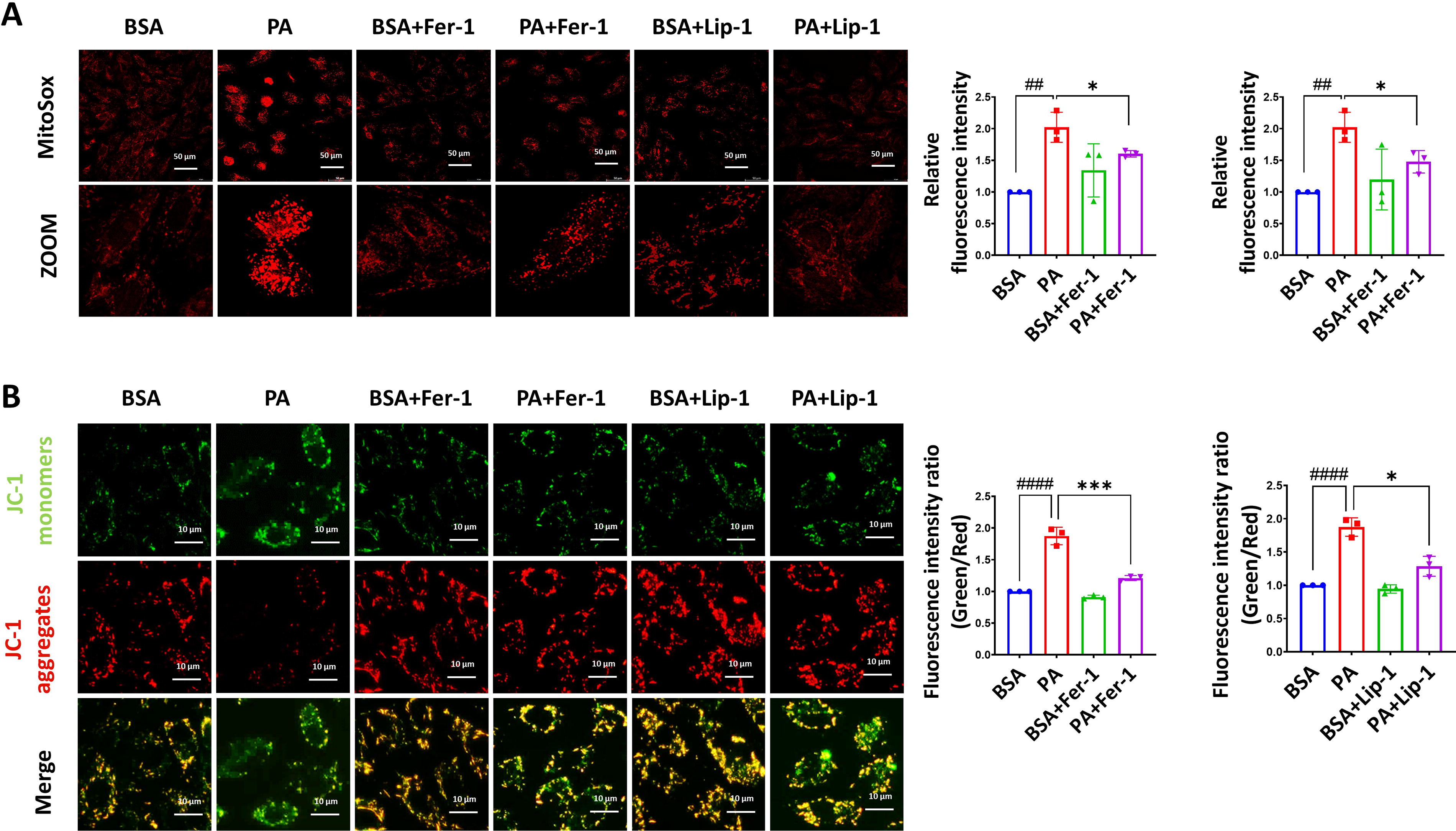
To assess mitochondrial dysfunction, we detected the mitochondrial membrane potential and mitochondrial reactive oxygen species (ROS) levels. The experimental results demonstrated that the PA group exhibited an increase in mitochondrial ROS levels (Fig. 3A), and an elevated green/red fluorescence ratio, indicating a loss of mitochondrial membrane potential (Fig. 3B), which suggests PA-induced mitochondrial dysfunction. In contrast, groups treated with ferroptosis inhibitors alongside PA preserved mitochondrial membrane potential and exhibited decreased mitochondrial ROS levels, suggesting that the ferroptosis inhibitors effectively alleviated PA-induced mitochondrial dysfunction.
STING pathway involves in PA-induced ferroptosis of myocardial cells and HFD-induced ferroptosis in rat myocardium
To explore the molecular mechanisms underlying lipotoxicity-induced myocardial ferroptosis, we established lipotoxic cell models by subjecting them to various durations of PA treatment (0, 3, 6, and 12 h). Subsequently, we quantified the alterations in proteins related to the implicated pathways. Emerging research has linked the STING pathway to several metabolic and cardiovascular diseases (Gao et al., 2023; Jin et al., 2023; Kong et al., 2022; Rech and Rainer, 2021; Xiong et al., 2021; Zhang et al., 2020). Herein, we attempt to explore the potential involvement of the STING pathway in myocardial cell ferroptosis induced by PA. Our study reveals the activation of STING pathway proteins, including STING, TBK-1, p-TBK-1, IRF3, p-IRF3, p65, and p-p65, upon exposure to PA (Fig. 4A). Following a 3- or 6-h exposure to PA, the expression levels of proteins associated with the STING pathway exhibited substantial increases in myocardial cells. Notably, these expression levels remained unaltered in the presence of the ferroptosis inhibitor Fer-1. These findings suggest a potential connection between the activation of the STING pathway and PA-induced ferroptosis in myocardial cells.
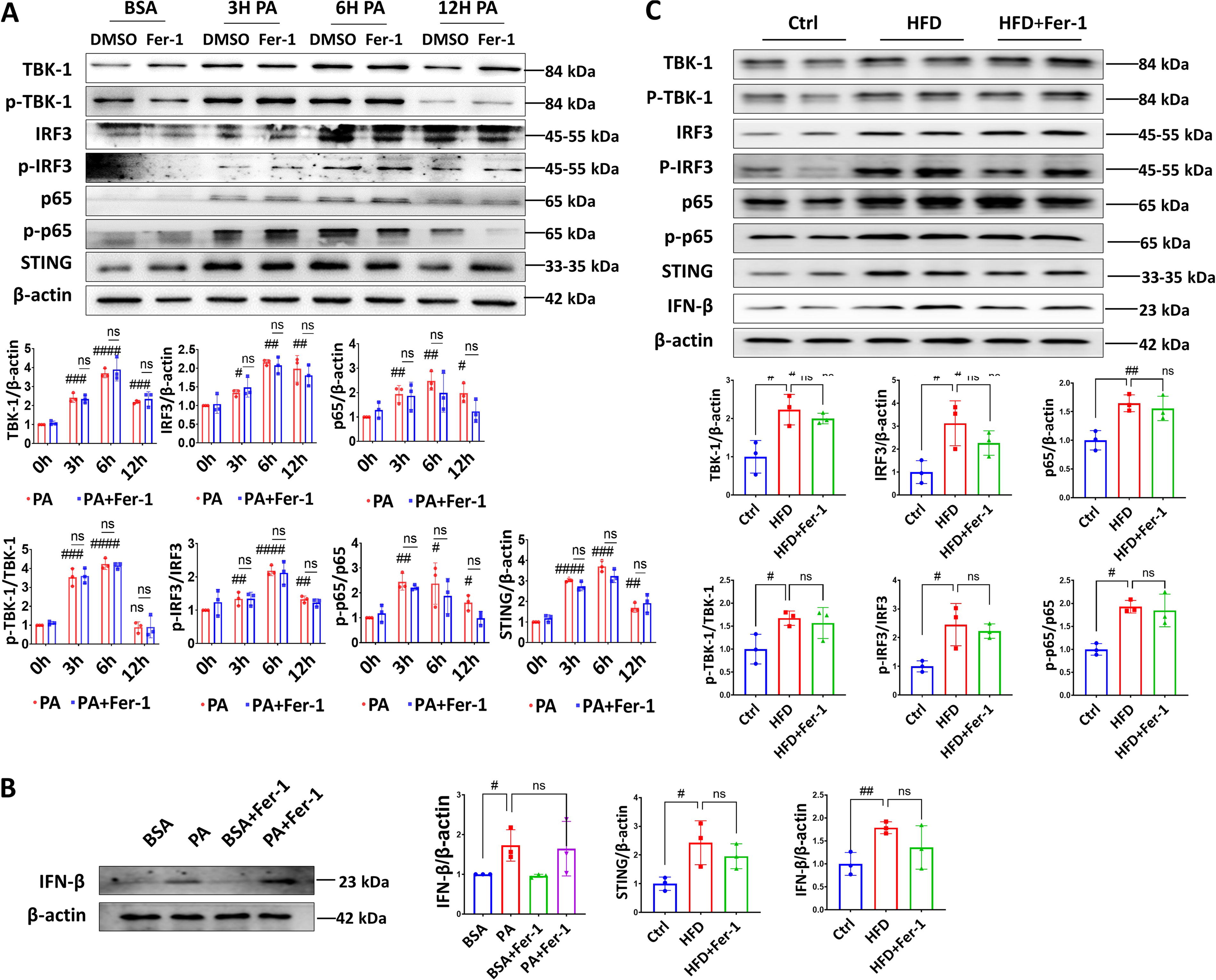
In addition, the analysis of interferon-β (IFN-β) expression was determined to assess the STING pathway activation and downstream signaling events. The group exposed to PA exhibited a significant upregulation of IFN-β expression compared with the control group (Fig. 4B), which indicates activation of the STING pathway and downstream IFN-β signaling in response to PA-induced stress. Interestingly, the upregulation of IFN-β expression induced by PA was not suppressed by the ferroptosis inhibitors, indicating a lack of effect of the ferroptosis inhibitors on PA-induced IFN-β activation. The observed increase in IFN-β expression in response to PA exposure highlights the potential involvement of the STING pathway in the cellular response to lipotoxic stress.
In vivo, consistent with the in vitro findings, the expression levels of STING-related pathway proteins in the myocardial tissue of rats in the HFD group were elevated (Fig. 4C), including higher levels of the downstream signaling molecule, IFN-β. However, despite the administration of a ferroptosis inhibitor, there was no significant reduction observed in the activation or subsequent effects of the STING pathway.
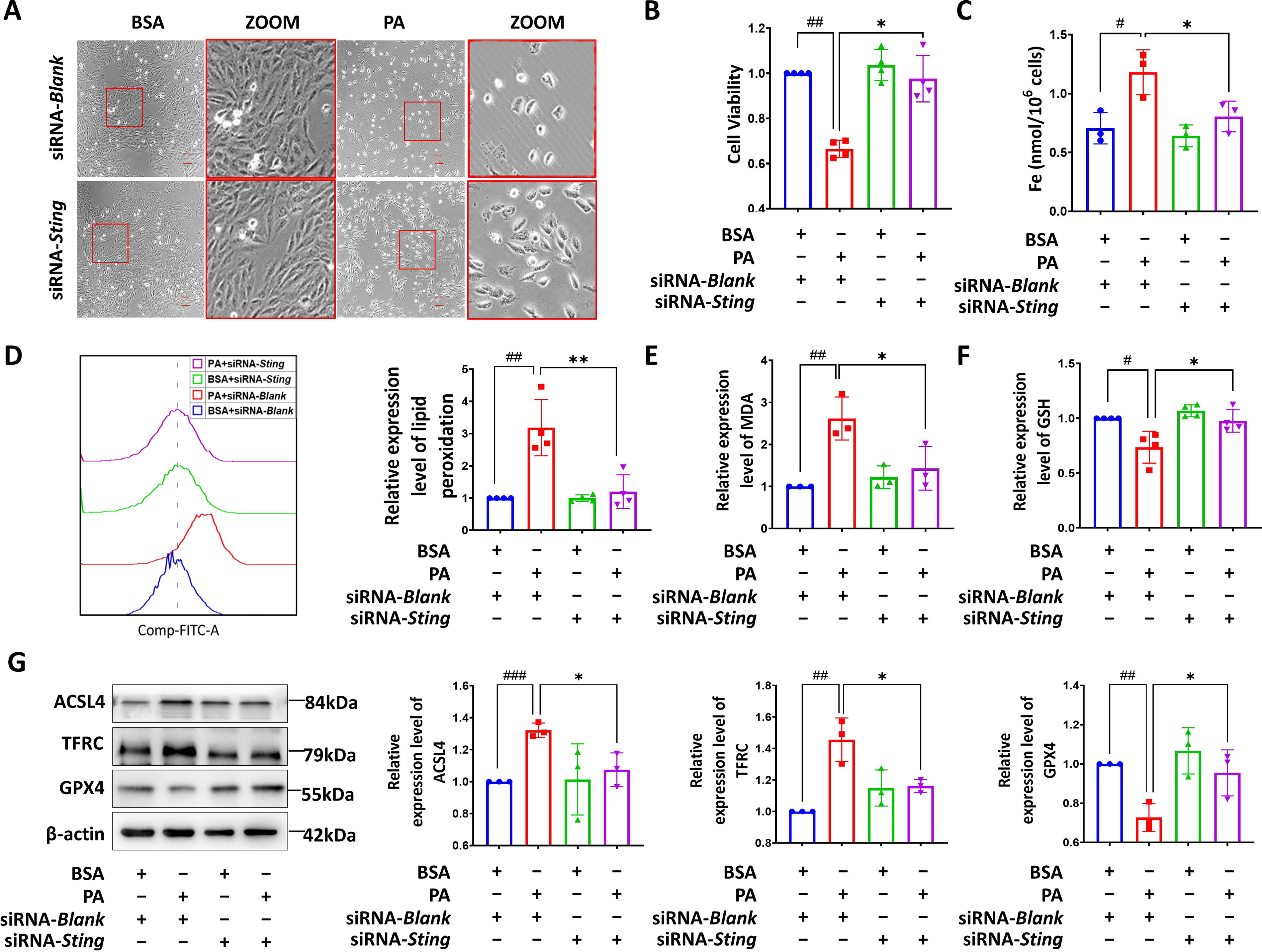
To validate the involvement of the STING pathway in PA-induced cardiomyocyte ferroptosis, we successfully inhibited the expression of STING in H9c2 cardiomyocytes by 50% using siRNA-Sting transfection (Supplementary Figure S4). We found that the downregulated STING could blunt the decreased cell number in cells with PA treatment, accompanied by a transition from a round to a regular elongated spindle shape (Fig. 5A). In addition, a notable improvement in cell viability was observed in STING-deficient cells with PA challenge (Fig. 5B). Inhibition of STING effectively prevented the elevation of iron content (Fig. 5C) and lipid peroxidation levels (Fig. 5D and E), and the depletion of GSH levels (Fig. 5F) induced by PA in cardiomyocytes. In addition, loss of STING significantly reduced the upregulation of ACSL4 and TFRC and rescued the decreased in the expression of GPX4 in H9c2 cells with PA treatment (Fig. 5G). Collectively, these findings strongly support the involvement of STING in PA-mediated cardiomyocyte ferroptosis.
Downregulation of STING ameliorates lipotoxicity-induced myocardial ferroptosis and cardiac dysfunction in HFD rats
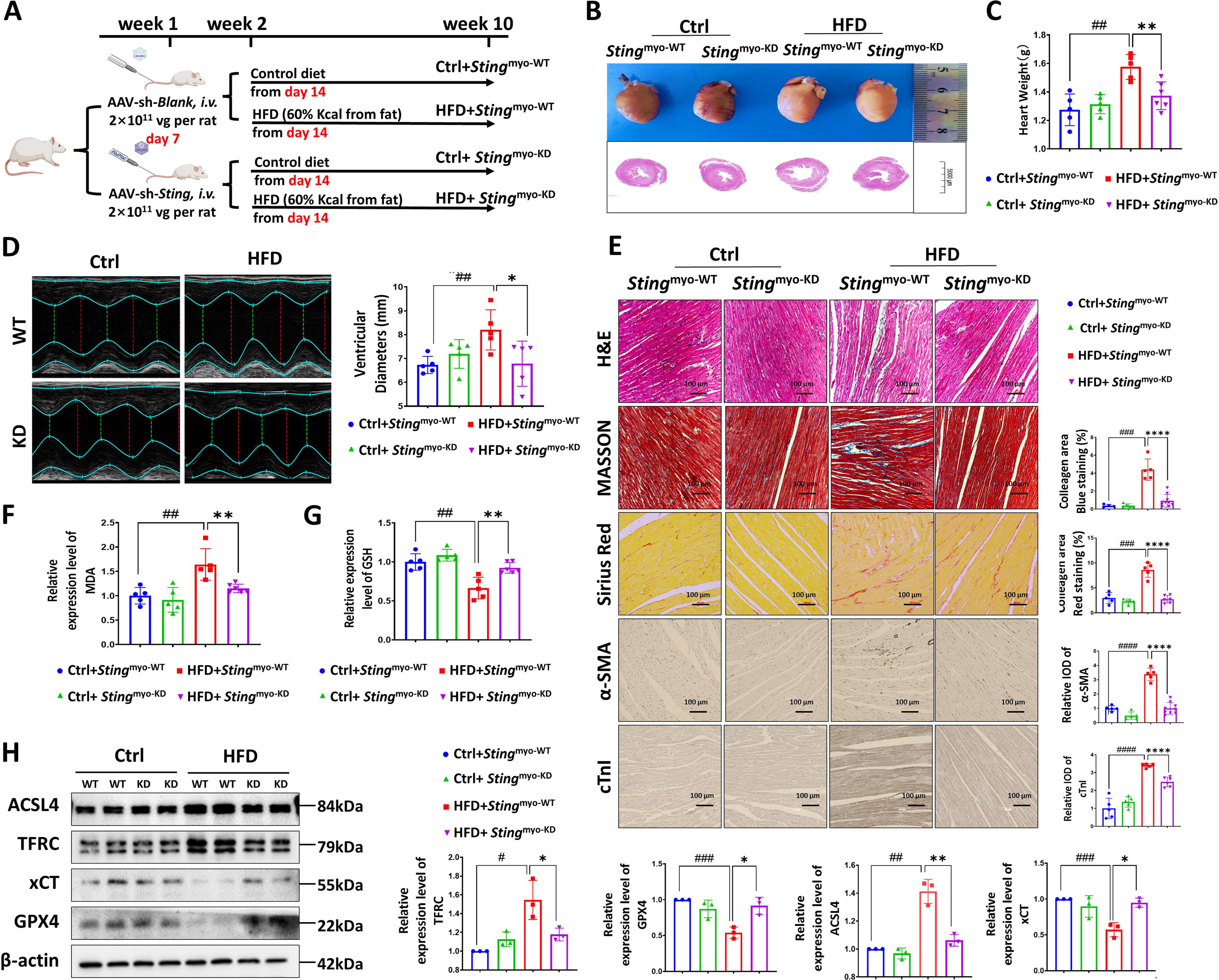
To investigate the impact of myocardial STING on lipotoxicity-induced myocardial ferroptosis in vivo, we induced HFD animal models in myocardial-specific STING knockdown rat (Sting myo-KD) by AAV-sh-Sting tail vein injection (Fig. 6A). As shown in Supplementary Figure S5, we performed the immunohistochemical (Supplementary Figure S5A), Western blot (WB) (Supplementary Figure S5B), and quantitative PCR (qPCR) (Supplementary Figure S5C) analysis with the rats’ myocardial tissue and found that the STING protein and mRNA levels were markedly downregulated in the heart from Sting myo-KD rat with or without HFD. We assessed cardiac injury and myocardial ferroptosis in rats and found that HFD Sting myo-KD rats exhibited decreased heart weight and ventricular inner diameter (Fig. 6B–D), reduced myocardial fiber rupture and disorder, and alleviated troponin level and fibrosis compared with HFD Sting myo-WT rats (Fig. 6E). In addition, the HFD Sting myo-KD rats had lower levels of MDA (Fig. 6F) and higher levels of the antioxidant substance GSH (Fig. 6G). This suggests a reduction in oxidative stress and an enhanced antioxidant defense in the hearts of HFD Sting myo-KD rats. WB results revealed that the expression of ferroptosis-related proteins ACSL4 and TFRC decreased, while the expression of GPX4 protein increased in HFD Sting myo-KD rats compared with HFD Sting myo-WT rats (Fig. 6H). These results suggest that knocking down myocardial STING can mitigate lipotoxicity-induced myocardial ferroptosis and reduce myocardial injury.
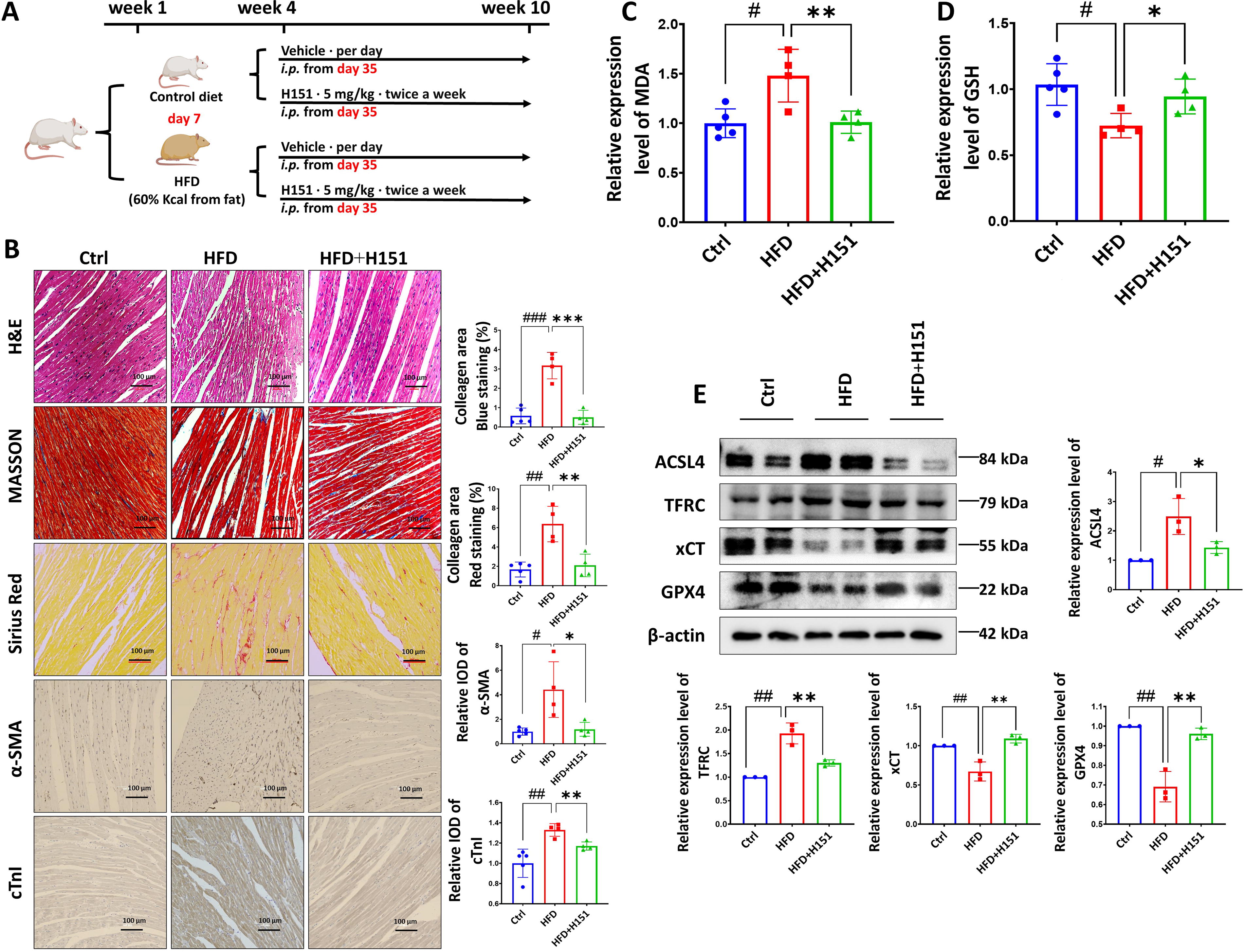
In addition, we utilized intraperitoneal injection of the STING inhibitor H151 (5 mg/kg twice a week), a highly potent and selective small-molecule antagonist of STING, for 4 weeks to inhibit systemic STING expression in rats (Fig. 7A). Remarkably, our findings demonstrate that this intervention led to a reduction in myocardial injuries and fibrosis (Fig. 7B) as well as MDA levels (Fig. 7C) and an increase in the content of the antioxidant substance GSH (Fig. 7D) in lipotoxic rats. Furthermore, the administration of H151 resulted in a significant decrease in the expression of ferroptosis-related proteins ACSL4 and TFRC, while the expression of GPX4 and xCT protein increased (Fig. 7E). Taken together, these results highlight the potential of H151, a STING inhibitor, to suppress lipotoxicity-induced ferroptosis in rat cardiomyocytes and mitigate myocardial injury.
Discussion
Lipotoxicity contributes to the development and progression of cardiac dysfunction in individuals with metabolic disorders; however, the underlying mechanisms have not been fully elucidated. Our current study provides strong evidence supporting a critical role of ferroptosis in lipotoxicity-induced cardiomyocyte damage. Our results also show that ferroptosis inhibitors targeting various pathways effectively reduce cell death and lipid peroxidation levels and restore GSH/GPX4 antioxidant activity in lipotoxic cardiomyocytes (Fig. 8).
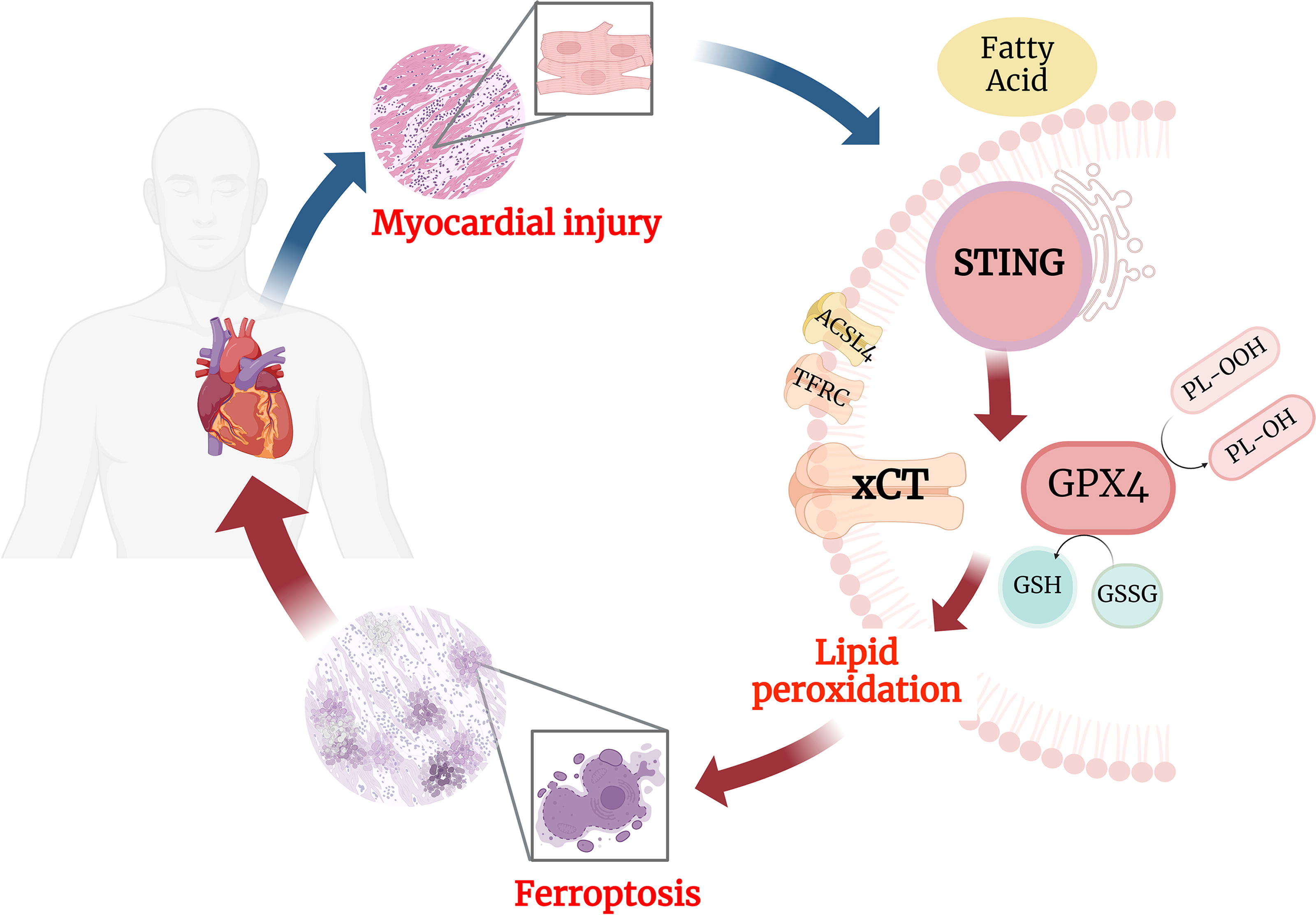
Ferroptosis, a type of cell death, plays a crucial role in cardiovascular diseases. Numerous studies have demonstrated its involvement in the pathogenesis of various cardiac diseases, including ischemic heart disease, heart failure, arrhythmia, and cardiomyopathy (Cai et al., 2023; Chen et al., 2019; Dai et al., 2022; Fang et al., 2019; Jang et al., 2021). In obesity and diabetes, dysregulated glucose and lipid metabolism promotes lipotoxicity, oxidative stress, and antioxidant system damage, triggering ferroptosis in myocardial cells. This process disrupts cardiac structure and function, ultimately leading to diabetic cardiomyopathy or obesity-related cardiomyopathy (Fang et al., 2023; He et al., 2023; Sha et al., 2021). Abnormal glucose and lipid metabolism in diabetes led to the downregulation of solute carrier family 11 member 2 (SLC11A2) expression via the AMPK/NRF2 pathway, exacerbating myocardial ferroptosis (Wang et al., 2022). Pei et al. (2021) demonstrated that ACSL4 mediates cellular ferroptosis induced by a high-fat diet, resulting in impaired cardiac contractile function and structural remodeling. Wang et al. (2021) confirmed in their study that PA induces lipid peroxidation, regulates gene expression related to iron metabolism, and disrupts iron homeostasis, leading to ferroptosis of myocardial cells. Aligned with previous finding, we established both an in vivo model using HFD-induced lipotoxic rats and an in vitro model using PA-induced lipotoxic cells to confirm the crucial role of myocardial cell ferroptosis in inducing lipotoxic injury within the heart (Pei et al., 2021; Wang et al., 2021).
Mitochondrial oxidative respiration is essential for cardiomyocytes, supplying the energy needed for heart function (Calabrese et al., 2010). Ferroptosis-induced myocardial injury stems from an overaccumulation of ROS, resulting in oxidative stress that harms cellular components and ultimately triggers ferroptosis. Studies have showed antioxidants such as 7-DHC and curcumin protect against ferroptosis, offering new treatment potentials for related diseases, including cancer and ischemia–reperfusion injury (Concetta Scuto et al., 2019; Concetta Scuto et al., 2019; Foroutan et al., 2024). Our research indicates that PA treatment significantly increases mitochondrial dysfunction in cardiomyocytes, but ferroptosis inhibitors Fer-1 and Lip-1 mitigate this damage, linking lipotoxic cardiomyocyte ferroptosis to mitochondrial dysfunction. Hence, oxidative stress plays a central role in the initiation and progression of ferroptosis in lipotoxicity-induced cardiomyocyte injury.
STING is an important molecule in innate immune response, which plays an immune defense role in pathogen infection and malignant tumors (Zhang et al., 2020). Recent studies have found that the STING pathway also involved in metabolic homeostasis (Oduro et al., 2022; Vila et al., 2022). Abnormal glycolipid metabolism can induce mitochondrial damage and activate the STING pathway to mediate chronic inflammatory states, which lead to insulin resistance, sterile inflammation, and related metabolic dysfunction. This results in target organ damage, including endothelial injury (Wen et al., 2023), the inhibition of angiogenesis (Yuan et al., 2017), ectopic fat deposition, lipid-related renal damage (Yang et al., 2019), obesity-related lung dysfunction (Qi et al., 2023), and non-alcoholic fatty liver disease (Luo et al., 2018). Emerging research has linked the STING pathway to lipotoxicity-induced cardiac injury (Bai et al., 2017; Gong et al., 2020; Ma et al., 2023). Several studies have found that inflammation and cardiomyocyte apoptosis in obesity-related cardiac dysfunction (Bai et al., 2017; Gong et al., 2020) and models of diabetic cardiomyopathy (Ma et al., 2023) are associated with the activation of the STING pathway. In the present study, HFD-fed rats exhibited activation of the STING pathway and increased levels of its downstream molecule, IFN. Notable increases in myocardial rupture and disarray were observed, along with elevated cTnI content and fibrosis. These effects could be attenuated by inhibiting the STING pathway. This further confirms the findings of previous studies that showed STING activation induces lipotoxic myocardial injury. However, it remains uncertain whether the STING pathway is involved in the ferroptosis of cardiomyocytes induced by lipotoxicity. We used adenovirus AAV-sh-Sting to specifically inhibit STING expression in the rat myocardium, in combination with intraperitoneal injection of H151 to systemically inhibit STING and provide further evidence for the possible involvement of STING in lipotoxic-induced cardiomyocyte ferroptosis, ultimately leading to cardiac lipotoxic injury. Notably, we confirmed that lipotoxicity can also lead to the upregulation of IFN-β, a signaling molecule downstream of the STING pathway, indicating the possible role of IFN signaling in STING-mediated ferroptosis. However, further investigations are required to fully elucidate the underlying mechanisms.
Accumulated studies have found a connection between the STING pathway and ferroptosis, but the mechanisms involved are not yet clear (Gao et al., 2023; Kong et al., 2022; Zhang et al., 2022). Zhang et al. (2022) found that activation of the cGAS-STING signaling pathway induces mitochondrial lipid peroxidation and ROS production, ultimately leading to ferroptosis in tumor cells. Gao et al. (2023) revealed that STING directly interacts with ACSL4 at specific amino acid residues D53 and K412, and this interaction triggers renal inflammation and fibrosis through ACSL4-mediated ferroptosis. In addition, Jin et al. (2023) found that STING-mediated ferritin autophagy, regulated by NCOA4, contributes to iron-dependent cell death during ischemic acute kidney injury. In a model of septic cardiomyopathy, activation of the STING pathway promotes the autophagic degradation of GPX4, resulting in reduced GPX4 levels, excessive lipid peroxidation, and subsequent myocardial ferroptosis (Kong et al., 2022). In summary, activation of the STING pathway enhances ferroptosis through mechanisms that primarily focus on the regulation of iron homeostasis and lipid peroxidation. Interestingly, Jia et al. (2020) found that GPX4 deficiency results in elevated levels of lipid peroxidation, leading to the carbonylation of STING and hindering its translocation from the endoplasmic reticulum to the Golgi complex. However, our study confirmed that lipotoxicity-induced stress leads to STING activation, accompanied by GPX4 downregulation and increased lipid peroxidation. This leads us to hypothesize that an intracellular deficiency of GPX4, by enhancing excessive lipid peroxidation, might act as a regulatory brake on STING to avert further cellular disturbance. Such a dualistic mechanism unveils a complex interplay where the dysregulation of GPX4 and lipid peroxidation possesses the capacity to modulate STING activation in a bidirectional manner.
While our study provides valuable insights into the role of the STING pathway in lipotoxicity-induced ferroptosis, it also raises several important questions and opportunities for further investigation. First, further investigation is warranted to explore the precise mechanisms and molecular interactions involved in the lipotoxicity-STING-ferroptosis axis. Second, the pivotal role of STING signaling in the immune response against tumors has made it an attractive therapeutic target for cancer immunotherapy. Currently, several STING agonists with promising preclinical benefits have been reported, and some have entered clinical trials. However, the cardio-adverse effects for these STING agonists treating cancer should be carefully considered. Third, regarding experimental methods, to ensure the use of standardized protocols and the attainment of stable results, we opted for the rat H9c2 cell model to conduct an extensive investigation into the in vitro system of lipotoxicity. However, it is crucial to acknowledge that the biological characteristics of this model significantly diverge from those of tissue cells in vivo. Thus, an alternative approach that involves extracting primary myocardial cells from rat hearts for subsequent experiments proves to be a promising alternative. Finally, the myocardial-specific Sting knockout rat should be used to strongly provide the evidence for the present finding.
Materials and Methods
Animal studies
Specific pathogen-free (SPF) male Sprague-Dawley rats aged 8 weeks were acquired from Yancheng Biotechnology (Guangzhou, China) and housed individually in a temperature- and humidity-controlled environment with a 12-h light/dark cycle at the College of Animal Science of South China Agricultural University following a protocol approved by the Institutional Animal Care and Use Committee. The rats were acclimated for 1 week with normal chow diet and water ad libitum before being fed ad libitum with a high-fat diet (HFD) that contained 60% kcal from fat (Si Pei Fu, Beijing, China) for 8 weeks to develop the rat model of lipotoxicity-induced cardiomyopathy (Feriani et al., 2021; Marín-Royo et al., 2019). Rats fed ad libitum with a standard chow diet were used as controls (Ctrl). Body weight of rats was monitored weekly. After 8 weeks of HFD feeding and all treatments, rats were euthanized, and serum samples and heart tissues were harvested for further evaluation.
Adeno-associated viral infection
To obtain myocardium-specific knockdown of STING in rats, we used adeno-associated virus 9 (AAV9) to deliver the STING-shRNA genetic code, which was synthesized, purified, and cloned into the PGMAAV9-egGFP vector provided by Genomeditech (Shanghai, China; rat STING shRNA sequence: shRNA5′-GCCACCUCAAUAUGUAGCACATT-3′). The produced vectors had viral titers of 1 × 1012 vg/mL for the rAAV9-sh-STING vector and 2 × 1012 vg/mL for the AAV9-control vector. Male Sprague-Dawley rats were tail vein-injected with 2 × 1011 vg particles of AAV9 in 100 µL of phosphate-buffered saline (PBS) per rat (Palomeque et al., 2007; Wu et al., 2022). Then, rats were treated followed by 8 weeks of HFD or standard chow fed as described above.
Reagent administration
To generate HFD rats with inhibited ferroptosis, HFD rats were intraperitoneally injected with ferrostatin-1 (Fer-1, MedChemExpress, USA) at a dose of 0.5 mg/kg daily for 4 weeks (Scarpellini et al., 2023). To generate HFD rats with inhibited STING, HFD rats were intraperitoneally injected with H151 (STING inhibitor, MedChemExpress, USA) at a dose of 5 mg/kg twice a week for 4 weeks to inhibit systemic STING activation in rats (Gong et al., 2021; Kobritz et al., 2023). Control HFD mice were administered with equivalent volume injections of PBS.
Echocardiography
Echocardiography was conducted to assess rat cardiac structure and function using the Vevo 3100 Imaging System (VisualSonics, Toronto, Canada). Rats were anesthetized with isoflurane (RDW Biotech, Shenzhen, China), with heart rate remaining stably around 450 beats/min. A probe with a frequency of 21 MHz was utilized to collect echocardiographic parameters through 2-dimensional targeted M-mode images in the parasternal long-axis view. The left ventricular diameter was measured and calculated, and each parameter was measured triple times.
Histology
The hearts of rats were fixed in 10% formalin for 12 h followed by a gradient dehydrated and embedded in paraffin for histological evaluation. Samples of 3-µm-thick myocardial sections were stained with H&E (hematoxylin–eosin), Masson, and Sirius Red.
For immunohistochemical staining, the sections were incubated overnight at 4°C with a primary antibody, then incubated with the corresponding secondary antibody next day for 45 min at room temperature, followed by hematoxylin and 3,3’-diaminobenzidine staining. The antibodies used in the study were anti-cTnI and anti-α-SMA (1:50; Servicebio, Wuhan, China).
At least five images per heart were randomly captured using the automatic Digital Slide Scanning System (Carl Zeiss AG, Oberkochen, Germany).
Cell culture and treatment
H9c2 cells (Procell Biotech, Wuhan, China) were used for in vitro studies and were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin antibiotics (Gibco, Waltham, MA, USA). Cells were cultured at 37°C in a 5% CO2 environment. To simulate lipid toxicity in cardiomyocytes in vitro, H9c2 cells at 70% confluence were treated with PA (PA, Sigma-Aldrich, USA) or bovine serum albumin (BSA, Sigma-Aldrich, USA) for 24 h in culture medium containing 5% FBS.
RNA interference
To knock down STING gene, H9c2 cells were transfected with STING small interfering RNA (siRNA-Sting, sequence: siRNA5′-GCAUCAAGGAUCGGGUUUATT-3′, GenePharma Co., Ltd, Suzhou, China) or scramble siRNA (siRNA-Blank) (GenePharma, Suzhou, China) for 12 h using the Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, Waltham, USA) following the manufacturer’s instructions. Cells were stimulated as directed after transfection (24 h).
Cell viability assay
Cell viability was examined using a Cell Counting Kit-8 (CCK8, Dojindo, Japan) assay. CCK-8, 10 μL, solution was added to each group in 96-well plates, which were then placed in a 5% CO2 atmosphere at 37°C for 2 h. The survival rate was calculated based on the absorbance values measured at 450 nm using the automated multimode microplate reader (BioTek, Vermont, USA).
The Calcein-AM/PI Cell Viability/Cytotoxicity Assay Kit (Beyotime Biotechnology, China) was utilized to determine live and dead cells. H9c2 cells were stained with calcein-AM or PI according to the instructions provided with the kit. After extensive washing, the cells were then incubated with the dying solution composed of 2 μM calcein-AM, 4.5 μM PI, and detection buffer at a volume ratio of 1:1000 for 30 min at 37°C in the dark. The average fluorescence intensity was detected by flow cytometry with an excitation filter at 490/515 nm and 535/617 nm (BD Biosciences, New Jersey, USA).
Lipid peroxidation measurement
BODIPY™ 581/591 C11 (Thermo Fisher Scientific, Waltham, USA), a fluorophore that responds to lipid peroxidation, was used to measure the level of lipid peroxides. BODIPY™ 581/591 C11 is lipophilic, allowing it to easily incorporate into cellular membranes or lipid-containing structures within cells. When the cellular lipid peroxidation occurs, the BODIPY™ 581/591 C11 transitions from emitting red fluorescence (presenting a nonoxidized state) to emitting green fluorescence (presenting lipid peroxidation). Cells were cotreated with test compounds (prepared in media containing BODIPY™ 581/591 C11 at a final concentration of 2.5 μM) for 30 min. Cells were washed and maintained with PBS before measuring fluorescence at 565/600 nm (excitation/emission) and 477/525 nm using an inverted microscope (Nikon, Tokyo, Japan). Besides, cells were washed and collected by PBS and mean fluorescence intensity was measured by flow cytometry at 565/600 nm for red fluorescence and 477/525 nm for green fluorescence (BD Biosciences, New Jersey, USA). The level of lipid peroxidation was presented as the ratio of green to red C11 BODIPY fluorescence.
The level of MDA, the end product of lipid peroxidation, in the rat myocardium and cardiomyocyte was measured by the thiobarbituric acid reactive substances Assay Kit (Cayman Chemical, Michigan, USA). The myocardium homogenate or cell lysis was added to the plate before generating MDA by a reaction between arachidonic acid (100 μM) and ammonium iron (II) sulfate (20 μM). Thiobarbituric acid (TBA) was then added to the plate and incubated at 90°C for 1 h to generate MDA-TBA adducts. Following incubation, the contents of the samples and positive control were measured with the absorbance values at 530/540 nm using an automated multimode microplate reader (BioTek, Vermont, USA).
QuantiChrom GSH assay kit
The level of GSH in cells and heart tissue was determined by the QuantiChromTM GSH Assay Kit (BioAssay Systems, CA, USA). Cells and tissues were washed with cold PBS, sonicated in 1–2 mL of buffer containing 50 mM MES or phosphate (pH 6–7) and 1 mM ethylenediaminetetraacetic acid, and the supernatant was mixed well with Reagen A. The mixture was then transferred to a 96-well plate and mixed with Reagen B. After incubation at room temperature for 25 min, the mixture was measured with the absorbance values at 412 nm using an automated multimode microplate reader (BioTek, Vermont, USA).
ROS production measurement
To detect mitochondrial ROS accumulation, the MitoSOX™ Red fluorescent probe was used (MedChemExpress, New Jersey, USA). At the end of the treatment, cells were incubated in the presence of 5 μM MitoSOX mitochondrial superoxide indicator fluorescent probe for 25 min at 37°C protected from light. Once washed with PBS, the image of the cells was observed by a fluorescence microscope (Nikon, Tokyo, Japan). Upon oxidation by superoxide, MitoSOX Red emits red fluorescence, allowing for the visualization and quantification of mitochondrial ROS levels through fluorescence microscopy.
Mitochondrial membrane potential (ΔΨm) measurement
Mitochondrial membrane potential was detected by the JC-1 kit (Beyotime, Shanghai, China), which accumulates in mitochondria. After treatment, H9c2 cells were incubated with the JC-1 dye for 30 min at 37°C protected from light, and then washed with PBS three times. Then images were captured immediately with a fluorescence microscope (Nikon, Tokyo, Japan). JC-1 forms aggregates in healthy polarized mitochondria (red fluorescence), while in depolarized mitochondria, it remains in a monomeric form (green fluorescence). By measuring the ratio of green to red fluorescence, changes in the mitochondrial membrane potential can be assessed.
WB analysis
H9c2 cells or heart tissue homogenates were lysed using WB Super RIPA Lysis Buffer (HaiGene, Harbin, China) containing 1% phenylmethylsulfonyl fluoride, 1% protease inhibitor cocktail, 1% phosphatase inhibitor cocktail (MedChemExpress, New Jersey, USA), and 0.25% benzonase nuclease (HaiGene, Harbin, China) on ice. The protein concentration was measured using the BCA Protein Assay Kit (Beyotime, Shanghai, China). Fifteen micrograms of total protein was separated using 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel and transferred to 0.45 μm polyvinylidene fluoride membranes for 1.5 h. Then the membranes were blocked with Tris-buffered saline containing 5% BSA and incubated with primary antibodies overnight at 4°C. Secondary antibodies were added and incubated for 1 h at room temperature. The signal was detected using the chemiluminescence image analysis system (Tanon, Shanghai, China). All protein quantifications were normalized to the expression level of β-actin. The antibodies used in the study were anti-TFRC, anti-ACSL4, anti-XCT, anti-GPX4 (1:5000; Abcam, Cambridge, UK), anti-STING, anti-p65, anti-p-p65, anti-TBK1, anti-p-TBK1, anti-IRF3, anti-p-IRF3, anti-β-actin (1:1000; Cell Signaling Technology, Danvers, MA, USA), and anti-IFN-β (1:1000; Affinity Biosciences, OH, USA).
Statistical analysis
All the experiments were repeated at least three times, and results were presented as mean ± SD. Comparisons between groups were performed using the paired or unpaired Student’s t-test or post hoc analysis with one-way analysis of variance. All statistical analyses were performed with the aid of SPSS Version 22 (SPSS, Chicago, USA). Graphs were drawn using GraphPad Prism 9.3 (GraphPad Software, San Diego, USA). A p value of < 0.05 was indicated as statistically significant. Electronic laboratory notebook was not used.
Footnotes
Authors’ Contributions
X.T. and S.L. designed the experiments and article; Q.C., Y.W., J.W., X.O., Y.H., Z.H., and B.Z. were responsible for the cell and animal experiments. J.Z. and L.P. instructed the technology for the experiments. Q.C., Y.W., and J.W. contributed to the draft article. X.T. and S.L. critically revised the article. All authors read and approved the final article.
Data Availability Statement
All data generated or analyzed during this study are included in the article/Supplementary Data S1.
Author Disclosure Statement
The authors declare no conflict of interests.
Funding Information
This study was funded by the
Supplementary Material
Supplementary Data S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.