
Abstract
Aims:
Tumor microenvironment (TME) plays a crucial role in sustaining cancer stem cells (CSCs). 4-hydroxynonenal (4-HNE) is abundantly present in the TME of colorectal cancer (CRC). However, the contribution of 4-HNE to CSCs and cancer progression remains unclear. This study aimed to investigate the impact of 4-HNE on the regulation of CSC fate and tumor progression.
Methods:
Human CRC cells were exposed to 4-HNE, and CSC signaling was analyzed using quantitative real-time polymerase chain reaction, immunofluorescent staining, fluorescence-activated cell sorting, and bioinformatic analysis. The tumor-promoting role of 4-HNE was confirmed using a xenograft model.
Results:
Exposure of CRC cells to 4-HNE activated noncanonical hedgehog (HH) signaling and homologous recombination repair (HRR) pathways in LGR5+ CSCs. Furthermore, blocking HH signaling led to a significant increase in the expression of γH2AX, indicating that 4-HNE induces double-stranded DNA breaks (DSBs) and simultaneously activates HH signaling to protect CSCs from 4-HNE-induced damage via the HRR pathway. In addition, 4-HNE treatment increased the population of LGR5+ CSCs and promoted asymmetric division in these cells, leading to enhanced self-renewal and differentiation. Notably, 4-HNE also promoted xenograft tumor growth and activated CSC signaling in vivo.
Innovation and Conclusion:
These findings demonstrate that 4-HNE, as a signaling inducer in the TME, activates the noncanonical HH pathway to shield CSCs from oxidative damage, enhances the proliferation and asymmetric division of LGR5+ CSCs, and thereby facilitates tumor growth. These novel insights shed light on the regulation of CSC fate within the oxidative TME, offering potential implications for understanding and targeting CSCs for CRC therapy. Antioxid. Redox Signal. 42, 265–279.
Innovation
4-HNE, a lipid peroxidation product, is enriched in the TME of CRC. However, the impact of 4-HNE overloading on CRC progression remains unclear. In the present study, we demonstrate that 4-HNE activates a noncanonical HH signaling pathway, which protects colorectal CSCs against 4-HNE-induced oxidative damage. Importantly, 4-HNE increases the LGR5+ CSC population (i.e., self-renewal) and also promotes asymmetric division (i.e., differentiation) of LGR5+ CSCs, leading to CRC growth and progression. These novel findings shed light on the regulation of CSC fate by the oxidative TME and may help develop new strategies for CRC treatment.
Introduction
The tumor microenvironment (TME) is a complex architecture that comprised various types of cells, extracellular matrix, growth factors, cytokines, and fatty acids (Anderson and Simon, 2020). TME plays crucial roles in facilitating tumor growth, progression, and metastasis, as well as maintaining the cancer stem cell (CSC) niche. CSCs, a subset of cancer cells within the tumor mass, possess self-renewal and multidirectional differentiation abilities. CSCs are pivotal for tumor heterogeneity, metastasis, and recurrence. Furthermore, CSCs are equipped with robust DNA damage repair systems and exhibit resistance to apoptosis, leading to cancer resistance against chemotherapeutic drugs and radiation therapy (Abad et al., 2020). Consequently, the development of drugs targeting TME, particularly CSCs, represents a promising area for cancer therapy (Saygin et al., 2019).
Fast-proliferating cancer cells exhibit heightened lipid metabolism, leading to an accumulation of lipid metabolic by-products. Moreover, TME is characterized by hypoxia, acidity, and oxidative stress, which result in elevated levels of reactive oxygen species/reactive nitrogen species (ROS/RNS). These free radicals can trigger lipid peroxidation, generating toxic compounds such as 4-hydroxynonenal (4-HNE) and malondialdehyde. Specifically, 4-HNE is a highly reactive aldehyde generated by peroxidation of ω−6 polyunsaturated fatty acids. It reacts with DNA, RNA, and proteins leading to DNA cross-links, mutations, and functional alterations in proteins (Feng et al., 2003; Huang et al., 2010). Previous studies have demonstrated that 4-HNE derived from M1-polarized macrophages induces gene mutations, chromosomal instability, stemness, and neoplastic transformation, leading to colorectal cancer (CRC) (Wang et al., 2015; Wang et al., 2013; Wang et al., 2012). Furthermore, 4-HNE-adducts are strongly expressed in the TME of human-invasive CRC and murine colitis-associated cancer (Ma et al., 2023; Yang et al., 2016). However, the impact of 4-HNE accumulation in the TME on CSC fate remains uncertain.
Hedgehog (HH) signaling plays a crucial role in colorectal carcinogenesis and CSC expansion (Hanna and Shevde, 2016; Varnat et al., 2009). The canonical HH signaling pathway is triggered by binding of HH ligands to a 12-pass transmembrane receptor PTCH1, leading to the release of smoothened (SMO). SMO activates glioma-associated oncogene homolog 1 (GLI1) that is translocated to the nucleus and subsequently activates transcription of downstream genes. In contrast, noncanonical activation of HH signaling, independent of ligand overexpression or receptor malfunction, is crucial for colorectal CSC survival (Regan et al., 2017). In addition, HH signaling contributes to DNA damage repair mechanisms, including nucleotide excision repair (NER), base excision repair (BER), and nonhomologous end joining (NHEJ) repair (Kudo et al., 2012; Lama-Sherpa et al., 2020). Recent studies have highlighted a role of HH signaling in regulating chemoresistance (Chen et al., 2021). However, the specific contribution of HH signaling-mediated DNA damage response to CSC survival in the oxidative TME remains unclear.
In the present study, we explored the impact of 4-HNE on the activation of HH signaling and its influence on CSC fate determination in human CRC cells. We observed that CRC cells and TME substantially produced 4-HNE. Exogenous 4-HNE induced double-stranded DNA breaks (DSBs) in CRC cells and, concurrently, activated a noncanonical HH signaling pathway that initiates DNA repair through the homologous recombination repair (HRR) mechanism. Furthermore, 4-HNE enhanced both self-renewal and differentiation of LGR5+ CSCs, thereby promoting tumor growth. These findings offer novel insights into the role of 4-HNE within the TME in sustaining CSCs and facilitating CRC progression.
Results
Colon cancer cells and TME generate 4-HNE
Previous studies have shown that 4-HNE mediates the microbiota-induced bystander effect (MIBE) leading to CRC in murine models. In addition, 4-HNE-adducts are present in human and murine CRC (Ma et al., 2023; Wang et al., 2012; Yang et al., 2016). To determine the source of 4-HNE in CRC, we initially stained 4-HNE-adducts in HCT116 cells. Immunofluorescent (IF) staining showed positive staining for 4-HNE-adducts in HCT116 cells, indicating autocrine production of 4-HNE by CRC cells in vitro (Supplementary Fig. S1A). We next stained 4-HNE-adducts in HCT116-derived xenograft tumors and showed positive staining for 4-HNE and colocalization with COX-2 in cancerous cells (Supplementary Fig. S1B), supporting the notion that 4-HNE may be derived from COX-2 signaling as previously reported (Wang et al., 2013). Notably, F4/80+ macrophages also displayed positive staining for 4-HNE-adducts (Supplementary Fig. S1C). We observed intensive staining of 4-HNE-adducts in both M1- and M2-polarized macrophages (Supplementary Fig. S1D and E). Furthermore, IF staining indicated that CD3+ T cells, but not neutrophils (Elastase+) and B cells (CD19+), were partially responsible for producing 4-HNE in the TME (Supplementary Fig. S1F–H). Collectively, these results suggest that CRC cells have the capacity to generate 4-HNE in an autocrine manner, and that the TME is enriched with 4-HNE.
4-HNE activates noncanonical HH signaling pathway in CRC cells
Because HH signaling plays a crucial role in modulating CSC fate and behavior (Ruiz i Altaba, 2011), we determined the impact of 4-HNE on the HH signaling pathway. Quantitative real-time polymerase chain reaction (qRT-PCR) demonstrated that treatment with 4-HNE led to a tendency of decreased expression of sonic HH (SHH), a canonical ligand of HH signaling, in HCT116 and HT29 cells compared with untreated controls (p = 0.055 and p < 0.05, respectively; Fig. 1A and B). However, the expression of PTCH1, the receptor of SHH, remained unchanged in both cell lines following 4-HNE treatment (Fig. 1A and B). Interestingly, the expression of the HH signaling transducer SMO significantly increased in HCT116 cells (p < 0.05) but decreased in HT29 cells (p < 0.01). Remarkably, the transcription factor GLI1 significantly increased in both HCT116 and HT29 cells compared with untreated controls (p < 0.05; Fig. 1A and B). Further analysis using fluorescence-activated cell sorting (FACS) demonstrated that the proportion of SMO- and GLI1-positive cells significantly increased for HCT116 cells treated with 4-HNE compared with control (28.70 ± 1.66 vs. 19.87 ± 2.52 and 38.08 ± 2.31 vs. 26.78 ± 2.22 for SMO and GLI1, respectively, Fig. 1C and D). Furthermore, IF staining confirmed increased expression of SMO and GLI1 in 4-HNE-treated HCT116 cells compared with untreated control (Fig. 1E). As a control experiment, expression of Smo and Gli1 in a primary rat intestinal epithelial cell line (IEC-6) was assessed. Unlike CRC cells, IEC-6 cells constitutively expressed Smo and Gli1, whereas 4-HNE treatment had no effect on the expression of Smo and Gli1 in these cells (Supplementary Fig. S2). Taken together, 4-HNE activates HH signaling in CRC cells independently of the SHH/PTCH1 cascade, suggesting a noncanonical mechanism of HH signaling activation.

HH signaling protects CRC cells from 4-HNE-induced DNA damage
4-HNE is an endogenous mutagen leading to DSBs and G2/M arrest (Wang et al., 2012). Consistent with our previous findings, exposure of HCT116 and HT29 cells to 4-HNE remarkably increased the expression of γH2AX, a biomarker for DSBs, compared with untreated controls (Fig. 2A–C). To explore the impact of HH signaling on 4-HNE-induced DNA damage, HCT116 and HT29 cells were treated with 4-HNE in combination with GANT61, a specific inhibitor of GLI1, and DNA damage response was analyzed. As depicted in Figure 2A–C, GANT61 inhibited GLI1 expression and exacerbated γH2AX expression compared with cells treated with 4-HNE alone, indicating a protective role of HH signaling for 4-HNE-induced DNA damage. Moreover, 4-HNE treatment also increased the expression of phosphorylated ATM (p-ATM), phosphorylated CHK2 (p-CHK2), and cyclin B1, while inhibition of HH signaling further exacerbated 4-HNE-induced p-ATM, p-CHK2, and cyclin B1 (Fig. 2A and B). Notably, although 4-HNE-induced γH2AX and GLI1 were observable independent of the cell cycle, cyclin B1-positive cells significantly increased in 4-HNE-treated HCT116 cells compared with untreated controls (Supplementary Fig. S3), further supporting G2/M arrest caused by 4-HNE (Wang et al., 2012). IF staining confirmed an increase in γH2AX foci in HCT116 cells treated with 4-HNE in combination with GANT61 compared with those treated with 4-HNE alone (Fig. 2D and E). These results suggest that 4-HNE-activated HH signaling protects CRC cells against 4-HNE-induced DNA damage in a feedback manner.
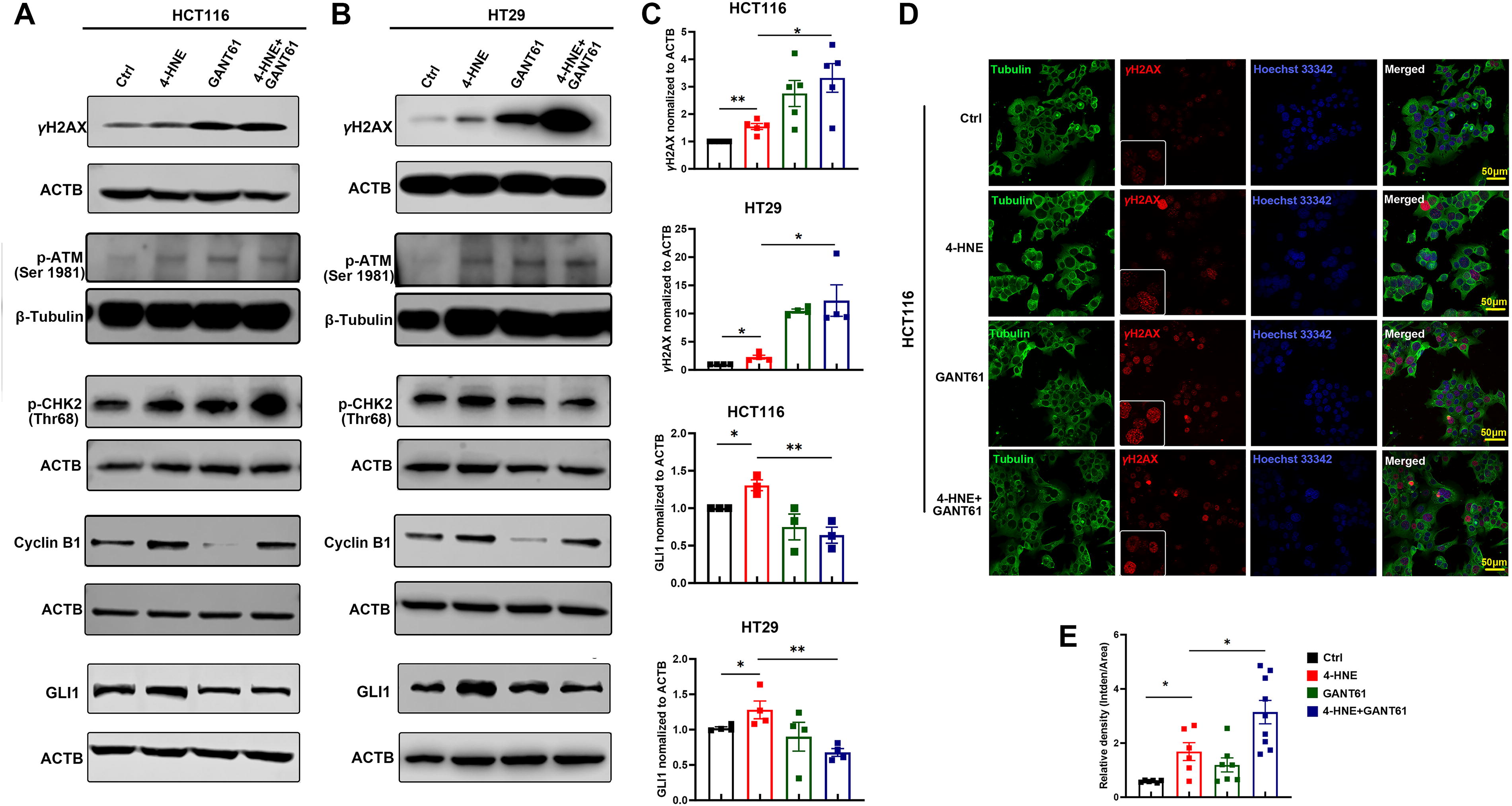
Activation of HH signaling is associated with self-renewal of LGR5+ CSCs
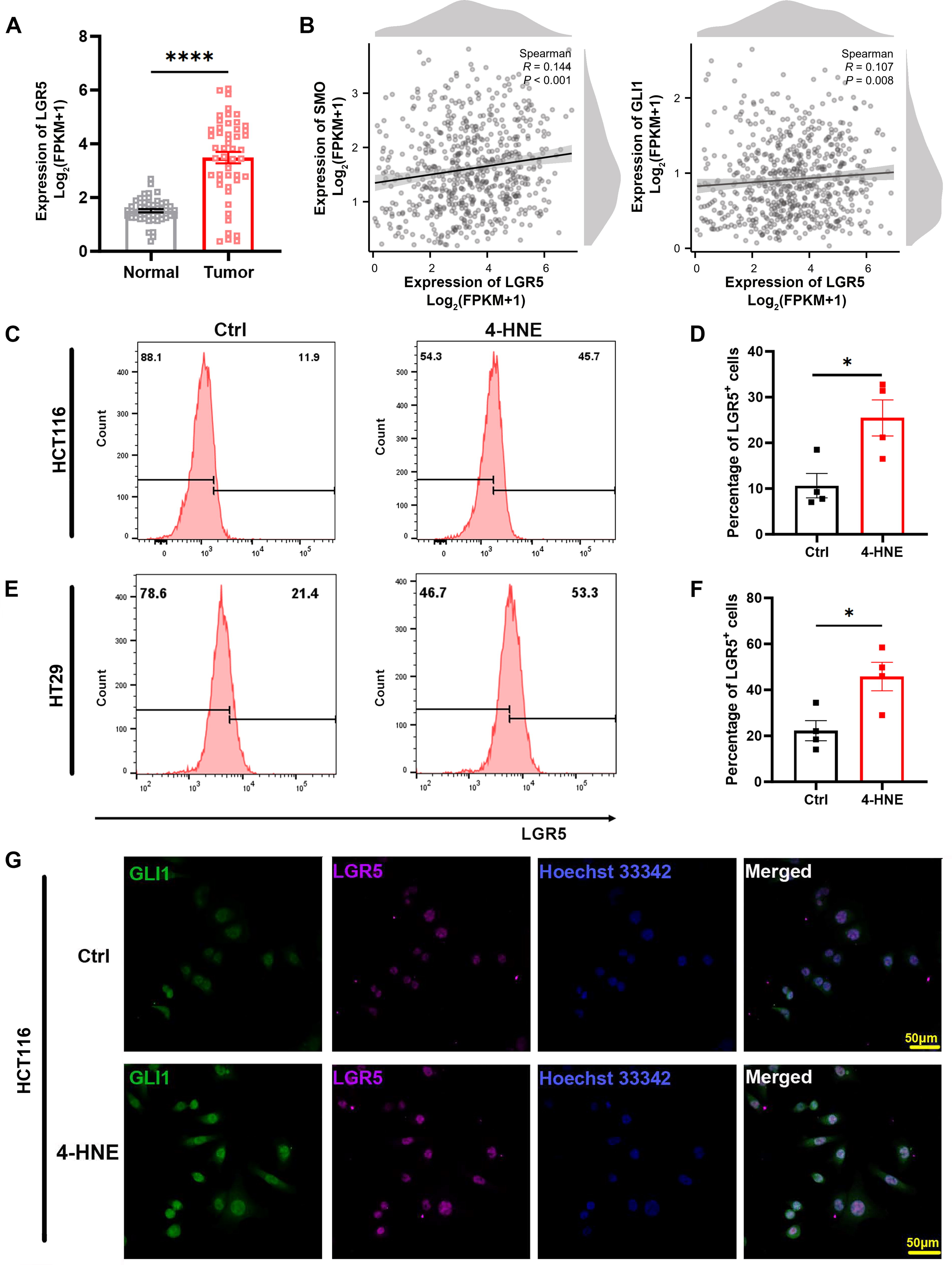
HH signaling is crucial for CSC self-renewal (Zhao, 2014). To investigate the impact of 4-HNE-induced HH signaling on colorectal CSCs, we analyzed the expression of LGR5, an intestinal stem cell marker, in human CRC cells and the adjacent normal colon tissues. Analysis of The Cancer Genome Atlas (TCGA) data showed significantly elevated LGR5 expression in CRC cells compared with matched normal tissues (p < 0.0001; Fig. 3A). Furthermore, LGR5 expression was positively correlated with the expression of SMO (R = 0.144, p < 0.001) and GLI1 (R = 0.107, p < 0.01) in CRC cells (Fig. 3B). To validate these findings, we treated HCT116 and HT29 cells with serially diluted 4-HNE and analyzed LGR5 expression. Western blots showed that 4-HNE treatment enhanced LGR5 expression independently of 4-HNE concentration in both cell lines (Supplementary Fig. S4A and B). Furthermore, we analyzed the percentage of LGR5-positive (LGR5+) cells in 4-HNE-treated HCT116 and HT29 cells using FACS assay. Treatment with 4-HNE significantly increased the proportion of LGR5+ cells in both cell lines compared with untreated controls (25.48 ± 3.95 vs. 10.65 ± 2.66, p < 0.05 and 45.83 ± 6.19 vs. 22.28 ± 4.36, p < 0.05 for HCT116 and HT29 cells, respectively; Fig. 3C–F). IF staining of LGR5 and GLI1 further corroborated elevated expression in 4-HNE-treated cells compared with untreated controls (Fig. 3G and Supplementary Fig. S4C). Notably, colocalization of GLI1 with LGR5 supported the activation of HH signaling in LGR5+ CSCs, linking it to self-renewal of colorectal CSCs.
4-HNE activates HRR pathway in CSCs
CSCs, akin to adult stem cells, possess robust DNA damage repair mechanisms that confer resistance to oxidative damage from TME and therapies (Vitale et al., 2017). We next investigated the DNA repair mechanisms in response to 4-HNE-induced DNA damage. qRT-PCR demonstrated significantly increased expression of the HRR genes BRCA1 and RAD51 in 4-HNE-treated HCT116 and HT29 cells compared with untreated controls (Fig. 4A–B). IF staining further revealed enhanced BRCA1 and nuclear-localized RAD51 foci in 4-HNE-treated HCT116 cells compared with untreated control (Supplementary Fig. S5A). FACS analysis confirmed a higher proportion of RAD51-positive cells in 4-HNE-treated HCT116 cells compared with untreated cells (74.45 ± 3.31% vs. 62.53 ± 3.07%, p < 0.05; Fig. 4C). The proportion of BRCA1-positive cells also had a tendency to increase after 4-HNE treatment, but no statistical significance was reached compared with untreated control (2.67 ± 0.27% vs. 1.98 ± 0.17%, p = 0.07; Fig. 4D).

Furthermore, TCGA data analysis also showed significantly elevated BRCA1 and RAD51 expression in human CRC cells compared with normal tissues (Fig. 4E). Interestingly, BRCA1 expression correlated positively with LGR5 expression (R = 0.137, p < 0.001), whereas no significant correlation was noted between RAD51 and LGR5 (R = −0.059, p = 0.143) in human CRC cells (Fig. 4F). Notably, the expression of RAD51 showed a significant correlation with the expression of two other colorectal CSC markers, CD44 (R = 0.131, p < 0.01) and ALDH1A1 (R = 0.114, p < 0.01, Supplementary Fig. S5B and C), highlighting the protective role of HRR pathways in CSCs. Finally, IF staining confirmed colocalization of RAD51 with LGR5 in 4-HNE-treated HCT116 and HT29 cells (Fig. 4G and Supplementary Fig. S5D), indicating activation of HRR pathway in LGR5+ CSCs in response to 4-HNE-induced DNA damage.
4-HNE induces pluripotency and asymmetric division
Previous studies have shown that 4-HNE can induce expression of CSC markers and pluripotency transcription factors (Wang et al., 2017; Wang et al., 2015). Hence, the expression of OCT4, KLF4, SOX2, and MYC was determined in 4-HNE-treated HCT116 and HT29 cells. qRT-PCR showed upregulated expression of MYC and OCT4 in 4-HNE-treated cells compared with untreated controls, while SOX2 expression decreased after 4-HNE treatment (Fig. 5A and B), suggesting partial activation of pluripotency transcription factors in CRC cells. In addition, spheroid forming assays showed that 4-HNE treatment increased the size of spheroids, indicative of enhanced anchorage-independent growth of CRC cells (Fig. 5C).

Because CSCs possess the capacity of asymmetric division (Mukherjee et al., 2015), we analyzed asymmetric division in HCT116 and HT29 cells using IF staining. Rare asymmetrically dividing cells were observed in both HCT116 and HT29 cells, while an overwhelming majority of symmetrically dividing cells were observed in these cells (Fig. 6). The number of CSCs undergoing asymmetric division (LGR5+/LGR5−) significantly increased in 4-HNE-treated HCT116 cells compared with control (19.67 ± 3.28 vs. 6.00 ± 1.16 per 500 dividing cells, p < 0.01; Fig. 6A and B). Likewise, the asymmetric division events also increased in 4-HNE-treated HT29 cells compared with control (17.67 ± 1.76 vs. 6.67 ± 0.89 per 500 dividing cells, p < 0.05; Fig. 6C and D), further supporting that 4-HNE influences both self-renewal and differentiation of LGR5+ CSCs, thereby promoting cancer growth.

4-HNE promotes xenograft tumor growth and CSC phenotype
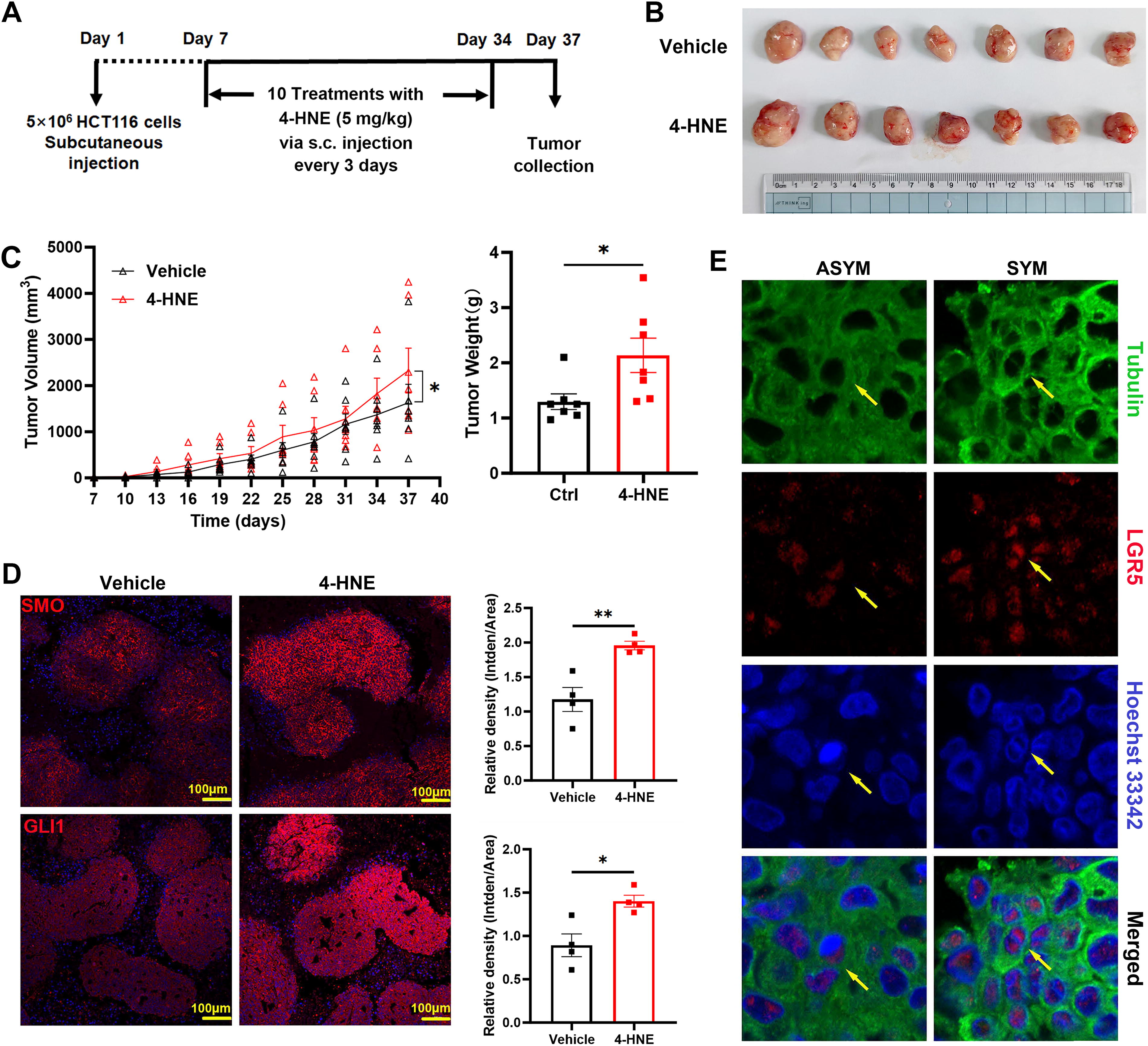
Finally, we investigated the impact of 4-HNE on CSC fate and tumor growth in vivo (Fig. 7A). Treatment with 4-HNE significantly increased xenograft tumor size and weight compared with untreated controls (Fig. 7B and C). IF staining demonstrated markedly increased expression of SMO and GLI1 in tumors from 4-HNE-treated mice compared with untreated mice (Fig. 7D), indicating activation of HH signaling in vivo. Moreover, both symmetric and asymmetric distributions of LGR5 were observed in xenograft tumors (Fig. 7E). These results support that 4-HNE activates HH signaling, influences CSC fate determination, and promotes tumor growth.
Discussion
The TME is an oxidative environment enriched with ROS due to the excessive growth and metabolism of cancer cells (Martinez-Reyes and Chandel, 2021). ROS have multiple effects on stem cell physiology, including proliferation, differentiation, and reprogramming (Cieślar-Pobuda et al., 2017). 4-HNE has been shown to reduce the growth and survival of breast CSCs and adult salivary stem cells (Cipak et al., 2010; Viswanathan et al., 2022). Furthermore, it has been observed that low levels of 4-HNE can prompt the differentiation of breast CSCs, while high levels promote epithelial–mesenchymal transition (Čipak Gašparović et al., 2019). Our previous studies have shown that 4-HNE induces dedifferentiation and reprogramming of colon epithelial cells (Wang et al., 2015).
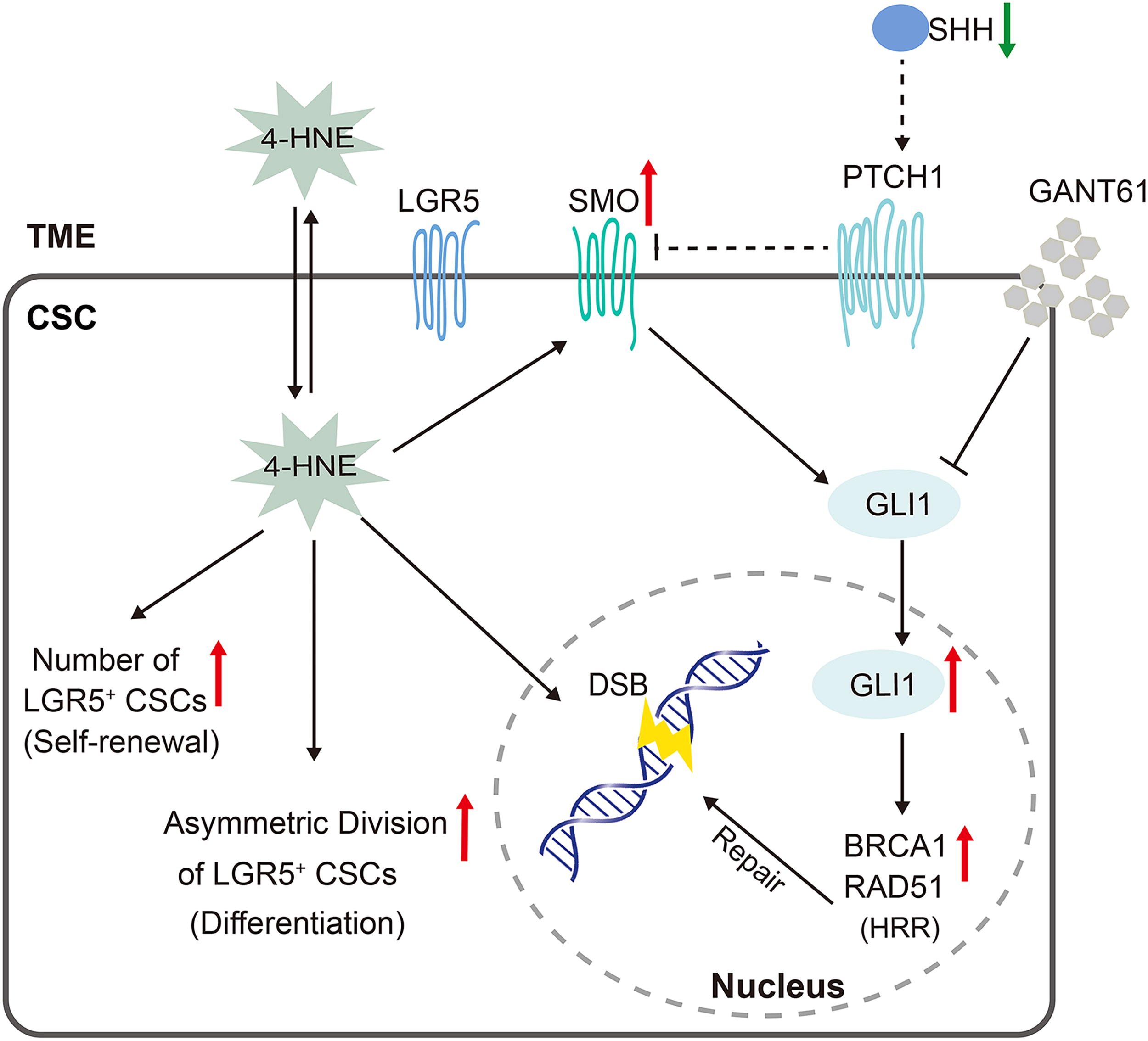
In the present study, we show that CRC cells substantially produce 4-HNE in an autocrine manner, and both M1- and M2-polarized macrophages and T cells in the TME also generate 4-HNE. These findings are consistent with previous reports showing 4-HNE-adducts in murine and human CRC cells (Wang et al., 2012; Yang et al., 2016). Overloading of 4-HNE in the TME induces DSBs and activates the noncanonical HH signaling pathway, which in turn activates DNA damage response in CSCs. Moreover, 4-HNE enhances LGR5+ CSC self-renewal and differentiation by increasing asymmetric division. Finally, 4-HNE promotes tumor growth and activates HH signaling in a xenograft tumor model, indicating that the accumulation of 4-HNE in the TME activates the HH signaling pathway in colorectal CSCs to repair 4-HNE-induced DNA damage and promote CSC self-renewal, differentiation, and tumor growth (Fig. 8).
As a signaling inducer, 4-HNE can regulate cancer cell proliferation, differentiation, and cell death through the activation of cancer-related signaling pathways such as NF-κB, Wnt/β-catenin, and AP-1 (Gasparovic et al., 2017; Wang et al., 2017; Yang et al., 2016). However, the regulation of HH signaling by 4-HNE has not been extensively investigated. Our study indicates noncanonical activation of HH signaling in CRC cells following 4-HNE treatment, which is supported by our observations that 4-HNE induces GLI1 expression and nuclear translocation, independent of SHH and PTCH1 activation, as previously reported (Geyer and Gerling, 2021; Pietrobono et al., 2019). Despite the decrease of SMO expression in 4-HNE-treated HT29 cells, GLI1 expression still increased in the treated cells.
In the colon, canonical activation of HH signaling often occurs in a paracrine manner with a tumor-suppressing role, whereas noncanonically activated HH signaling may play a tumor-promoting role (Geyer and Gerling, 2021). Although mutations in SHH gene have been reported in some cancers (Couve-Privat et al., 2004), gene mutation database searching confirmed no SHH gene mutation in both HCT116 and HT29 cells. Other HH ligands, including Indian hedgehog (IHH) and desert hedgehog, have much lower activity in PTCH1 activation (Pathi et al., 2001), and 4-HNE did not induce PTCH1 expression (Fig. 1A and B). In addition, deletion of Ihh in Apc −/− mice causes accumulation of Lgr5+ stem cells (Westendorp et al., 2021). As such, IHH may not be related to 4-HNE-induced HH signaling, which warrants further investigation.
In addition to regulating the CSC phenotype, HH signaling also participates in DSB repair (Lama-Sherpa et al., 2020). 4-HNE has been shown to cause DSBs, mutations, and chromosomal instability, while compromising DNA damage repair mechanisms such as the NER and BER pathways (Feng et al., 2004; Wang et al., 2015; Wang et al., 2012; Winczura et al., 2014). HRR can faithfully repair DSBs in dividing cells. Our results demonstrate that 4-HNE activates the HRR pathway in response to 4-HNE-induced DNA damage. ATM and RAD51 are the functional enzymes and indicators for HRR (Matsuoka et al., 2007; Schild and Wiese, 2010). While we did not analyze Holliday junction or perform radiation-induced HRR assay due to technical limitations, the significant increase in the expression of p-ATM and RAD51, following 4-HNE treatment, suggests the activation of HRR by 4-HNE. Taken together, 4-HNE activates noncanonical HH signaling and the HRR pathway that protect CSCs from oxidative damage in the TME, leading to tumor progression. Strategies targeting 4-HNE-induced signaling in CSCs and/or eliminating abundant 4-HNE from the TME may have implications for CRC therapy.
We have shown that 4-HNE induces the expression of CSC biomarkers and dedifferentiation through activating pluripotency transcription factors in YAMC primary mouse colonic epithelial cells (Wang et al., 2017; Wang et al., 2015). Although 4-HNE treatment increased the expression of MYC and OCT4 in CRC cells we have tested, no increased expression of KLF4 and SOX2 was observed in these cells. This may be due to the time point at which KLF4 expression did not respond to 4-HNE, and downregulation of SOX2 gene may be caused by hypermethylation often found in cancer cells (Otsubo et al., 2008). Indeed, reprogramming and dedifferentiation can be induced independent of SOX2 by alternative transcription factors or small molecules (Ichida et al., 2009; Li et al., 2011).
LGR5 is a marker of colon crypt stem cells that may serve as the cell origin of CRC cells (Barker et al., 2009). Asymmetric distribution of LGR5 was previously reported in CRC and hair follicle stem cells (Huh et al., 2011; Srinivasan et al., 2016). Interestingly and importantly, 4-HNE treatment increased the number of asymmetrically dividing LGR5+ cells, supporting LGR5 as a colorectal CSC marker. Asymmetric division is a mechanism that balances stem cell self-renewal and differentiation. Increased asymmetric division of CSCs can generate more differentiated progeny to satisfy the requirements of the cancer cell population for cancer growth while maintaining cancer heterogeneity and the CSC pool. Finally, Wnt signaling drives asymmetric division and expression of pluripotency transcription factors in embryoid stem cells (Habib et al., 2013). As such, 4-HNE may promote asymmetric division through Wnt signaling as 4-HNE does activate Wnt signaling and pluripotency transcription factors (Wang et al., 2017). Further studies are required for clarifying mechanisms of 4-HNE-induced asymmetric division.
In summary, our results show that CRC cells and the TME substantially produce 4-HNE that activates noncanonical HH signaling in CSCs in response to 4-HNE-induced DNA damage. Blocking HH signaling exacerbates 4-HNE-induced DSBs, while simultaneously activating the HRR pathway to repair 4-HNE-induced DSBs. This suggests a negative feedback mechanism that protects CSCs against oxidative damage from the TME. Furthermore, 4-HNE contributes to cancer growth and progression by promoting proliferation and asymmetric division of LGR5+ CSCs, reinforcing its role as a signaling inducer in promoting cancer progression and maintaining cancer cell plasticity.
Materials and Methods
Cell lines and reagents
Human colon cancer cell lines HCT116 and HT29 were purchased from the National Collection of Authenticated Cell Cultures of China (Shanghai, China). Rat small intestinal epithelial cell line IEC-6 was obtained from the National Infrastructure of Cell Line Resource (Beijing, China). 4-HNE was purchased from Abcam (Shanghai, China) and GLI1 inhibitor GANT61 was purchased from Picasso (Shanghai, China).
Tissue culture and treatment
HCT116 and HT29 cells were cultured in McCoy’5A (Pricella, Wuhan, China), and IEC-6 cells were cultured in DMEM (Gibco, Shanghai, China) supplemented with 10% fetal bovine serum (Servicebio, Wuhan, China), 100 units/mL penicillin (Gibco), and 100 μg/mL streptomycin (Gibco). Cells were maintained at 37°C in a humidified environment containing 5% CO2. Unless otherwise noted, HCT116 and HT29 cells were treated with 20 µM and 40 µM 4-HNE, respectively, for 48 h. For the HH/GLI1 blockage experiments, in addition to 4-HNE, HCT116 and HT29 cells were concurrently treated with 10 and 20 µM GANT61, respectively, for 48 h.
Xenograft assay
Animal study was approved by the Institutional Animal Care and Use Committee of Nantong University (Approval number: S20210912-301). Xenograft assay was performed as previously reported with modifications (Zhang et al., 2022). Briefly, 6-week-old specific pathogen-free male BALB/cJGpt-Foxn1nu/Gpt nude mice were randomly divided into 2 groups, with 7 mice per group. Mice were subcutaneously injected with HCT116 cells (5 × 106 cells/site) into the left flank. One week after grafting, a group of mice were subcutaneously injected with 4-HNE (5 mg/kg body weight in 200 μL sterile saline) every 3 days for 10 treatments. The other group was subcutaneously injected with sterile saline as controls. Mice were euthanized by cervical dislocation under anesthesia with CO2 overdose and tumor removed for subsequent experiments.
TCGA data analysis
All data used in this study were obtained from the TCGA database (https://tcga-data.nci.nih.gov/tcga/). Gene expression data of LGR5, SMO, and GLI1 were analyzed in 50 human CRC cells and 50 paired adjacent normal colon tissues (Supplementary Table S1), and gene expression correlation was analyzed using 622 CRC cells (Supplementary Table S2). The detailed demographic, genetic alteration, and stage of disease information for patients can be found on the TCGA website (https://portal.gdc.cancer.gov/). Bioinformatic analysis was performed using an online analysis tool (https://www.xiantaozi.com/). This online platform provides a user-friendly interface for analyzing bioinformatic data sets. Data were processed using R program (v3.6.3) and visualized with the ggplot2 (v3.3.3). The Spearman’s rank correlation coefficient test was used for correlation analysis.
IF staining
Cells were grown on tissue culture coverglasses in 24-well plates and subjected to treatments. Following treatment, cells were rinsed with PBS, fixed with 4% paraformaldehyde, and blocked with 5% goat serum and 0.3% Triton™ X-100 in PBS at room temperature for 1 h. Primary antibodies (Supplementary Table S3) were diluted in sterile PBS containing 1% bovine serum albumin (BSA) and 0.3% Triton™ X-100. Cells were incubated with the primary antibodies at 4°C overnight. After washing with PBS, cells were incubated with a secondary antibody (Supplementary Table S4) for 1 h at room temperature. Nuclei were counterstained with Hoechst 33342, and fluorescent images were captured using a confocal laser scanning microscope (Nikon, NY, USA).
For IF staining of paraffin-embedded tissue sections, tissue sections were deparaffinized, rehydrated, and subjected to epitope retrieval by boiling tissue sections for 10 min in glycine buffer (100 mM, pH 9.0). Tissue sections were then blocked with PBS containing 5% normal goat serum, 5% goat anti-mouse IgG serum (Solarbio, Beijing, China), and 0.3% Triton X-100 at room temperature for 1 h. The subsequent staining procedures were as described above.
Cell division symmetry assay
To analyze cell division symmetry, cells were treated with 4-HNE or PBS for 24 h. Cell division was synchronized by adding 2 mM of thymidine (Solarbio) to the medium and incubating for 16 h as previously reported (Wang et al., 2008). Cells were continuously treated with 4-HNE for 8 h in fresh medium free of thymidine. Subsequently, the cells were fixed, blocked, and stained with an antibody against the stem cell marker LGR5 (Affinity Biosciences, Liyang, China). Nuclei were counterstained by Hoechst 33342, and images were captured using a confocal laser scanning microscope (Nikon). A total of 500 dividing cells were captured for each group, and the number of asymmetric and symmetric divisions was counted.
Quantitative real-time polymerase chain reaction
Total RNA was isolated from both HCT116 and HT29 cells using the FastPure® Cell/Tissue Total RNA Isolation Kit v2 (Vazyme, Nanjing, China) and then reverse transcribed using HiScript III RT SuperMix (Vazyme) according to the manufacturer’s instruction. qRT-PCR was carried out using AceQ qPCR SYBR® Green Master Mix (Vazyme) on the LightCycler 96 (Roche, Shanghai, China). The relative gene expression was calculated using β-actin (ACTB) as a reference gene. The primers used are listed in Supplementary Table S5.
Fluorescence-activated cell sorting
Cells were harvested by trypsinization and washed with PBS after treatment. Cells (1.5 × 106) were fixed by 4% paraformaldehyde and permeabilized with ice-cold 90% methanol at 4°C for 15 min each. The cells were incubated with primary antibodies (Supplementary Table S3) for 1 h at 4°C, followed by incubation with a fluorescence-conjugated secondary antibody for 1 h at room temperature. FACS analysis was carried out using the DxFLEX Flow Cytometer (Beckman Coulter, Suzhou, China) and data analyzed using FlowJo software (BD Biosciences, Shanghai, China).
Western blotting
Whole cell lysates were collected using the cell lysis solution (Beyotime, Shanghai, China). Protein was separated by SDS–PAGE and transferred to PVDF membrane (Merck, Beijing, China). Blots were blocked with 5% skim milk in tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 h at room temperature and then incubated with a primary antibody at 4°C overnight. After washing with TBST, the blots were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. The primary and secondary antibodies are listed in Supplementary Tables S3 and S4. Signal was generated by an enhanced chemiluminescence substrate (Biosharp, Hefei, China) and captured using the Odyssey Fc system (LI-COR Biosciences, Lincoln, USA). Grayscale analysis was performed using ImageJ software.
Spheroid formation assay
Anchorage-independent growth was assessed by spheroid formation using the soft agar spheroid formation assay. Briefly, HCT116 cells (2.5 × 103) were embedded in 0.5% molten upper agarose (Sangon Biotech, Shanghai) and plated in 1% agarose-coated 12-well tissue culture plates. Cells were incubated in complete McCoy’s 5A medium at 37°C for 1 week to allow spheroid formation. The spheroids were then treated with 20 µM 4-HNE or DMSO as control, in complete McCoy’s 5A medium for an additional 7 days. The spheroid size was measured and analyzed using the ImageJ software.
Statistical analysis
Statistical analysis was performed using GraphPad version 8 (GraphPad Software, Beijing, China). Unpaired two-tailed Student’s t test or two-way analysis of variance was used for comparisons between groups. Chi-square test was used for comparisons of symmetric/asymmetric divisions between groups. Data are expressed as means ± standard errors of the means (SEMs). A p value of < 0.05 was considered statistically significant. An electronic laboratory notebook was not used.
Footnotes
Authors’ Contributions
X.H. and L.H.: study design, data acquisition, analysis, and interpretation, and drafting the article; C.M., M.H., L.X., and Y.J.: data acquisition and analysis and interpretation; H.L. and Y.W.: data analysis and critical review of the article; X.W.: conception and study design, analysis and interpretation of data, supervising the study, writing the article, and final approval of the version to be submitted.
Author Disclosure Statement
The authors declare that they have no conflicts of interest.
Funding Information
This study was supported by the
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.