
Abstract
Aims:
Succinate, a metabolite in the tricarboxylic acid cycle, is increasingly recognized to play essential roles in inflammation by functioning either as an intracellular or extracellular signaling molecule. However, the role and mechanisms of succinate in inflammation remain elusive. Here, we investigated the mechanism underlying the effects of succinate on neuroinflammation in intracerebral hemorrhage (ICH) models.
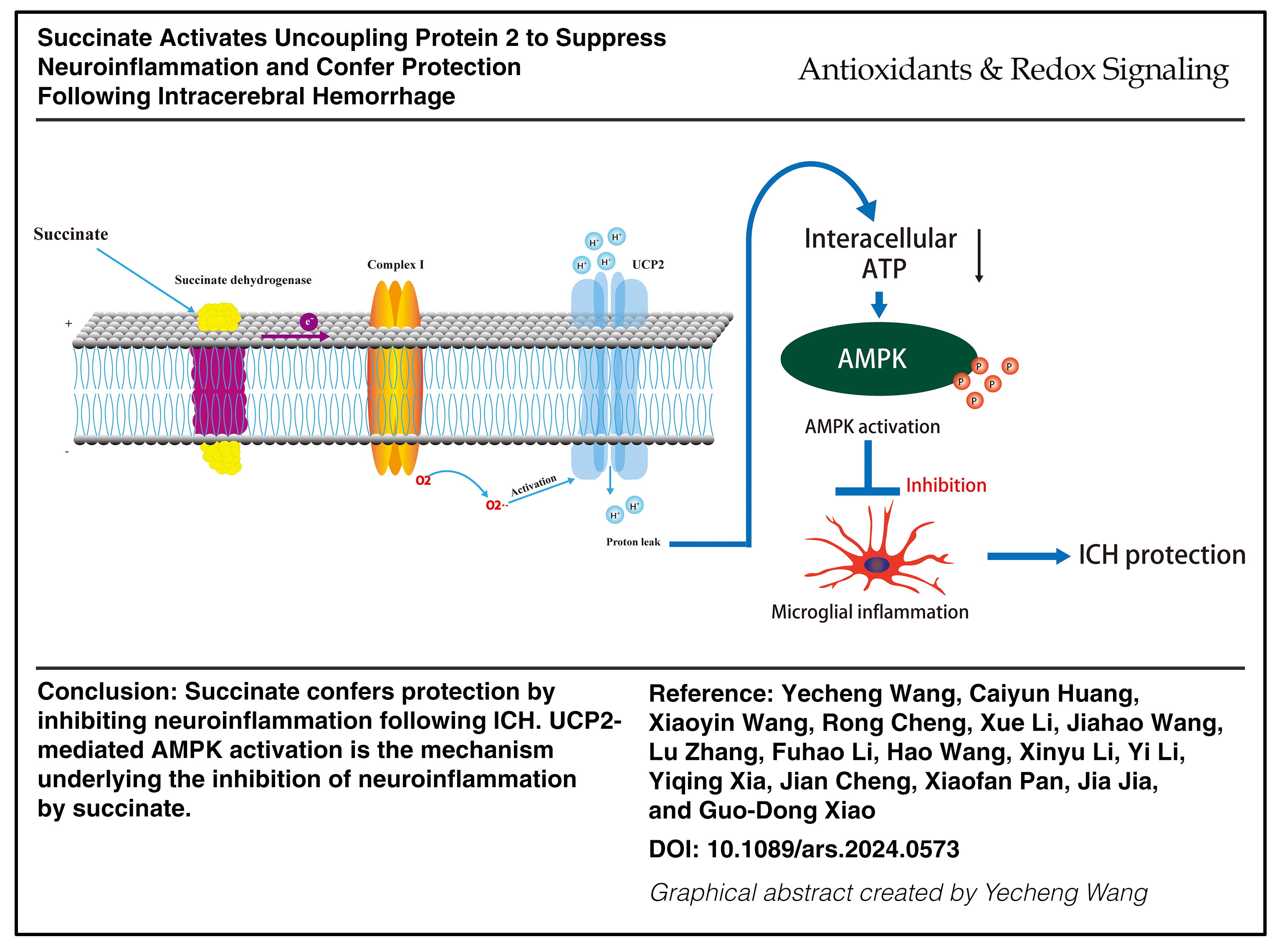
Results:
We unexpectedly found that succinate robustly inhibited neuroinflammation and conferred protection following ICH. Mechanistically, the oxidation of succinate by succinate dehydrogenase (SDH) drove reverse electron transport (RET) at mitochondrial complex I, leading to mitochondrial superoxide production in microglia. Complex I-derived superoxides, in turn, activated uncoupling protein 2 (UCP2). By using mice with specific deletion of UCP2 in microglia/macrophages, we showed that UCP2 was needed for succinate to inhibit neuroinflammation, confer protection, and activate downstream 5′-adenosine monophosphate-activated protein kinase (AMPK) following ICH. Moreover, knockdown of SDH, complex I, or AMPK abolished the therapeutic effects of succinate following ICH.
Innovation and Conclusion:
We provide evidence that driving complex I RET to activate UCP2 is a novel mechanism of succinate-mediated intracellular signaling and a mechanism underlying the inhibition of neuroinflammation by succinate. Antioxid. Redox Signal. 42, 687–710.
Introduction
Succinate is a metabolic intermediate in the tricarboxylic acid (TCA) cycle and an essential component of oxidative phosphorylation (Fernandez-Veledo et al., 2021). Succinate is at the crossroads of a variety of cellular metabolic pathways, including heme synthesis, ketone utilization, and macrophage-specific synthesis of itaconate (Abdullah et al., 2023). Notably, succinate also acts as a signaling transmitter both extracellularly and intracellularly (Abdullah et al., 2023; Fernandez-Veledo et al., 2021). In response to ischemic and proinflammatory stimuli, succinate accumulates in mitochondria. Then, succinate is oxidized by succinate dehydrogenase (SDH) to trigger intracellular signaling via mitochondrial reactive oxygen species (ROS) (Chouchani et al., 2014; Mills et al., 2016; Murphy and O’Neill, 2018). In contrast, intracellular succinate can be transported outside of cells, where it signals through succinate receptor 1 (SUCNR1) (Fernandez-Veledo et al., 2021). The wide expression pattern of SUCNR1 implies that the activation of SUCNR1 by extracellular succinate is a common signaling mechanism of succinate (Fernandez-Veledo et al., 2021; He et al., 2004; Krzak et al., 2021). As an extracellular signaling molecule, succinate plays a dual role in inflammation. Extracellular succinate either boosts the inflammatory response or resolves acute inflammation by facilitating alternative polarization of macrophages (Fernandez-Veledo et al., 2021; Keiran et al., 2019).
Innovation
Although the role and mechanisms of succinate in inflammation remain elusive, succinate is increasingly recognized to play essential roles in inflammation by functioning as either an intracellular or an extracellular signaling molecule. Here, we reported that succinate acts as a novel UCP2 activator to inhibit microglia/macrophage-mediated neuroinflammation and confer neuroprotection following ICH. This study provides a potentially new and practical pharmacological approach to treat ICH by activating UCP2 with succinate-based therapies. Moreover, we provide new evidence that driving complex I RET to activate UCP2 is a novel mechanism of cellular signaling by succinate.
Intracellular succinate also directly modulates inflammation. Succinate is a well-established inducer of reverse electron transport (RET) at mitochondrial complex I (complex I RET) (Chouchani et al., 2014; Mills et al., 2016). Intracellular succinate accumulates mainly via glutamine-dependent anaplerosis in lipopolysaccharide (LPS)-treated macrophages (Mills et al., 2016). Then, accumulated succinate is oxidized by SDH, leading to complex I RET. Consequently, complex I RET enhances the production of ROS in mitochondria. ROS, in turn, induce the expression of the proinflammatory cytokine interleukin (IL)-1β by inhibiting the prolyl hydroxylase domain protein to stabilize hypoxia-inducible factor 1 (Mills et al., 2016; Tannahill et al., 2013). These results suggest that intracellular succinate exacerbates inflammation. In contrast, recent publications have shown that succinate suppresses microglia/macrophage-mediated inflammation via a SUCNR1-independent mechanism or by reducing ROS generation (Harber et al., 2020; Wang et al., 2021). Moreover, intracellular succinate accumulates in various tissues due to the reversal of SDH function during ischemia. Then, accumulated succinate is rapidly oxidized by SDH, thereby driving complex I RET to generate high levels of ROS (Abdullah et al., 2023; Chouchani et al., 2014; Chouchani et al., 2016b). This likely represents a common mechanism underlying tissue injury following ischemia and traumatic injury (Chouchani et al., 2016b).
Intracerebral hemorrhage (ICH) is triggered by the rupture of cerebral vessels. Subsequently, the entry of blood components into the cerebral parenchyma induces multiple pathogenic cascades, including microglia/macrophage-mediated neuroinflammation (Wang et al., 2014; Zhou et al., 2014b). Since ROS exacerbate microglia/macrophage-mediated inflammation and injury following injury (Abdullah et al., 2023; Chouchani et al., 2014; Chouchani et al., 2016b; Mills et al., 2016), we initially assumed that succinate, an inducer of complex I RET, would exacerbate brain injury by enhancing the generation of mitochondrial ROS following ICH. However, unexpectedly, we found that supplementation with succinate ameliorated brain injury and suppressed microglia/macrophage-mediated neuroinflammation following ICH. This indicates that the roles of succinate in inflammation as well as the mechanisms underlying succinate signaling are complex. Mitochondria play essential roles in neurodegenerative diseases (Wang et al., 2019a; Wang et al., 2019b). Notably, we recently reported that the mitochondrial superoxide anion (O2 −) radical derived from complex I RET activated mitochondrial uncoupling protein 2 (UCP2). UCP2 activation consequently inhibited microglia-mediated neuroinflammation and ameliorated brain injury following ICH (Jia et al., 2020; Yan et al., 2022b). Thus, we tested the hypothesis that succinate acts through UCP2 activation to inhibit neuroinflammation and confer protection following ICH. Our study revealed a novel intracellular signaling pathway and an unexpected protective role of succinate following ICH.
Results
Succinate suppresses microglia/macrophage-mediated neuroinflammation and confers protection following ICH
To mimic ICH-induced activation of microglia in the in vitro neuroinflammation model, we treated BV2 microglia with lysates (LYs) of red blood cells (RBCs), as reported previously (Jia et al., 2020; Wang et al., 2014; Yan et al., 2022b). Moreover, we used a more cell-permeable analog, diethyl succinate (Suc), in the cellular model since it is commonly used in vitro (Abdullah et al., 2023; Mills et al., 2016; Wang et al., 2021). Compared with the vehicle treatment, RBC LY treatment increased the protein levels of IL-1β, tumor necrosis factor (TNF)-α, and IL-6 in the culture medium of BV2 cells (Fig. 1A–C), suggesting that RBC LY induced neuroinflammation in microglia. Suc at a concentration of 5 mM exacerbates macrophage-mediated inflammation upon LPS stimulation (Mills et al., 2016). To our surprise, Suc at 5 mM suppressed neuroinflammation in BV2 microglia treated with RBC LY, as evidenced by the reduction in the protein levels of proinflammatory cytokines (IL-1β, TNF-α, and IL-6) secreted from microglia treated with RBC LY (Fig. 1A–C). Since these results contrast with those of the study by Mills (Mills et al., 2016), we investigated whether the inconsistency can be attributed to the use of different inflammation models. Thus, we examined the effects of succinate on LPS-treated microglia. Succinate also inhibited inflammation in LPS-treated microglia at the same concentration as that used by Mills (Supplementary Figure S1A–C).
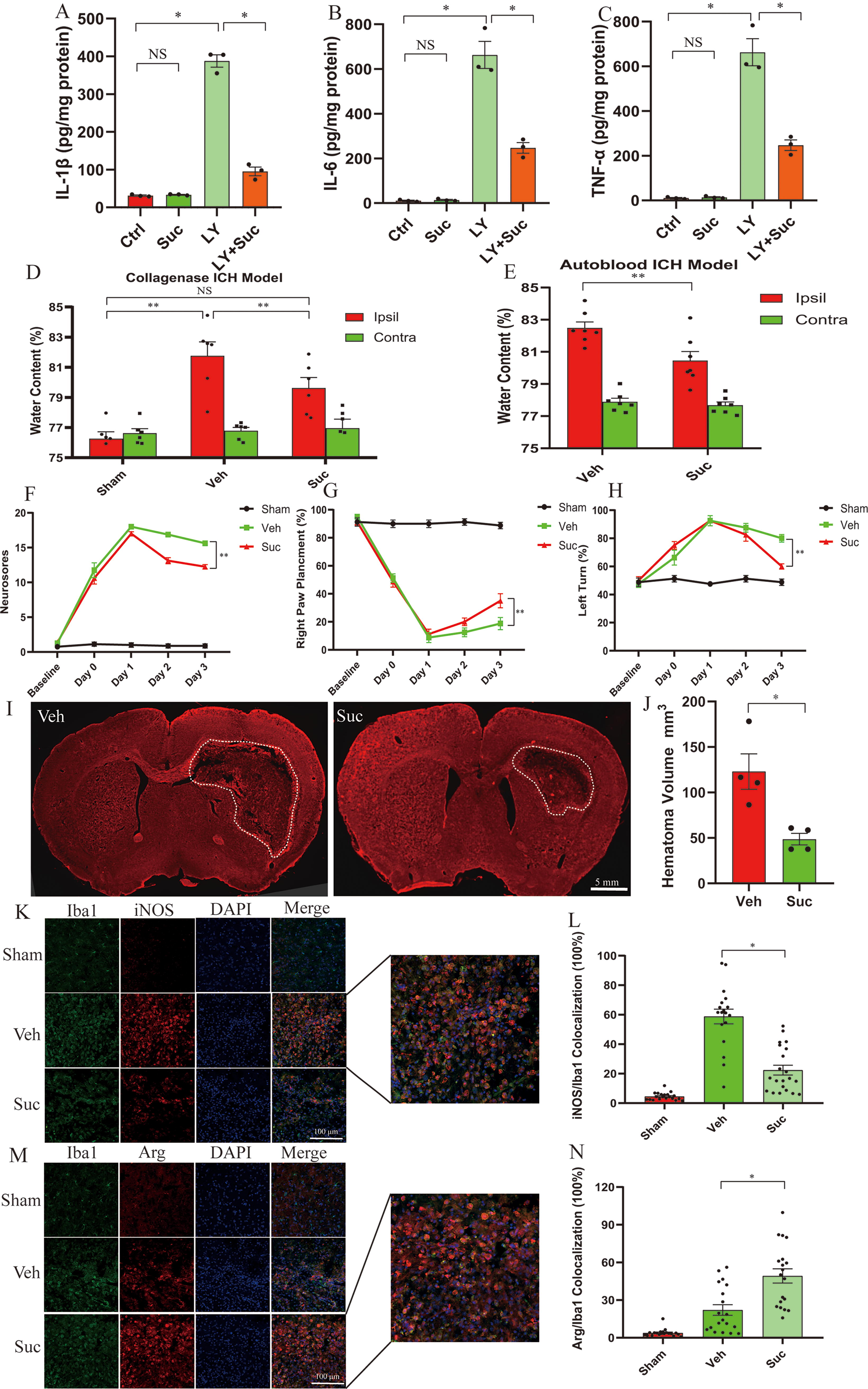
Next, we investigated the effects of succinate on the in vivo ICH models. Succinic disodium perfused into mice not only penetrates cells but is also oxidized by mitochondrial SDH to generate ROS (Jalloh et al., 2017; Mills et al., 2018). We administered succinic disodium via daily intraperitoneal injection starting 3 h after the induction of ICH. First, we found that succinate dose-dependently reduced brain edema (20–80 mg/kg/day) at 3 days following collagenase-induced ICH, as indicated by the water content of the striatum (Supplementary Figure S2A). Based on these results, we used succinate at 40 mg/kg/day in subsequent in vivo research. We found that the endogenous levels of succinate both in the serum and in the brain were not altered following ICH (Supplementary Figure S2B, C). Notably, succinate levels in the serum and brain were significantly greater in the mice that received an intraperitoneal injection of succinate (40 mg/kg/day) than in the vehicle-injected mice at 3 days following collagenase-induced ICH or in the sham-operated mice (Supplementary Figure S2B, C). We found that succinate (40 mg/kg/day) conferred protection at 3 days following ICH, as evidenced by reduced brain edema in both the collagenase-induced and autologous blood-induced ICH models (Fig. 1D, E). Moreover, compared with the vehicle administration, succinate administration improved behavioral performance in three neurobehavioral tests (neurological deficits, the vibrissae-elicited forelimb placement test, and the corner test) following collagenase-induced ICH (Fig. 1F–H). To further show the therapeutic effects of succinate, we histologically examined hemorrhagic lesions by identifying the destruction of myelin architectures as previously reported (Chang et al., 2014; Yan et al., 2022b). Mice receiving succinate displayed smaller hemorrhagic lesions than mice receiving vehicle at 3 days following collagenase-induced ICH (Fig. 1I, J).
We further assessed the effect of succinate on neuroinflammation following ICH. The enzyme-linked immunosorbent assay (ELISA) results suggested that ICH enhanced the expression of IL-1β, TNF-α, and IL-6 in the hemorrhagic striatum but not in the contralateral striatum at 3 days post-ICH, and this effect was markedly inhibited by succinate in both the collagenase-induced and autologous blood-induced ICH models (Supplementary Figure S2D–I). We further characterized the M1 and M2 polarization of microglia/macrophages by using inducible nitric oxide (iNOS) and arginase (Arg), respectively, as specific markers of M1 and M2 polarization (Hu et al., 2015). Compared with that in the sham-operated brain, iNOS expression in the hemorrhagic striatum was markedly increased, and iNOS was predominantly colocalized with the microglia/macrophage marker Iba1 3 days after ICH. ICH-enhanced iNOS expression in the hemorrhagic striatum was markedly attenuated in succinate-treated mice (Fig. 1K, L). In contrast, compared with vehicle treatment, succinate treatment markedly elevated the expression of the M2 marker Arg in the hemorrhagic striatum (Fig. 1M, N). In conclusion, succinate conferred protection and suppressed neuroinflammation following ICH.
Succinate drives complex I RET to activate UCP2 in microglia
The hallmarks of complex I RET are the enhanced production of mitochondrial superoxides and the blockade of this enhancement by inhibitors of complex I (Jia et al., 2020; Mills et al., 2016; Robb et al., 2018). As indicated by the enhanced colocalization of MitoSOX red fluorescence and MitoTracker green fluorescence (Jia et al., 2020), diethyl succinate (Suc) enhanced mitochondrial superoxide levels in BV2 microglia in a dose-dependent manner (Supplementary Figure S3A, B). Elevation of mitochondrial superoxides was evident in microglia treated with 5 mM Suc, while Suc concentrations <5 mM did not significantly enhance superoxide generation in BV2 microglia (Supplementary Figure S3A, B). Consistently, a publication by Mills also showed that succinate at 5 mM induced mitochondrial generation of superoxides via complex I RET in macrophages (Mills et al., 2016). Thus, we used 5 mM Suc in the in vitro studies. We found that the Suc-induced increase in superoxide generation was abrogated by the complex I inhibitor rotenone (Rot) (Fig. 2A). Overall, these results suggested that succinate induced the mitochondrial production of superoxides by driving complex I RET in microglia.

The activation of UCP2 leads to mitochondrial uncoupling. The hallmarks of mitochondrial uncoupling are an enhanced oxygen consumption rate (OCR) in the presence of the ATP synthase inhibitor oligomycin and a reduction in intracellular ATP levels and mitochondrial membrane potential (ΔΨm) (Jia et al., 2020; Tao et al., 2014; Yan et al., 2022b). Compared with the vehicle treatment, diethyl succinate (Suc) enhanced the OCR in the presence of oligomycin (Fig. 2B, C), decreased the intracellular ATP levels, and increased the ADP-ATP ratio (Fig. 2D, E). Moreover, Suc reduced ΔΨm of BV2 microglia 15 min after Suc treatment (Fig. 2F, G). Thus, by assessing these properties, we confirmed that succinate induced mitochondrial uncoupling in BV2 microglia. Mitochondrial uncoupling induced by complex I RET-derived superoxides can be blocked by Rot, an inhibitor of complex I (Jia et al., 2020; Yan et al., 2022b). Consistently, the increase in the OCR induced by Suc in the presence of oligomycin was blocked by Rot (Fig. 2B, C). Moreover, Rot, a complex I inhibitor, significantly reduced ΔΨm and intracellular ATP levels (Fig. 2D–G). Succinate also reduced the ΔΨm and intracellular ATP levels (Fig. 2D–G). Strikingly, Rot markedly reversed the decrease in intracellular ATP levels and ΔΨm induced by succinate in BV2 microglia at 30 min after succinate and Rot cotreatment (Fig. 2D, E, H, I). Overall, we showed that Rot blocked succinate-induced mitochondrial uncoupling, suggesting that Suc induced uncoupling via complex I RET.
To further show that succinate-induced complex I RET leads to uncoupling via mitochondrial superoxides, we used the mitochondrion-targeted antioxidant Mito-Tempo (Mills et al., 2016). Mito-Tempo treatment abolished the succinate-induced mitochondrial superoxide burst in BV2 microglia (Supplementary Figure S3C). Importantly, Mito-Tempo also abrogated Suc-induced uncoupling, as indicated by the fact that Mito-Tempo reversed the reduction in ΔΨm in Suc-treated BV2 microglia (Supplementary Figure S3D & E). Collectively, the results suggested that succinate induced mitochondrial uncoupling via complex I RET-derived mitochondrial superoxides.
The production of mitochondrial superoxides via complex I RET strongly depends on high ΔΨm, and even a slight decrease in ΔΨm dramatically reduces superoxide generation via complex I RET (Robb et al., 2018). Theoretically, succinate cannot induce superoxide generation via complex I RET after the induction of mitochondrial uncoupling since the reduction in ΔΨm induced by uncoupling blocks complex I RET. Notably, treatment of microglia with succinate for 10 min markedly elevated ΔΨm (Fig. 2F, G), which preceded the succinate-induced decrease in ΔΨm 15 min after succinate treatment in BV2 microglia (Fig. 2F, G). The results suggested that succinate elevated ΔΨm, a prerequisite for superoxide generation via complex I RET, before succinate induced a decrease in ΔΨm. Consistently, slow-releasing H2S donors that induce uncoupling via complex I RET also increase ΔΨm before it decreases the ΔΨm (Jia et al., 2020; Yan et al., 2022b). Moreover, similar to these slow-releasing H2S donors, succinate continually elevated mitochondrial superoxide generation after the induction of uncoupling (30 min after Suc treatment). The elevation in mitochondrial superoxides after the drop in ΔΨm was also blocked by the complex I inhibitor Rot (Fig. 2A). Superoxide generation via deactive complex I is inhibited by Rot (Hernansanz-Agustin et al., 2017; Roberts and Hirst, 2012). Thus, similar to slow-releasing H2S donors, succinate possibly enhanced superoxide generation via deactive complex I after uncoupling induced a decrease in ΔΨm.
Finally, we investigated whether succinate induced mitochondrial uncoupling via a UCP2-dependent mechanism. As indicated by the OCR in the presence of oligomycin, small interfering RNA (siRNA)-mediated knockdown of UCP2 abolished the mitochondrial uncoupling induced by succinate in BV2 microglia (Fig. 2J, K). Genipin specifically inhibits mitochondrial ROS-induced activation of UCP2 (Zhang et al., 2006). Consistently, genipin also reversed the decrease in the ΔΨm induced by succinate (Fig. 2L, M). To conclude, succinate activates endogenous UCP2 by driving complex I RET to generate mitochondrial superoxides, consequently leading to mitochondrial uncoupling.
Microglia/macrophage UCP2 is needed for succinate to suppress neuroinflammation and confer protection following ICH
Based on the above results, we hypothesized that microglia/macrophage UCP2 is required for the suppression of neuroinflammation and protection by succinate following ICH. By assessing the protein levels of proinflammatory cytokines (IL-1β, TNF-α, and IL-6) secreted by BV2 microglia treated with RBC LY, we found that cell-permeable diethyl succinate (Suc) inhibited neuroinflammation in the microglial ICH model (Fig. 3A–C). siRNA-mediated knockdown of UCP2 abolished the inhibition of neuroinflammation by Suc in BV2 microglia treated with RBC LY (Fig. 3A–C). To further investigate whether microglia/macrophage UCP2 mediates succinate effects in vivo, we generated mice with microglia/macrophage-specific deletion of UCP2 (Cx3cr1Cre: UCP2fl/fl). Immunoblot analysis revealed that UCP2 was efficiently ablated in primary microglia derived from Cx3cr1Cre: UCP2fl/fl mice compared with the control mice (UCP2fl/fl) and wild-type mice (Fig. 3D, E). Moreover, Cx3cr1Cre: UCP2fl/fl mice did not display any overt abnormalities. In the control mice (UCP2fl/fl), succinate suppressed the expression of proinflammatory mediators (IL-1β, TNF-α, and IL-6) in the ipsilateral, hemorrhagic striatum (Fig. 3F–H), attenuated brain edema (Fig. 3I), and improved neurological deficits at 3 days post-ICH (Fig. 3J–L). The suppressive effects of succinate on the expression of proinflammatory mediators in the hemorrhagic striatum were abolished by microglia/macrophage-specific deletion of UCP2 at 3 days following ICH (Fig. 3F–H). Moreover, by assessing brain edema and neurodeficits (Fig. 3I–L), we further showed that microglia/macrophage-specific deletion of UCP2 abolished the protection conferred by succinate following ICH. Collectively, the results suggested that succinate acted through microglia/macrophage UCP2 to suppress neuroinflammation and confer protection following ICH.
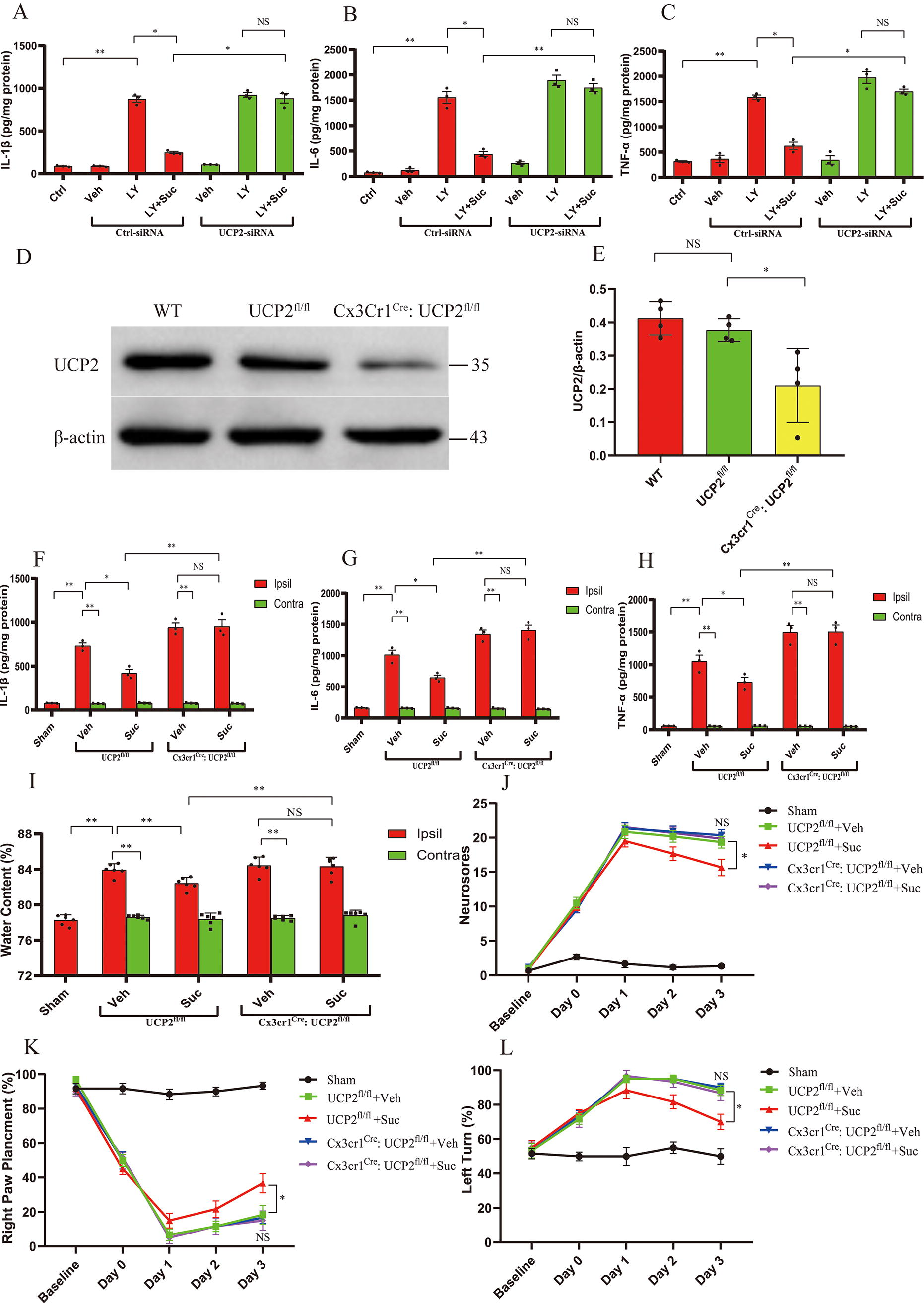
The above results suggested that succinate drove complex I RET to activate UCP2. Thus, we further investigated whether complex I was indispensable for the effects of succinate following ICH. To do this, we knocked down the endogenous expression of NADH dehydrogenase (ubiquinone) Fe-S protein 3 (NDUFS3), an essential subunit of complex I, via an siRNA approach. The effectiveness of the approach has been confirmed by our previous publications (Jia et al., 2020; Yan et al., 2022b). By assessing the protein levels of proinflammatory cytokines (IL-1β, TNF-α, and IL-6) secreted by BV2 microglia treated with RBC LY, we found that cell-permeable diethyl succinate (Suc) inhibited neuroinflammation in the microglial ICH model. In contrast, siRNA-mediated knockdown of NDUFS3 abolished the inhibitory effect of Suc on neuroinflammation in BV2 microglia treated with RBC LY (Fig. 4A–C). To knock down the endogenous expression of NDUFS3 in the hemorrhagic striatum, we injected lentiviruses coexpressing NDUFS3-shRNA and eGFP into the left striatum 14 days before ICH. Compared with the control lentivirus, the lentivirus expressing NDUFS3-shRNA decreased the endogenous expression of NDUFS3 in the striatum ipsilateral to the lentivirus (Fig. 4D, E). Immunohistochemical results further showed that the virus-injected brain sections displayed widespread expression of GFP from the injection site in the striatum (Supplementary Figure S4A). In addition, magnified images confirmed that GFP was colocalized with Iba 1, suggesting that lentivirus transduced microglia (Supplementary Figure S4B). Notably, striatal knockdown of NDUFS3 abrogated the suppressive effects of succinate on neuroinflammation in the hemorrhagic brain at 3 days following ICH (Fig. 4F–H). Moreover, the protective effect of succinate was also abrogated by striatal knockdown of NDUFS3 following ICH, as evidenced by exacerbated brain edema and neurological deficits (Fig. 4I–L). Taken together, the complex I RET-UCP2 cascade mediated the suppressive effects of succinate on neuroinflammation and the protective effects of succinate following ICH.

SDH is indispensable for succinate-induced uncoupling, suppression of neuroinflammation, and protection following ICH
SDH-mediated oxidation of succinate is a prerequisite for succinate-induced complex I RET (Abdullah et al., 2023; Chouchani et al., 2016a; Chouchani et al., 2014). Thus, we further investigated whether SDH was needed for the effects of succinate. We used the tetramethylrhodamine methyl ester perchlorate (TMRM) fluorescent probe to assess ΔΨm as an indicator of mitochondrial uncoupling. Consistent with this hypothesis, siRNA-mediated knockdown of SDHA, a subunit of SDH, abolished the diethyl succinate (Suc)-induced decrease in ΔΨm in BV2 microglia (Fig. 5A, B). Western blot results showed that, compared with the control siRNA, SDHA-siRNA significantly reduced the endogenous expression of SDHA. This indicated that SDH was needed for the mitochondrial uncoupling induced by succinate. By measuring the protein levels of proinflammatory cytokines (IL-1β, TNF-α, and IL-6) secreted from microglia treated with RBC LY, we further showed that siRNA-mediated knockdown of SDHA abolished the inhibition of neuroinflammation by succinate in BV2 microglia treated with RBC LY (Fig. 5E–G). To determine the role of SDH in vivo, we injected lentiviruses expressing SDHA-shRNA into the left striatum 14 days before ICH. Compared with the control lentivirus, the lentivirus expressing SDHA-shRNA decreased the endogenous expression of SDHA in the striatum ipsilateral to the lentivirus (Fig. 5H, I). Moreover, striatal knockdown of SDHA abrogated the suppressive effects of succinate on neuroinflammation in the hemorrhagic striatum at 3 days following ICH (Fig. 5J–L). Moreover, the protective effect of succinate was also abrogated by the striatal knockdown of SDHA following ICH, as evidenced by exacerbated brain edema and neurological deficits (Fig. 5M–P). Taken together, these results suggested that SDH was needed for succinate-induced mitochondrial uncoupling, suppression of neuroinflammation, and protection following ICH.
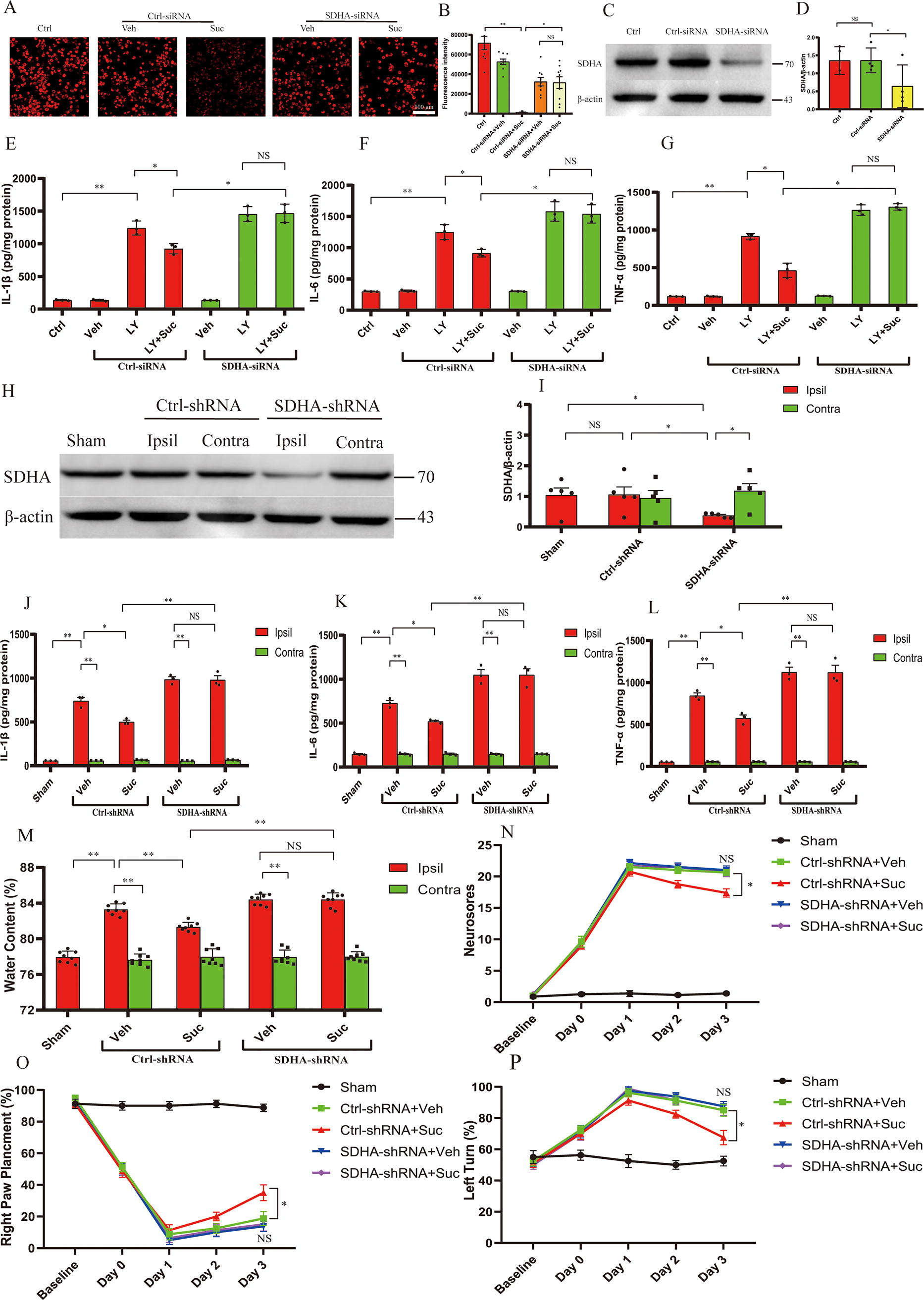
SDH is indispensable for succinate-induced uncoupling, suppression of neuroinflammation, and protection following ICH.
Succinate-induced UCP2 activation mediates the suppression of inflammation and protection conferred by succinate via AMPK activation following ICH
Mitochondrial uncoupling is a major mechanism underlying 5′-adenosine monophosphate-activated protein kinase (AMPK) activation (Tao et al., 2014). Notably, AMPK activation is essential for the inhibition of inflammation following stroke (Jia et al., 2020; Zhang et al., 2017; Zhou et al., 2014a; Zhu et al., 2015). Thus, we tested the hypothesis that succinate-induced UCP2 activation activated AMPK to suppress inflammation and confer protection. Compared with the vehicle treatment, Suc enhanced AMPK activation in BV2 microglia treated with RBC LY, as evidenced by enhanced AMPK phosphorylation (Fig. 6A, B). Importantly, the Suc-mediated increase in AMPK activation in BV2 microglia treated with RBC LY was attenuated by siRNA-mediated knockdown of UCP2, suggesting that UCP2 plays an indispensable role in succinate-induced AMPK activation. Consistent with the finding that succinate-induced UCP2 activation was dependent on SDH oxidation of succinate and complex I RET, knockdown of SDHA or the complex I subunit NDUFS3 also abrogated succinate-enhanced AMPK activation in BV2 microglia treated with RBC LY (Fig. 6C–F). More importantly, knockdown of AMPK in BV2 microglia abolished the inhibitory effects of succinate on RBC LY-induced expression of proinflammatory mediators in BV2 microglia (Fig. 6G–I). Collectively, the in vitro results suggested that succinate suppressed microglia-mediated neuroinflammation by acting through the SDH–complex I RET–UCP2 cascade to activate AMPK.
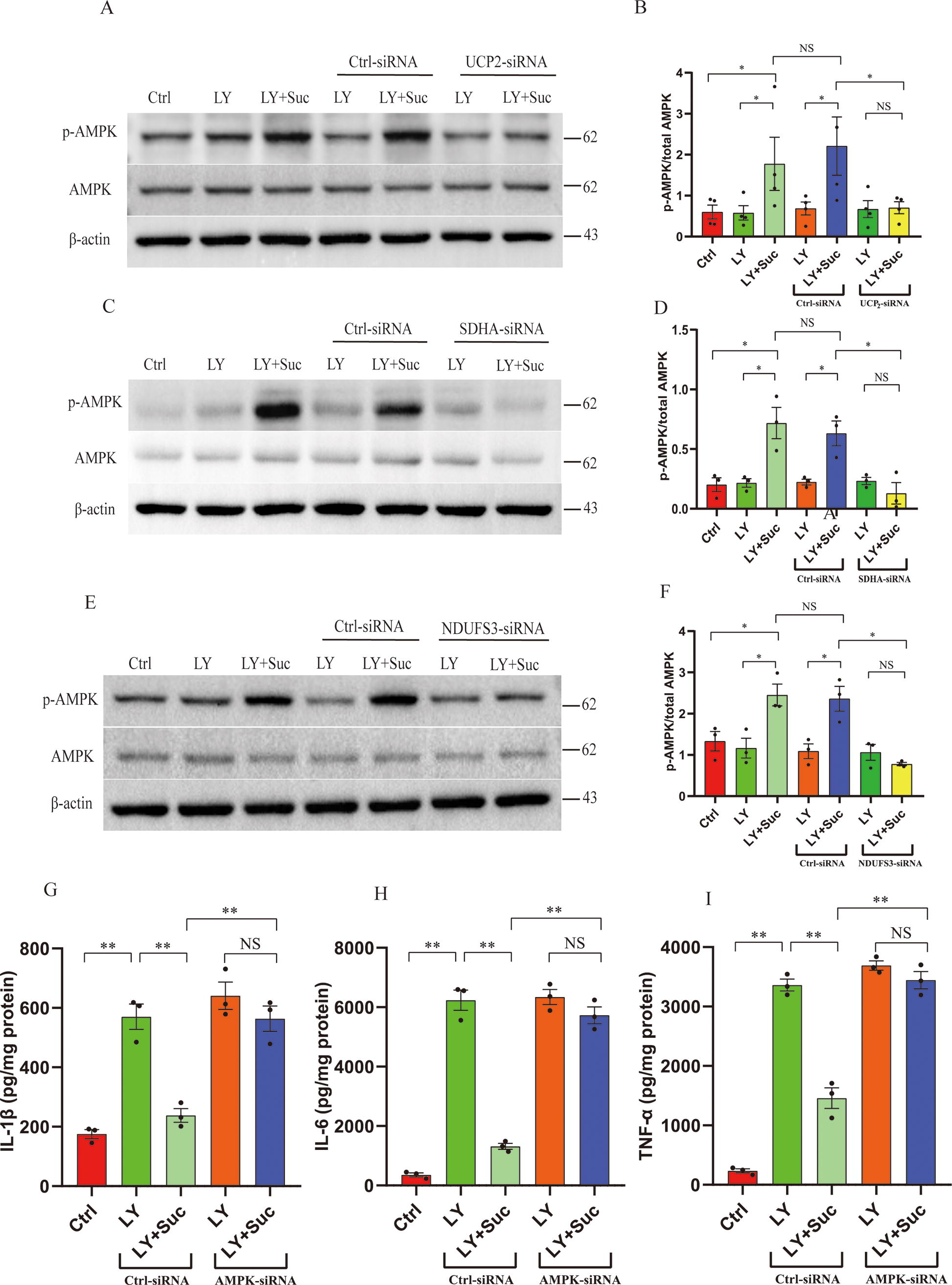
We further tested this hypothesis in an in vivo ICH model. Compared with the vehicle treatment, succinate markedly enhanced AMPK activation (phosphorylation) in the ipsilateral/hemorrhagic striatum as well as in the contralateral striatum in control mice (UCP2fl/fl) at 3 days following ICH. As expected, succinate-enhanced AMPK activation in both the ipsilateral and contralateral striatum was markedly attenuated in mice with microglia/macrophage-specific deletion of UCP2 (Cx3cr1Cre: UCP2fl/fl) at 3 days following ICH (Fig. 7A, B). To knock down the endogenous expression of SDHA or the complex I subunit NDUFS3 in vivo, we injected lentiviruses expressing SDHA-shRNA or NDUFS3-shRNA into the left striatum of mice 14 days before ICH was induced. Lentiviral knockdown of endogenous SDHA or NDUFS3 also attenuated succinate-enhanced AMPK activation in the ipsilateral hemorrhagic striatum (Fig. 7C–F).

Succinate-induced UCP2 activation mediates the suppression of inflammation by succinate via AMPK activation in the mouse ICH model.
To further investigate whether AMPK activation was needed for the suppression of neuroinflammation by succinate and protection by succinate following ICH, we injected a lentivirus expressing AMPK-shRNA into the left striatum, as we previously reported (Yan et al., 2022b; Zhang et al., 2017). By measuring proinflammatory mediators in the ipsilateral/hemorrhagic stratum, we found that striatal knockdown of AMPK abrogated the suppressive effects of succinate on neuroinflammation at 3 days following ICH, as evidenced by the protein levels of proinflammatory mediators in the hemorrhagic striatum (Fig. 7G–I). Moreover, the protective effect of succinate was also abrogated by striatal knockdown of AMPK following ICH, as evidenced by exacerbated brain edema and neurological deficits (Fig. 7J–M). Taken together, these results suggested that succinate-induced UCP2 activation mediated the suppressive effects of succinate on inflammation and protection by succinate via AMPK activation following ICH.
Finally, we explored whether succinate acts through nuclear factor erythroid 2-related factor 2 (Nrf2) to regulate redox homeostasis and suppress inflammation following ICH. We found that the administration of succinate did not affect the keap-Nrf2 signaling pathway, as evidenced by the fact that succinate had no impact on the activation (nuclear translocation) of Nrf2 in the hemorrhagic striatum at 3 days following ICH (Supplementary Figure S5A). Moreover, exogenously administered succinate did not alter the levels of reduced or oxidized glutathione (GSH) in the hemorrhagic striatum following ICH (Supplementary Figure S5B, C). These results suggested that succinate is unlikely to act through the Nrf2-GSH pathway to confer protection following ICH.
Discussion
This study provides novel findings about the link between succinate and inflammation as well as the intracellular signaling cascade of succinate. We found that succinate inhibited proinflammatory responses and conferred protection following ICH. Moreover, we showed that the protective role of succinate and the inhibition of inflammation by succinate were related to UCP2 activation in microglia/macrophages. Specifically, we proposed a working model for succinate intracellular signaling and its inhibitory effects on neuroinflammation (Fig. 8). Oxidation of succinate by SDH drove RET at mitochondrial complex I, leading to enhanced production of mitochondrial ROS in microglia. Complex I-derived O2 −, in turn, activated UCP2, leading to mitochondrial uncoupling and a consequent reduction in cellular ATP levels. Consequently, the succinate-induced reduction in ATP activated AMPK to suppress inflammation following ICH (Fig. 8). In conclusion, we provide new evidence that succinate, an intracellular signaling molecule, inhibits neuroinflammation and confers protection by functioning as an activator of UCP2.
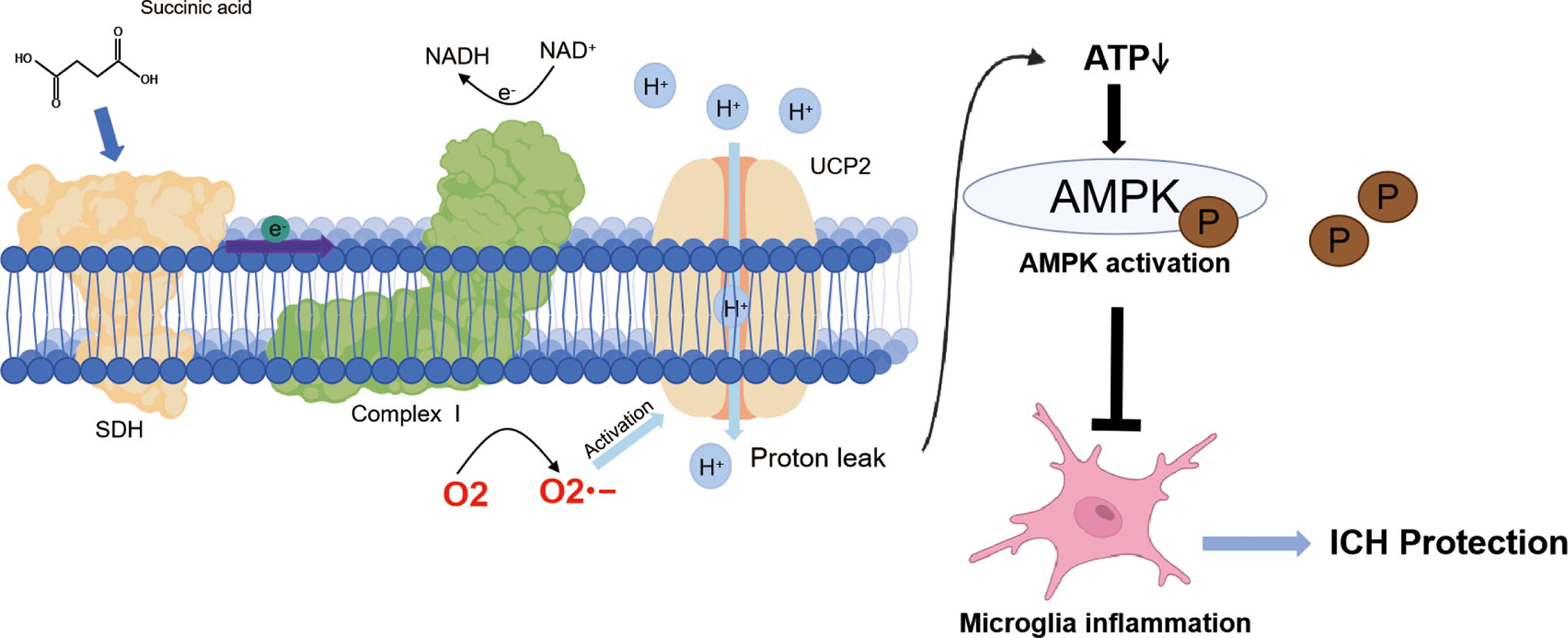
Graphic illustration for a new intracellular signaling mechanism of succinate and inhibitory effects of succinate on neuroinflammation. Oxidation of succinate by microglial SDH drives RET at mitochondrial complex I (complex I RET), leading to enhanced production of superoxides anion radical in mitochondria. Complex I-derived superoxides, in turn, activate UCP2, and succinate-induced UCP2 activation mediates the suppression of microglial inflammation via AMPK activation following ICH. Color images are available online.
Succinate is synthesized from succinyl-coenzyme A by succinyl-coenzyme A synthetase (Tretter et al., 2016). Extracellular and/or cytosolic accumulation of succinate is observed under various physiological and pathological conditions (Abdullah et al., 2023; Chouchani et al., 2014; Mills et al., 2016; Mills et al., 2018). Although initially identified as an essential mediator of the TCA cycle, succinate plays important roles not only in energy metabolism but also in inflammation by functioning as an intracellular or extracellular signaling molecule (Krzak et al., 2021; Tretter et al., 2016). However, the role of succinate in inflammation remains elusive. LPS treatment induces succinate accumulation in macrophages. The accumulation of succinate is considered a proinflammatory modulator since it enhances the production of mitochondrial ROS via complex I RET (Mills et al., 2016). Elevated intracellular levels of succinate have also been shown to augment the production of the proinflammatory mediator IL-1β by stabilizing hypoxia-induced factor 1α in LPS-treated macrophages (Tannahill et al., 2013). In sharp contrast, a recent study showed that intracellular succinate inhibited inflammation in microglia by reducing ROS production (Wang et al., 2021).
Extracellular succinate acts through SUCNR1 to trigger cellular signaling and plays a dual role in inflammation (Fernandez-Veledo et al., 2021). Succinate may boost the inflammatory response in classically activated macrophages (Littlewood-Evans et al., 2016; Mills et al., 2016) or contradictorily promote alternative polarization of macrophages to counteract acute inflammation (Fernandez-Veledo et al., 2021; Keiran et al., 2019). SUCNR1 is widely expressed on the cell membrane of various types of immune cells, including macrophages, microglia, and dendritic cells (Fernandez-Veledo et al., 2021; Rubic et al., 2008). These findings indicate that the activation of SUCNR1 by succinate is an important mechanism underlying the modulation of inflammatory responses. For instance, in synergy with Toll-like receptors, extracellular succinate acts through SUCNR1 to enhance the production of proinflammatory cytokines in immature dendritic cells (Rubic et al., 2008). In contrast, extracellular succinate excreted from cancer cells promotes macrophage polarization into tumor-associated macrophages and consequently contributes to tumor development (Wu et al., 2020). To conclude, the roles of succinate in inflammation as well as the underlying mechanisms involved remain elusive.
In this study, we showed that treating microglia with cell-permeable diethyl succinate suppressed neuroinflammation in a cellular ICH model. Moreover, supplementing mice with succinate also suppressed neuroinflammation and conferred protection following ICH. Importantly, these therapeutic effects were dependent on SDH, complex I, and UCP2, all of which are located in mitochondria. These results suggested that extracellular succinate can enter mitochondria and that the oxidation of succinate by SDH drives complex I RET to generate mitochondrial ROS. Consequently, succinate functions as an activator of UCP2 to suppress neuroinflammation.
Our results are in sharp contrast to those of Mills (Mills et al., 2016), who showed that mitochondrial succinate acts as a proinflammatory modulator by driving complex I RET to enhance the mitochondrial production of ROS in LPS-treated macrophages. This contradiction cannot be attributed to the use of different inflammatory models, since we found that succinate not only reduced neuroinflammation in in vivo and in vitro ICH models but also inhibited inflammation in LPS-treated microglia at the same concentration as that used by O’Neill (Mills et al., 2016) (Supplementary Figure S1). Notably, Mills used bone marrow cells taken from mouse leg bones, and we used BV2 microglia in this study. In addition, Mills used DMEM supplemented with 10% fetal bovine serum (FBS) and 20% L929 cells to culture bone marrow-derived macrophages (Mills et al., 2016). We used DMEM containing 10% FBS to culture BV2 microglia. Whether the discrepancy between our and Mills’ results stems from the use of different types of cells and different in vitro culture conditions warrants further investigation. Nevertheless, the finding that succinate inhibited neuroinflammation in the in vivo model strongly suggested that succinate is an inhibitory modulator of inflammation following ICH.
The results presented here were consistent with our previous publications (Jia et al., 2020; Yan et al., 2022b), which showed that some slow-releasing hydrogen sulfide donors inhibited inflammation by inducing complex I RET to activate UCP2 via mitochondrial superoxides. Based on the results presented here and our previous publication (Jia et al., 2020; Yan et al., 2022b), we propose that the generation of mitochondrial superoxides via complex I RET is a common mechanism underlying UCP2 activation and the inhibition of inflammation. Although membrane SUCNR1 reportedly mediates the inhibitory effects of succinate on macrophage inflammatory responses (Keiran et al., 2019), we found that SDH, mitochondrial complex I, and UCP2 were indispensable for the inhibition of neuroinflammation by succinate. This suggested that SDH oxidation of succinate, complex I RET, and UCP2 activation mediated the succinate-mediated inhibition of inflammation. Consistently, a recent publication also showed that succinate suppressed the secretion and expression of inflammatory mediators in macrophages via a SUCNR1-independent mechanism (Harber et al., 2020). However, further research is warranted to determine the role of SUCNR1 in ICH models.
This study provided evidence that succinate likely induces intracellular signaling by functioning as an activator of UCP2. UCP2 is abundantly expressed in microglia (Kim et al., 2019; Yan et al., 2022b). It is a long-held view that mitochondrial uncoupling impacts microglia/macrophage inflammation (Emre and Nubel, 2010). However, the role of UCP2 in inflammation remains elusive. For instance, UCP2 has been reported to exacerbate microglia/macrophage inflammation induced by sepsis and a high-fat diet (Kim et al., 2019; Moon et al., 2015). In contrast, our recent research suggested a suppressive role of UCP2 activation in neuroinflammation (Jia et al., 2020; Yan et al., 2022b). UCP2 is considered an uncoupling protein based on its homology to UCP1 (Rousset et al., 2004). However, UCPs are weak uncoupling proteins unless they are activated by mitochondrial ROS (Echtay et al., 2002a; Echtay et al., 2002b; Rousset et al., 2004). Indeed, succinate has been reported to activate UCP1 by generating ROS via a mechanism independent of mitochondrial complex I (Mills et al., 2018). In contrast, we showed here that complex I RET and subsequent ROS production were needed for UCP2 activation by succinate. Consistently, we have also reported that H2S-induced complex I RET leads to uncoupling by generating mitochondrial ROS to activate UCP2 (Jia et al., 2020; Yan et al., 2022b). Collectively, these results suggested that driving complex I RET to enhance mitochondrial ROS production represents a common mechanism underlying UCP activation.
Using mice with a specific deletion of UCP2 in macrophages/microglia, we showed that UCP2 was required for the suppressive effects of succinate on inflammation. To further show that succinate-mediated inhibition of neuroinflammation involves complex I RET, we knocked down the complex I subunit NDUFS3 in the striatum. We have shown that knockdown of NDUFS3 inhibits mitochondrial functions, as indicated by the reduction in OCRs under basal conditions (Jia et al., 2020). Succinate is oxidized by complex II (SDH). Theoretically, knockdown of NDUFS3 does not affect basal succinate activity. In this study, we found that Rot, an inhibitor of complex I, reversed the reduction in the mitochondrial membrane potential induced by succinate. This finding was consistent with the hypothesis that succinate leads to mitochondrial uncoupling via complex I RET. Moreover, knockdown of NDUFS3 abrogated the inhibitory effect of succinate on neuroinflammation following ICH. Overall, we provide the first line of evidence that driving complex I RET to activate UCP2 is a novel mechanism underlying the cellular signaling mediated by succinate. Moreover, we found that the activation of UCP2 is a novel mechanism underlying the inhibition of inflammation by succinate.
The downstream mechanisms through which UCP2 suppresses neuroinflammation might be complex. Mitochondrial uncoupling reduces cellular ATP levels, which is an essential mechanism underlying the activation of AMPK (Tao et al., 2014). We have reported that AMPK activation is an essential mechanism underlying the inhibition of macrophage/microglia-mediated inflammation following ICH (Jia et al., 2020; Yan et al., 2022b). Consistently, succinate enhanced AMPK activation in a microglial ICH model as well as in the hemorrhagic brain of mice following ICH, and this effect was dependent on microglia/macrophage UCP2, complex I, and SDH. More importantly, knockdown of AMPK in the hemorrhagic brain abolished the suppressive effects of succinate on neuroinflammation as well as the protection conferred by succinate in mice following ICH. AMPK activation inhibits the activity of mTOR (Laplante and Sabatini, 2012; Liu et al., 2018; Zhu et al., 2020), and mTOR inhibition is an essential mechanism through which macrophage-mediated inflammation is suppressed (Han et al., 2019; Ip et al., 2017). Moreover, the activation of AMPK inhibits inflammation by suppressing SATA3 (Gong et al., 2020). Thus, further research is warranted to explore the mechanisms through which succinate-induced AMPK activation inhibits neuroinflammation following ICH.
Succinate, increased by the genetic depletion of succinyl-CoA synthetase or by direct administration, results in
This study has several limitations. First, systemic inflammation and circulating inflammatory cells play essential roles following ICH. Since succinate was injected intraperitoneally in this study, we cannot exclude the possibility that circulating inflammatory cells play a role in mediating the effects of succinate following ICH. In particular, the data obtained from mice in which UCP2 was deleted in microglia and macrophages suggested that both brain-resident microglia and peripheral macrophages play essential roles in mediating the effects of succinate. Moreover, succinate can also affect the cellular or mitochondrial metabolism of other types of cells, such as endothelial cells. Thus, further research is needed to determine whether other cell types also mediate the effects of succinate. Second, by examining the dose-dependent effects of succinate in vivo and in vitro, succinate at a 5 mM concentration was used in the cellular model, and succinate at 40 mg/kg/day was used in the in vivo models. However, further research is needed to investigate how the in vitro dose is biologically relevant to physiological succinate. Third, by knocking down SDHA in the striatum, we showed that SDH is required for the inhibition of inflammation by succinate. It was recently reported that SDH deficiency significantly alters the cellular levels of TCA-related metabolites (Gobelli et al., 2023). For instance, SDH deficiency reduces itaconate levels, accompanied by decreases in citrate and malate levels. Whether succinate-induced alterations in metabolism contribute to the suppression of inflammation by succinate warrants further investigation.
In conclusion, the results presented in this study suggest that succinate confers protection by inhibiting microglia/macrophage-mediated neuroinflammation following ICH. Moreover, UCP2-mediated AMPK activation represents a novel mechanism underlying succinate-mediated intracellular signaling and the inhibition of neuroinflammation by succinate.
Materials and Methods
Antibodies and reagents
The following antibodies were used for Western blotting: anti-SDHA (1:5000, Protein Tech, 14865-1-AP), anti-UCP2 (1:200, Santa Cruz Biotechnology, sc-390189), antiphosphorylated AMPK (1:1000, Cell Signaling, 2535), anti-NDUFS3 (1:400, sc-374282, Santa Cruz Biotechnology), antitotal AMPK (1:1000, Cell Signaling, 2532), anti-β-actin (Fudebio, Hangzhou, FD0060), goat antirabbit immunoglobulin G (IgG) (Fudebio, FDR007), and goat antimouse IgG (Fudebio, FDM007).
The following agents were purchased from Gibco: DMEM (high glucose, C11995500BT), FBS (10099-141), GlutaMAX (35050-061), and penicillin–streptomycin (15140122). DMEM/F12 (SH3002301) was purchased from HyClone. The following agents were purchased from Sigma: dimethyl sulfoxide (D4540), sodium hydrosulfide (161527), diethyl succinate (112402), peroxidase from horseradish (P8375), Rot (R8875), an ADP-ATP ratio assay kit (MAK135), TMRM (T5428), and MitoTEMPO (SML0737). The following reagents were purchased from Thermo Fisher Scientific: MitoSOX Red (M36008), MitoTracker Green (M7514), protein ladder (26616), Pierce™ BCA Protein Assay Kit (23227), enhanced chemiluminescent Western blotting reagents (SuperSignal West Pico, Pierce, 34077), and High Capacity cDNA Reverse Transcription Kit (4368813). Mouse IL-1β uncoated ELISA kit (88-7013-88), mouse TNF-α uncoated ELISA kit (88-7324-88), and mouse IL-6 uncoated ELISA kit (88-7013-88) were purchased from Invitrogen. Nonfat powdered milk (A600669-0250) was purchased from BBI Life Sciences. Genipin (G101204) was purchased from Aladdin. Superoxide dismutase (S0086), RIPA lysis buffer (P0013B), and a mitochondrial membrane potential assay kit with JC-1 (C2006-1) were purchased from Beyotime. The RNAprep Pure Micro Kit (DP420) and RNAprep Pure Tissue Kit (DP431) were purchased from TIANGEN Biotech. SYBR qPCR Master Mix (Q331-02) was purchased from Vazyme. Lipofectamine RNAiMAX was purchased from Invitrogen (13778-150). The Seahorse XF Cell Mito Stress Test Kit (containing oligomycin and antimycin A, 103015-100), Seahorse XFe24 FluxPaks Kit (102342-100), XF Calibrant Solution Kit (100840-000), and XF Base Medium Kit (102353-100) were obtained from Agilent Technologies.
BV2 microglial cultures and LPS treatment
The BV2 cell line was originally generated by infecting primary mouse microglia with a retrovirus (J2) carrying a v-raf/v-myc oncogene. BV2 microglia have been characterized as an alternative cellular system for primary microglial cultures. BV2 microglia were cultured in DMEM supplemented with 10% (v/v) FBS and high glucose at 37°C and 5% carbon dioxide (CO2), as we previously reported (Yan et al., 2022b; Zhang et al., 2017). BV2 microglia were pretreated for 3 h with diethyl succinate (Suc; 5 mM) before being stimulated with LPS (100 ng/mL) for 24 h. The protocol was the same as that used in Mills’ study (Mills et al., 2016).
Measurement of ATP levels and the ADP-ATP ratio
A commercial kit (Sigma–Aldrich, MAK135) was used to assess the intracellular ATP and ADP levels. BV2 microglia were seeded in 96-well plates (105 cells/well) overnight and then treated with cell-permeable diethyl succinate (5 mM) for 30 min. In some experiments, BV2 microglia inoculated in 96-well plates were cotreated with diethyl succinate (5 mM) and Rot (10 nM) for 30 min. Then, the ADP-ATP ratio was measured according to the manufacturer’s instructions. ATP levels are expressed as a percentage of ATP levels relative to those in vehicle-treated control cells.
Measurement of mitochondrial membrane potential (ΔΨm)
The fluorescent probe TMRM (Sigma–Aldrich, T5428) was used for ΔΨm assessment. To investigate the role of SDH in succinate-induced uncoupling, BV2 microglia were transfected with siRNA against SDHA for 48 h and then treated with diethyl succinate (5 mM) for the indicated times. In some experiments, BV2 microglia were cotreated with diethyl succinate (5 mM) and Rot (10 nM) or the UCP2 inhibitor genipin (20 nM) for 30 min. After treatment, the BV2 cells were washed with phosphate-buffered saline (PBS), followed by staining with 100 nM TMRM for 10 min. Fluorescence images were captured via confocal microscopy (Zeiss LSM700) using constant parameters.
Cellular oxygen consumption (OCR)
BV2 microglia were seeded in 24-well plates (105 cells/well) overnight, and one well per row containing only culture medium served as a background control. After washing with XF experimental buffer, the cells were incubated with XF experimental buffer and kept in a CO2-free incubator for 60 min. Diethyl succinate (5 mM), oligomycin (Oligo) and antimycin (AA), and Rot were added to the cells as indicated. The OCR (pmol O2/min) was measured in real time with a Seahorse XF-24 system. In particular, the nonmitochondrial OCR, measured as the OCR in the presence of Rot and AA, was subtracted from the original data of the basal OCR and those of the OCR in the presence of Oligo or succinate. To minimize the effects of basal OCR variations, OCR values in the presence of Oligo and/or succinate were normalized to basal values and expressed as percentages of basal OCR per well for statistical analysis, as we previously reported (Jia et al., 2020).
Assessment of mitochondrial superoxide generation in cultured cells
BV2 microglia were inoculated at a density of 105/well in 24-well plates and cultured overnight. The cells were incubated with 50 nM MitoTracker Green. The cells were then treated with diethyl succinate (5 mM). In some experiments, BV2 microglia were cotreated with diethyl succinate (5 mM) and Rot (10 nM). Then, the cells were washed and incubated with 5 μM MitoSOX and Hoechst 33342 (10 μM) for 15 min. Fluorescence images were captured under a confocal microscope (Zeiss LSM 700) with constant parameters.
Generation of mice with microglia/macrophage-specific deletion of UCP2
UCP2 conditional knockout mice on a C57BL/6 background (UCP2fl/+), in which an exon of UCP2 was flanked by loxP sites, were generated using the CRISPR/Cas9-mediated genome engineering technique by Cyagen and crossed on a C57BL/6 background over 10. Cx3crCre mice, in which the Cx3cr1 promoter drives the expression of Cre recombinase in microglia/macrophages, were purchased from the Jackson Laboratory (stock number 025524). Mice heterozygous for loxP-flanked (floxed; fl) UCP2 alleles (UCP2fl/+) were crossed with the heterozygous Cx3crCre line. F1 offspring, which were heterozygous floxed and Cre positive (Cx3crCre: UCP2fl/+), were crossed with mice homozygous for loxP-flanked UCP2 alleles (UCP2fl/fl). The experimental genotypes were Cx3cr1Cre: UCP2rfl/fl and the floxed control (UCP2fl/fl). The littermates were used for the experiments, and the genotypes were confirmed via polymerase chain reaction (PCR).
Generation of primary microglia from mice with specific deletion of UCP2 in microglia/macrophages
To prepare UCP2-deleted primary microglia (Cx3cr1Cre: UCP2fl/fl), we crossed mice that were homozygous floxed and Cre positive (Cx3crCre/Cre:UCP2fl/fl) with homozygous floxed mice (UCP2fl/fl). Then, newborn F1 offspring (Cx3crCre:UCP2fl/fl) or newborn control mice (UCP2fl/fl) aged 1–2 days were used for the preparation of primary microglia, as we previously reported (Jia et al., 2020). Briefly, cortical tissue was harvested, kept in ice-cold DMEM/F12 medium, and digested with ethylenediaminetetraacetic acid–trypsin for 10 min. DMEM/F12 supplemented with 10% FBS was used to terminate the digestion. The cell suspension was filtered through a 50 μm cell strainer to remove tissue debris. Cells were plated in poly-
In vitro neuroinflammation model associated with ICH
To mimic ICH-induced neuroinflammation in vitro, cultured BV2 microglia were treated with RBC LYs as reported previously (Wang et al., 2014; Yan et al., 2022b). Blood (0.5 mL) was drawn from the mice and centrifuged at 3000 g for 5 min, after which the plasma and buffy coat were discarded. Packed RBCs were frozen in liquid nitrogen for 5 min, followed by thawing at 37°C. The process was repeated three times to lyse RBCs. Ten microliters of the RBC LY was added to microglia inoculated on 24-well plates, as we reported (Yan et al., 2022b).
Transfection of siRNA into BV2 microglia
The sequences of the siRNAs used to target mouse UCP2, NDUFS3, and AMPK are listed in Table 1. The effects of mouse UCP2-siRNA, NDUFS3-siRNA, and AMPK-siRNA have been validated in our published study (Jia et al., 2020; Pan et al., 2020; Yan et al., 2022a; Yan et al., 2022b). SDHA-siRNA was designed, and the knockdown efficiency was determined experimentally. A nonsense siRNA (Ctrl-siRNA) that does not target any mammalian DNA sequences was used as a negative control (Ctrl). siRNAs were synthesized by GenePharma. siRNA (0.5 μg/mL) was transfected into BV2 microglia inoculated in 24-well plates with Lipofectamine RNAiMAX according to the manufacturer’s instructions (Invitrogen, 13778-150). The cells were collected and used for further experiments at 48 h post-transfection. Knockdown efficiency was assessed by Western blot at 48 h post-transfection.
siRNA and shRNA Sequences
AMPK, 5′-adenosine monophosphate-activated protein kinase; NDUFS3, NADH dehydrogenase (ubiquinone) Fe-S protein 3; UCP2, uncoupling protein 2.
Mouse ICH models
All animal protocols were conducted according to the regulations of the Animal Care and Use Committee of Soochow University. Animal research was reported according to the ARRIVE guidelines (Percie du Sert et al., 2020). Male wild-type ICR mice, UCP2 knockout mice, or control littermates aged 2 months were subjected to ICH. ICH was induced by injecting collagenase VII-S (Sigma-Aldrich) into the left striatum (Wang et al., 2014; Yan et al., 2022a). Briefly, mice were anesthetized with 2.5% 2,2,2-tribromoethanol (intraperitoneal 125 mg/kg, Sigma), as previously reported (Papadopoulos and Verkman, 2005). Then, a cranial burr hole was drilled, and a needle (26-s gauge) was stereotaxically inserted into the left striatum at the following coordinates: 2 mm lateral to the midline, 0.5 mm anterior to the bregma, and 3.5 mm below the skull. Collagenase (0.03 units/μL saline) was infused at 0.1 μL/min using a microinfusion pump (RWD Instruments). To induce ICH with autologous blood, autologous blood (15 μL) drawn from the caudal artery was injected with a 30-gauge needle at 2 μL/min. Saline was infused for the sham surgery. To prevent reflux, the needle remained in place for 10 min. After ICH induction, the mice were randomized to receive daily injections of vehicle (saline) or disodium succinate (40 mg/kg/day) via the intraperitoneal route for three consecutive days beginning 3 h after collagenase injection.
Pain-relieving drugs were not given after surgery. The use of nonsteroidal anti-inflammatory drugs (NSAIDs) and opioids in experimental stroke models is problematic. Considerable evidence indicates that ICH triggers inflammation, which contributes to brain injury and worsening of neurological outcomes. Consequently, the use of NSAIDs or corticosteroids in ICH models could bias the experimental results. However, we used a published behavioral scoring system based on subjective and objective measures for pain evaluation (Kirsch et al., 2002). Animals displaying indications of severe pain will be evaluated by a veterinarian to determine whether the animal can remain in study or should be euthanized. In this study, no animals were euthanized before endpoint detection due to severe pain. Skin incisions will be checked daily for evidence of postsurgical infections (i.e., redness, swelling, and exudates) or dehiscence/herniation. A veterinarian will be consulted if there is evidence of postoperative infection or other complications (i.e., seizures). Prophylactic antibiotic treatment was not used in the study since none of the mice showed indications of infection.
Assessment of neurological deficits
Neurological deficits were blindly assessed, as we reported (Pan et al., 2020). For assessing neuroscores, mice were given points based on the following tests: circling behavior, symmetry in the front limbs, body symmetry, gait, climbing, compulsory circling, and whisker response. These scores were added for each mouse. Higher scores indicate more severe deficits. In the forelimb placement test, the tentacles of each mouse were gently brushed against the corner edge of a countertop. Normal mice quickly placed the forelimb ipsilateral to the stimulated vibrissae on the countertop. Depending on the severity of the injury following brain hemorrhage, the placement of the forelimb contralateral to the brain injury was impaired. Each mouse was tested 10 times. For each animal, the number of trials in which the forelimb was appropriately placed was expressed as a percentage of the total number of trials. Each animal represents an independent biological replicate, and six mice were included for statistical analysis. The corner-turning test was performed by forcing the mice into a corner at a 30° angle. The choice of orientation was recorded when the mice turned. Intact mice had no preference, while hemorrhagic mice preferentially turned toward the undamaged (right) side. Each mouse was tested 10 times. For each mouse, the number of right turns was expressed as a percentage of the total number of trials. Each animal represents an independent biological replicate, and six mice were included for statistical analysis.
Lentiviral knockdown of target genes in the brain
Lentiviruses were produced by GeneChem. Briefly, siRNA sequences targeting mouse SDHA, NDUFS3, the AMPK subunits α1 and α2, and a negative control sequence were synthesized as short hairpin RNAs (Table 1) and inserted into a GV493 lentiviral vector containing an U6 promoter upstream of the restriction sites (AgeI and EcoRI) and a CBh-driven EGFP receptor gene. The SDHA-shRNA sequences were designed based on the siRNA sequences validated in the study. We validated and successfully used NDUFS3-shRNA and AMPK-shRNA to knock down target genes in the striatum in our published study (Jia et al., 2020; Pan et al., 2020; Yan et al., 2022a; Yan et al., 2022b). All constructs were confirmed by sequence analysis. Recombinant lentivirus was produced by transfecting 293T cells. Viral titers were determined by measuring GFP expression in 293T cells and are expressed as transducing units (TUs) per milliliter. The final titer was > 1 × 108 TU/mL. Lentivirus was injected stereotactically into the left striatum, as we reported previously (Yan et al., 2022a; Yan et al., 2022b). Briefly, mice were anesthetized and placed on a stereotactic device. Lentiviruses expressing SDHA-shRNA, NDUFS3-shRNA, AMPK-shRNA, or a nontargeting short hairpin RNA (Ctrl-shRNA) were injected into the striatum through two sites of the left striatum at a rate of 0.15 μL/min using a microinjector (1.5 μL of 1 × 108 TU/mL lentivirus per injection site). The coordinates of the two injection sites were as follows: anterior 0.5 mm bregma, lateral 2.0 mm midline, and 3.5 mm below the skull and anterior 1 mm cranial, lateral 1.5 mm midline, and 3.2 mm below the skull. The in vivo knockdown efficiency was analyzed in mouse striatum tissue collected at 14 days after lentivirus injection. Mice were subjected to ICH on day 14 after lentivirus injection.
Assessment of cerebral edema after intracranial hemorrhage
Brains were collected, and the left and right striatum were dissected 3 days after ICH. The wet weight of the striatal tissue was determined. Dry weight was measured after the tissue was kept in a 75°C oven for 48 h. Edema weight was calculated according to the following formula: [(wet weight-dry weight)/wet weight]× 100%.
Enzyme-linked immunosorbent assay
The protein concentrations of IL-1β, TNF-α, and IL-6 in the culture medium and mouse striatal tissue were assayed using commercial kits. The culture medium was collected from BV2 microglia for ELISA, and the cells were harvested for protein concentration assays with a BCA kit [Pierce™ BCA Protein Assay Kit (23227)]. The ipsilateral striatum and contralateral striatum were harvested from mice 3 days after ICH. Protein concentrations were determined using a BCA kit. The protein levels of the inflammatory factors IL-1β, IL-6, and TNF-α were determined according to the manufacturer’s instructions.
Western blot
Tissues or cells were homogenized in RIPA buffer containing a protein phosphatase inhibitor mixture and a protease inhibitor cocktail. Then, the samples were collected by centrifugation. The protein concentrations of the LYs were determined with a commercial BCA kit (Pierce™ BCA Protein Assay Kit [23227]). LY containing 40 μg of protein was mixed with loading buffer and boiled at 99°C for 8 min. After SDS-PAGE, the proteins were transferred onto polyvinylidene difluoride membranes and incubated with Tris-buffered saline containing 10% nonfat milk and 0.1% (v/v) Tween-20 for blocking. The membranes were incubated with primary antibodies at 4°C overnight, followed by incubation with HRP-conjugated secondary antibodies at room temperature for 2 h. The protein bands were detected with a ChemiDoc MP Imaging System using hypersensitive chemiluminescence reagents. The densities of the protein bands were semi-quantified by ImageJ software and are expressed as the ratio of the target protein to the internal reference protein or total protein.
Quantitative real-time PCR
BV2 microglia seeded onto 24-well plates were treated with LPS and/or cell-permeable dimethyl succinate for 24 h. Then, the cells were harvested, and total RNA was isolated using an RNAprep Pure Tissue Kit (TIANGEN). The total RNA concentration was quantified using a Nanodrop 2000, and cDNA was reverse transcribed from 1 µg of total RNA using a cDNA synthesis kit (Vazyme). The SYBR Green technique was used, and real-time PCR was performed with cDNA on a real-time PCR system (7900HT, Applied Biosystems). The thermal cycling parameters for qPCR were as follows: incubation at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The mRNA levels of the target genes were normalized to the 18S mRNA levels. The following qPCR primers were used to assess the mRNA levels of IL-1β and GADPH. The sequence of the IL-1β forward primer was 5′-GGCTCTGCAGCTCTTCCAGAG-3′, and that of the IL-1β reverse primer was 5′-GGCTCTGCAGCTCTTCCAGAG-3′. The sequence of the GAPDH forward primer was 5′-GAAGGTCGGTGTGAACGGAT-3′, and that of the GAPDH reverse primer was 5′-AATCTCCACTTTGCCACTGC-3′. The sequence of the 18S forward primer was 5′-TCAACACGGGAAACCTCAC-3′, and that of the 18S reverse primer was 5′-CGCTCCACCAACTAAGAAC-3′.
Immunohistochemistry
Mice were euthanized and transcardially perfused with 0.05 M PBS, followed by fixation with 4% paraformaldehyde in 0.1 M phosphate buffer. The brains were collected, postfixed in 0.1 M phosphate buffer containing 4% paraformaldehyde at 4°C for 20 h, and then kept in 0.05 M PBS containing 30% sucrose. Serial coronal sections (20 μm thick) were cut on a cryomicrotome at 4°C. For immunohistochemical analysis, 1 out of every 10 sections was collected from the hemorrhagic regions, and a total of 4 sections were collected for each brain. The sections were rinsed with PBS and then treated with a solution containing 3% bovine serum and 0.2% Triton X-100 for 2 h at room temperature. The sections were rinsed with PBS and then treated with a solution containing 3% bovine serum and 0.2% Triton X-100 for 2 h at room temperature. The sections were washed with PBS and incubated with primary antibodies against Iba1 (1:500, Oasia, OB-PGP049), Nrf2 (1:500, HUABIO, ER1706-41), INOS (1:1000, BD Pharmingen, BD-610330), Arg (1:500, Protein Tech, 16001-1-AP), and DIPA Fluoromount-G (SouthernBiotech, 0100-20) overnight. After rising, the sections were further incubated with fluorescent dye-conjugated secondary antibodies conjugated to Alexa Flour 488 (1:500, Invitrogen, A11008), Alexa Flour 546 (1:2000, Invitrogen, A21432), or Alexa Flour 647 (1:500, Oasis, OB-GP647) for 2 h at room temperature.
Measurement of hemorrhagic lesions
To histologically evaluate brain injury following ICH, mouse brain sections were incubated in a solution containing 3% normal bovine serum and 0.2% Triton X-100 at 37°C for 1 h. Then, the myelin structure was stained by incubating the brain sections with a solution containing fluoromyelin (1:300, Invitrogen, F34652). Hemorrhagic damage, as indicated by the destruction of myelin structures, was assessed via fluorescence microscopy.
Measurement of succinate levels in the mouse serum and brain following ICH
At 3 days following ICH, blood was drawn from the hearts of the ICH mice or sham-operated mice. After the blood was allowed to clot, the clot was removed by centrifugation at 300 g for 2 min, and the resulting serum was collected. Before the brains were harvested, the mice were perfused transcardially with 0.1 mol/L PBS. Then, the brains were harvested, washed with cold PBS, and dissected into the contralateral and ipsilateral sides. A succinate assay kit (Abcam, ab204718) was used to assess succinate levels in the brain and serum. Briefly, brain tissues were homogenized in PBS and centrifuged at 1000 g for 3 min. The supernatants were collected, and protein concentrations were determined with a commercial BCA kit (Pierce™ BCA Protein Assay Kit [23227]). Brain tissues (40 mg) or serum (10 μL) was resuspended in succinate assay buffer. The mixtures were incubated at 37°C for 30 min in the dark. The absorbance was measured at 450 nm. The levels of succinate were calculated from a standard curve made from succinate and adjusted with total protein levels for brain tissues.
GSH measurement
At 3 days after ICH, the mice were perfused transcardially with 0.1 mol/L PBS. Then, the brains were collected and washed with cold PBS. The ipsilateral hemispheres were dissected and collected. Brain tissues were homogenized in RIPA buffer and then centrifuged at 1000 g for 3 min. The supernatants were collected, and protein concentrations were determined with a commercial BCA kit (Pierce™ BCA Protein Assay Kit [23227]). Thirty milligrams of brain tissue was used for the measurement of reduced GSH and oxidized GSSH with a commercial GSH and GSSG Assay Kit (Beyotime Biotechnology, S0053). The levels of GSH and oxidized GSSH were calculated from a standard curve made from standard samples and adjusted for total protein levels in brain tissues.
Statistical analysis
The data are expressed as the means ± standard error of the mean. One-way analysis of variance (ANOVA) was applied for multiple comparisons, and two-tailed Student’s t tests were used for pairwise comparisons using SPSS Statistics 17.0. For ANOVA, the homogeneity of variance was evaluated with Levene’s test. If the results were similar, post hoc Tukey’s analysis was performed. Otherwise, the data were analyzed by the Games–Howell test.
Footnotes
Authors’ Contributions
G.-D.X., J.J., X.P., and. J.C. conceived the concept, designed the research, interpreted the data, and wrote the article; Y.L. designed the research and interpreted the data; all other authors conducted the research and/or analyzed the data.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by the National Natural Science Foundation of China (82271315, 81971119, 82071469, and 32271033), Suzhou Clinical Key Disease Diagnosis and Treatment Technology Special Project (To G.-D.X., No. LCZX202306), Priority Academic Program Development of the Jiangsu Higher Education Institutions (PAPD), the Suzhou Clinical Research Center of Neurological Disease (Szzx201503), a Jiangsu Key Laboratory Grant (BM2013003), the Jiangsu Provincial Health Commission Research Project (to XF. P, No. H2023064), the Wuxi Municipal Health Commission Research Project (to X.P., No. M202232), and Suzhou International Joint Laboratory for Diagnosis and Treatment of Brain Diseases.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.