
Abstract
Significance:
Cholesterol plays a crucial role in the brain, where it is highly concentrated and tightly regulated to support normal brain functions. It serves as a vital component of cell membranes, ensuring their integrity, and acts as a key regulator of various brain processes. Dysregulation of cholesterol metabolism in the brain has been linked to impaired brain function and the onset of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease, and Huntington’s disease.
Recent Advances:
A significant advancement has been the identification of astrocyte-derived apoliprotein E as a key regulator of de novo cholesterol biosynthesis in neurons, providing insights into how extracellular signals influence neuronal cholesterol levels. In addition, the development of antibody-based therapies, particularly for AD, presents promising opportunities for therapeutic interventions.
Critical Issues:
Despite significant research, the association between cholesterol and neurodegenerative diseases remains inconclusive. It is crucial to distinguish between plasma cholesterol and brain cholesterol, as these pools are relatively independent. This differentiation should be considered when evaluating statin-based treatment approaches. Furthermore, assessing not only the total cholesterol content in the brain but also its distribution among different types of brain cells is essential.
Future Direction:
Establishing a causal link between changes in brain/plasma cholesterol levels and the onset of brain dysfunction/neurodegenerative diseases remains a key objective. In addition, conducting cell-specific analyses of cholesterol homeostasis in various types of brain cells under pathological conditions will enhance our understanding of cholesterol metabolism in neurodegenerative diseases. Manipulating cholesterol levels to restore homeostasis may represent a novel approach for alleviating neurological symptoms. Antioxid. Redox Signal. 41, 1051–1072.
Introduction
Cholesterol is a 27-carbon polycyclic lipid molecule characterized by three primary domains: a hydrophilic end featuring the beta-hydroxyl group (C3), a hydrophobic end comprising an 8-carbon aliphatic tail, and a central 4-ring structure with an unsaturated double bond (C5–C6), rendering the molecule extremely rigid. Serving as an indispensable lipid constituent of cell membranes, cholesterol also serves as a precursor for steroid hormones, bile acids, and vitamin D (Ikonen, 2008). Within the brain, cholesterol performs a myriad of functions, including the maintenance of structural integrity, regulation of lipid fluidity and transport, modulation of membrane permeability to ions and metabolites, and provision of insulation for efficient action potential propagation (Gonzalez-Guevara et al., 2020). Moreover, cholesterol exerts regulatory influence on neuronal function by modulating signal transduction, gene transcription, and post-translational modification (Schumacher and DeBose-Boyd, 2021, Vance, 2012).
Despite constituting only ∼2% of total body weight, the human brain harbors up to 20% of the body’s total cholesterol. Predominantly existing in an unesterified form (>99.5%), brain cholesterol is distributed across myelin (70%), glial cells (20%), and neurons (10%) (Dietschy and Turley, 2001). Notably, a significant proportion of body cholesterol resides within myelin sheaths, exhibiting a slow turnover rate with a half-life of ∼5 years, whereas cholesterol turnover in astrocytes and neurons occurs at a relatively faster pace, with a half-life of 5–10 months (Petrov et al., 2016).
The maintenance of cholesterol homeostasis in the brain is intricately regulated through a dynamic equilibrium involving de novo synthesis, transport, uptake, catabolism, and storage. Increasing evidence links dysregulation of cholesterol homeostasis in the brain to the pathogenesis of neurodegenerative diseases, including Alzheimer’s disease (AD) (Liu and Zhang, 2014), Huntington’s disease (HD) (Karasinska and Hayden, 2011), Parkinson’s disease (PD) (Garcia-Sanz et al., 2021), and Niemann–Pick type C (NPC) disease (Dai et al., 2021). In this review, we provide an overview of current knowledge regarding cholesterol synthesis, transport, catabolism, and storage in the brain. In addition, we examine how disruptions in cholesterol metabolism contribute to the onset and progression of neurodegenerative disorders and evaluate the potential efficacy of cholesterol-targeted treatments in mitigating these diseases.
Cholesterol Biosynthesis in the Brain
The brain primarily relies on de novo synthesis of cholesterol because of the presence of the blood–brain barrier (BBB), which restricts the transfer of cholesterol from the periphery to the brain (Dietschy and Turley, 2004; Pfrieger and Ungerer, 2011; Zhang and Liu, 2015). Cholesterol synthesis occurs in the endoplasmic reticulum (ER), where precursor acetyl-coenzyme A (CoA) undergoes conversion into cholesterol through a series of reactions involving over 20 catalyzing enzymes (Luo et al., 2020). The de novo synthesis of cholesterol involves the rapid transportation of newly synthesized cholesterol from the ER to the plasma membrane (PM) (Lusa et al., 2003). This process contributes to maintaining low sterol content within the ER.
Cholesterol synthesis is an energy-intensive process, requiring large amounts of adenosine triphosphate (ATP), oxygen, and the reducing factors nicotinamide-adenine dinucleotide phosphate (NADPH) and nicotinamide adenine dinucleotide, in addition to precursor acetyl-CoA (Luo et al., 2020). The initial step of cholesterol synthesis, known as the mevalonate pathway, begins with the conversion of acetyl-CoA into 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) by 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1). Subsequently, HMG-CoA reductase (HMGCR) mediates the transformation of HMG-CoA into mevalonate, a 30-carbon linear molecule (Fig. 1). This step is NADPH-dependent and is considered the rate-limiting step (Cortes et al., 2014). HMGCR is a key enzyme in cholesterol biosynthesis and is also the therapeutic target for cholesterol-lowering drugs, such as statins.

Following continuous catalytic reactions by mevalonate kinase (MVK), phosphomevalonate kinase (PMVK), and mevalonate 5-diphosphate decarboxylase (MDD), mevalonate is converted into isopentenyl diphosphate (IPP). IPP is then catalyzed to form farnesyl pyrophosphate (FPP) by farnesyl pyrophosphate synthase (FDPS), which catalyzes a chain elongation reaction and controls the first branching point of the mevalonate pathway (Thulasiram and Poulter, 2006). Squalene synthase, also known as farnesyl diphosphate farnesyl transferase (FDFT1), catalyzes the conversion of two molecules of FPP into squalene in a reaction that is also NADPH dependent (Fig. 1).
Linear squalene undergoes a series of oxygenation and cyclization processes to form lanosterol, a four-ring compound (Chen et al., 2023). Squalene monooxygenase (SM) catalyzes the initial oxygenation step, converting squalene to 2,3(S)-mono-oxysqualene (MOS), the nonsterol precursor of lanosterol (Gill et al., 2011). Alongside HMGCR, SM is considered the second rate-limiting enzyme in the cholesterol synthesis pathway (Hidaka et al., 1990). Subsequently, lanosterol synthase (2,3-oxidosqualene-lanosterol cyclase) further converts MOS to lanosterol, marking another rate-limiting step in cholesterol biosynthesis (Zhao et al., 2015). The conversion of lanosterol into cholesterol is a complex, multistep process necessitating additional catalyzing enzymes (Ershov et al., 2021).
Cholesterol biosynthesis predominantly occurs in glial cells (over 95%) in the adult brain, with less than 5% taking place in neuronal cells (Petrov et al., 2016). Lanosterol, the precursor, undergoes two distinct pathways to yield cholesterol, known as the Bloch pathway and the Kandutsch–Russell pathway, resulting in desmosterol or 7-dehydrocholesterol, respectively (Nieweg et al., 2009). It is proposed that neurons primarily synthesize cholesterol via the Kandutsch–Russell pathway, given their abundance of sterol precursors such as lathosterol and 7-dehydrocholesterol, whereas astrocytes predominantly employ the Bloch pathway, as they primarily contain sterol precursors such as desmosterol (Zhang and Liu, 2015). In the Bloch pathway, 3β-hydroxysterol Δ7-reductase (DHCR7) and 3β-hydroxysterol Δ24-reductase (DHCR24) sequentially catalyze the conversion of zymosterol to desmosterol, ultimately yielding cholesterol.
Cholesterol biosynthesis is crucial for embryonic development. Notably, the loss of key catalyzing enzymes leads to embryonic lethality in mice: loss of HMGCR results in embryonic lethality at the preimplantation stage, loss of MVK leads to embryonic lethality at an early developmental stage, and loss of FDFT1 results in embryonic lethality at a later developmental stage (Hager et al., 2007; Ohashi et al., 2003; Tozawa et al., 1999). Mutations in the DHCR24 gene cause desmosterolosis, a rare autosomal recessive disorder characterized by multiple neurological problems, including brain abnormalities and developmental delay (Waterham et al., 2001). Patients with desmosterolosis exhibit elevated levels of the cholesterol precursor desmosterol in both plasma and tissues. Remarkably, deficiencies in terminal enzymes appear to manifest much milder phenotypes compared with deficiencies in the initial enzymes in the cholesterol biosynthesis pathway, likely because of the presence of alternate cholesterol biosynthesis pathways in the absence of terminal enzymes.
Cholesterol biosynthesis is active in developing neurons. Conditional knockout (KO) of FDFT1 in neuronal progenitor cells results in reduced brain size and perinatal lethality because of apoptosis of newly generated neurons (Saito et al., 2009). These observations imply that neurons heavily depend on autonomous cholesterol synthesis during development. This inference is further supported by in vitro studies of cultured neurons, commonly regarded as immature, which primarily rely on autonomously produced cholesterol. Interestingly, the amount of cholesterol produced by cultured neurons surpasses that produced by cultured astrocytes (Genaro-Mattos et al., 2019). Approximately 70% of brain cholesterol is found in myelin, making the cholesterol biosynthesis rate highest during active myelination. Conditional inactivation of FDFT1 in myelin-forming oligodendrocytes significantly reduces the myelination rate in mutant mice. However, mutant oligodendrocytes are able to use cholesterol produced by the neighboring cell types to compensate for the deficiency in cholesterol biosynthesis (Saher et al., 2005). These findings suggest that cholesterol transport can partially overcome cholesterol deficiency.
Neurons exhibit a higher demand for cholesterol to develop and maintain axons, dendrites, and synaptic connections (Nieweg et al., 2009). Although adult neurons are not efficient in producing cholesterol, they rely on cholesterol transported from astrocytes. Mice with conditional KO of FDFT1 in adult neurons display phenotypes indistinguishable from control mice, indicating that cholesterol biosynthesis is dispensable for adult neurons (Funfschilling et al., 2007). Nevertheless, neurons retain the capacity to produce cholesterol, with de novo cholesterol biosynthesis increasing upon stimulation by the brain-derived neurotrophic factor (BDNF) (Numakawa et al., 2010). These findings underscore the delicate maintenance of cholesterol homeostasis through the regulation of cholesterol biosynthesis and transport among different types of brain cells.
Regulation of Cholesterol Biosynthesis
Cholesterol biosynthesis undergoes strict regulation to maintain the balance of cholesterol metabolism. Sterol regulatory element-binding protein 2 (SREBP2) plays a crucial role in regulating cholesterol biosynthesis by stimulating the transcription of key genes involved, such as HMGCR, HMGCS, and MVK (Brown and Goldstein, 1997, Horton et al., 2002).
Initially synthesized SREBP2 is localized to the ER as a membrane protein. Subsequently, it undergoes proteolytic cleavage to become a functional form within the Golgi apparatus (Sato, 2009). This cleavage process is controlled by the sterol-sensing SREBP cleavage-activating protein (SCAP) and insulin-inducing gene (INSIG) protein, both of which are membrane proteins of the ER. When ER cholesterol levels decrease, SCAP escorts premature SREBP2 to the Golgi, where sequential proteolytic processing by site-1 protease (S1P) and site-2 protease (S2P) occurs, releasing mature forms of SREBP2. These mature forms then enter the nucleus, where they enhance the transcription of cholesterol biosynthesis genes, including HMGCR, by binding to the sterol regulatory element (SRE) on their promoter regions (Brown et al., 2018; Ikonen and Zhou, 2021; Irisawa et al., 2009). Conversely, when ER cholesterol levels increase, SREBP2 remains in the ER, and no active form of SREBP2 is generated for transcriptional regulation.
SREBP2 is widely expressed in developing embryos, highlighting its critical role in embryonic development. Deficiency of SREBP2 results in embryonic lethality in mice, underscoring the essential nature of cholesterol biosynthesis during development. Mice with partial loss of SREBP2 function can survive development but exhibit significant gender dimorphism (Vergnes et al., 2016). Notably, mice with SREBP2 KO in astrocytes display impaired brain development, leading to abnormal memory function and defective carbohydrate and lipid metabolism both systemically and within the brain (Ferris et al., 2017). Loss of astrocytic SREBP2 functionally impairs neuronal dendrite outgrowth in vivo cocultures (Ferris et al., 2017), further emphasizing the necessity of astrocyte-derived cholesterol in maintaining normal neuronal function.
Cholesterol biosynthesis genes are tightly regulated at various stages, including transcriptional, translational, and post-translational levels (Goldstein and Brown, 1990). When cholesterol levels rise, the transcription of HMGCR is inhibited by sterols, while the translation of HMGCR mRNA is hindered by nonsterol products derived from mevalonate (Nakanishi et al., 1988). Consequently, both sterols and nonsterol products work together to decrease HMGCR levels. Furthermore, the stability and activity of HMGCR can be modulated at the posttranslational level. Accumulation of oxysterols and lanosterol leads to the activation of INSIG1 and INSIG2, which facilitate the degradation of HMGCR by binding to it and inducing its ubiquitylation, followed by proteasome-mediated degradation (Song et al., 2005). In addition, the levels of squalene epoxidase, another crucial enzyme in the cholesterol biosynthesis pathway, are also regulated both transcriptionally and post-translationally (Tan et al., 2019; Zelcer et al., 2014). These regulatory mechanisms collectively ensure the coordinated regulation of cholesterol biosynthesis by multiple genes at the transcriptional, translational, and post-translational levels.
Cholesterol Transport in the Brain
Cholesterol in the brain is predominantly synthesized in astrocytes and subsequently transferred to neurons for utilization (Poirier et al., 1993). Owing to its insolubility in water, cholesterol primarily exists in the form of lipoproteins (Koch et al., 2017; Vance et al., 2006; Vitali et al., 2014). Astrocyte-produced cholesterol associates with apoliprotein E (ApoE), a lipoprotein primarily synthesized in astrocytes, forming high-density lipoprotein (HDL)-like particles. These particles are then secreted through the interaction with ATP-binding cassette (ABC) transporters (Hayashi et al., 2004; Mauch et al., 2001). Neurons uptake these secreted HDL-like ApoE particles via lipoprotein receptors, predominantly the low-density lipoprotein receptor (LDLR) and LDLR-related protein 1 (LRP1) (Zhang and Liu, 2015). Within the late endosome/lysosome, the endocytosed ApoE particles undergo hydrolysis, releasing free cholesterol for utilization (Fig. 2). This process of cholesterol transport serves as a crucial mechanism for transferring cholesterol between different types of brain cells, ensuring that neurons have access to the necessary cholesterol required for their specialization in action potentials.
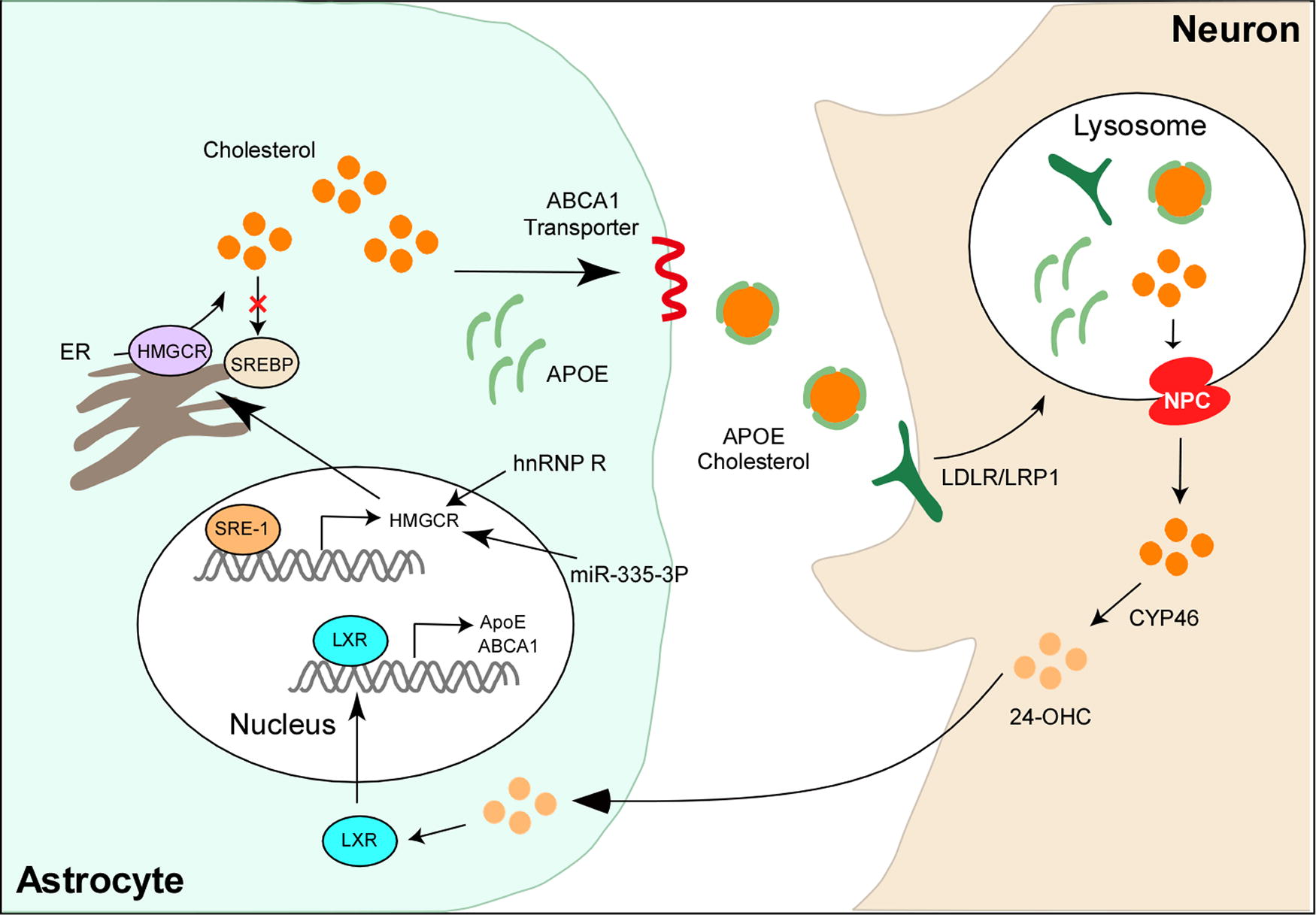
ApoE
ApoE is a glycoprotein, and its C-terminal domain (residues 206–299) contains lipid-binding sequences responsible for lipid binding, whereas its N-terminal domain (residues 1–191) harbors sequences for binding to lipoprotein receptors, facilitating the uptake of ApoE by members of the LDLR family (Chen et al., 2011). In the brain, ApoE serves as a major cholesterol carrier protein, capable of being transported into neurons through interactions with the LDLR, thereby facilitating neuronal uptake (Fig. 2) (Holtzman et al., 2012). Cholesterol and ApoE derived from astrocytes are crucial for maintaining proper neuronal function, including growth, repair, and synaptogenesis (Lanfranco et al., 2020).
ApoE is primarily synthesized in glial cells, predominantly astrocytes, although microglia and oligodendrocytes also contribute to its production (Grehan et al., 2001; Polazzi et al., 2015; Xu et al., 2006). Under certain circumstances, such as during stress conditions within the brain such as excitotoxic or ischemic injury, neurons can also produce ApoE (Xu et al., 2006) although at lower levels (Xu et al., 1999). The expression of ApoE increases in response to such stress conditions, as observed both in cultured neurons and in mice (Xu et al., 2006). At a young age, ApoE KO mice do not exhibit learning and memory deficits. However, these deficits manifest in aged ApoE KO mice, coinciding with decreased cholesterol content and synaptic loss in the brain (Nunes et al., 2018). Metabolomic analysis reveals a reduced content of desmosterol, campesterol, and 24-hydroxycholesterol (24-OHC) in the brains of ApoE KO mice compared with wild-type (WT) mice, indicating that ApoE deficiency reduces both cholesterol biosynthesis and catabolism in the brain (Nunes et al., 2018). These findings suggest that ApoE plays a crucial role in maintaining proper levels of cholesterol by simultaneously regulating its biosynthesis and catabolism. A specific splicing variant of ApoE mRNA, characterized by intron 3 retention, is detected exclusively in neurons. This variant localizes to the nucleus and its expression is decreased in degenerative neurons in mice. It appears that this variant functions as a noncoding RNA, negatively regulating the transcription of ApoE in neurons (Xu et al., 2008).
In addition to its role as a cholesterol carrier, ApoE also plays a crucial role in regulating neuronal cholesterol metabolism. Astrocyte-derived ApoE particles carry numerous miRNAs, which are transported into neurons and extensively bind to the 3′-untranslated region (UTR) of genes involved in cholesterol biosynthesis, thereby suppressing their expression (Li et al., 2021). Consequently, there is an accumulation of acetyl-CoA, a precursor for cholesterol biosynthesis, leading to nuclear acetyl-CoA accumulation, histone acetylation, and the subsequent activation of genes related to memory formation. This process enhances memory consolidation in mice (Li et al., 2021) (Fig. 2), indicating that astrocytic ApoE can regulate neuronal functions by reshaping cholesterol metabolism and altering epigenetic modifications.
The human APOE gene has three isoforms: ε2, ε3, and ε4, differing by a single amino acid (arginine or cysteine) at residues 112 or 158. The three main ApoE isoforms are ApoE2 (Cys-112, Cys-158), ApoE3 (Cys-112, Arg-158), and ApoE4 (Arg-112, Arg-158) (Mahley and Rall, 2000). Allelic combinations produce three homozygous (APOE2/2, APOE3/3, and APOE4/4) as well as three heterozygous (APOE2/3, APOE2/4, and APOE3/4) genotypes. ε4 carriers exhibit lower ApoE levels than ε3 carriers, whereas ε2 carriers exhibit higher ApoE levels in both cerebrospinal fluid (CSF) and plasma (Cruchaga et al., 2012).
The lipidation state of ApoE is also isoform-dependent, with ApoE4 being poorly lipidated compared with ApoE2 and ApoE3 (Zhao et al., 2017). This lipidation state correlates with differential efficiency in cholesterol efflux, as ApoE4 demonstrates the lowest efficiency, whereas ApoE2 exhibits the highest efficiency in cholesterol efflux (Michikawa et al., 2000). Internalized cholesterol dissociates from ApoE in late endosomes, followed by ApoE recycling in early endosomes (Yassine and Finch, 2020). ApoE4 has a lower recycling capacity because of its higher affinity for cholesterol binding, resulting in reduced efficiency in cholesterol efflux (Yassine and Finch, 2020). Single-cell transcriptomic profiling of postmortem human brains reveals that ApoE4 significantly alters signaling pathways associated with cholesterol homeostasis and transport (Blanchard et al., 2022). These findings underscore the isoform-dependent regulation of cholesterol homeostasis by ApoE.
ABC transporters
ABC transporters are prevalent in various types of cells in the brain, including neurons, glial cells, and capillary endothelial cells, where they play crucial roles in brain cholesterol transport (Kim et al., 2006). This superfamily includes ABCA1, ABCG1, and ABCG4. ABCA1 facilitates cholesterol efflux to lipid-free ApoE, forming lipoproteins, whereas ABCG1 and ABCG4 promote the further lipidation of these nascent lipoproteins (Wang et al., 2004) (Fig. 2).
ABCA1 is the most abundantly expressed ABC transporter in the brain (Dean et al., 2001; Martin et al., 2014; Schmitz et al., 2000), present in both neuronal and glial cells, with the highest expression observed in neurons and microglia. The expression of ABCA1 in astrocytes is only 25% of that in neurons (Kim et al., 2008). Mice lacking ABCA1 show poorly lipidated ApoE in both the brain and CSF (Burgess et al., 2008). This impairment in lipidation adversely affects cholesterol transport to neurons, resulting in defective neuronal function and memory behavior in KO mice (Hirsch-Reinshagen et al., 2004; Li et al., 2022). Notably, although total cholesterol levels exhibit only a mild reduction in ABCA1 KO brains compared with WT brains, mice with ABCA1 KO specifically in astrocytes display unaltered total cholesterol levels in the brain, suggesting that ABCA1-mediated changes in cholesterol levels are likely attributed to neuronal ABCA1 (Li et al., 2022).
Similarly, ABCG1 and ABCG4 are specific membrane transporters for cholesterol efflux. Analysis of human brain cells indicates that ABCG1 is most abundantly expressed in microglia but is also present in oligodendrocytes, neurons, and astrocytes (Kim et al., 2006). Mice lacking ABCG1/4 exhibit reduced cholesterol contents comparable with ABCA1 KO mice and also display deficits in learning and memory function (Li et al., 2022). In addition to measuring total cholesterol levels in the brain, assessing the distribution of cholesterol among different types of brain cells may provide a more accurate reflection of the precise changes occurring within the brain.
ApoE receptors
LDLR and LRP1 serve as the two major ApoE receptors in the brain, with high expression levels observed in neurons (Fuentealba et al., 2010; Liu et al., 2010). The binding affinity of these receptors to ApoE is largely dependent on the conformation and lipidation state of ApoE (Zhao et al., 2018). Astrocyte-derived ApoE particles exhibit higher affinity for LDLR than LRP1, whereas CSF-isolated HDL particles preferentially bind to LRP1 (Bu, 2009). Upon binding to neuronal receptors, ApoE particles undergo endocytosis, with core cargo lipids and a portion of ApoE delivered to lysosomes for degradation, while the remainder is recycled (Heeren et al., 2006).
Mice lacking LDLR exhibit increased levels of ApoE in both the brain and CSF, yet their brain cholesterol levels remain comparable with those of WT mice. Conversely, mice with LDLR overexpression in the central nervous system display a reduction in soluble ApoE levels in the brain (Kim et al., 2009). LDLR KO mice exhibit spatial memory deficits, likely attributable to hippocampal neuron apoptosis and synapse deficits (Wang et al., 2014). Conditional knockout of neuronal LRP1 in adult mice results in reduced cholesterol levels in the brain, accompanied by progressive age-dependent synapse loss and neurodegeneration (Liu et al., 2010).
In summary, cholesterol transport is crucial for maintaining synaptic and memory function in the brain, underscoring the importance of assessing cholesterol distribution among different types of brain cells rather than solely measuring the total cholesterol levels in the brain.
Liver X receptors
Liver X receptors (LXRs) are nuclear receptors actively involved in maintaining cholesterol homeostasis (Liang et al., 2004; Okabe et al., 2013; Waterham et al., 2001; Wang et al., 2016). In the brain, both isoforms of LXRs, namely LXRα and LXRβ, are expressed (Moutinho and Landreth, 2017). Upon activation by endogenous oxysterols, LXRs form heterodimers with retinoid X receptors (RXRs). These LXR/RXR heterodimers bind to the LXR response element on the promoter regions of target genes, thereby stimulating their transcription (Liang et al., 2004). Numerous genes implicated in cholesterol metabolism, such as SREBP1, several ABC transporters, and ApoE, are known to be regulated by LXRs.
Functioning as cholesterol sensors, LXRs modulate the expression of genes that govern cholesterol transport and catabolism. Various oxysterols, including 24-OHC, 22(R)-hydroxycholesterol, 24(S), 25-epoxycholesterol, and 27-hydroxycholesterol (27-OHC), serve as activators of LXRs (Wang et al., 2002). Of these, 24-OHC emerges as the primary metabolite of cholesterol catabolism within the brain. Neurons produce and release 24-OHC, which is then taken up by astrocytes. Within astrocytes, 24-OHC not only inhibits cholesterol synthesis but also boosts the levels of ABCA1, ABCG1, and ApoE to promote cholesterol transport through LXR activation (Abildayeva et al., 2006).
In contrast, 27-OHC, predominantly generated peripherally by 27-hydroxylase (CYP27A1), traverses the circulation to reach the brain (Czuba et al., 2017). Although neurons, astrocytes, and oligodendrocytes possess the capability to produce 27-OHC to a limited extent (Petrov et al., 2016), its levels within the brain remain notably low, accounting for less than 10% of the levels observed for 24-OHC (Wu et al., 2022).
In cultured astrocytes, LXR agonists enhance cholesterol efflux; however, they do not exert a similar effect in cultured neurons (Whitney et al., 2002). Mice lacking both LXRα and LXRβ display brain abnormalities characterized by excessive lipid deposition, disorganized myelin sheaths, and neuronal loss (Wang et al., 2002). Specific knockout of LXRβ mirrors lipid accumulation similar to that observed in double LXR KO mice. In addition, these mice exhibit motor neuron degeneration and astrogliosis (Andersson et al., 2005).
Cholesterol Catabolism and Excretion from the Brain
The brain lacks pathways for cholesterol degradation, relying instead on export mechanisms for its removal. Cholesterol is exported from the brain primarily in the form of 24-OHC, which is specifically synthesized within the brain. This molecule traverses the BBB through diffusion or via the anion transporting polypeptide 2, eventually entering the circulation. 24-OHC binds to LDL and is subsequently taken up by hepatocytes, where it is excreted in bile salts (Russell et al., 2009) (Fig. 2).
Cellular cholesterol handling in the brain is intricately linked to catabolic pathways of lysosomes and autophagosomes. Upon binding to receptors, ApoE particles are endocytosed into neurons. Within the endosome, ApoE may disassociate from lipid components and be recycled to the cell surface or transported to lysosomes for degradation (Kanekiyo et al., 2014). Within lysosomes, cholesterol esters are cleaved by acid lipase, releasing free cholesterol for cellular needs or storage. The cholesterol export process relies on NPC intracellular cholesterol transporter 1 (NPC1) and NPC2 (Trinh et al., 2020; Lu et al., 2022). Loss of NPC protein function leads to lysosomal accumulation of cholesterol (Hoglinger et al., 2019).
Insoluble cholesterol is converted into soluble 24-OHC by 24-hydroxylase (CYP46A1), representing the primary form of excreted cholesterol in the brain (Bjorkhem, 2006). CYP46A1 is predominantly expressed in the cell bodies and dendrites of neurons, it is also detectable in non-neuronal cells, such as astrocytes and microglia, particularly under stress conditions (Anchisi et al., 2012). As an oxysterol, 24-OHC activates LXR and induces the expression of genes involved in cholesterol biosynthesis and transport (Dietschy, 2009), promoting both cholesterol synthesis and transport to neurons.
Mice lacking CYP46A1 in their brain exhibit a substantial reduction in 24-OHC levels, whereas cholesterol levels remain relatively stable because of decreased biosynthesis (Russell et al., 2009). Conversely, mice overexpressing CYP46A1 produce elevated levels of 24-OHC, with no detectable alterations in brain cholesterol levels owing to increased biosynthesis (Hudry et al., 2010). These findings suggest a potential feedback mechanism through which cholesterol catabolism influences the rate of cholesterol biosynthesis.
A small fraction of cholesterol is excreted from the brain in lipoprotein particles via the BBB. Neuronal ABCA1 facilitates the efflux of excess cholesterol to lipid-poor ApoA1, which are then transported through the BBB via LRP1 and scavenger receptor class B type 1 (SR1B) (Gosselet et al., 2014; Pfrieger and Ungerer, 2011). Therefore, the expression of neuronal ABCA1 impacts cholesterol efflux (Hayashi, 2011). In summary, cholesterol is either converted into cholesterol esters and stored in the cytoplasm or converted into 24-OHC by CYP46A1 for excretion in the brain (Lund et al., 2003).
Lipid Storage in the Brain
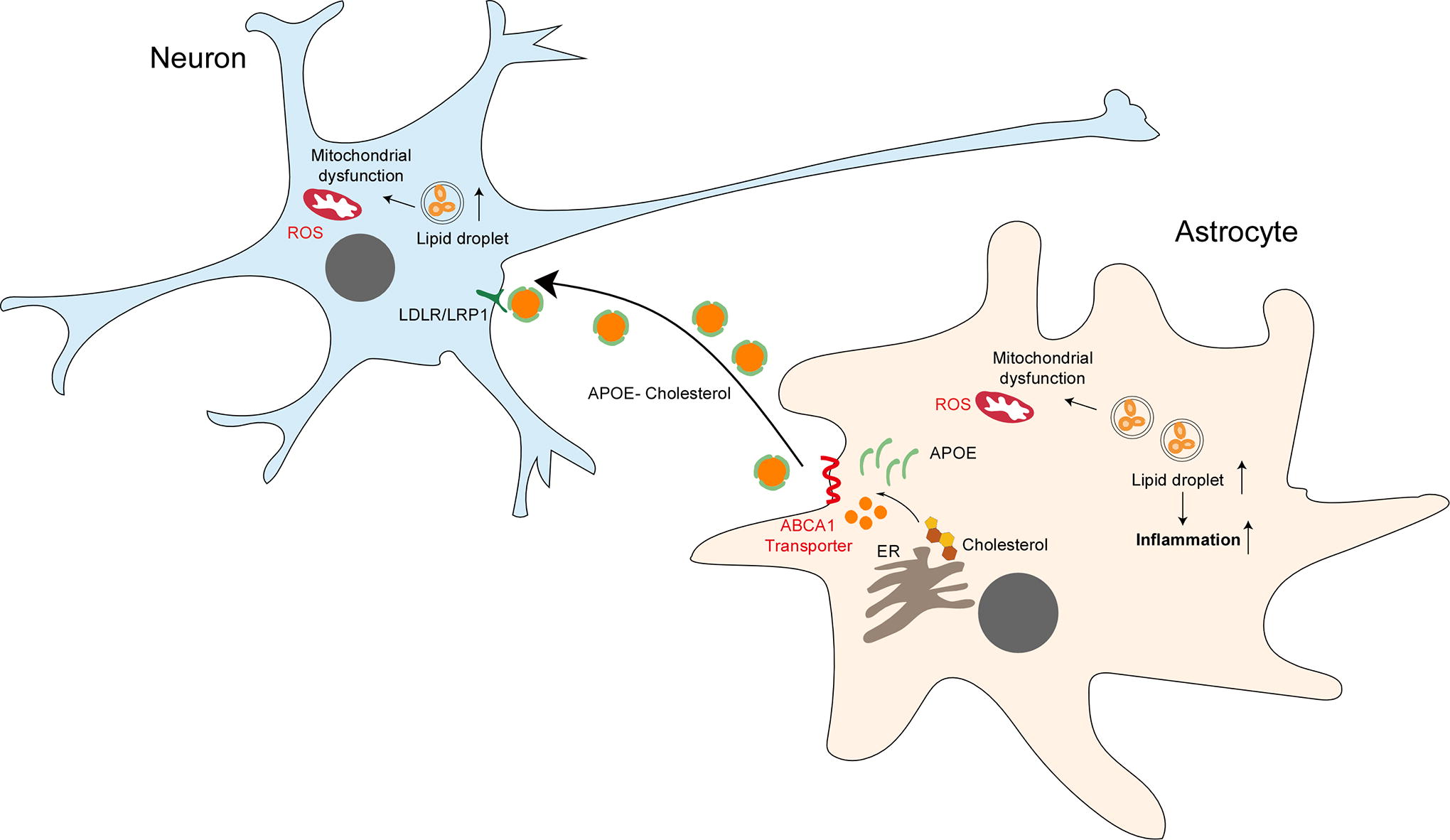
Lipid droplets constitute lipid-storing organelles containing neutral lipids, notably cholesterol and triglycerides, enveloped by a phospholipid monolayer (Bozza and Viola, 2010). They are ubiquitously present across various cell types within the brain, predominantly in glial cells (Ralhan et al., 2021). Lipid droplets manifest in both the developing and diseased brain, including instances of aging and neurodegenerative conditions (Ioannou et al., 2019; Liu et al., 2017; Marschallinger et al., 2020; Smolic et al., 2021).
Formation of lipid droplets is prompted by fluctuations in nutrient levels and cellular stressors, encompassing mitochondrial dysfunction, ER stress, hypoxia, and inflammatory stimuli (Boren and Brindle, 2012; Chitraju et al., 2017; de la Rosa Rodriguez et al., 2021; Karagiannis et al., 2020). Particularly during hypoxia, lipid droplets serve as buffers for specific fatty acids such as ceramides and acyl-carnitines, mitigating lipid-induced cytotoxicity and apoptosis (Ackerman et al., 2018). This role potentially shields cells from lipid peroxidation during oxidative stress, thus averting ferroptosis, a prevalent form of cell death observed in neuropathologies (Bao et al., 2021, Magtanong and Dixon, 2018). Lipid droplet turnover primarily occurs through two mechanisms: lipolysis and lipophagy. Lipolysis entails sequential triglyceride hydrolysis mediated by lipases, while lipophagy involves the engulfment of lipid droplets by autophagosomes, subsequently fusing with lysosomes for triglyceride breakdown (Ralhan et al., 2021). Notably, significant interplay exists between these two pathways (Cohen, 2018).
The distribution and lipid composition of lipid droplets vary among distinct brain cell types (Fitzner et al., 2020). Neurons exhibit limited capacity for fatty acid catabolism, with neuronal lipids predominantly transferred to neighboring glial cells for storage (Liu et al., 2017; Liu et al., 2015). Consequently, neuronal lipid droplet formation is restricted (Wat et al., 2020). By assimilating lipids derived from neurons, glial cells safeguard against lipid-induced toxicity. ApoE plays a pivotal role in lipid transfer, as evidenced by diminished lipid transfer and the subsequent lipid droplet formation in glial cells upon ApoE knockdown (KD) in either neurons or astrocytes (Ioannou et al., 2019; Liu et al., 2017). In addition, neuronal transporters such as ABCA1 and ABCA7 also contribute to lipid transfer and lipid droplet formation in glial cells (Moulton et al., 2021).
Non-neuronal cells, notably astrocytes and microglia, respond to cellular stressors, including hypoxia, metabolic perturbations, inflammation, or oxidative stress by forming lipid droplets (Smolic et al., 2021). This adaptive mechanism enables non-neuronal cells to sequester excess lipids generated during cellular stress within lipid droplets, thereby conferring protection (Fig. 3). Collectively, glial cells exhibit superior capability in fatty acid catabolism and lipid droplet formation compared with neurons, with neuronal lipids transferred to glial cells to mitigate oxidative stress-induced toxicity.
Cholesterol in Neurodegenerative Disorders
Cholesterol plays a vital role in the growth of neurites, the formation of synapses and dendrites, as well as in the guidance of axons. Disruptions in cholesterol metabolism can perturb neurotransmission, synaptic plasticity, and neurofilament integrity, ultimately culminating in neurodegeneration. The dysregulation of cholesterol homeostasis is intricately linked with various neurodegenerative disorders, including AD, PD, HD, amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS). In this segment of the review, we provide a comprehensive overview of the current understanding regarding the association between dysregulated cholesterol metabolism and neurodegenerative diseases.
AD
AD represents a prevalent form of neurodegenerative illness characterized by distinct hallmarks. These include the accumulation of extracellular plaques primarily comprising aggregated amyloid-β (Aβ) and intracellular tangles constituted of hyperphosphorylated Tau protein (Long and Holtzman, 2019). Concurrently, there is often an abnormal elevation or alteration in neurofilaments, accompanied by neurite atrophy, astrogliosis, and/or microglial activation. These pathological changes collectively contribute to the loss of synapses and/or neurons, ultimately leading to brain atrophy, a hallmark feature of AD (Long and Holtzman, 2019).
Cholesterol
The association between serum cholesterol levels and the risk of AD development is contentious. The age at which high serum cholesterol occurs appears to have a distinct impact on the risk of AD development. High serum cholesterol in midlife appears to increase the risk of AD in later life, while high serum cholesterol in later life seems to decrease the risk of AD (Solomon et al., 2009; Mielke et al., 2005). However, there are scattered reports claiming no association between serum cholesterol and AD risk (Li et al., 2005). Consequently, the effect of cholesterol-lowering drugs on the prevalence of AD remains inconclusive. Some reports suggest a beneficial effect, whereas others argue against any beneficial effect at all (Loera-Valencia et al., 2019).
Neuronal cholesterol increases the risk of AD development by influencing the production of Aβ. Aβ is produced through the proteolytic cleavage of amyloid precursor protein (APP). In the amyloid genesis pathway, APP is sequentially cleaved by β-secretase (BACE1) and γ-secretase (presenilin 1, PS1) to generate Aβ. In the nonamyloid genesis pathway, APP is cleaved by α-secretase (ADAM10/17) to release a soluble APP α fragment (sAPPα) (Fig. 4). Reduced neuronal cholesterol leads to preferential cleavage of APP by α-secretase, resulting in decreased Aβ production (Wang et al., 2019). Conversely, increased cholesterol can enhance β- and γ-secretase activity, leading to increased Aβ production (Wang et al., 2019).

Adult neurons primarily rely on astrocytes for cholesterol supply, and astrocyte-derived cholesterol also serves as a key regulator of neuronal Aβ metabolism. ApoE uses astrocyte-derived cholesterol to transport APP to lipid clusters, where APP interacts with β- and γ-secretase to generate Aβ. Inhibition of cholesterol synthesis in astrocytes reduces cholesterol levels in neurons, causing APP to leave lipid rafts and produce sAPPα through interaction with α-secretase. Importantly, targeted inhibition of cholesterol synthesis in astrocytes reduces both amyloid and Tau burden in AD mouse models (Wang et al., 2021b; Zhang et al., 2023). Cholesterol’s impact on Aβ metabolism is closely associated with its distribution within the cell. An increase in cholesterol content in lipid rafts promotes Aβ aggregation, whereas an increase in cholesterol content in neuronal membranes promotes Aβ fibrillation (Fig. 4).
In addition to its impact on Aβ metabolism, cholesterol acts on Tau aggregation independently of APP processing or Aβ formation (Maccioni et al., 2010). Neurons treated with methyl-β-cyclodextrin, a chelating agent that removes cholesterol from membranes, exhibit increased Tau entry and intracellular Tau aggregation. Similar observations are obtained in cultured neurons with disrupted cholesterol trafficking because of depletion of NPC1 (Tuck et al., 2022). These findings indicate that dysregulation in cellular cholesterol levels, cholesterol distribution within cells, and cholesterol trafficking contribute to AD pathology by affecting Aβ formation and/or Tau aggregation.
Oxysterols
In addition to cholesterol, oxysterols, which are cholesterol oxidation products, also play a role in AD pathology (Poli et al., 2013). Among them, 24-OHC is the most abundant oxysterol in the brain. In the early stages of AD, levels of 24-OHC remain relatively stable, with a possible minor increase attributed to neuronal destruction and the release of free sterols. However, in more advanced stages, there is a significant decline in 24-OHC levels because of the selective loss of neurons expressing high levels of CYP46A1 (Brown et al., 2004; Testa et al., 2016).
CSF, being in direct contact with the brain’s extracellular space, reflects biochemical changes occurring within the brain. Consistent with observations in the brain, a positive correlation has been established between 24-OHC concentration and p-Tau levels in the CSF of patients with AD, whereas no such correlation is observed in control individuals (Popp et al., 2013). Elevated levels of 24-OHC in CSF may result from increased cholesterol turnover during neurodegeneration, occurring at both the early and advanced stages (Gamba et al., 2021). However, contradictory evidence suggests no change or even a reduction in CSF 24-OHC levels in subjects with AD (Gamba et al., 2021). Furthermore, over 90% of the 24-OHC produced in the brain enters circulation by crossing the BBB, suggesting that plasma 24-OHC levels could serve as an indicator of cholesterol homeostasis in the brain. In the absence of neurodegenerative diseases, plasma 24-OHC levels are relatively low, but they increase in the presence of neurodegeneration. These findings suggest a potential role for 24-OHC as a biomarker for AD diagnosis and a target for therapeutic intervention.
The regulatory function of 24-OHC in AD pathology is complex. On one hand, 24-OHC exhibits several beneficial effects against AD progression, including the suppression of Aβ production (Urano et al., 2013) and Tau hyperphosphorylation (Testa et al., 2018). On the other hand, 24-OHC can also promote neuroinflammation, oxidative stress, and cell death (Gamba et al., 2015; Gamba et al., 2011; Yamanaka et al., 2011). The regulatory effects of 24-OHC appear to be concentration-dependent; lower concentrations seem to induce an adaptive and neuroprotective response in the human brain, whereas higher concentrations may lead to increased oxidative damage (Cigliano et al., 2019; Okabe et al., 2013).
In addition to 24-OHC, the levels of other oxysterols, such as 26-hydroxycholesterol (26-OHC), 25-hydroxycholesterol (25-OHC), and 7-oxycholesterol, are elevated in the brains extracted from late-stage AD patients. Specifically, both 25-OHC and 7-oxycholesterol levels are increased in AD patients, whereas 26-OHC levels remain unchanged (Dias et al., 2022; Wong et al., 2020). Particularly, 25-OHC is known to be associated with an increasing risk of neuroinflammation (Toral-Rios et al., 2024; Wong et al., 2020).
Furthermore, 27-OHC, the primary circulating oxysterol, demonstrates increased levels in both the CSF and brain of patients with AD compared with control individuals (Heverin et al., 2004; Mateos et al., 2011; Testa et al., 2016; Wang et al., 2016). In addition, elevated serum levels of 27-OHC and the ratio of 27-OHC to cholesterol are associated with reduced cognitive performance in patients with mild cognitive decline and elderly ApoE4 carriers (Liu et al., 2016; van den Kommer et al., 2009). The heightened levels of 27-OHC in AD may contribute to pathology by enhancing the production of Aβ through the promotion of APP processing and/or increasing the susceptibility of neurons to Aβ toxicity (Prasanthi et al., 2009). Notably, mice overexpressing CYP27A1, an enzyme involved in 27-OHC synthesis, exhibit elevated levels of 27-OHC alongside impaired memory function, accompanied by impaired neuronal branching and reduced synaptic density (Mateos et al., 2011).
Clinically, understanding these mechanisms could facilitate the identification of new biomarkers for early diagnosis and potential therapeutic targets. This includes a focus on modulating oxysterol levels and their effects on inflammation to mitigate the progression of AD.
ApoE
The ApoE gene plays a pivotal role in cholesterol metabolism and is closely linked with AD pathology. Specifically, the ApoE4 isoform significantly increases the risk of developing AD, while the ApoE2 isoform is associated with a decreased risk (Gomez-Isla et al., 1996; Corder et al., 1993; Strittmatter et al., 1993). As individuals age, the presence of the ε4 allele correlates with an elevated risk of cerebral amyloid angiopathy and age-related cognitive decline (Liu et al., 2013).
ApoE4 exacerbates the pathology of Aβ by promoting its production, deposition, and aggregation, while also hindering its degradation in neurons (Chai et al., 2021; Jiang et al., 2008). In addition, ApoE4 contributes to Tau pathology, neuroinflammation, and Tau-mediated neurodegeneration, irrespective of Aβ pathology. Studies in tauopathy mouse models reveal that mice expressing ApoE4 exhibit elevated Tau levels, extensive Tau redistribution from soma to dendrites, and heightened neuroinflammation compared with those expressing other ApoE isoforms or lacking ApoE altogether (Shi et al., 2017). In vitro experiments further demonstrate that microglia expressing ApoE4 exhibit increased innate immune reactivity, leading to elevated secretion of tumor necrosis factor-α and reduced neuronal viability when cocultured with Tau-expressing neurons, in comparison with ApoE2- or ApoE3-expressing or ApoE KO glial cells (Shi et al., 2017). ApoE4 also contributes to neuronal apoptosis and synaptic loss, thereby impairing neuronal viability and synaptic integrity (Zhao et al., 2020). Notably, depletion of ApoE specifically in astrocytes reduces Aβ accumulation and Tau-mediated neurodegeneration in mouse models of AD (Wang et al., 2021a; Wang et al., 2021b).
Human brains carrying the ApoE4 genotype exhibit abnormal cholesterol deposition in oligodendrocytes, a phenomenon observed in ApoE4/4 isogenic iPSC-derived cells and ApoE4 target-replacement mice as well (Blanchard et al., 2022). In addition, ApoE4 diminishes glucose uptake capacity in human astrocytes (Williams et al., 2020). Given that the brain primarily relies on glucose for meeting its high energy demands, alterations in lipid and glucose metabolism mediated by ApoE isoforms contribute to the varying risk for AD.
These findings underscore the detrimental role of ApoE4 in AD pathology, whereas its absence confers protection against the disease. Understanding these mechanisms provides insights into the development of targeted therapies aimed at mitigating the impact of ApoE4 on AD progression.
ABCA1 and ABCG1
Various findings underscore the regulatory roles of ABCA1 and ABCG1 in AD pathology (Hirsch-Reinshagen et al., 2005; Kim et al., 2007; Tansley et al., 2007; Nordestgaard et al., 2015; Vitali et al., 2014). Human genetic investigations have unveiled that mutations leading to loss of function in ABCA1 correlate with elevated risk of AD (Nordestgaard et al., 2015). Furthermore, genome-wide association studies have pinpointed ABCA1 as a gene associated with AD risk (Bossaerts et al., 2022). Notably, research demonstrates that ABCA1 diminishes Aβ deposition in a mouse model of amyloidosis (Sun et al., 2003), while the absence of ABCA1 markedly accelerates Aβ accumulation (Hirsch-Reinshagen et al., 2005; Wahrle et al., 2008). Similarly, ABCG1 emerges as a candidate linked to AD risk in genetic studies (Wollmer et al., 2007). ABCG1-mediated regulation of APP trafficking likely contributes to AD pathogenesis (Tansley et al., 2007). Nevertheless, conflicting evidence suggests a potentially neuroprotective role of ABCG1 in AD, as it substantially curtails Aβ production in cells expressing APP (Kim et al., 2007; Sano et al., 2016).
LRP1 and LDLR
LRP1 has garnered significant attention for its pivotal role in Aβ clearance within the brain (Deane et al., 2004; Zlokovic et al., 2010). Enhanced expression of functional LRP1 facilitates the trafficking of Aβ to lysosomes, whereas KD of LRP1 diminishes neuronal uptake of Aβ (Fuentealba et al., 2010). Depletion of LRP1 results in elevated Aβ plaques in the cerebral cortex and hippocampus of APP/PS1 mice, thereby exacerbating memory impairments (Kanekiyo et al., 2013). In addition, silencing of LRP1 diminishes Tau uptake in glioma cells and induced pluripotent stem cells (iPSC) derived neurons.
In P301S tauopathy mice, overexpression of LDLR leads to decreased levels of ApoE in the brain, consequently ameliorating Tau pathology and neurodegeneration (Shi et al., 2021). Conversely, mice lacking LDLR exhibit heightened susceptibility to the neurotoxic effects induced by Aβ (de Oliveira et al., 2014). Collectively, these findings suggest that neuronal receptors play a beneficial role in AD pathology by modulating lipid metabolism, as well as Aβ and Tau pathologies.
Lipid droplets
Lipid droplets are generally considered beneficial for cell physiology under typical conditions, yet they may exacerbate cytotoxicity and disease progression in specific pathological contexts (Fig. 4). Glial cells harboring the ApoE4 exhibit heightened accumulation of lipid droplets compared with those carrying ApoE3 or ApoE2 (Qi et al., 2021; Victor et al., 2022). This surplus lipid production and diminished lipid breakdown in ApoE4 glial cells likely contribute to the augmented lipid droplet accumulation in these cells (Victor et al., 2022). These observations propose a lipid droplet-centered mechanism for ApoE4’s association with increased risk of AD pathology.
In AD patients, glial cells demonstrate elevated lipid droplet formation (Liu et al., 2015, Shimabukuro et al., 2016). This phenomenon is also evident in the brains of triple-transgenic AD model mice (Hamilton et al., 2015). Notably, lipid droplet accumulation precedes the appearance of senile plaques and neurofibrillary tangles in AD model mice. Incorporation of cholesterol into lipid droplets amplifies the levels of both Aβ and p-Tau, which can be reversed by inhibiting cholesterol biosynthesis (van der Kant et al., 2019). However, inhibition of glial lipid droplets exacerbates neurodegenerative phenotypes (Liu et al., 2015). Overall, the accumulation of lipid droplets may represent an adaptive response to neurotoxicity, and further investigations are warranted to elucidate whether lipid droplets exert detrimental or beneficial effects in AD.
AD Diagnosis and Treatment
Advancements in biomarkers for Aβ and Tau pathology, along with magnetic resonance imaging (MRI) measurements of atrophy, have led to the evolution of diagnostic criteria, enabling earlier and more specific diagnosis of AD. Neuroimaging techniques crucial for AD diagnosis encompass MRI assessment of medial temporal lobe atrophy and posterior cingulate, as well as positron emission tomography (PET) with 2-deoxy-2-[fluorine-18]fluoro-D-glucose (18FDG-PET ) for temporoparietal hypometabolism. A more precise diagnosis involves evaluating cortical Aβ deposition through amyloid-PET imaging (Scheltens et al., 2021). MRI plays a vital role in functionally identifying both AD and mild cognitive impairment (MCI). Distinct MRI patterns reveal brain damage distinguishing AD from other brain disorders, alongside brain abnormalities associated with MCI risk (Chandra et al., 2019). Although not yet standard in clinical practice, more advanced MR techniques such as diffusion tensor imaging, arterial spin labeling, magnetic resonance spectroscopy, and functional MRI (fMRI) offer promising avenues for further refining diagnostic accuracy.
Statins, known as HMG-CoA reductase inhibitors, are widely used for lowering cholesterol levels and are primarily recognized for their cardiovascular benefits in preventing ischemic events. However, there is growing interest in their potential effects on AD. A population-based case–control study suggests that early statin use may slow the progression of AD in patients with mild-to-moderate stages of the disease (Lin et al., 2015). Nevertheless, evidence from preclinical studies regarding the protective effects of statins remains inconclusive. Some clinical trials indicate that taking different statins, such as lovastatin and pravastatin, could significantly reduce the risk of AD (Wolozin et al., 2000), whereas others report no association between statin therapy and reduced dementia risk (Rea et al., 2005). Discrepancies among studies may stem from variations in disease severity among participants and differences in evaluation criteria. Furthermore, the distinct physical and chemical properties of statins should be considered: lipophilic statins such as simvastatin may more efficiently cross the BBB and lower cholesterol levels in the brain, whereas hydrophilic statins such as pravastatin may not exhibit such effects (Min et al., 2023). These findings suggest the potential of statins in AD treatment, but further clinical evidence is needed to establish their beneficial effects conclusively.
In recent years, more targeted therapeutic approaches have emerged for addressing AD, with a primary focus on reducing the abnormal accumulation of Aβ in the brain. One direct method involves administering antibodies designed to bind specifically to Aβ, facilitating its clearance and reducing plaque burden. Several antibodies targeting Aβ have advanced to clinical trial stages. Notably, aducanumab (marketed as Aduhelm by Biogen), a monoclonal antibody targeting amyloid plaques, received approval from the U.S. Food and Drug Administration (FDA) in 2021. However, the approval of aducanumab has sparked considerable debate because of its therapeutic effects being primarily based on biomarker changes, particularly the reduction in Aβ plaque, rather than robust clinical benefit evidence (Knopman et al., 2021). Moreover, the use of aducanumab has been associated with various side effects, including headaches, falls, diarrhea, and confusion. More concerning adverse effects, such as brain edema and microhemorrhage, have also been observed in clinical trials.
Gantenerumab, a humanized monoclonal antibody targeting Aβ, shows promise in promoting the elimination of Aβ and reducing its cerebral accumulation (Bateman et al., 2022). However, its impact on mitigating cognitive decline in AD remains uncertain, and it has not yet received approval from regulatory agencies for AD treatment. Clinical trials have reported certain side effects of gantenerumab, including headaches, injection-site reactions, and potential immunological responses.
Another therapeutic approach targets the Tau protein with tilavonemab, a monoclonal antibody designed to inhibit Tau protein aggregation, facilitate its degradation, or impede inter-neuronal transmission. While promising, the efficacy of tilavonemab in mitigating cognitive decline in AD is still being investigated.
In addition to Aβ and Tau, therapeutic strategies targeting ApoE have been developed. Anti-ApoE antibodies used in peripheral circulation have shown to reduce Aβ deposition even before the appearance of Aβ plaques (Patel et al., 2021). Furthermore, treatment with LXR agonists has demonstrated increased Aβ degradation and reduced intracellular Aβ42 levels (Jiang et al., 2008). Administration of LXR agonists in AD model mice (hAPPswe) has resulted in decreased plaque pathology and improved memory function (Jiang et al., 2008). Moreover, innovative therapeutic modalities, including gene and stem cell therapies, are being explored. However, their efficacy and safety profiles require thorough examination before clinical application. These advancements represent promising avenues in the pursuit of effective treatments for AD (Fig. 6).
PD
PD is a complex and the second-most common age-related progressive neurodegenerative disease. It is the most common form of movement disorders characterized by the development of tremor, rigidity, and bradykinesia (Berg et al., 2014). The main neuropathological hallmarks of PD are the Lewy bodies, which contain α-synuclein, and the loss of dopaminergic neurons in the substantia nigra. As PD progresses, Lewy body pathology spreads to the neocortical and cortical regions (Simon et al., 2020). It has long been considered that dopaminergic neurons in the substantia nigra are dead by the time clinical motor symptoms develop (Surmeier, 2018). The most common nonmotor symptoms associated with PD include autonomic dysfunctions, cognitive abnormalities, psychiatric symptoms such as anxiety, depression, and apathy, and sleep disorders, with high rates of insomnia and rapid eye movement disorder (Picillo et al., 2014). Defects in lysosomes and mitochondria are suspected to be the main cause of PD, but the definite cause of PD onset remains unknown in most cases. Increasing understanding of the genetic and environmental risk factors for PD will probably elucidate the cause of this disease in the future (Fig. 5).

Although measuring cholesterol levels in the brain would provide an overall assessment of cholesterol changes in PD pathology, no such studies have been reported so far. However, it has been reported that CSF 24-OHC is increased in the brains of PD patients, whereas CSF 24-OHC is correlated with the progression of the disease (Bjorkhem et al., 2013; Schonknecht et al., 2002), indicating that cholesterol catabolism could be enhanced in PD. 24-OHC could serve as an indicator to follow disease progression.
It has been reported that isopentenyl diphosphate isomerase, a cholesterol-synthesizing enzyme, is identified in Lewy bodies, suggesting that cholesterol metabolism participates in α-synuclein aggregation (Nakamura et al., 2015). In addition to this synthesizing enzyme, cholesterol itself also colocalizes with α-synuclein, supporting a role of cholesterol in Lewy body formation. Cholesterol accumulation promotes the formation of α-synuclein oligomers in PD model mice, and the oligomeric α-synuclein can be taken up in a prion-like manner, leading to the formation of α-synuclein aggregates (Garcia-Sanz et al., 2021). Moreover, the content of oxidative cholesterol metabolites is increased in Lewy body dementia, and these metabolites seem to accelerate α-synuclein aggregation (Bosco et al., 2006).
α-Synuclein exhibits structural similarity with ApoE, and it also contains two cholesterol-binding domains (Fantini et al., 2011). Transgenic mice expressing α-synuclein display increased cholesterol content in their brain (Koob et al., 2010), whereas depletion of neuronal α-synuclein results in a reduction in brain cholesterol (Fig. 5). The aggregation of α-synuclein is enhanced in the presence of lower concentrations of ApoE and reduced in the presence of higher concentrations of ApoE (Emamzadeh et al., 2016). Depletion of LRP1 in human iPSC-derived neurons (LRP1-KO iPSNs) decreases the uptake of various forms of α-synuclein, including monomers, oligomers, and preformed fibrils. Similarly, LRP1 KO mice also exhibit reduced α-synuclein spread. 27‐OHC promotes α-synuclein expression by binding to LXR, which subsequently binds to the LXR response element on the α-synuclein promoter region, thereby enhancing α-synuclein transcription (Schommer et al., 2018). In addition, 27-OHC may elevate α-synuclein protein levels by inhibiting proteasomal activity in human dopaminergic neurons (Schommer et al., 2018).
Approximately 10% of PD cases are familial, with several genetic factors associated with familial PD being implicated in cholesterol metabolism. Mutations in the GBA1 gene, which encodes the lipid-metabolizing lysosomal enzyme glucocerebrosidase, are correlated with PD development. GBA1 mutations promote lysosomal cholesterol accumulation, thereby facilitating α-synuclein polymerization in the synaptic membrane. The most common genetic cause of PD involves autosomal dominant mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) (Bakshi et al., 2019). LRRK2 knockout rats exhibit elevated plasma cholesterol levels (Galper et al., 2022), yet further investigation is necessary to understand how genetic variations in lipid metabolism in genes contribute to PD risk.
The diagnosis of PD primarily relies on clinical evaluation of patients’ symptoms and brain imaging modalities such as MRI, ultrasound, and PET scan. MRI plays a crucial role in diagnosing and distinguishing neurodegenerative diseases resembling PD. High-field MRI and optimized sequences enable the identification of specific MRI signs of PD and facilitate early disease detection (Aludin and Schmill, 2021). Specific single-photon emission computerized tomography scans, such as dopamine transporter scans, aid in identifying suspected PD cases, but disease symptoms and neurological assessments remain the gold standard for diagnosis.
Lipid-modulating drugs, including statins, may confer protection against PD (Fig. 6). Studies have suggested that statins prevent dopaminergic neuron degeneration and suppress α-synuclein aggregation in vitro and in animal models of PD (Bar-On et al., 2008). A recent clinical trial indicated a trend toward reduced progression of motor symptoms in patients with PD treated with lovastatin based on preclinical and observational data (Lin et al., 2021). However, conflicting reports suggest that simvastatin might have detrimental effects on baseline nigrostriatal dopamine degeneration and long-term prognosis in patients with PD (Jeong et al., 2021). Further investigation is warranted to unravel the intricate relationship between statin therapy and PD.
ALS
ALS is a neurodegenerative disorder characterized by the progressive loss of motor neurons in the brain and spinal cord, leading to paralysis and eventual respiratory failure typically within 3–5 years of symptom onset. This condition shares many pathobiological features with frontotemporal dementia (van Es et al., 2017). Although the exact cause of ALS remains incompletely understood, mutations in genes such as superoxide dismutase 1 (SOD1), fused in sarcoma (FUS), TAR DNA-binding protein (TARDBP), and chromosome 9 open reading frame 72 (C9orf72) are commonly observed in patients with ALS (Chakraborty and Diwan, 2022).
One of the primary pathological hallmarks of ALS is the aggregation of proteins within the cytoplasm of motor neurons, with excessive deposition of TDP-43 (encoded by the TARDBP gene) being the most prevalent (Scotter et al., 2015, Blokhuis et al., 2013). TDP-43 cytoplasmic inclusions are predominantly found in regions such as the pyramidal, frontal, and temporal cortex, as well as the hippocampus of the brain. Notably, individuals with mutations in the C9orf72 gene are at a significantly higher risk of developing cognitive impairment compared with those without this mutation (Chio et al., 2019). Mutations in the SOD gene are implicated in both familial and sporadic cases of ALS (Dominov et al., 2023).
Genome-wide studies of DNA methylation have identified numerous differentially methylated positions associated with ALS, particularly within pathways related to cholesterol biosynthesis. This suggests a potential causal relationship between cholesterol metabolism and ALS pathogenesis (Hop et al., 2022). In addition, patients with ALS have been reported to exhibit abnormal elevations in cerebrospinal fluid cholesterol levels and its precursors, such as desmosterol (Abdel-Khalik et al., 2017).
TDP-43 is a DNA/RNA-binding protein involved in various cellular processes, including gene transcription, mRNA splicing, and translation regulation (Warraich et al., 2010). Moreover, TDP-43 appears to modulate cholesterol levels by impairing the transcriptional activity of SREBP2 and consequently reducing cholesterol biosynthesis (Egawa et al., 2022). Depletion of TDP-43 results in decreased expression of SREBP2 and LDLR, leading to reduced cholesterol levels both in vivo and in vitro (Ho et al., 2021). Furthermore, the SOD1 G85R mutation has been linked to increased cholesterol synthesis in spinal cord motor neurons (Dominov et al., 2023), underscoring the significance of cholesterol dysregulation in ALS pathology.
HD
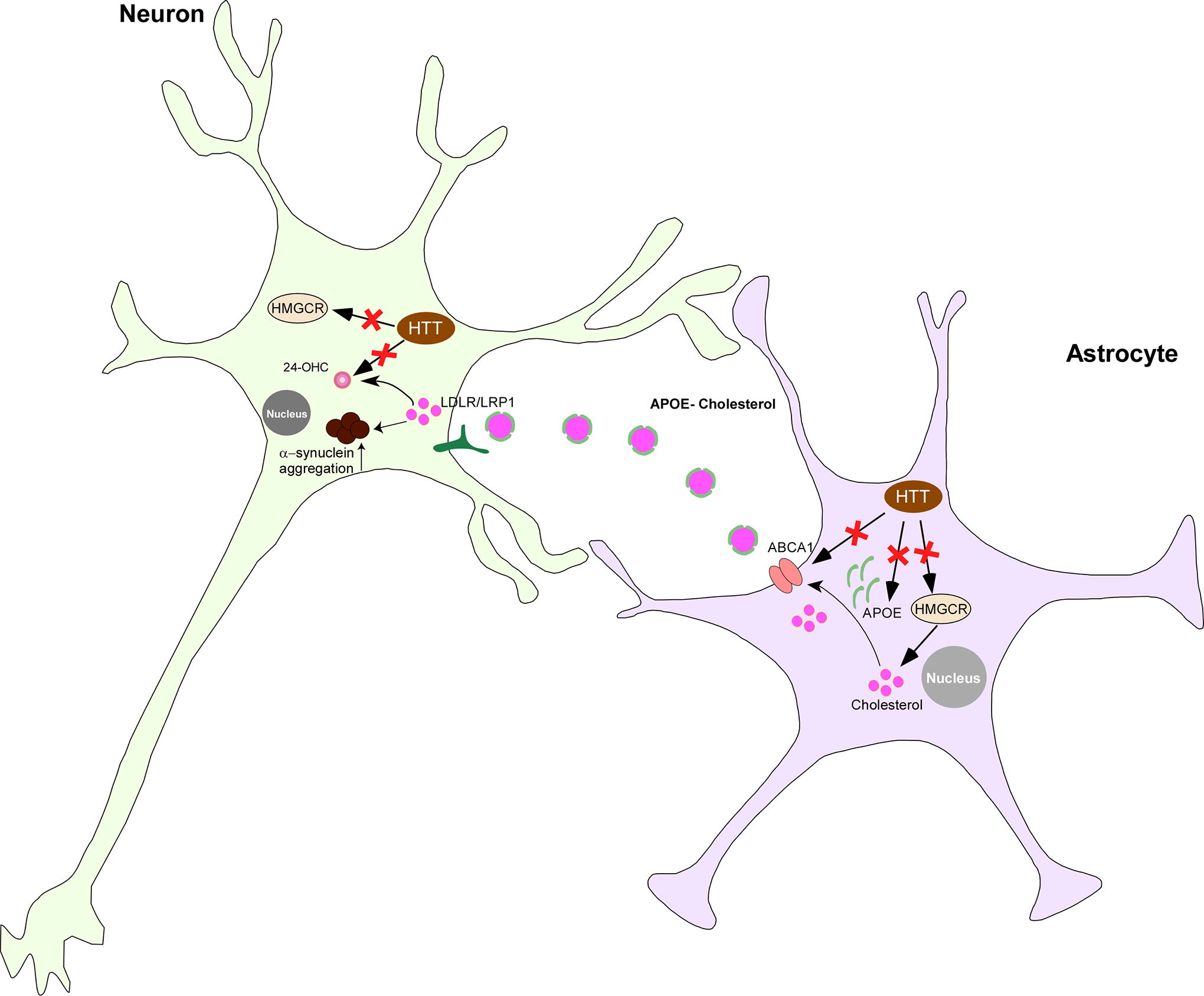
HD is a hereditary neurodegenerative disorder characterized by three main symptoms: movement, cognition, and psychiatric disturbances. It arises from an inherited CAG trinucleotide repeat expansion within the huntingtin gene on chromosome 4. This expansion leads to the production of a mutant huntingtin (mHTT) protein with an abnormal expansion of polyglutamine on the N-terminus of the huntingtin protein (HTT). Individuals with more than 39 repeats on the huntingtin gene are almost certain to develop the disease, whereas those with repeats between 36 and 39 exhibit reduced penetrance (McColgan and Tabrizi, 2018). mHTT protein primarily affects the striatum and cortex in the early stages of the disease, gradually spreading to other brain structures. Striatal atrophy can begin up to 15 years before the predicted onset of symptoms (Aylward et al., 2011).
Under normal physiological conditions, HTT acts as a coactivator for LXR, enhancing cholesterol biosynthesis (Kacher et al., 2022). Functional loss of HTT reduces LXR activity, resulting in decreased expression of ApoE and ABCA1, leading to less efficient cholesterol transport (Kacher et al., 2022). In HD, mHTT decreases the expression of peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), a transcription factor that promotes the expression of HMGCS1 and HMGCR, thereby reducing cholesterol biosynthesis (Xiang et al., 2011).
In the YAC128 HD mouse model, mHTT binds to the SREBP2/importin β complex, causing accumulation of mature cleaved SREBP2 in the cytoplasm (Birolini et al., 2021). This may explain the reduction in expression of HMGCR, CYP51, DHCR7, and DHCR24 observed in the HD brain and in primary cultured astrocytes (Kacher et al., 2022). Overexpression of active SREBP2 restores normal cholesterol biosynthesis and improves behavioral deficits in HD mouse models (Birolini et al., 2021).
Given the alterations in cholesterol biosynthesis observed in HD pathology, it is not surprising to find consistent reductions in cholesterol content across patients with HD, animal models, and cultured cells (Gonzalez-Guevara et al., 2020). Infusion of cholesterol into the striatum of HD model mice leads to dose-dependent behavioral improvements (Birolini et al., 2020). Consequently, levels of 24-OHC are reduced in the brain and plasma of patients with HD, as well as in HD model mice (Kreilaus et al., 2016). These findings suggest a decrease in cholesterol content because of a reduction in cholesterol biosynthesis rates during HD pathology.
The clinical diagnosis of HD primarily relies on family history, symptomatology (including involuntary movements, cognitive decline, and behavioral alterations), and physical examinations. Genetic testing for the HD-causing gene variant can further confirm the diagnosis. Although brain imaging is not mandatory for HD diagnosis, it may reveal characteristic abnormalities in brain regions affected by the disease.
Treatment for HD typically involves a multidisciplinary approach, incorporating specialized neurology, speech therapy, occupational therapy, physical therapy, nutritional support, and palliative care. Currently, there are no available disease-modifying drugs for HD. However, there is considerable interest in silencing the expression of the mutant huntingtin gene. Cholesterol has emerged as a potential therapeutic target for HD. Evidence suggests that statins may offer benefits for HD treatment (Patassini et al., 2008) by reducing neuroinflammation, oxidative stress, mitochondrial deficits, and excitatory toxicity in affected individuals (Kacher et al., 2022). Atorvastatin and simvastatin have demonstrated the potential to ameliorate HD-like symptoms and the associated cognitive dysfunction in rat models (Fig. 6). While there are currently no ongoing clinical trials evaluating the effects of statins on HD, initiating such trials appears promising.
MS
MS stands out as the most prevalent chronic inflammatory disease, characterized by the progressive loss of myelin in the brain and spinal cord, leading to subsequent axonal degeneration that impairs the physical mobility of patients. The signs and symptoms of MS exhibit wide variation among patients and hinge up on the location and severity of nerve fiber damage in the central nervous system (Dobson and Giovannoni, 2019). Remarkably, about 50% of patients with MS exhibit disease-associated cognitive deficits.
Cholesterol, a fundamental structural component of cell membranes and myelin, emerges as a pivotal player in MS pathology owing to its dysregulation. Notably, studies have underscored that elevated levels of circulating LDL cholesterol and total cholesterol correlate with unfavorable clinical outcomes in MS. In addition, investigations have highlighted the potential of oxysterols, cholesterol precursors, and ApoE as biomarkers for specific disease processes in MS (Zhornitsky et al., 2016). The mutation of the NR1H3 gene, encoding LXRα, has been associated with an augmented risk of developing MS. Encouragingly, correction in cholesterol dysregulation bears the potential to ameliorate cognitive outcomes in patients with MS (Kiani, 2023), underscoring the contribution of dysregulated cholesterol homeostasis to MS pathology.
Despite the pressing need, there remains a scarcity of biomarkers for both MS diagnosis and disease progression. At present, MRI emerges as the most reliable and routinely used diagnostic tool for MS. T2-weighted MRI images effectively discern white matter and gray matter lesions, whereas gadolinium-enhancing T1 lesions serve as markers for MS recurrence and active inflammation. Furthermore, magnetic transfer imaging offers valuable insights into disease severity by distinguishing MS lesions and monitoring the evolution of acute lesions (Yang et al., 2022). Notably, neurofilaments released from damaged axons into the CSF and bloodstream hold promise as diagnostic markers for MS (Kuhle et al., 2015). Indeed, serum neurofilament (sNfL) light chain levels are elevated in patients with MS and exhibit a robust correlation with pertinent clinical and neuroimaging outcomes (Canto et al., 2019), thereby advocating sNfL as a potential surrogate for MS diagnosis and prognosis.
At present, effective treatment modalities for multiple sclerosis remain elusive, with current interventions primarily aimed at hastening recovery from an attack, reducing clinical relapses, and slowing disease progression. Noteworthy, in experimental autoimmune encephalomyelitis mouse models, treatment with the statin drug atorvastatin demonstrates immunomodulatory effects. Conversely, patients with MS treated with simvastatin exhibit slower disability progression. Although the causal relationship between MS and abnormal cholesterol metabolism remains incompletely understood, advancements in cholesterol metabolism regulation hold promise for enhancing comprehension of MS pathophysiology and identifying novel therapeutic targets and strategies.
Conclusion
In the brain, cholesterol not only serves as an indispensable constituent for cell membranes but also exerts regulatory influence on neuronal function. Cholesterol homeostasis is delicately maintained through the regulation of cholesterol biosynthesis, transport, catabolism, and storage. This homeostasis is maintained not only within each individual type of brain cells but also among different types of brain cells. The brain primarily depends on de novo synthesis of cholesterol because of the presence of BBB. Cholesterol biosynthesis predominantly occurs in astrocytes, and cholesterol is subsequently transported to neurons for utilization. Thus, cholesterol transport serves as a crucial mechanism for transferring cholesterol between different types of brain cells. In addition to transfer of cholesterol, cholesterol carrier conveys other molecules that regulate neuronal functions by reshaping their gene expression. Excessive cholesterol either undergoes catabolism for breakdown in neurons or storage in the form of lipid droplet mainly in astrocytes, both processes could protect neurons from oxidative stress-induced toxicity.
Dysregulation in cholesterol homeostasis in the brain is tightly associated with the pathogenesis of multiple neurodegenerative diseases. However, cell-type-specific assessment of cholesterol metabolism remains elusive. Further studies should focus on assessing the distribution of cholesterol, its precursors, and metabolites within each type of brain cells to more accurately reflect the precise changes occurring within the brain. The involvement of cholesterol dysregulation in disease pathogenesis is incontrovertible; however, cholesterol-lowing drugs, statins, demonstrate a rather inconclusive therapeutic effect for the treatment of neurodegenerative disorders, owing to inconsistencies in clinical efficacy and a lack of efficient delivery systems to the brain. Therapeutic approaches targeting pathological hallmarks such as Aβ and Tau appear to be more promising. Some of the antibody-based strategies have received approval from the FDA, although controversies have arisen because of the severe adverse events and lack of robust clinical benefits. The efficacy of other antibody-based strategies targeting Tau or ApoE4 is still under investigations. Further investigation is warranted to unravel the precise role of cholesterol in neurovegetative diseases, and to develop a valuable diagnosis biomarker as well as personalized medicine.
Authors’ Contributions
Q.L., D.L., J.Z., and K.H. conceptualization and writing. Z.Z. and S.W. critical reading.
Author Disclosure Statement
The authors declare no competing interests.
Footnotes
Funding Information
This research was supported by the National Key R&D Program of China (2021YFA0804900 and 2020YFA0509300), the