
Abstract
Significance:
Lipids, which constitute the highest portion (over 50%) of brain dry mass, are crucial for brain integrity, energy homeostasis, and signaling regulation. Emerging evidence revealed that lipid profile alterations and abnormal lipid metabolism occur during normal aging and in different forms of neurodegenerative diseases. Moreover, increasing genome-wide association studies have validated new targets on lipid-associated pathways involved in disease development. Myelin, the protective sheath surrounding axons, is crucial for efficient neural signaling transduction. As the primary site enriched with lipids, impairments of myelin are increasingly recognized as playing significant and complex roles in various neurodegenerative diseases, beyond simply being secondary effects of neuronal loss.
Recent Advances:
With advances in the lipidomics field, myelin lipid alterations and their roles in contributing to or reflecting the progression of diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and others, have recently caught great attention.
Critical Issues:
This review summarizes recent findings of myelin lipid alterations in the five most common neurodegenerative diseases and discusses their implications in disease pathogenesis.
Future Directions:
By highlighting myelin lipid abnormalities in neurodegenerative diseases, this review aims to encourage further research focused on lipids and the development of new lipid-oriented therapeutic approaches in this area. Antioxid. Redox Signal. 00, 000–000.
Introduction
Neurodegenerative diseases
Neurodegenerative disorders, which involve the gradual impairment and death of neurons, pose significant challenges in the realms of neuroscience and public health. These disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS), are characterized by their gradual onset and prolonged progression, which result in substantial illness and death. Neurodegenerative disorders impact millions of individuals globally. The current emphasis in treating neurodegenerative disorders is on managing symptoms, as there is no definitive cure available. This presents a major issue worldwide. The goal of research is to comprehend the intricate pathophysiological mechanisms of these diseases, including genetic, molecular, and environmental components, to develop therapies that can alter disease advancement. Current research explores novel approaches such as drugs, stem cell treatments, and lifestyle modifications aiming at reducing the risk or delaying the development of disease. However, these tactics are all focused on enhancing the quality of life for patients, there are currently no definitive cures available (Yiannopoulou and Papageorgiou, 2020).
Myelin/white matter abnormalities in neurodegenerative diseases
The inadequate comprehension of nonconventional yet crucial pathologies, such as myelin/white matter (WM) impairments, contributes to the existing constraints in the treatment of neurodegenerative disorders. These impairments have been increasingly recognized as key contributors to the advancement of different neurodegenerative disorders. In AD, WM abnormalities frequently coexist with amyloid plaques and tau tangles and positively correlate with cognitive decline (Bartzokis et al., 2003), which indicates a direct correlation between WM abnormalities and disease progression. Likewise, with PD, there are observable WM abnormalities in multiple areas of the brain (Chondrogiorgi et al., 2019), which are directly related to the severity of both motor and nonmotor symptoms (Zhang and Burock, 2020). These findings suggest that in addition to the degeneration of dopaminergic neurons, the presence of WM pathology plays a substantial role in the clinical symptoms of PD (Blume et al., 2017). HD, although mostly linked to degeneration of the basal ganglia, also exhibits substantial alterations in WM, which are associated with various physical and cognitive impairments (Odish et al., 2015). ALS, predominantly recognized for its influence on motor neurons (Hardiman et al., 2017), also exhibits alterations in WM. Recent research indicates that ALS is characterized by extensive neurodegenerative mechanisms, such as the weakening of myelin sheath, which impairs motor neuron function (Philips and Rothstein, 2014). MS, on the contrary, is a classic example of a disease that primarily affects central nervous system (CNS) WM, due to the majority loss of myelin (demyelination) and destruction of nerve cells. The demyelination observed in MS leads to neurological impairments and correlates with cognitive decline (Nelson et al., 2011), highlighting the crucial need for preserving the integrity of WM for brain functioning (Trapp and Nave, 2008). Despite different myelin-related pathologies occur in various neurodegenerative diseases, oxidative stress might serve as one of the important players in regulating myelin physiology/pathology. It is shown that excessive oxidative stress has a negative impact on myelin homeostasis, in decreasing oligodendrocyte-precursor cell proliferation and maturation, oligodendrocyte numbers, and myelin-related genes and proteins both in vitro and in vivo (Maas et al., 2017).
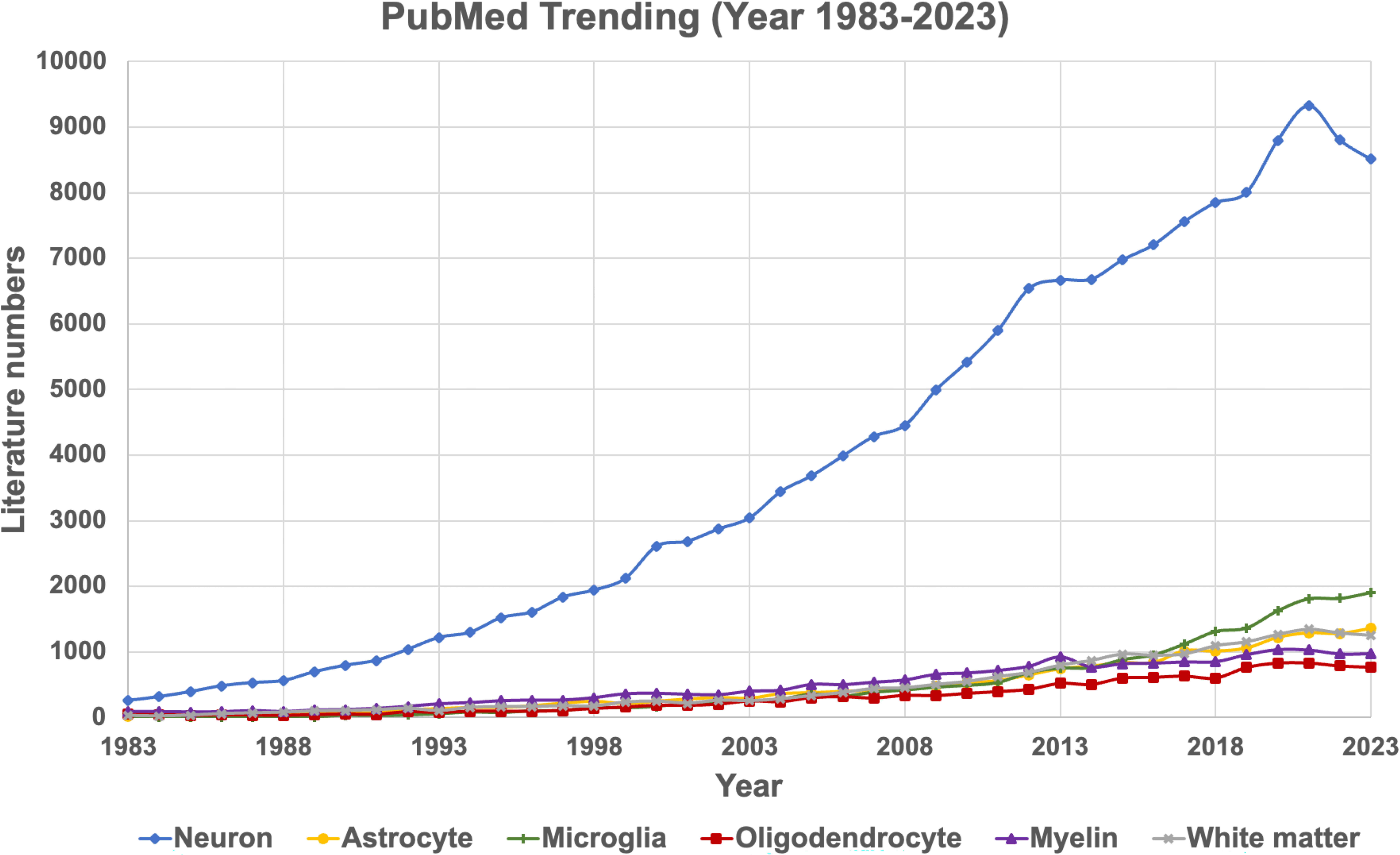
All aforementioned myelin/WM abnormalities are frequently observed but often overlooked in different neurodegenerative disorders. Meanwhile, a literature search in the PubMed database over the past 40 years showed an obvious lack of publications that focus on oligodendrocyte/myelin/WM related to neurodegenerative diseases, which is highly in contrast to the traditional emphasis on neuronal research (Fig. 1). It is crucial to acknowledge and comprehend the full scope of myelin dysfunction to devise more efficacious therapeutic approaches. Future therapeutic strategies must target both established characteristics of these diseases and the related deterioration of myelin/WM, which significantly influences the advancement of disease and its symptoms.
Myelin lipids in health and neurodegeneration
The dry weight of CNS is primarily composed of lipids, making up 70%–85% of the total weight (Poitelon et al., 2020). Majority of these lipids are contributed by myelin, an extended membrane structure of oligodendrocytes (as opposed to Schwann cells in the peripheral nervous system). Myelin is a lipid-rich substance that acts as an insulating sheath around axons. It is essential for the fast transmission of electrical impulses in the nervous system. Thus, investigating myelin lipid content and metabolism is crucial for understanding the myelin/WM impairments that occur in various neurodegenerative diseases.
The role of myelin lipids is complex and diverse. They not only serve as structural elements but also actively engage in biological functions of myelin. Myelin lipids can be classified into three major categories. First, glycerophospholipids, which include phosphatidylethanolamine, phosphatidylcholine, phosphatidylserine, phosphatidylinositide, and others, make up ∼40% of the myelin lipid content (Poitelon et al., 2020). Myelin relies heavily on them for its bilayer structure, and they also have important functions in cell signaling (Chrast et al., 2011). Second, cholesterol, which makes up ∼27% of the overall lipid content of myelin (Jackman et al., 2009), plays a crucial role in preserving the structural integrity and fluidity of myelin (Saher et al., 2005). Third, sphingolipids, such as sphingomyelin (∼6% of myelin in rat [Norton and Poduslo, 1973]), play a role in the creation of lipid rafts and are crucial for signal transduction and protein transport in myelin (Bieberich, 2018; Gault et al., 2010). Other more complex glycosphingolipids, including cerebroside (HexCer), sulfatide, and ganglioside, also are prominent structural lipids (∼31% [Jackman et al., 2009]) found in myelin. Among these, galactoylceramide (GalCer), which is crucial for the synthesis and stability of myelin, makes up around 22% of the lipids found in myelin (Coetzee et al., 1996). Sulfatide, which is the sulfated derivative of GalCer, has a major role in maintaining the structural integrity of myelin and facilitating connections between myelin and axons (Ishibashi et al., 2002). Ganglioside is enriched in WM tracts and is important for axon-myelin stability and axon regeneration (Arends et al., 2022). Facilitated by the presence of these lipids, myelin not only facilitates nerve impulse transmission but also plays a crucial role in the upkeep and regrowth of nerve fibers.
The complex relationship between lipids and neurodegenerative diseases has been evidenced by various studies at different levels. In AD, improper regulation of cholesterol metabolism in myelin is linked to the transport of a major genetic risk factor for AD (i.e., apolipoprotein E [ApoE] allele 4, APOE4), and the abnormal processing of amyloid precursor protein (APP) and the subsequent buildup of Aβ plaques (Marquer et al., 2011). In PD, phosphatidylcholine is one of the major lipid classes that was found to be altered (Wood et al., 2018), which also shows binding affinity with alpha-synuclein (Fanning et al., 2020). Disruptions of sphingolipid metabolism, namely sphingomyelin and ceramide, are linked to neuroinflammation and neuronal death in various neurodegenerative diseases (Cutler et al., 2004; Hannun and Obeid, 2018). Sulfatide has also been found to regulate immune cell activity in MS (Jeon et al., 2008) and was found significantly lost in the early stage of brains of patients with AD (Han et al., 2002). Additionally, the abnormal dynamics and metabolism of lipid rafts, which are enriched with cholesterol and sphingolipids, are known to be involved in various neurodegenerative diseases (Simons and Ehehalt, 2002). Although cholesterol and glycerophospholipids are enriched in the myelin sheath, they are also abundant in almost all other CNS cells and matters. Thus, it is hard to determine whether there are myelin/WM-specific abnormalities even if their content is altered in different diseases. Similarly for plasmalogen, which is not only presented in myelin sheath but also commonly found in all cellular membranes, it is hard to distinguish the source even if its level is found to be altered in disease conditions. Herein, we will only focus on specific sphingolipids-cerebroside, sulfatide, and ganglioside GM1 whose specificities with myelin or WM have been well-documented. These will be referred to as myelin-specific or myelin-enriched lipids (hereafter, myelin lipids) in this review. Detailed structures and related synthetical pathways of myelin-enriched sphingolipids are illustrated in Figure 2.
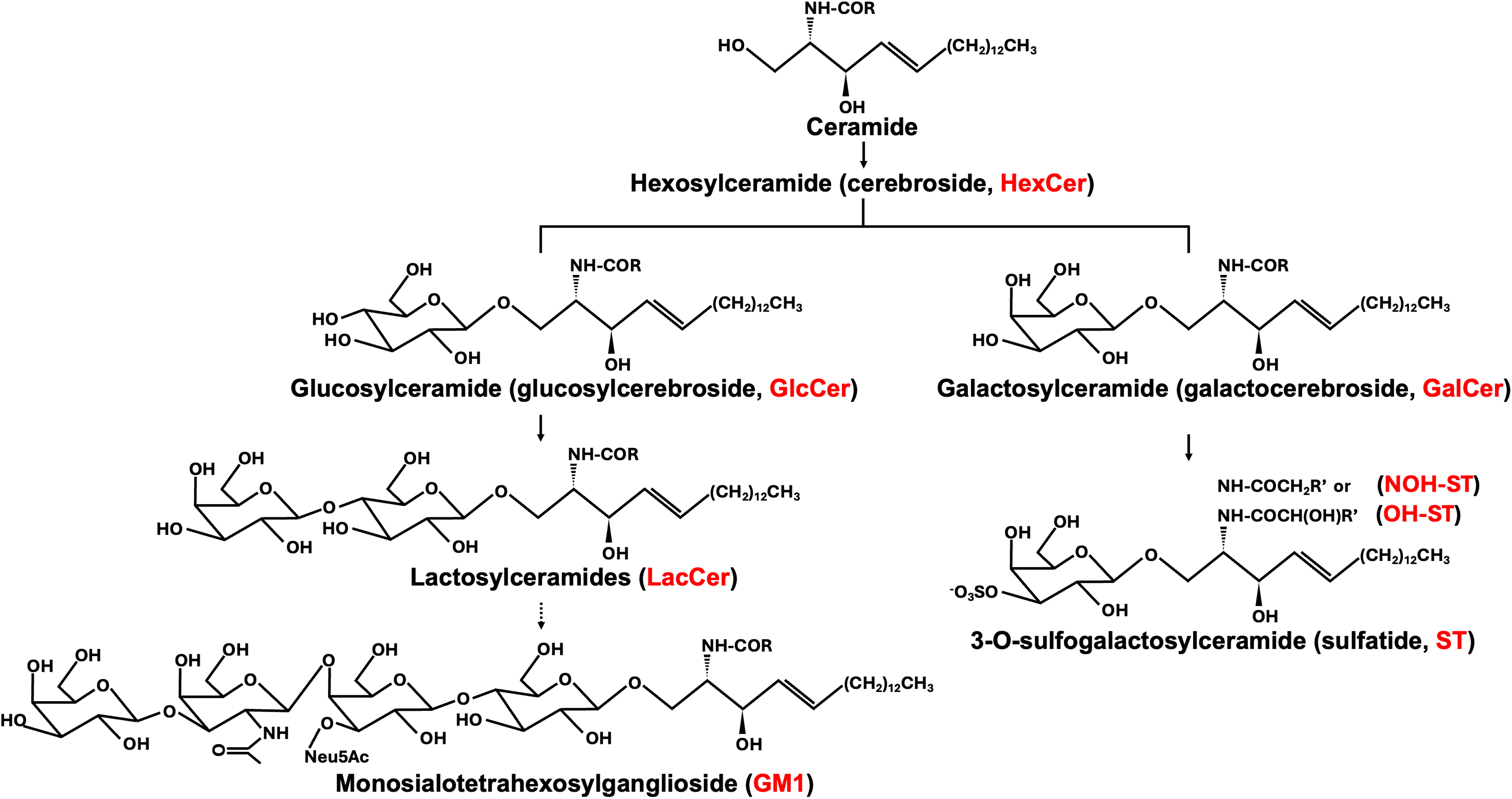
Major myelin lipid structures and pathways.
Lipidomics for studying myelin lipids in neurodegeneration
The significance of lipid metabolism in biological studies has been acknowledged for more than a century, yet understanding its mechanisms was challenging due to technical limitations in identifying and quantifying specific lipid molecules. The launching of lipidomics was made possible thanks to the fundamental developments of soft ionization (electrospray ionization [ESI] and matrix-assisted laser desorption/ionization [MALDI]) and fragmentation methods from the 1980s to the early 1990s (Han and Gross, 2022). Later in 2003, Han and Gross (2003) first coined the field of lipidomics, which nowadays has become an important constitution of systems biology along with genomics and proteomics. In 2005, the fund of the LIPID MAPS consortium greatly flourished in the growth and development of lipidomics by promoting systematic lipid classification, lipid standard preparation for quantification, and database development (Han and Gross, 2022). Early in 2002, a pioneer study (Han et al., 2002) published by Han et al. was the first to show myelin lipid abnormalities in AD postmortem brains using ESI mass spectrometry. This study made an indelible contribution to the field, by not only giving strong evidence to support the occurrence of WM abnormalities in AD from a lipidomics perspective but also inspiring and pushing the lipidomics application into the neurodegeneration field, especially those with myelin defects.
In summary, this review aims to (i) summarize the recent findings on alterations of CNS myelin-specific/enriched lipids in various neurodegenerative diseases, (ii) discuss the therapeutic potential of myelin lipid-based manipulations, and (iii) discuss in detail about the role of a key class of myelin-specific lipid, sulfatides, in the pathogenesis of AD.
Alzheimer’s Disease
General introduction
AD represents the most common form of dementia, posing significant challenges to healthcare systems worldwide (Anonymous, 2023). This progressive neurodegenerative disorder is primarily characterized by the gradual decline in cognitive function and memory, severely impacting the quality of life in elderly populations. Aging is considered the major risk factor for AD development. The pathophysiology of AD is complicated. Traditionally, it has been viewed as a gray matter disease since the two primary pathological hallmarks, the accumulation of Aβ plaques and tau tangles, are found mainly in gray matter. However, emerging evidence suggests that WM abnormalities also play critical roles in AD.
Myelin/WM abnormalities in AD
Neuroimaging studies have found that a group of patients diagonalized with mild cognitive impairments (MCI), a prodromal stage of AD, has widespread WM abnormalities that precede any detectable gray matter defects (Agosta et al., 2011). Specifically, WM hyperintensities, which represent lesions in WM, are considered to be more associated with preclinical AD than cognitive and other markers of neurodegeneration (Garnier-Crussard et al., 2021; Kandel et al., 2016). In addition, WM axonopathy (Stokin et al., 2005) and demyelination (Desai et al., 2009; Mitew et al., 2010) are also reported to occur before the presence of Aβ plaques and neurofibrillary tangles. Myelin impairments have gained growing attention in AD studies. It has been reported that Aβ plaques tend to occur in focal demyelinating areas (Mitew et al., 2010), and others have shown a negative correlation between myelin integrity and clinical Tau-PET score (Rubinski et al., 2022). Advanced imaging techniques, such as magnetic resonance imaging (MRI), have also been used to detect myelin loss in patients with AD (Dean et al., 2017). These imaging findings also correlate with the severity of cognitive impairment. These findings further support the critical and complex roles of myelin in AD pathogenesis.
Myelin lipid disorder in AD
At the molecular level, several studies have reported alterations in lipid metabolism in AD. These changes include the dysregulation of cholesterol metabolism, alterations in the levels of sphingolipids and phospholipids, and imbalances in fatty acid composition. Cholesterol has been shown to influence the processing of APP (Rudajev and Novotny, 2023). Abnormal cholesterol levels can lead to increased production of Aβ, a hallmark of AD pathology. Oxidative stress, common in AD, can lead to lipid peroxidation, resulting in damaged cell membranes and impaired neuronal function. Lipid peroxidation products have been found at higher levels in the brains of patients with AD, which correlates with the loss of endogenous antioxidant lipids (i.e., plasmalogen) (Han et al., 2001). The ApoE4 allele, a well-known genetic risk factor for AD, is involved in lipid transport. Individuals with ApoE4 variant have altered myelin lipid metabolism which may contribute to the development and progression of AD. A recent study revealed the unexpected APOE4 toxicity on cholesterol metabolism in oligodendrocytes, which further impairs myelin property (Blanchard et al., 2022).
As mentioned earlier, myelin is enriched with lipids and is the most lipid-abundant component in the CNS. Lots of abnormal lipid metabolism are myelin-specific or myelin-enriched, including sulfatide, cerebroside, ganglioside GM1, and others. One of the key lipid markers for myelin is sulfatide since it is exclusively synthesized by oligodendrocytes and is the essential maintenance material for myelin sheath. Studies have found significant sulfatide loss in postmodern brains in patients with very early stages of AD (Cheng et al., 2013; Han, 2007; Han et al., 2002). This finding is supported by other studies in AD populations with various disease stages and brain regions using different techniques (Dill et al., 2010; Gonzalez de San Roman et al., 2017; Jackson et al., 2005; Svennerholm and Gottfries, 1994; Zimmer et al., 2024). While the majority of these studies reported changes in the whole class level, one study showed species-specific and brain region-dependent alterations of sulfatide in patients with AD. Using MALDI-MSI, the investigators found 24:0 (OH)- and 26:0 (OH)-containing sulfatide species are lost in the dentate gyrus, while 26:0 (OH) species is increased in hippocampus WM (Yuki et al., 2011). Consistent with human data, sulfatide is also reduced in various AD mouse models that display amyloidosis (Cheng et al., 2010; Hong et al., 2016; Kaya et al., 2020b; Zhang et al., 2024), although some studies showed only mild decline or stable levels of sulfatide in APP/PS1 using conventional HPTLC (Barrier et al., 2010a; Barrier et al., 2010b). In addition to these well-established AD models, ApoE4 overexpression could also lead to a marked decrease of sulfatide levels in AD mouse brains (Han, 2010; Han et al., 2003).
While studies have been exploring the impact of amyloidosis on myelin lipid metabolism, less has been done on tau pathology mouse models. The only study that reported sulfatide level is on the 3xTg mouse model, which harbors three mutations, two of which are on Aβ production (APP KM670/671NL and Psen1 M146V) and one is on MAPT (P301L). Yet, the sulfatide level remains stable in the hippocampus and corpus callosum of the 3xTg mouse brains using MALDI-MSI in this study (Gonzalez de San Roman et al., 2021). This could be interpreted as AD-associated tauopathy having the opposite impact on sulfatide or other myelin lipids compared to amyloidosis, thus neutralizing such a loss observed in other amyloidosis mouse models as aforementioned. Another related evidence did show inactivation of the myelin synthesis enzyme, ceramide synthase 2, occurs before the onset of tau pathologies in the cortical regions of patients with AD (Couttas et al., 2016), which may imply the loss of myelin lipids precedes AD-associated tauopathies. However, the exact impact of AD-associated tauopathies on myelin lipids, and the role of myelin lipid deficiency in inducing AD-associated tauopathies remain elusive. All these data suggest that myelin-specific sulfatide deficiency is a converged pathology that occurs in AD and it could play an important role in AD pathogenesis. We will further discuss this topic in “Sulfatide and AD” section.
As the precursor of sulfatide, cerebroside is also enriched in myelin, yet with more contradicting alterations in AD. Some found that total cerebroside (HexCer) levels remain rather stable despite the reduction of sulfatide (Han, 2005; Han et al., 2002) in the cortex, others reported a reduction in the WM regions of frontal lobe (Svennerholm and Gottfries, 1994). In one study, although a reduction of total GalCer in the hippocampus was observed, the 2-hydroxylated fatty acids-containing species were increased in the same brain region (Hejazi et al., 2011). The ratio of 2-hydroxy/nonhydroxy cerebroside was found to decrease in brains with AD (Cherayil, 1968). Hence, the variances among brain regions and disease progression stages might contribute to the disparate findings noted across various studies, with lipid species potentially exerting an additional influence. Limited studies have looked into cerebroside levels in AD mouse models with inconsistent observations. In one study, the total GalCer was found to be unchanged in the cortex, hippocampus, and cerebellum of APP/PS1 mice (Barrier et al., 2010a). Yet, another study reported a sex difference where males showed stable levels but females had a significant loss of total GalCer in the cerebral cortex of APP/PS1 mouse (Barrier et al., 2010b). In more detail, decreased Glc/GalCer (d18:1/24:1) was reported in both 5xFAD (Zhang et al., 2020) and APP/PS1 mice (Kaya et al., 2020b).
Another class of myelin-enriched lipid is ganglioside GM1 (Ledeen et al., 1980; Marconi et al., 2005; Vajn et al., 2013). GM1 is important for maintaining axon-myelin stability (Schnaar, 2010), it also binds to Aβ and forms a GM1-Aβ complex (Yanagisawa, 2015). Although early studies (Fukami et al., 2017; Kracun et al., 1992; Kracun et al., 1991) reported a reduction of GM1 in AD brains, the majority of studies found that, consistent with the accumulation of Aβ, GM1 also accumulates in the brain (Gylys et al., 2007; Molander-Melin et al., 2005; Pernber et al., 2012; Svennerholm and Gottfries, 1994) and cerebrospinal fluid (CSF) (Blennow et al., 1991) of patients with AD. This accumulation of GM1 is also observed in several AD mouse models (Gylys et al., 2007; Kaya et al., 2020a; Wang et al., 2023). Other supporting studies have found that GM1 inhibition shows rescue effects on neurodegeneration and improves spatial memory of 5xFAD mice (Herzer et al., 2018; Herzer et al., 2016). A detailed summary on myelin lipid alterations in AD is included in Table 1.
Summary of Myelin Lipid Alterations in AD
\: Nonspecified or unavailable.
AD, Alzheimer’s disease; APP, amyloid precursor protein; CBM, cerebellum; CC, corpus callosum, CCx, cerebral cortex; Cx, cortex, DESI-MS, desorption electrospray ionization coupled mass spectrometry; DG, dentate gyrus; EO, early onset; ESI, electrospray ionization; FCx, frontal cortex; FL, frontal lobe; GM, gray matter; HexCr, hexosylceramide; HP, hippocampus; HPLC, high-performance liquid chromatography; HPTLC, high-performance thin-layer chromatography; IHC, immunohistochemistry; LO, late onset; MALDI, matrix-assisted laser desorption/ionization; MALDI-MSI, MALDI mass spectrometry imaging; MS, multiple sclerosis; NBM, nucleus basalis of Meynert; NCx, neocortex; PL, parietal lobe; TCx, temporal cortex; TL, temporal lobe; WM, white matter.
Risk factors and therapeutics
The precise etiology of AD remains largely elusive, although genetic, environmental, and lifestyle factors are known contributors. Age is the most significant risk factor for AD. The likelihood of developing AD increases significantly as one gets older, particularly after the age of 65. The major risk genes associated with AD include APP, PSEN1, PSEN2, APOE4, Trem2, SORL1, and ABCA7. Other risk factors include cardiovascular impairments, physical and cognitive inactivity, high-fat high-sugar diet, smoking, alcohol overconsumption, sleep disorder, mental disorder, social isolation, head injuries, and environmental toxins. More recently, studies have shown that COVID-19 infection is associated with an increased risk of cognitive impairment (Blackhurst and Funk, 2023; Mizrahi et al., 2023).
Current therapeutic strategies encompass pharmacological treatments aimed at mitigating symptoms, and emerging therapies focusing on the underlying disease mechanisms. There are currently three classes of the U.S. Food and Drug Administration (FDA)-approved medications for AD treatments: acetylcholinesterase inhibitors (AChEIs) (donepezil, rivastigmine, and galantamine) (Orr et al., 2017), N-methyl-
Parkinson’s Disease
General introduction
PD is a progressive neurological disorder primarily affecting movement and is characterized by a wide range of both motor and nonmotor symptoms. The motor symptoms of PD include bradykinesia, resting tremor, muscle rigidity, and postural instability. The nonmotor symptoms, which can sometimes precede motor symptoms, include cognitive changes, mood disorders, sleep disturbances, autonomic dysfunction, and sensory symptoms. PD is characterized by the loss of dopamine-producing neurons in the substantia nigra, a brain region that controls movement. Some other brain regions such as the superior frontal and striatum are also reported to be affected. The reason for this neuronal loss is not fully understood but involves a combination of genetic and environmental factors, including oxidative stress (Puspita et al., 2017). Additionally, mutations in genes such as LRRK2, PARK7, PINK1, PRKN, and SNCA have been linked to familial forms of PD.
More and more hits involved in lipid-associated pathways in PD development have been highlighted by genome-wide association studies (Klemann et al., 2017). The GBA1 variant is one of the most prevalent risk genes for developing PD (Neumann et al., 2009; Sidransky et al., 2009). While homozygous GBA1 mutation is linked with a lysosomal storage disorder called Gaucher disease (Grabowski, 2008), the risk of developing PD increases by over five times if carrying heterozygous GBA1 mutations (PD+GBA) (Sidransky et al., 2009). GBA1 gene encodes for glucocerebrosidase (GCase, GBA), which is a major lysosomal enzyme that catabolizes glucosylceramide (GlcCer) into ceramide and glucose. GBA deficiency-resulted GlcCer accumulation is found to destabilize α-synuclein, promote its aggregation, and lead to cellular toxicity (Kim et al., 2018; Suzuki et al., 2015; Taguchi et al., 2017; Xu et al., 2011; Zunke et al., 2018). In turn, α-synuclein can inhibit GBA activity (Mazzulli et al., 2011) and lead to increased GlcCer. Interestingly, cerebroside tends to accumulate during normal aging in wild-type mice (Hallett et al., 2018). All aforementioned data highlight the importance of sphingolipid metabolism in PD pathogenesis. Not only that, α-synuclein, the key pathological feature of PD, has been proposed to be a lipid-binding protein that can physiologically interact with various lipid classes including phospholipids, fatty acids, and others (Fanning et al., 2020). The polyunsaturated fatty acyl chain-containing phosphatidylcholines (34:5, 36:5, and 38:5) were reduced in the brains of patients with PD (Seyfried et al., 2018; Wood et al., 2018; Zhang et al., 2017), while others (44:6 and 44:5) were increased in plasma of patients with PD (Stoessel et al., 2018). Changes in fatty acids in the frontal cortex lipid rafts, including 20:4 and 22:6 fatty acids, have also been associated with PD (Xicoy et al., 2019).
Myelin/WM abnormalities in PD
Other than the traditionally characterized pathologies, growing evidence suggests significant involvement of WM beyond these well-known features. Increased expression of myelin-associated genes was detected in the frontal cortex of patients with PD (Hentrich et al., 2020). Diffusion tensor imaging (DTI), an advanced neuroimaging technique that allows the visualization of the water molecule diffusion in the brain and provides insights into the microstructural integrity of WM (Alexander et al., 2007), has been employed in detecting and characterizing WM changes in PD. WM abnormalities have been reported in brains with PD from individuals with dementia, especially in the corpus callosum, corona radiata, and cingulum regions (Chondrogiorgi et al., 2019), and have been linked to cognitive impairments (Yang et al., 2023; Zhang and Burock, 2020). The CNS myelin-producing cells, oligodendrocytes, also have been found to harbor disease-associated phenotypes including increased PD-associated gene expression, decreased oligodendrocyte differentiation marker OPALIN, enhanced inflammatory response and protein folding stress, and myelination abnormalities (Bae et al., 2023).
Myelin lipid disorder in PD
The aforementioned myelin/WM abnormalities can also be reflected by its lipid content. For example, sulfatide is found to accumulate in the plasma (Kurup and Kurup, 2003; Xiao et al., 2013), visual cortex (Cheng et al., 2011), superior frontal and cerebellar (Cheng et al., 2003), and substantia nigra (male-specific) (Seyfried et al., 2018) of patients with PD. The metabolism of sulfatide involves three major pathways: synthesis, transport, and degradation. While there is no clear evidence that the synthesis and transport pathways are altered under PD conditions, abnormal activities of the degradation enzyme, Arylsulfatase A (ARSA), have been reported repeatedly. In an early study published in 1994 (Martinelli et al., 1994), ARSA activity was found significantly reduced in patients diagnosed with parkinsonism or essential tremor compared with healthy controls and neurological patients without movement disorders. A case study also reported that partial ARSA mutation has a high recurrence of parkinsonism among siblings (Antelmi et al., 2014). A more detailed regional and species-specific alteration of sulfatide in an experimental 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD primate model was reported in a recent study using spatial lipidomics by mass spectrometry imaging, where certain long-chain hydroxylated sulfatide was reduced within motor control-related brain regions in animals, while certain long-chain nonhydroxylated sulfatide was increased on the contrary within the same brain regions (Kaya et al., 2023). In summary, the majority of the evidence supports that sulfatide accumulates in PD, although it is disease stage-, brain region-, and lipid species-dependent.
The importance of cerebroside metabolism has been highlighted in the introduction, and its accumulation correlates with high disease risk. It is consistently increased in the plasma of patients with PD (Chan et al., 2017; Guedes et al., 2017; Kurup and Kurup, 2003; Mielke et al., 2013). Yet, the brain cerebroside level is more region- and specie-dependent, similar to what was observed for sulfatide. The overall GlcCer level was found no change in the temporal cortex (Boutin et al., 2016), putamen, or cerebellum (Gegg et al., 2015) in GBA1 mutation-containing or sporadic PD cases. One additional study did report a sex difference regarding the total cerebroside (HexCer) level detected in brains with PD, which only increased in the substantia nigra of male patients with PD (Seyfried et al., 2018). This is consistent with the prevalence of PD where men are more likely to develop PD than women (Cerri et al., 2019). Whether the cerebroside level difference could be one of the explanations for this sex difference will need further validation, but we can certainly see a correlation there. At a more detailed species level, it was found that both cerebroside (HexCer, 24:1) and LacCer (18:1) were decreased in the frontal cortex of patients with PD (Wood et al., 2018).
As for ganglioside, its accumulation in the plasma of patients with PD has been observed in most studies (Chan et al., 2017; Kurup and Kurup, 2003; Zhang et al., 2017). However, the ganglioside GM1 was found with either no change (Gegg et al., 2015; Seyfried et al., 2018) or even decreased (Hadaczek et al., 2015; Wu et al., 2012) in brains with PD. Although the direct evidence of GM1 decline in brains with PD is obscure, studies showed that GM1 supplement exerts disease-modifying benefits for both patients with PD (Schneider, 1998; Schneider et al., 2015; Schneider et al., 2013; Schneider et al., 2010; Schneider et al., 1998; Schneider et al., 1995b) and mouse models (Wu et al., 2012; Wu et al., 2011). Long-term administration of GM1 to MPTP-induced nonhuman primate model has positive effects on cognition and motor functions (Pope-Coleman and Schneider, 1998). Yet, how exactly GM1 mediates these benefits is still under investigation. Several studies have shown that GM1 supplements promote dopamine innervation and tyrosine hydroxylase expression (Fazzini et al., 1990; Gupta et al., 1990; Hadjiconstantinou et al., 1989; Kastner et al., 1994; Rothblat and Schneider, 1998; Schneider, 1992; Schneider and Yuwiler, 1989; Schneider et al., 1995a; Schneider et al., 1992) in MPTP lesion models. It also reduced inflammation (Ba, 2016) and promoted microglial phagocytosis of α-synuclein (Park et al., 2009). Some controversial evidence also pointed to certain regulations between GM1 and α-synuclein. While some claimed GM1 promotes α-synuclein aggregation (Grey et al., 2015; Suzuki et al., 2015), others argued α-synuclein fibril formation is regulated by GM1 in a dose-dependent manner (Bartels et al., 2014; Martinez et al., 2007). In summary, sphingolipid metabolism seems to play nonnegligible roles in mediating PD pathogenesis. More evidence is needed to support if GM1 is a promising lipid candidate for PD treatment. Detailed myelin lipid alterations in PD are summarized in Table 2.
Summary of Myelin Lipid Alterations in PD
\: Nonspecified or unavailable.
GlcCer, glucosylceramide; GP, globus pallidus; iPD, idiopathic PD; LC-MS/MS, liquid chromatography with tandem mass spectrometry; MRM, multiple reaction monitoring; PD, Parkinson’s disease; PUT, putamen; SFG, superior frontal gyrus; SN, substantia nigra; SNpr, substantia nigra pars reticulata; sPD, sporadic PD; UPLC, ultra-performance liquid chromatography.
Huntington’s Disease
General introduction
HD is a progressive neurodegenerative genetic disorder characterized by a combination of motor, cognitive, and psychiatric symptoms. The most prominent motor symptom is chorea, which is characterized by jerky, uncontrollable dance-like movements, while other movement disorders such as rigidity, bradykinesia (slowness of movement), and dystonia (muscle spasms) can also occur. HD is also associated with gradual cognitive decline, memory loss, impaired judgment, difficult in planning and organizing, and language problems, which all lead to progressive dementia. In addition, patients with HD may suffer from psychiatric disturbances including depression, anxiety, irritability, personality changes, and occasionally obsessive-compulsive behaviors. Symptoms typically start between ages 30 and 50 but can begin at any age. The disease progresses gradually over 10–25 years, leading to increasing disability. Oxidative stress has long been considered one of the key players in disease progression, although no confirmative conclusions can be drawn (Kumar and Ratan, 2016).
Myelin/WM abnormalities in HD
While traditionally known for its gray matter loss, primarily caused by the loss of basal ganglia, WM abnormalities are increasingly recognized as playing a significant role in HD progression. HD is caused by a hereditary fault in a specific gene (HTT) located on chromosome 4. This gene defect involves an abnormal expansion of a DNA segment known as a CAG repeat. The number of CAG repeats is associated with the onset and severity of the disease. Surprisingly, this genetic risk factor (CAG repeat length) and disease severity have shown associations with cross-sectional WM abnormalities (Della Nave et al., 2010; Novak et al., 2014; Phillips et al., 2013).
Multiple groups have reported severe brain atrophies in cortical and WM regions (Georgiou-Karistianis et al., 2013; Rosas et al., 2003; Tabrizi et al., 2009) in patients with HD across different disease stages using MRI-based volumetric approaches. A two-year longitudinal TRACK-HD study (Odish et al., 2015) was able to show WM abnormalities in premanifest gene carriers (preHD) and early manifest HD compared to controls using DTI analysis. Later, the multisite PADDINGTON study also showed that premanifest gene carriers (preHD) show progressive WM abnormalities compared to controls (Gregory et al., 2015). In addition, increased myelin breakdown and iron levels were detected in HD (Bartzokis et al., 2007). Oligodendrocytes contribute to the majority of CNS iron production, highlighting the role of myelin dynamics in HD development. WM pathologies were observed in the YAC128 mouse model of HD (Garcia-Miralles et al., 2016) as well.
Myelin lipid disorder in HD
Emerging research suggests that changes in lipid metabolism, particularly involving sphingolipid metabolism, might play a role in the disease progression. Some studies reported increased overall ceramides in the brains of patients with HD compared with healthy controls (Di Pardo et al., 2017a), while the other showed that one particular ceramide species dihydroceramide (C18:0) showed a reduction in HD, along with the reduction of two other myelin-enriched lipids dihydrosphingosine and dihydrosphingosine-1-phospahte (Di Pardo et al., 2017b). Another myelin-enriched lipid class ganglioside GM1 was shown to be decreased in HD mouse models (Alpaugh et al., 2017; Desplats et al., 2007). More detailed brain regional studies have shown other lipid alterations in HD. It was found that sulfatide was lost in the subventricular zone in patients with HD using MRI (Hunter et al., 2018). This sulfatide reduction was also observed in the cerebellum of an HD mouse model, the R6/1 transgenic (Tg) mice, along with the reduction of its synthesis precursor, cerebroside (HexCer) (Denny et al., 2010). An early study published in 1978 has shown extracellular deposits of cerebroside (HexCer) in postmortem HD frontal cortex and caudate (Den Hartog Jager, 1978). This could be one of the explanations for sulfatide reduction in HD due to the loss of its synthesis precursor intracellularly. Yet, studies in mouse models showed a more detailed and complex landscape. One study showed an overall reduction of cerebroside (HexCer) in the cerebellum of R2/1 mice (Denny et al., 2010), while another study showed only HexCer (C18:1) is reduced in striatum and cortex of R2/2 mice with increased HexCer (C18:0), HexCer (C24:1), and GlcCer (Pepe et al., 2023). The latest study has paid additional attention to the species-specific alterations of those myelin-enriched glycosphingolipids, where they found that patients with HD harbor more long-chain (C13–C21) over very-long-chain (C22–C26) containing sphingolipids, including ceramides, sphingomyelins and lactosylceramides (Phillips et al., 2022). Similar trends were carried out resulting in a reduction of C22–C24 sulfatide (Phillips et al., 2022). All these data suggest myelin lipids in HD exert brain-regional and species-dependent alterations, which require attention in interpreting the data. Detailed myelin lipid alterations in HD are summarized in Table 3.
Summary of Myelin Lipid Alterations in HD
\: Nonspecified or unavailable.
CAU, caudate; HD, Huntington’s disease; SVZ, subventricular zone.
Risk factors and therapeutics
Ongoing research aims to unravel the disease mechanisms and explore novel treatments, including targeting the mutant HTT protein and investigating neuroprotective strategies. While there is no cure, treatments focus on symptom management, utilizing medications, and supportive therapies like physical and occupational therapy. Yet, very little has been done to understand how those treatments affect myelin lipid metabolism. However, a study did show that GM1 administration resulted in decreased levels of mutant huntingtin and brought a wide array of beneficial effects that include changes in levels of DARPP32, ferritin, Iba1, and GFAP, modulation of dopamine and serotonin metabolism, and restoration of neurotransmitters back to normal levels (Alpaugh et al., 2017).
Amyotrophic Lateral Sclerosis
General introduction
ALS, also known as motor neuron disease or Lou Gehrig’s disease, is a progressive neurodegenerative disorder that primarily affects motor neurons, the nerve cells that are responsible for controlling voluntary muscle movements (Wijesekera and Leigh, 2009). The disease pathologies start with stiff muscle stiffness, gradually increasing weakness, and muscle wasting, and eventually, develop into paralysis. There are two types of ALS depending on the lower or upper motor neurons being affected first: Limb-onset or bulbar-onset ALS, respectively. Limb-onset ALS begins with weakness in the arms or legs, while bulbar-onset ALS begins with difficulty in speaking or swallowing (Hardiman et al., 2017). Only 5%–10% of patients with ALS are inheritable and caused by certain gene variants, thus being characterized as familial ALS (Goutman et al., 2022). The remaining ALS cases (∼90%–95%) are sporadic, with unknown causes, although studies have seen that, in addition to environmental factors, certain gene variants also occur in sporadic cases, including SOD1 and TARDBP (Wingo et al., 2011). The majority of the patients with ALS have more or less cognitive impairments (Van Es et al., 2017). Although the exact pathogenic mechanism is uncertain, multiple lines of evidence have pointed to the involvement of oxidative stress (Ferrante et al., 1997; Pedersen et al., 1998), glutamate receptors (Carriedo et al., 1996; Kruman et al., 1999; Rothstein et al., 1995), and apoptosis (Pedersen et al., 2000).
Myelin/WM abnormalities in ALS
Although ALS is traditionally characterized by motor neuron deficits, the insulating myelin sheath plays an essential role in ensuring its proper signal transduction. Myelin-enriched WM regions have been reported to show deficits at the early stages of ALS, even before motor neuron structural changes (Ciccarelli et al., 2006; Matsusue et al., 2007; Prudlo et al., 2012; Rafałowska and Dziewulska, 1996; Sach et al., 2004; Sarro et al., 2011; Verstraete et al., 2011; Zhang et al., 2018). These WM abnormalities in ALS also exert a sex-specific dimorphism. Besides shared lesions in the corpus callosum in both sexes, females showed greater WM involvement in the right dorsolateral prefrontal cortex and forceps major compared with males in a DTI-based study (Bede et al., 2014). This might provide a potential explanation and mechanism for the sex difference in the ALS incidence (male:female ratio 3:1) (Cacabelos et al., 2016). Besides the structural changes of WM, more detailed neuropathological changes also involve axonal degeneration and myelin-producing oligodendrocyte loss with ALS (Fischer et al., 2004; Nonneman et al., 2014). Evidence also showed that ALS oligodendrocyte degeneration and impaired maturation occur before disease onset and might be the trigger for subsequent motor neuron death through a superoxide dismutase (SOD) 1-dependent mechanism (Ferraiuolo et al., 2016), supporting a direct correlation between demyelination and ALS progression (Barateiro et al., 2016; Kang et al., 2013; Nonneman et al., 2014).
Myelin lipid disorder in ALS
The most common risk gene that links with both familial and sporadic cases of ALS is the antioxidant enzyme SOD1. This mutation leads to abnormal aggregation of mutant SOD1 and causes reactive oxygen species (ROS) overproduction and mitochondrial dysfunction (Parakh et al., 2013; Taylor et al., 2016; Vandoorne et al., 2018). Since both ROS and mitochondrial function are highly involved in lipid metabolism, lipid alterations have been well documented in ALS cases. Studies have shown some plasmalogen species, which are believed to be the major antioxidant lipids, were reduced in ALS (Area-Gomez et al., 2021; Chaves-Filho et al., 2019). The accumulation of ceramides and GlcCer in CSF (Blasco et al., 2017) and GM1 in serum (Salazar-Grueso et al., 1990) of patients with ALS was reported, whereas it was also found that higher circulating triglyceride and cholesterol levels correlate with increased survival in patients with ALS (Dorst et al., 2011; Dupuis et al., 2008). However, a subsequent study (Kollewe et al., 2015), using antibody-based analysis, found no evidence of increased levels of gangliosides (including GM1) in the blood of patients with ALS, nor did it find any association between ganglioside levels and ALS pathologies or patient survival. This suggests a more detailed analysis is needed.
Previous studies have also found that ceramides, sphingomyelins, and cholesterol esters accumulated in the spinal cord of both ALS and Cu/Zn-SOD mutant mice (a mostly studied ALS model [Taylor et al., 2016]), and blockage of sphingolipid synthesis using pharmacological intervention was able to attenuate the ROS toxicity posted upon motor neurons (Cutler et al., 2002). More specifically, myelin lipids, including several cerebroside species and ganglioside GM1, were found to accumulate in both gray matter and ventral WM of spinal cord samples of patients with ALS (Dodge et al., 2015). In the same study, only GlcCer (C24:1) accumulation was found in the late stage of the disease, while most of the cerebroside and ganglioside GM1 species were found reduced in the early and middle disease stages of Cu/Zn-SOD mutant mice. A most recent study has found an overt accumulation of certain myelin lipids, including GalCer and sulfatide, in the spinal cord of Cu/Zn-SOD G93A rat model in both asymptomatic and symptomatic stages (Chaves-Filho et al., 2019). Although ALS affects both the brain and spinal cord, current efforts in understanding the abnormal lipid metabolism are mainly focused on spinal cord samples. Very little has been done to investigate brain lipid changes under ALS conditions in either patients or animal models, not to mention myelin-specific/enriched lipids. Detailed myelin lipid alterations in ALS are summarized in Table 4.
Summary of Myelin Lipid Alterations in ALS
\: Nonspecified or unavailable.
ALS, amyotrophic lateral sclerosis; SOD, superoxide dismutase; Sp. Cd, spinal cord.
Risk factors and therapeutics
There is still no cure for ALS, and treatments primarily focus on managing symptoms and maintaining quality of life. This includes medications to slow disease progression, such as riluzole and edaravone, and supportive therapies such as physical therapy, occupational therapy, and speech therapy. Although no direct evidence shows if those treatments have an impact on overall lipidome in the CNS, lipid-targeting interventions exert protective effects against ALS pathologies. Studies showed that GM3 administration (Dodge et al., 2015), ganglioside catabolic enzymes glucosylceramidase beta 2 inhibition (Bouscary et al., 2019), and acid β-glucosidase inhibition (Henriques et al., 2017) attenuate disease progression and even increase the survival of ALS mice.
Multiple Sclerosis
General introduction
MS is a chronic autoimmune disorder, where the immune system spontaneously attacks the CNS, particularly the myelin sheath. This results in inflammation and scar tissue formation (sclerosis) that develops on the WM of the brain and spinal cord (Clanet, 2008), thus, MS is also considered the most common form of demyelinating disease. MS usually begins between the ages of 20 and 50 and is twice as common in women as in men (Milo and Kahana, 2010), which is the opposite of ALS, although there are lots of key characteristics shared between these two diseases (Zhou et al., 2017). The symptoms of MS are diverse and depend on the location and extent of nerve damage. They can include fatigue, muscle weakness, spasticity, problems with coordination and balance, numbness or tingling, vision problems, and cognitive and emotional changes (Compston and Coles, 2008).
Currently, there is still no single test for MS. Diagnosis is typically made based on medical history, clinical examination, and MRI of the brain and spinal cord, and sometimes includes spinal fluid analysis. There is a wide range of neurological symptoms that can vary greatly among individuals. Based on the patterns of progression, MS is classified into (i) relapsing-remitting MS (RRMS), characterized by clear relapses (worsening of symptoms) followed by periods of recovery; (ii) primary progressive MS (PPMS), characterized by a steady worsening of symptoms from the onset without relapses and remissions; (iii) secondary progressive MS (SPMS), which initially starts as RRMS and then transitions into a progressive form; and (iv) a clinically isolated syndrome (CIS), characterized by a single demyelination lesion found on the CNS using MRI. Patients who suffer from CIS may or may not develop MS, but from the study (Miller et al., 2005), 30–70% of patients with CIS eventually do.
Myelin/WM abnormalities in MS
As the leading demyelinating disease, myelin/WM abnormalities have been widely explored in MS. Traditionally, focal demyelination is considered a typical pathology for MS. These lesions could occur in WM regions of the optic nerve, brain stem, basal ganglia, and spinal cord, or WM tracts close to the lateral ventricles (Compston and Coles, 2008). Clinical diagnosis of MS is made based on lesion distribution, morphology, evolution, and signal abnormalities using MRI scans (Filippi et al., 2016).
As myelin-producing cells in the CNS, oligodendrocytes are lost in MS, which then leads to the thinning or complete loss of myelin (Compston and Coles, 2008). As myelin sheath provides essential isolation, protection, and support to axons, loss of myelin implicates axon breakdown as disease advances (Compston and Coles, 2002). At the early stage of MS, remyelination often occurs as a repair mechanism toward this pathological demyelination, during which oligodendrocytes try to rebuild myelin sheath (Chari, 2007). With disease relapsing, the efficacy of remyelination reduces. With repeated attacks, scar-like focal plaques occur around the damaged axons, especially in WM regions, resulting in sclerosis (Chari, 2007). The exact reason for the attack is unknown, but likely involves infiltration of peripheral T cells into the CNS which causes autoreactive immune responses (Compston and Coles, 2008; Compston and Coles, 2002). With deepened understanding of the disease, it is realized that microglia (Minagar et al., 2002; Ward and Goldman, 2022) and astrocytes (Minagar et al., 2002) are also likely involved in the immune attack and scar formation in MS. Oxidative stress is also believed to play roles in mediating inflammatory responses in MS (Ohl et al., 2016). In alignment with this, plasmalogens were found lost in serum of MS cases (Ferreira et al., 2021). Nowadays, more and more research has revealed that the changes in diffusely abnormal (Cairns et al., 2022) or even normal appearing (Abel et al., 2020) WM contribute significantly toward disease progression, highlighting the important role of myelin integrity/health itself in mediating MS, rather than a secondary consequences.
Myelin lipid disorder in MS
Since myelin is the most lipid-condensed material in the CNS, naturally, lots of attention has been paid to the abnormal lipid metabolism in MS. In the CNS, there were significant decreases in sphingolipids (Hayes and Ntambi, 2020), ceramides, and cholesterol in both gray matter and WM of brains of patients with MS, while an increase in phosphatidylethanolamine species was observed (Wheeler et al., 2008). The accumulation of C16:0- and C18:0-ceramides in MS WM was associated with oligodendrocyte apoptosis (Qin et al., 2010). Increased 4-HNE was only observed in WM (Wheeler et al., 2008), indicating lipid peroxidation primarily increases in WM of MS.
Myelin lipids, sulfatide (Marbois et al., 2000), cerebroside (Podbielska et al., 2011), and ganglioside (Bansal et al., 1994) were reduced in MS brains. In the normal-appearing WM cases, myelin lipids (e.g., sulfatide C20:0) were lost in both PPMS and SPMS cases (Pousinis et al., 2020). In addition, nervonic acid (C24:1), a major building block for myelin-enriched sulfatide and cerebroside, was sharply decreased in MS, while other saturated fatty acyl chain–containing species were increased (Hayes and Ntambi, 2020). In addition, a study has shown that GM1 improved oligodendrocyte-specific neurofascin155 (NF155) association with lipid rafts and consequently prevented myelin and axonal damage by stabilizing the paranodal junction (Zhang et al., 2011). In MS, along with GM1 reduction (Bansal et al., 1994), NF155 was reduced, which correlates with the latter occurring destruction of paranodal structure (Howell et al., 2006). Moreover, GM1 has also been shown to promote glycolysis in astrocytes (Finsterwald et al., 2021). Whether this is linked with astrogliosis observed in MS needs further study.
Compared to the limited studies on lipid changes in the CNS, lots of efforts were given to identify potential biomarkers for MS. From a lipid perspective, myelin lipids, sulfatide, cerebroside, and certain ganglioside are detectable and elevated in the circulating system, including serum (Acarin et al., 1996; Menge et al., 2005; Zaprianova et al., 2004), plasma (Sadatipour et al., 1998; Valentino et al., 2009), and CSF (Ilyas et al., 2003; Ryberg, 1978). Depending on the study performed, these alterations also showed species-dependent differences. For example, although total sulfatide was accumulated, sulfatide (C20:0) was lost in the CSF of patients with RRMS (Nogueras et al., 2019). Several cerebroside species (GlcCer C16:0 [Kurz et al., 2018], GlcCer C24:1 [Kurz et al., 2018], HexCer C16:0 [Checa et al., 2015; Vidaurre et al., 2014], and HexCer C20:0 [Amatruda et al., 2020]) were accumulated in the CSF and plasma of patients with MS, while others (LacCer C16:0 [Kurz et al., 2018]) were reduced. Detailed myelin lipid alterations in MS are summarized in Table 5.
Summary of Myelin Lipid Alterations in MS
\: Nonspecified or unavailable.
ELISA, enzyme-linked immunosorbent assay; NAWM, normal-appearing white matter; PPMS, primary progressive MS; RP-UPLC-TOF MSE, reversed-phase ultra-performance liquid chromatography time-of-flight mass spectrometry; SPMS, secondary progressive MS.
Risk factors and therapeutics
Although the cause of MS is unclear, the underlying mechanism is likely linked with immune system dysregulation and/or oligodendrocyte dysfunction (Nakahara et al., 2012), which also involves genetic and environmental factors (Aloisi and Cross, 2022; Ascherio and Munger, 2007; Compston and Coles, 2002; Ward and Goldman, 2022). There is still no cure for MS. Current treatments are mainly disease-modifying medications that either mitigate ongoing symptoms or prevent future attacks (Compston and Coles, 2002; McGinley et al., 2021). Some lipid-targeting approaches showed promising disease-modifying efficacy. Sulfatide administration was shown to ameliorate experimental autoimmune encephalomyelitis (Halder et al., 2007), likely through suppressing immune responses (Hamatani and Kondo, 2023; Hamatani et al., 2022).
Sulfatide and AD
Introduction
As one of the most prevalent and debilitating neurodegenerative diseases, great efforts have been made to investigate AD etiology, most of which were focused on traditional pathological markers (Aβ and Tau), yet limited progress has been made on AD prevention and therapy to date. Meanwhile, the current understanding of the involvement of myelin impairment in AD development opens a new era for tackling disease progression (Qiu et al., 2023b). Among several myelin lipids that are associated with AD, the involvement of sulfatide has been demonstrated to be the most promising.
Sulfatide, also known as 3-O-sulfogalactosylceramide, sulfated galactocerebroside, or SM4, is a class of sphingolipids that were originally isolated from human brain (Eckhardt, 2008). Structurally, sulfatide consists of a sphingosine backbone, a fatty acid chain with or without a hydroxy group in the α-position, and a polar head group with a sulfate (-SO4) moiety (Kyogashima et al., 2006; Stoffyn, 1966). The synthesis of sulfatide is carried out by two steps from ceramide counterparts in Golgi apparatus: (i) galactosyltransferase (CGT) mediates the transfer of galactose onto ceramide, forming GalCer intermediate molecule, and (ii) cerebroside sulfotransferase (CST, encoded by Gal3st1 gene) catalyzes the interaction of GalCer with phosphoadenosine-5′-phosphosulfate to produce sulfatide. Once synthesized, sulfatide can also be converted to GalCer by ARSA and its cofactor saposin B in lysosome (Takahashi and Suzuki, 2012). A brief myelin structure and a sulfatide metabolism pathways are illustrated in Figure 3.
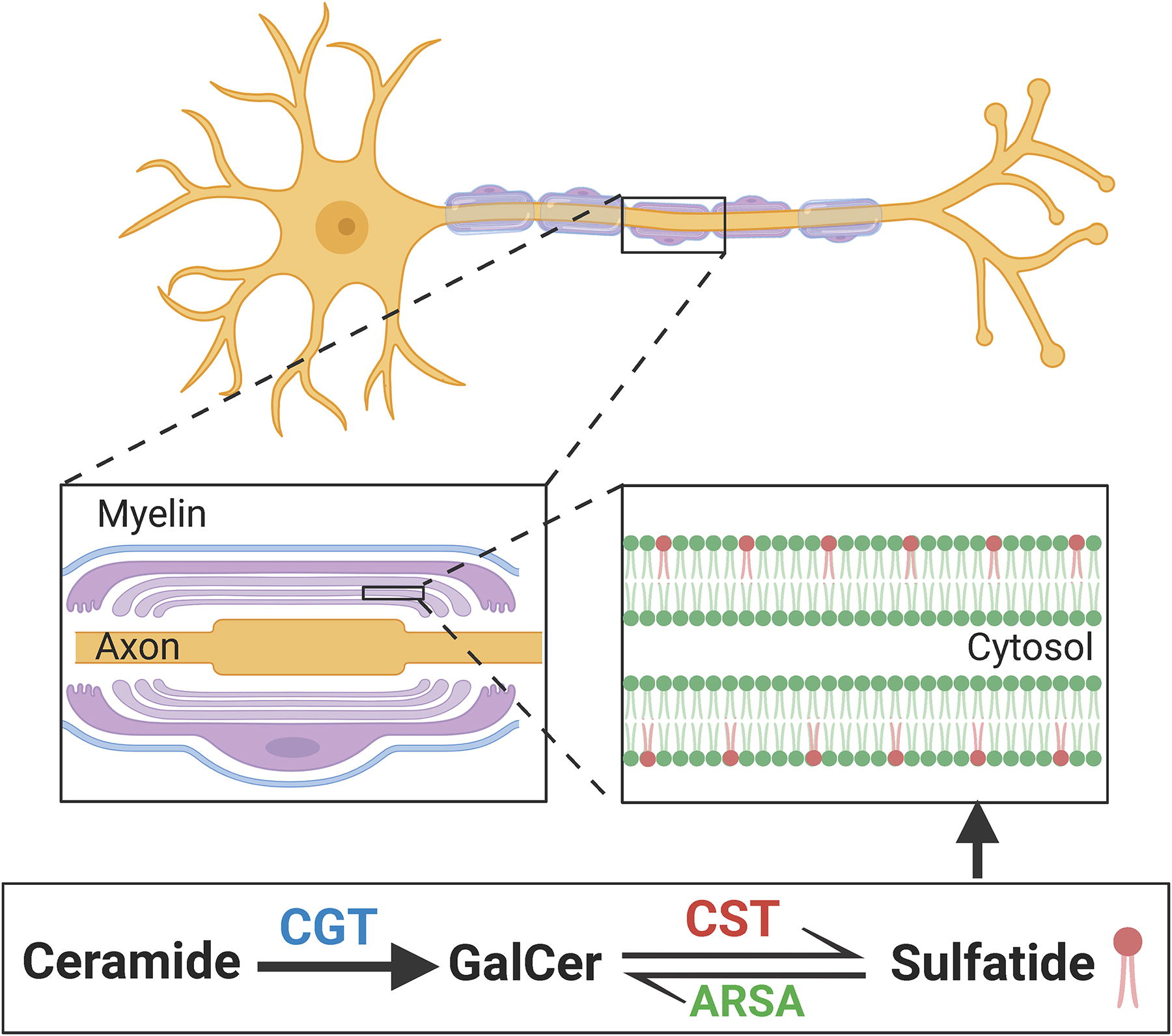
The myelin localization and metabolism of sulfatide.
Sulfatide has been detected in various tissues such as pancrease (Fredman et al., 2000), kidney (Marsching et al., 2014), and the circulation system (Marsching et al., 2014). Particularly, sulfatide is highly enriched in myelin sheath that surrounds axons in the nervous system. With their unique sulfate structure, sulfatide is involved in diverse functions in various biological processes. They act as cell signaling and adhesion factors that contribute to the stability of myelin membranes and are crucial for the proper functioning of neurons (Baba and Ishibashi, 2019). They are also involved in immune response (Su et al., 2021), insulin secretion (Holm et al., 2018), and blood clotting processes (Nakayama et al., 2020).
Sulfatide and AD risk factors
Given the drastic change of sulfatide in AD, many studies have investigated the relationship between sulfatide and several well-established AD risk factors, which helps to pinpoint the potential roles of sulfatide loss in AD.
As a lipid transport protein, the ε4 allele of ApoE is a major genetic risk factor for late-onset AD, accounting for over 95% of total cases (Aslam et al., 2023; Bertram and Tanzi, 2009; Cedazo-Minguez and Cowburn, 2001). Interestingly, our studies showed that sulfatide content was greatly and specifically elevated in the brain tissues of ApoE knockout mice, while human ApoE transgene led to decreased brain sulfatide content with ApoE4 causing more decrease than ApoE3 isoform (Han et al., 2003). Meanwhile, the decline of sulfatide was abolished in AD mouse models with ApoE deletion (Cheng et al., 2010). Further investigation using human CSF showed that sulfatide molecules were specifically associated with ApoE-containing HDL-like lipoproteins (Han et al., 2003). These findings strongly suggested that ApoE functions as a sulfatide transporter in the nervous systems. Under disease conditions, abnormal ApoE-mediated sulfatide trafficking and enhanced associated lipoprotein metabolic pathways contribute to sulfatide depletion (Han, 2007). Moreover, the observation that brain sulfatide content is ApoE isoform-dependent (in the rank order of ApoE4 < E3 < E2) provides a potential mechanistic link for a better understanding of the genetic association of ApoE with AD (Han, 2010).
Aβ accumulation and fibril formation are key pathological characteristics of AD. Abnormal Aβ peptide metabolism and accumulation of self-aggregating Aβ have also been proposed to be the cause of AD (Selkoe, 2006). Intriguingly, previous studies demonstrated the essential role of sulfatide in ApoE-facilitated clearance of extracellular Aβ peptides (Zeng and Han, 2008). Supplementation of sulfatide to the H4-APPwt cell culture media selectively reduced the concentration of Aβ42 in the media and enhanced Aβ peptide content in both lysosome- and endosome-enriched fractions. By employing a chemically well-defined vesicular model system, it was further demonstrated that sulfatide dramatically enhances the binding of Aβ peptides to ApoE-associated vesicles (Zeng and Han, 2008). In addition to regulating Aβ degradation, a recent study also suggested sulfatide participates in Aβ production processes with the observation that sulfatide incubation downregulated β-secretase and γ-secretase activity in a cell culture model (Zimmer et al., 2024). Conversely, studies suggest that the byproduct of Aβ production, APP intracellular domain, can decrease the sulfatide synthesis enzyme CST expression and down-regulate sulfatide production (Van Dyck et al., 2023). Collectively, these studies provide a strong link between ApoE, Aβ, and sulfatide, which established a foundation for elucidating AD mechanisms and developing potential therapeutic interventions for AD.
Accumulating evidence posits obesity as a risk factor for dementia (Flores-Cordero et al., 2022). Robust correlations have been established between obesity and AD features, including cognitive deficits, impaired long-term potentiation, synaptic plasticity, and decreased brain volume (Tsai et al., 2019). Additionally, the causal relationship between obesity and memory impairment has been substantiated by multiple studies. Notably, diet-induced obesity induces memory decline in 3xTg AD mouse model (Knight et al., 2014). Early-life high-fat diet treatment increases neuroinflammation, elevates Aβ deposition, and promotes cognitive decline in APP/PS1 AD mouse model (So et al., 2023). Nonetheless, the precise mechanisms governing obesity-related AD remain elusive. Intriguingly, a recent study investigating the lipidomic profile of central and peripheral nervous systems in diabetic mice models revealed significant alterations in myelin signature (Palavicini et al., 2020). Specifically, myelin-enriched lipid species such as sulfatide, GalCer, and plasmalogen phosphatidylethanolamine exhibited a significant loss in the obese cohort. Further temporal analysis suggested that the change of lipids preceded mitochondrial, myelin, and axonal structural/functional defects and were associated with obesity and hyperlipidemia rather than hyperglycemia (Palavicini et al., 2020). These findings strongly indicate that sulfatide loss may serve as an early indicator of obesity-related myelin impairment. Given the established causal connections between obesity, sulfatide loss, and dementia, it is highly plausible that myelin sulfatide decline acts as a mechanistic link between obesity and associated cognitive impairment. Subsequent studies are imperative to elucidate the potential contribution of sulfatide loss to obesity-related AD progression.
Impact of sulfatide deficiency on CNS function
Based on multiple lines of evidence that pointed to the potential roles of sulfatide in AD pathogeneses, a substantial number of studies have investigated the outcome of modulating sulfatide levels in vivo. Early studies using CGT-deficient mice that lack both GalCer and sulfatide documented progressive hindlimb paralysis and extensive vacuolation of the spinal cord ventral region (Coetzee et al., 1996). Later on, a CST(-/-) mouse model was established using gene targeting by Honke et al. (2002). The CST(-/-) mice began to display hindlimb weakness by 6 weeks of age and showed pronounced tremor and progressive ataxia subsequently. These phenotypes suggest that sulfatide plays a critical role in myelin maintenance/function. Further investigation in CST(-/-) mice showed an age-dependent increase of myelin abnormality (uncompacted, and degenerating myelin sheaths) as well as deteriorating nodal/paranodal structures (Marcus et al., 2006). Consistently, a global reduction of myelin lipids and a decrease of major myelin proteins including myelin basic protein (MBP) and proteolipid protein (PLP) were detected in CST-null mice (Palavicini et al., 2016). These findings indicated that both lipid and protein profile alterations may contribute to the phenotype of sulfatide-deleted mice.
Substantial reduction of sulfatide was found in the brain of patients with AD (Han et al., 2002) and animal models with AD (Cheng et al., 2010), raising the question of whether this reduction could affect disease progression. To answer this question, a CNS sulfatide-deficient adult-onset mouse model was established by specific deletion of the Gal3st1 gene in oligodendrocytes (Dustin et al., 2023; Qiu et al., 2021). Induction of Gal3st1 ablation in the adult stage achieved ∼50% loss of sulfatide content in cerebrum tissues without affecting oligodendrocyte/myelin homeostasis, recapitulating the pattern of sulfatide loss in the human AD brain. Interestingly, a deficiency of sulfatide led to cognitive impairment and an AD-like immune/inflammatory response characterized by the progressive activation of microglia and astrocytes (Qiu et al., 2021). A further investigation into the intricate relationship among ApoE and different glia cell types revealed that sulfatide loss-induced neuroinflammation was independent of ApoE, and ablation of microglia does not abolish sulfatide loss-induced astrocyte activation (Qiu et al., 2021). This suggests that independent mechanisms are responsible for sulfatide loss-related glial cell activation. To this end, Stat3 and PU.1/SPI1 have been proposed to be the drivers of astrocyte and microglia activation, respectively (Qiu et al., 2021).
In addition to neurobiological alterations, structural changes including cortical atrophy and ventricular enlargement were often found to be accelerated among patients with AD compared with that of MCI or normal individuals (Carmichael et al., 2007; Nestor et al., 2008) and are positively correlated with deterioration in cognitive performance. Using histological and MRI approaches, a comparison of ventricular volumes among human ApoE isoform knock-in mouse models was performed, which showed that mice with ApoE4 have larger ventricles compared to mice with ApoE2 isoform (Palavicini et al., 2022). Furthermore, sulfatide losses led to significant ventricular enlargement without impacting hippocampal or whole brain volumes (Palavicini et al., 2022). Given that no profound cell death was detected in sulfatide-deficient mice brain tissues, it was speculated that ventricle alterations in sulfatide-depleted condition were due to the production, exchange, or absorption of CSF, this is supported by the observation of elevated membrane channel protein Aquaporin4 expression in the circumventricular region of sulfatide-deficient mice brain (Palavicini et al., 2022). As an increase in ventricular size (Barron et al., 1976) and a decline in sulfatide (Couttas et al., 2018; Svennerholm et al., 1997; Svennerholm et al., 1994) were also observed in human brains with normal aging, it is likely that sulfatide deficiency may drive the ventricular enlargement in both AD and normal aging conditions.
Collectively, these findings have established the essential function of sulfatide in maintaining myelin homeostasis in the CNS and demonstrated that the loss of CNS sulfatide may act as a critical driver for developing AD such as behavioral, neurological, and cerebrum structural phenotypes.
Peripheral effects of sulfatide deficiency
Although classically viewed as a brain disorder, AD is well known to be associated with several physical and systemic manifestations beyond the CNS (Morris et al., 2014). For example, loss of bladder and bowel control often develops during the mid-late stage of AD, incidence of infection rises as the disease progresses, all of which play a major role in the mortality of patients with AD (Brunnstrom and Englund, 2009). However, the mechanisms of peripheral organ dysfunction in AD were not fully understood. With the knowledge that sulfatide loss led to a series of CNS phenotypes closely resembling AD, He et al. reported a striking phenotype of the enlarged bladder in aged sulfatide-deficient mice (He et al., 2023). Further investigation into the cause of such phenotype has excluded potential contribution by renal failure, bladder organ muscle dysfunction, and peripheral nerve damage. With lipidomics and transcriptomic evaluation, spinal cord dysfunction was identified as the major contributor to the neurogenic bladder phenotype. Importantly, sulfatide levels were found to be greatly decreased in AD spinal cord tissues compared to controls, moreover, downregulating CNS sulfatide in mice led to lipidome and gene expression profiles highly similar to that of human AD spinal cord. This suggests that sulfatide deficiency may function as a causal factor for spinal cord dysfunction and bladder dysregulation among patients with late AD.
AD is a multifactorial disease that affects both the CNS and periphery, this is also reflected by the rise of metabolic disturbance among patients with AD. Meanwhile, multiple studies have led to the hypothesis that peripheral processes such as obesity can function as a contributing factor to AD. Interestingly, Qiu et al. recently reported that adult-onset CNS sulfatide deficiency causes sex-dependent metabolic disruption in mice (Qiu et al., 2023a). Specifically, the investigators discovered an enhanced bodyweight gain and impaired glucose metabolism in aged sulfatide knockout mice, these phenotypes were found to be associated with elevated food intake and hypothalamic inflammation (Qiu et al., 2023a), suggesting CNS sulfatide loss may also promote the development of peripheral metabolic disorders through regulating the hypothalamic-food intake axis. Although additional studies are required to elucidate the exact events in the central–peripheral communication disruption in AD, these observations strongly indicate an essential role of sulfatide in between.
In addition to its highly enriched distribution in the CNS, sulfatide can also be detected in other peripheral organs. In the pancreas, the C16:0 sulfatide isoform exhibits chaperone activity to facilitate insulin production, while the C24:0 sulfatide isoform may help to prevent the development of type 1 diabetes by blocking autoimmune attacks (Buschard and Antvorskov, 2022). In the kidney, sulfatide is considered important for the osmotic stability of renal cells (Marsching et al., 2011), which is required for renal adaptation to chronic metabolic acidosis (Stettner et al., 2013). It is also reported that sulfatide can act as a lipid antigen, to be recognized by natural killer T cells, which then modulate dendritic cells to protect against autoimmune disease (Halder et al., 2007). Additionally, sulfatide was found to be an endogenous ligand for the TLR4-MD-2 complex, with the ability to induce or block inflammation depending on the receptor status (Su et al., 2021). Multiple lines of evidence have demonstrated the involvement of sulfatide in regulating both CNS and peripheral organ functions, which strongly supports the notion that sulfatide is a critical player in health and disease, targeting to maintain sulfatide levels may serve as a promising strategy for the disease therapy and prevention.
Therapeutic implications
The systemic investigation on sulfatide suggested potential benefits in maintaining sulfatide levels for preventing AD and other peripheral disorders. Yet, no specific effective method has been established for the upregulation of sulfatide in vivo to date. A few studies have suggested conditions/treatments that stimulate sulfatide levels. Despite no known mechanism, it was reported that vitamin K can increase the synthesis of brain sulfatide in mice (Popescu et al., 2018; Sundaram et al., 1996), however, as vitamin K plays numerous physiological roles in a variety of organs (including facilitating blood clots), it would be challenging to be used as a sulfatide enhancer. Nakajima et al. reported that PPARα fenofibrate stimulates Gal3st1 gene expression, but the effect was only detected in peripheral organs (kidney, heart, liver, and small intestine) with no impact in the brain (Nakajima et al., 2013).
Several enzyme-targeting strategies have been proven to be effective in downregulating sulfatide level during the past few decades. For example, studies have focused on stimulating ARSA, the enzyme that mediates sulfatide degradation for the treatment of metachromatic leukodystrophy (a disease with brain WM destruction caused by excessive myelin sulfatide accumulation). This can be achieved via (i) transplantation of gene-corrected hematopoietic stem progenitor cells (Biffi et al., 2006) or oligodendrocyte progenitors (Givogri et al., 2006), (ii) enzyme replacement therapy based on intravenous injections of the recombinant functional enzyme (coated with biodegradable nanoparticles) (Schuster et al., 2017), and (iii) AAV1-mediated gene therapy (Kurai et al., 2007) or intrathecal administration of an AAV9 vector encoding the enzyme (Miyake et al., 2021).
Using both whole-body Gal3st1 depletion and oligodendrocyte-specific Gal3st1 knockout mouse models, we and others demonstrated that targeting sulfatide synthesis enzymes can effectively modulate brain sulfatide levels. This suggests that it is possible to develop enzyme‐targeting strategies to manipulate sulfatide levels for disease treatment purposes. With current available knowledge, we expect future efforts on maintaining sulfatide levels to prevent AD may be focused on enhancing CST levels or activity in the brain, which could be achieved via cell/enzyme replacement therapy as well as AAV-mediated gene therapy.
Overall Summary and Limitations
There is a huge umbrella of neurodegenerative diseases, given the space limitation, here we only reviewed five major representative ones. Others such as Batten disease (Johnson et al., 2019), Creutzfeldt–Jakob disease (Liberski et al., 1989), and Frontotemporal dementia (Marian et al., 2023) also harbor myelin abnormalities, yet, whether there are associated lipid alterations needs further review and characterization. Given the scope of this review in the mechanistic involvements of myelin lipids in neurodegenerations in the CNS, the circulating lipidome (whole blood, plasma, and CSF) is not extensively reviewed either, although there are numerous studies focused on identifying the lipid-based biomarkers for neurodegenerative diseases using a lipidomics approach. Additionally, as mentioned before, the myelin lipids referred to in this review are restricted to cerebroside, sulfatide, and ganglioside GM1, given their well-established myelin specificities. While there are more myelin-related lipids (cholesterol, free fatty acids, glycerophospholipids, plasmalogens, etc.) altered under disease conditions, whether they can reflect the myelin changes needs careful interpretation. Thus, these lipids are not extensively discussed in this review.
Aging, the biggest risk factor for all sorts of neurodegenerative diseases, also leads to abnormal myelin lipid levels in the CNS. The three major myelin lipids we discussed in this review are sulfatide (Couttas et al., 2018; Svennerholm et al., 1997; Svennerholm et al., 1994), cerebroside (Svennerholm et al., 1997), and ganglioside GM1 (Guo, 2023; Svennerholm et al., 1997), all show an aging-dependent decline in human postmortem brains. These shared myelin lipid alterations occur during normal aging and in neurodegenerative diseases, especially AD, might raise the evidence on how aging contributes to not only AD but also other neurodegenerative diseases from a lipid perspective. Yet, these declines were not recapitulated in mouse aging, since one of the myelin lipids, cerebroside, showed accumulation (Hallett et al., 2018) during normal aging in mouse brain. While mice show limited cognitive decline with aging, the myelin lipid alteration differences should be appreciated as they might indicate some intrinsic biological difference in terms of aging in primates and rodents.
In summary, through highlighting the myelin lipid abnormalities in neurodegenerative diseases, this review hopes to serve as a resource to summarize the current understanding of myelin/WM defects in neurodegenerations through the scope of lipidomics and aims to stimulate further lipid-oriented investigation and drug discovery in the field.
Footnotes
Acknowledgments
The authors thank the support from all members of Xianlin Han laboratory. They thank the support provided by the Department of Medicine and Sam and Ann Barshop Institute for Longevity and Aging Studies in University of Texas Health Science Left San Antonio.
Authors’ Contributions
Z.X., S.H., and X.H. were involved in the conceptualization and content design of the article. Z.X., S.H., and M.M.B. wrote the first draft of the article. Z.X., S.H., and X.H. contributed to editing of the text. X.H. directed and provided resources for the work.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This study was partially supported by National Institute on Aging grants R01 AG085545 (X.H.), R01 AG061872 (X.H.), RF1 AG061729 (X.H.), P30 AG066546, P30 AG013319, and P30 AG044271, and UT Health SA intramural institutional research funds (X.H.), Methodist Hospital Foundation (X.H.), Cure Alzheimer’s Fund (X.H.), and William and Ella Owens Medical Research Foundation (X.H.).