
Abstract
Significance:
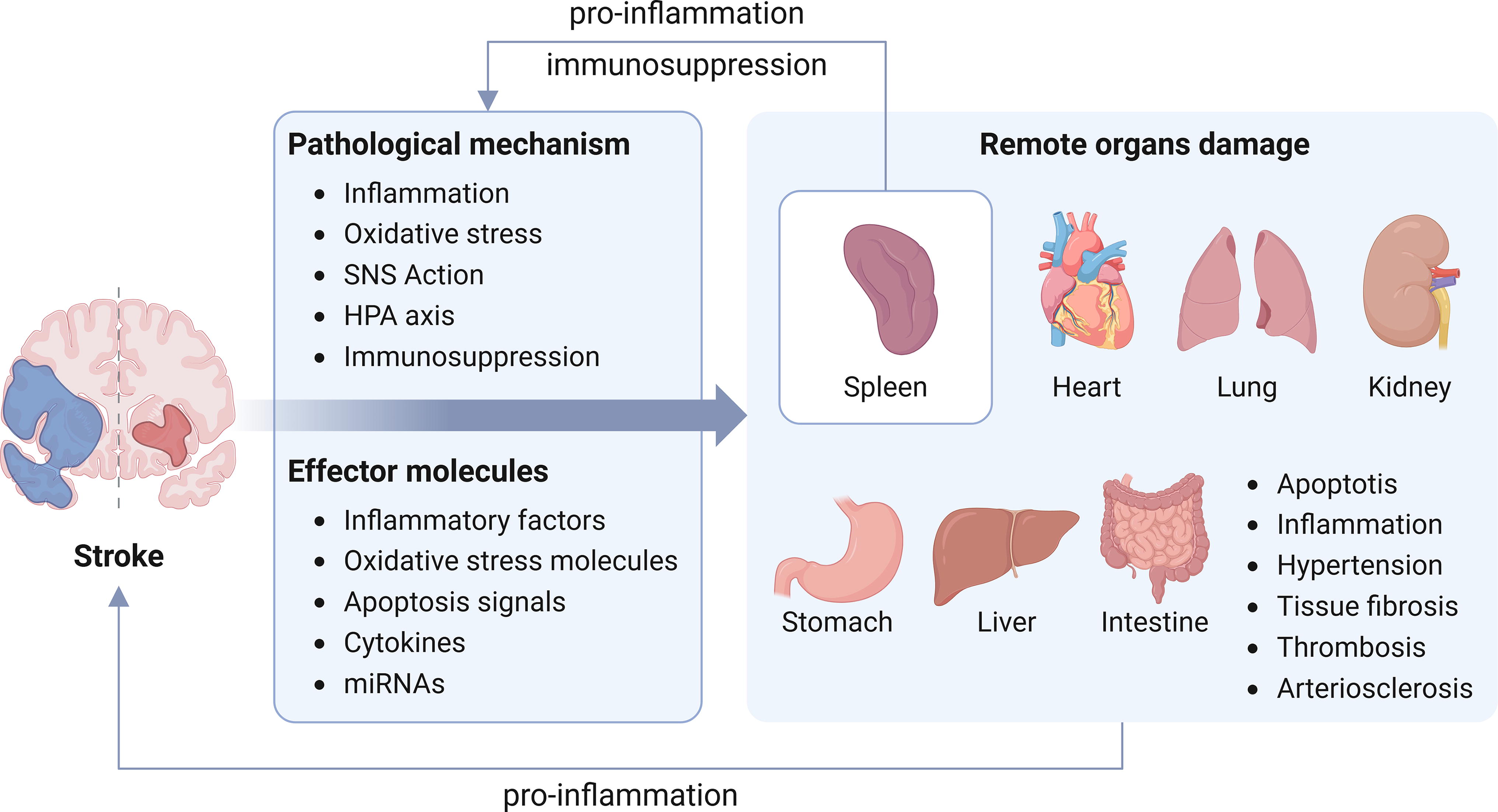
Damage after stroke is not only limited to the brain but also often occurs in remote organs, including the heart, lung, liver, kidney, digestive tract, and spleen, which are frequently affected by complex pathophysiological changes. The organs in the human body are closely connected, and signals transmitted through various molecular substances could regulate the pathophysiological changes of remote organs.
Recent Advances:
The latest studies have shown that inflammatory response plays an important role in remote organ damage after stroke, and can aggravate remote organ damage by activating oxidative stress, sympathetic axis, and hypothalamic axis, and disturbing immunological homeostasis. Remote organ damage can also cause damage to the brain, aggravating inflammatory response and oxidative damage.
Critical Issues:
Therefore, an in-depth exploration of inflammatory and oxidative mechanisms and adopting corresponding comprehensive intervention strategies have become necessary to reduce damage to remote organs and promote brain protection.
Future Directions:
The comprehensive intervention strategy involves multifaceted treatment methods such as inflammation regulation, antioxidants, and neural stem cell differentiation. It provides a promising treatment alternative for the comprehensive recovery of stroke patients and an inspiration for future research and treatment. The various organs of the human body are interconnected at the molecular level. Only through comprehensive intervention at the molecular and organ levels can we save remote organ damage and protect the brain after stroke to the greatest extent. Antioxid. Redox Signal. 42, 885–904.
Introduction
Stroke is a serious cerebrovascular disease and the second leading cause of death worldwide, causing great harm to the society (Hilkens et al., 2024; Lou et al., 2020). According to the form of occurrence, as shown in Figure 1, stroke could be divided into cerebral ischemic stroke (IS), intracranial hemorrhage (ICH), and subarachnoid hemorrhage (SAH) (Scott et al., 2022; Tu et al., 2023). The mechanism of brain damage caused by stroke is very complex. On the one hand, the occurrence of stroke promotes the release of damaging substances and the activation of inflammatory factors, which lead to oxidative stress, cell apoptosis, and homeostasis imbalance (Jelinek et al., 2021; Monsour et al., 2023). On the other hand, the interruption of oxygen and glucose supply to the brain leads to the death of neuronal cells. However, this damage is not only limited to the brain, but through a series of mechanisms, it may also cause certain damage to remote organs (Balch et al., 2020; Wang et al., 2022a; Wu et al., 2022). Related studies have shown that inflammatory mechanisms often affect this type of remote damage (Sun et al., 2021). Normally, there is a balance inside and outside the cell, and stroke leads to an imbalance of inflammatory mediators (Gelderblom et al., 2015). These inflammatory mediators can lead to blood–brain barrier (BBB) disruption, neuronal damage, and brain edema, promote neuronal apoptosis, and impair neural plasticity, ultimately aggravating neurological deficits (Alsbrook et al., 2023). In addition to direct damage to the brain, it could also be transmitted through the blood, disrupting the homeostasis of remote organs and affecting homeostatic cellular functions such as cell membranes, proteins, and nucleic acids (Li and Chen, 2023).

A thorough study of the inflammatory response in remote organ following stroke could enable us to better understand the impact of stroke upon peripheral organs. Further analysis of the association between inflammation and remote organ damage after stroke and strengthening the connection between various mechanisms can help us identify the essential target organ. In addition, it is particularly important to develop a comprehensive intervention strategy targeting the inflammatory response mechanism, including the regulation of inflammatory mediators, antioxidant treatment, and nutrition and nursing measures to reduce brain damage and remote organ damage after stroke.
This review aims to explore the relationship between stroke-induced remote organ damage and inflammation and discuss comprehensive intervention treatment measures by linking multiple mechanisms. By exploring this field, we hope to provide more effective treatment for stroke and its induced remote organ damage.
Classification and pathophysiology of stroke
Ischemic stroke
IS refers to the interruption of cerebral arterial blood flow, local brain tissue hypoxia, and ischemic necrosis and the corresponding neurological deficits, which usually account for the vast majority of strokes. According to the Trial of ORG 10172 in Acute Stroke Treatment’s classification, it can usually be divided into five subtypes as follows: LAAS (large-artery atherosclerosis), cardioembolism, small vessel disease, other determined etiology, and undetermined etiology (Medlin et al., 2015; Mendelson and Prabhakaran, 2021; Miceli et al., 2023). The most common type is LAAS, which is usually related to atherosclerosis, that is, plaque formation in the arteries causes the arteries to narrow or occlude, which, in turn, causes local blood flow to be reduced or completely interrupted (Puig et al., 2023). Common symptoms include sudden numbness or paralysis of the face and limbs, slurred speech, visual impairment, balance disorders, and headaches (Oliveira and Sampaio Rocha-Filho, 2019; Runchey and McGee, 2010). General treatments include thrombolysis, vascular recanalization, drug therapy, and surgery (Huo et al., 2023; Sarraj et al., 2024). The main treatment options include intravenous thrombolysis with alteplase or tenecteplase within 4.5 h and endovascular therapy in the 6-h and 6- to 24-h windows. There is still much controversy regarding mismatched treatment options, including the timing of the use of aspirin and clopidogrel (Lyden and Wold, 2022).
Intracranial hemorrhage
ICH refers to a type of disease in which cerebral blood vessels rupture and blood invades the central nervous system (CNS) (Feigin et al., 2018; Gross et al., 2019). It is usually divided into intracerebral and intraventricular hemorrhages according to anatomical locations (Fox et al., 2022; Sadegh et al., 2023). Common causes of ICH include hypertension, arteriovenous malformations, the use of anticoagulants and antiplatelet drugs, and brain trauma (Acosta et al., 2023; Ali et al., 2024). The most common site of cerebral hemorrhage is basal ganglia, often as a result of lesions of the lenticulostriate arteries after hypertension. Common symptoms may include nausea and vomiting, severe headache, impaired consciousness, convulsions, and neurological dysfunction (Runchey and McGee, 2010). Compared with IS, hemorrhagic diseases are more urgent and usually require emergency treatment (Kim et al., 2022a; Kim et al., 2016). Common treatments include surgery, drug therapy, and conservative treatment (Lin et al., 2021; Magid-Bernstein et al., 2022; Yang et al., 2022). With the change of technology, discussions about small bone window craniotomy and endoscopy have emerged. However, no matter which method is used, the latest surgical plans tend to be intelligent positioning and artificial intelligence assistance (Zhi et al., 2024).
Subarachnoid hemorrhage
SAH is a serious neurological emergency that refers to blood entering the subarachnoid space of the brain (Claassen and Park, 2022). Aneurysm rupture is the most common cause, accounting for about 85% of all cases (Macdonald and Schweizer, 2017). Common symptoms include sudden severe headache, meningeal irritation, nausea and vomiting, epilepsy, and changes in consciousness (Claassen and Park, 2022; Darkwah Oppong et al., 2023; Gaastra et al., 2022; Sonne et al., 2021). Treatments include craniotomy, clipping of the aneurysm, or interventional embolization of the aneurysm, as well as management of complications and supportive care (Buscot et al., 2022; Tawk et al., 2021). There has been considerable controversy regarding the choice of endovascular coiling (EC) and surgical clipping (SC). A study from Japan showed that EC in low-grade aneurysmal SAH is more effective. Treatment results were significantly better than SC (Ishikawa et al., 2023).
Pathophysiology
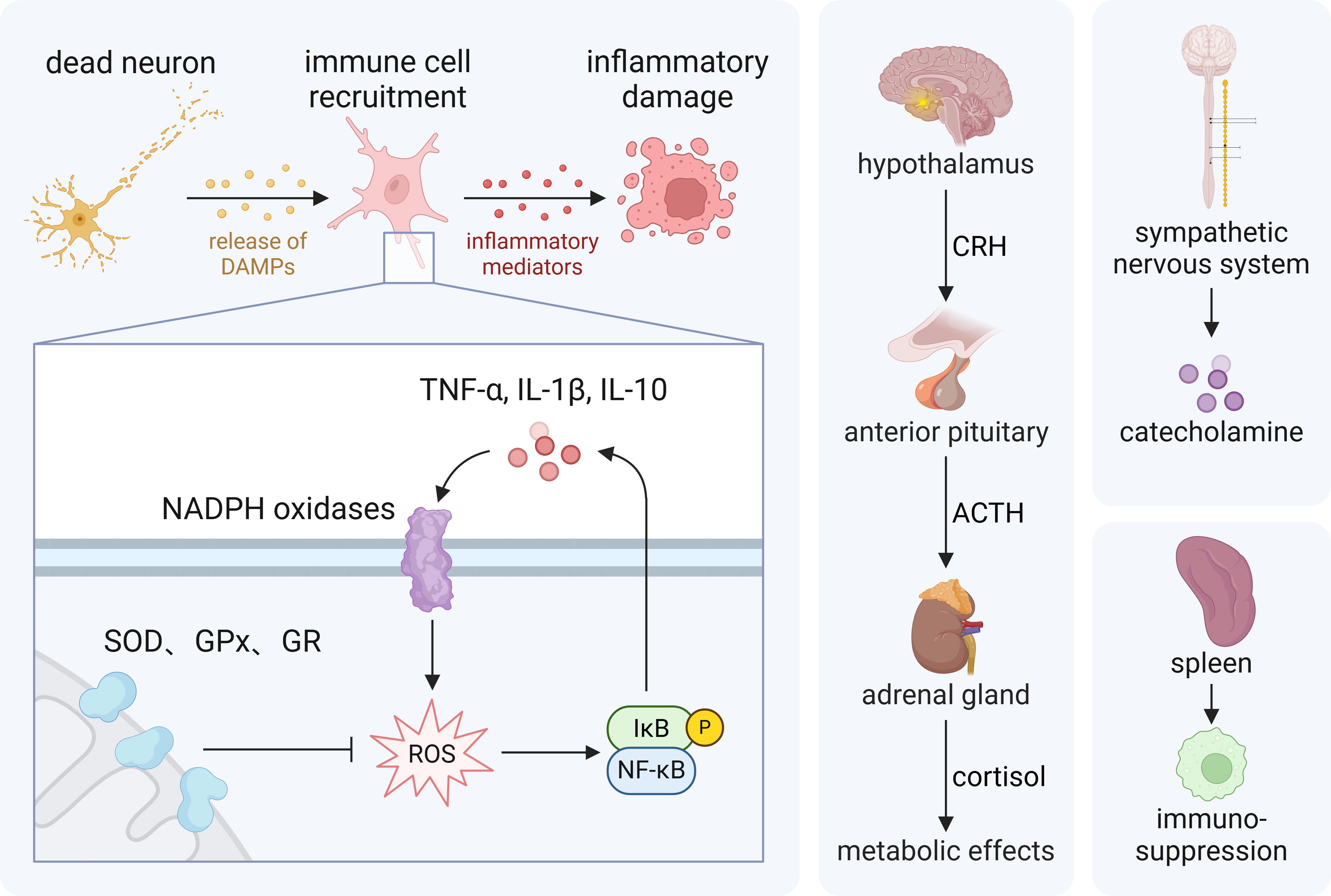
After stroke attack, due to varying degrees of lesion infarction, there is insufficient blood supply to maintain the physiological function of the brain, and the local neuronal cells are deprived of oxygen and glucose and other nutrients, leading to the occurrence of cell necrosis (Mathias et al., 2024; Wegener et al., 2024). In addition, due to vascular damage, blood infiltration, and inflammatory response, diffuse cerebral edema further increases intracranial pressure and aggravates the degree of neurological dysfunction (Cordonnier et al., 2018; Zhu et al., 2024). On the contrary, hematoma and diffuse blood distribution in the brain could activate microglia. Microglial activation inflames the process of inflammation, and the body further releases inflammatory mediators, causing local and systemic inflammatory responses, and then aggravates BBB disruption and CNS damage (Gao et al., 2024; Han et al., 2023; Raffaele et al., 2024). Inflammation mediates the generation of reactive oxygen species (ROS). The generation of ROS is a signature change of stroke. In comparison with other organs, the brain is more susceptible to ROS. After stroke, ROS consume a large amount of adenosine triphosphate (ATP), leading to lipid peroxidation and ferroptosis. At the same time, it also disrupts the electron transport chain (ETC), causing mitochondrial membrane damage, leading to mitochondrial dysfunction, and further initiating apoptosis cascades, aggravating neuronal damage (Bernoud-Hubac et al., 2024). Damaged neurons might also undergo apoptosis and autophagy, which play a vital role in the damage and repair of the nervous system (Beccari et al., 2023). ROS and inflammatory mediators can also lead to the occurrence of immunosuppression, and activation of the sympathetic nervous system (SNS) and the hypothalamus–pituitary–adrenal (HPA) axis, thereby producing a cascade effect and further causing damage to the body. The neural signal network is the most intuitive mode of action after a stroke, including the activation of the sympathetic nerves and the vagus nerve. As a response to stimulation, sympathetic nerve activation has the characteristics of a double-edged sword, and excessive activation will further aggravate nerve damage. The vagus nerve can sense inflammatory signals and then reduce inflammation by secreting inhibitory signals (Duan et al., 2024). After a stroke, the brain undergoes a variety of pathophysiological changes, leading to the occurrence of brain damage and repair processes, as shown in Figure 2.

Remote organ damage after stroke
After stroke attack, common remote organ damage includes the cardiovascular system, digestive system, respiratory system, urinary system, blood system, immune system, and so forth (Fig. 3) (Balch et al., 2020; Han et al., 2021; Wang et al., 2022a).

As stroke occurs, the body may experience complications such as arrhythmia, myocardial infarction (MI), and heart failure (Scheitz et al., 2018). The pathological changes that the heart encounters after a stroke are called stroke-heart syndrome (SHS) (Wang et al., 2024a). QT prolongation may occur in 20%–65% of poststroke patients, ST segment changes may occur in 15%–25% of patients, and inverted “Cerebral” T waves may occur in 2%–18% of patients (Hu et al., 2023). After SAH, more than half the patients develop left ventricular diastolic dysfunction (Park et al., 2016). After IS, one-third of patients may experience a reduced ejection fraction, and a left ventricular ejection fraction ≤40% has become a consensus for cardiac complications after stroke (McNamara et al., 2024). A global database survey showed that 15% of nontraumatic ICH had ≥1 cardiovascular disease diagnosis within 4 weeks, and ICH significantly increased the probability of major adverse cardiovascular events (acute MI, IS, all-cause mortality, and recurrent ICH) within 5 years (Hoad et al., 2024).
When a stroke occurs, digestive system manifestations such as digestive tract dysfunction, dysphagia, gastrointestinal bleeding (GIB), and delayed gastrointestinal emptying may also occur (Camilleri, 2021). Dysphagia is a major complication after an acute stroke that affects most patients and is associated with an increased risk of mortality (Labeit et al., 2023). GIB is another common complication after a stroke (Du et al., 2020). The incidence of GIB after stroke is 1.5%−7.8%, which may be related to the subtype of stroke (O’Donnell et al., 2008). Advanced age, impaired consciousness, severe neurological dysfunction, infection, and posterior circulation infarction are independent risk factors for GIB in patients with acute stroke (Fu, 2019). Another reason of GIB is the use of antiplatelet drugs. The SOCRATES trial evaluated the safety of ticagrelor and aspirin in acute IS or transient ischemic attack, focusing on the possibility of major bleeding (ICH and GIB) (Easton et al., 2017). The results showed that the antiplatelet effect of ticagrelor is similar to the bleeding effect of aspirin (Easton et al., 2017).
Stroke patients are prone to immunosuppression, among which the most common complication is stroke-associated pneumonia (SAP) (Liu et al., 2018). Approximately 1/10 of stroke patients developed pneumonia during the acute phase (Badve et al., 2019). Two-thirds of cases of pneumonia in the first 3 months after stroke occur in the first week, with the highest incidence on the third day (de Jonge et al., 2022). The occurrence of SAP is also associated with dysphagia. In a prospective observational study, researchers found that patients with a lower abundance of Streptococcus were more likely to develop SAP, and an increase in specific taxa in the phylum Actinobacteriota was more likely to lead to adverse outcomes (Ren et al., 2023). In addition, neurogenic pulmonary edema (NPE) is also one of the prevailing pulmonary complications after stroke, which further worsens the prognosis of patients after stroke (de Jonge et al., 2022; Zhao et al., 2020).
Acute kidney injury (AKI) is one of the common complications after stroke and may lead to renal failure (Dulam et al., 2024; Qureshi et al., 2020). AKI after stroke is associated with higher mortality and worse functional outcomes. Hypertension, diabetes, increased plasma osmolality, and use of loop diuretics are risk factors for AKI after stroke (Jiang et al., 2019; Wang et al., 2022a). After a stroke, approximately 35% of patients develop chronic kidney damage (CKD), characterized by elevated levels of protein in the urine (proteinuria) and decreased glomerular filtration rate (Dulam et al., 2024). Related studies have shown that urinary biomarkers of tubular injury are independently associated with the development of AKI and 90-day mortality in patients with acute IS treated in the stroke care unit (Shimoyama et al., 2020).
When a stroke occurs, a severe inflammatory cascade occurs in the brain (Wang et al., 2023b). Due to the action of chemokines and cytokines, the brain recruits many spleen-derived immune cells to the site of brain injury to fight against the inflammatory response (Chen et al., 2018). Some studies have shown that shortly after a stroke, the spleen shrinks dramatically and the number of spleen cells decreases accordingly (Seifert and Offner, 2018). Some studies have also shown that splenocytes migrate specifically to the site of primary brain injury. Spleen size is also inversely correlated with infarct volume, with more severe spleen atrophy associated with larger infarct volume (Han et al., 2021; Yu et al., 2021).
In the blood system, the whole blood viscosity (WBV) of stroke patients increased, which further leads to thrombosis, resulting in microcirculatory congestion and reduced cerebral perfusion (Gyawali et al., 2024). Anemia is also one of the risk factors for IS and an independent prognostic factor affecting poststroke mortality. The anemia group has a significantly higher risk of death than the nonanemia group (Heo et al., 2021). In addition, due to the massive release of inflammatory mediators after stroke, leukocyte recruitment increases, and the blood is rich in immune factors and proinflammatory mediators, which further enhances the systemic inflammatory response and mediates remote organ damage (Kumari et al., 2024).
Mechanisms of remote organ damage after stroke
Autonomic and neuroendocrine dysregulation
After a stroke, brain damage occurs, including the centers that regulate the autonomic nervous system (ANS), such as the hypothalamus and brain stem (Dorrance and Fink, 2015; Mo et al., 2019). This damage can directly stimulate the activity of the SNS, thereby triggering the activation of the sympathetic nerve axis (Kang et al., 2024). In addition, brain edema and increased intracranial pressure caused by stroke also further activate the sympathetic system (Hasegawa et al., 2022). After stroke, vagus nerve stimulation (VNS) reduces neuroinflammation and plays a neuroprotective role. The afferent vagus nerve is a major component of the HPA axis and also serves as a key part of neuroendocrine and immune function (Jelinek et al., 2024).
Stroke mediates the release of many inflammatory mediators that circulate in the blood, which can first activate the SNS (Winklewski et al., 2014). Neutrophil chemotaxis, activation, and phagocytosis are dependent on norepinephrine (NE) (Nicholls et al., 2018). Many activated inflammatory mediators can enter the CNS through the BBB and further activate the SNS (Dorrance and Fink, 2015). Stroke also causes massive release of monocytes and neutrophils from the spleen and other organs (Bao et al., 2010). In addition, the occurrence of stroke will also destroy the balance between the body’s SNS and para-SNS, and the remodeling of damaged neural circuits is also very likely to lead to abnormal excitement of the ANS (Sposato et al., 2020).
As a stress response, the occurrence of stroke activates the HPA axis in a short period of time (Fan et al., 2024). As a regulator of the endocrine system, the HPA axis not only plays a role in adjusting emotions, stress, and metabolism, but is also the target and production organ of the neuroendocrine system, and has an important connection with the activation of the ANS (Zhou et al., 2022). After a stroke occurs, the HPA axis is activated, mediating the release of corticotropin releasing hormone from the paraventricular nucleus of the hypothalamus, thereby promoting the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. ACTH can act on the adrenal glands to cause the release of the steroid hormone cortisol, further adjusting psychological factors such as anxiety, fear, and stress (Ben Assayag et al., 2017; Wang et al., 2022b). Chronic elevated cortisol is potentially neurotoxic and is associated with increased mortality after stroke. Serum cortisol levels are associated with stroke severity and insular damage and are a potential predictor of stroke outcome (Fan et al., 2024). Activation of the hypothalamic axis also results in a significant increase in catecholamines and may serve as a key factor in predicting stroke prognosis (Du et al., 2021; Mracsko et al., 2014). ACTH can promote the synthesis of catecholamines by increasing the expression and activity of tyrosine hydroxylase and then play a relevant role in complex neuroendocrine regulation. In short, by activating the complex ANS and HPA axis, the relevant pathways after stroke are connected to the body, which further has a certain impact on remote organs (Fig. 4) (Kim et al., 2022b; Lichlyter et al., 2023; Mracsko et al., 2014).
Inflammation
After stroke, the BBB is damaged, and inflammatory cells such as neutrophils and macrophages are recruited to the injured area (Bai et al., 2023; Wanrooy et al., 2021). Microglia, an immune cell population within the CNS that phagocytoses cellular debris and regulates immune responses through cytokine signaling, is the first cell population to be activated (Wicks et al., 2022). After a stroke occurs, the internal environment is disturbed, ions are unbalanced, depolarization spreads, and microglial morphology and phenotype change, inducing proinflammatory properties. Macrophages are then recruited and migrate to the necrotic site, surrounded by microglia (Planas, 2024). Jordi showed that ischemia-induced reprogramming of the gene expression profile of CD163+ macrophages has a rapid impact on leukocyte chemotaxis and BBB integrity and promotes neurological damage in the acute phase of stroke (Pedragosa et al., 2018). Damaged cells, through necrosis or apoptosis, release damage-associated molecular patterns (DAMPs), such as high mobility group box-1 protein (HMGB1), heat shock proteins, and ATP, which are recognized by surrounding immune cells and initiate an inflammatory response (Maehara et al., 2021; Singh et al., 2016b). Inflammation promotes metabolic shifts that favor glycolysis, pentose phosphate shunting, and lipid synthesis, a process that favors microglial proliferation and repair of damage. However, some microglial lipid accumulation can aggravate damage (Planas, 2024).
During activation, inflammatory cells produce many ROS, such as superoxide anions (O2 −), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH), leading to increased oxidative stress (Forrester et al., 2018). Inflammatory mediators such as tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), and IL-6 can activate a variety of oxidases, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), which also produce ROS and aggravate oxidative stress (Coveney et al., 2022; Fiadeiro et al., 2024; Hernandes et al., 2022). Oxidative stress activates multiple inflammation-related signaling pathways, such as the nuclear factor κB (NF-κB) pathway (Xu et al., 2024). Activation of NF-κB induces the expression and release of multiple inflammatory mediators, thereby enhancing and maintaining the inflammatory response (Lu et al., 2024). After stroke, NOD-like receptor protein 3 (NLRP3) inflammasome activation leads to caspase-1-dependent maturation and release of IL-1β, IL-18, and gasdermin D, ultimately leading to cell apoptosis. On the contrary, the activation and phosphorylation levels of janus kinase 2/signal transducer and activator of transcription 3 increase, thereby promoting the release of proinflammatory factors and the expression of NLRP3 (Koyama and Shichita, 2023).
There is a positive feedback relationship between inflammation and oxidative stress, as shown in Figure 4 (Cao et al., 2023). ROS produced by inflammation lead to oxidative stress, which in turn further stimulates inflammation through various mechanisms. This positive feedback mechanism forms a vicious cycle, leading to persistent inflammation and oxidative damage, and delaying tissue repair and functional recovery.
Oxidative stress
After stroke occurs, the blood supply is suddenly interrupted, resulting in brain tissue hypoxia and energy metabolism disorder (Dunn and Isaacs, 2021). When reperfusion occurs, the generation and release of neuronal oxidative metabolites will generate a large number of oxygen free radicals and ROS (Orellana-Urzua et al., 2020). In addition to increased ROS production, increased NO production mediated by nitric oxide synthase (NOS) in neurons and endothelial cells (ECs) can lead to increased reactive nitrogen species (RNS). These ROS and RNS can damage brain cell membranes, proteins, and nucleic acids, leading to cell death, BBB disruption, and neurological dysfunction (Li et al., 2005). ROS are a characteristic product of stroke, representing the destruction of the antioxidant system. The brain is also very sensitive to ROS. Excessive ROS consume a large amount of ATP and promote lipid peroxidation and ferroptosis (Bernoud-Hubac et al., 2024).
Lipid peroxidation refers to the process by which lipids are oxidized to ROS, especially hydroxyl radicals, which can target and attack polyunsaturated fatty acids (PUFAs) in lipid membranes. Fe2+ increases hydroxyl radicals by triggering the Fenton and Haber–Weiss reactions. PUFA radicals react with molecular oxygen to form lipid peroxyl radicals. This peroxyl radical can in turn react with another PUFA, resulting in chain lipid peroxidation (Liu et al., 2024).
ROS generated during oxidative stress, such as O2 −, •OH, and H2O2, may generate highly harmful hydroxyl anions via the Fenton reaction or superoxide radicals, and can directly or indirectly cause mitochondrial damage (Duan et al., 2022; Mattson et al., 2008). These ROS can enter the mitochondria through different pathways and react with lipids, proteins, and nucleic acids in the mitochondria to cause ETC disruption, protein oxidation, and DNA damage in the mitochondrial membrane. After reperfusion, the input of oxygen exacerbates ROS generation, and electrons leak prematurely and interact with molecular oxygen. This leakage mainly occurs in complexes I and III of the respiratory chain, leading to excessive production of superoxide anions (Chavda and Lu, 2023). The antioxidant defense system in the mitochondria includes superoxide dismutase (SOD), glutathione peroxidase (GPX), glutathione reductase (GR), and the peroxiredoxin and thioredoxin system, which can remove ROS to a certain extent (Fan et al., 2023; Li et al., 2024c). However, when oxidative stress increases significantly, it may not be able to completely offset the toxic effects of ROS.
NOX and xanthine oxidase (XO) can be activated after stroke and produce superoxide. NOX, as a complex oligomeric enzyme, undergoes conformational changes after stroke, transferring electrons from cytosolic NADPH to molecular O2 on the outside of the cell membrane, ultimately leading to the formation of superoxide (Briones-Valdivieso et al., 2024). XO is a purine metabolic enzyme that, after a stroke, can catalyze the conversion of hypoxanthine and xanthine into uric acid and produce superoxide free radicals and hydrogen peroxide (Briones-Valdivieso et al., 2024; Yu et al., 2023).
When oxidative stress occurs, microglia can be activated and release proinflammatory factors, such as TNF-α and IL-1β, to trigger an inflammatory response (Cai et al., 2019; Coveney et al., 2022). This stimulation triggers the classical NF-κB pathway multimer, which then promotes the release of NF-κB into the nucleus and initiates gene transcription. The nonclassical pathway involves another NF-κB family member, P100, which specifically activates the signaling pathway by producing the P52 subunit. Excessive NF-κB activation can lead to apoptosis, further causing nerve damage (Xu et al., 2024). The cross talk between inflammation and oxidative stress aggravates the neurological damage of stroke and causes remote organ damage through signal transduction and molecular pathways.
Immunosuppression
In the early stages of stroke, ruptured cells release DAMPs, which activate the innate immune system and trigger local and systemic inflammatory responses, resulting in worsening outcomes of primary injury (Maehara et al., 2021). DAMPs are released from dead cells to living cells, activating pattern recognition receptors in immune cells. The activated immune cells produce various inflammatory cytokines and chemokines, triggering sterile brain inflammation in the ischemic brain, and leading to neuronal death (Sakai and Shichita, 2023; Santos Samary et al., 2016). Although inflammatory responses occur at the earliest stage, immunosuppression follows (Wang et al., 2024b). The main process leading to immunosuppression is a shift from the lymphocyte phenotype T helper (Th)1 to the Th2 phenotype, with a decrease in lymphocyte counts and natural killer (NK) cells in the blood and spleen, and impaired defense mechanisms of neutrophils and monocytes (Faura et al., 2021). Manifestations of immunosuppression include lymphopenia, and the resulting impaired antimicrobial defense renders stroke patients susceptible to infection (Westendorp et al., 2022).
Following stroke, oxidative stress and inflammatory responses can activate the HPA axis, releasing steroids (such as cortisol) (Fan et al., 2024). Corticosteroids have a wide range of immunosuppressive effects, including inhibition of T cell proliferation and function, reduction of cytokine production, and suppression of antigen-presenting cell activity (Lu et al., 2022). However, some previous studies have shown that hydrocortisone can restore hemodynamic stability and modulate the immune response to stress in a different way through anti-inflammatory rather than immunosuppressive means (Keh et al., 2003). Likewise, sympathetic activation will suppress the peripheral immune system, reducing the activity of immune cells such as T cells and NK cells (Deng et al., 2016; Sribnick et al., 2022).
Oxidative stress can induce immunosuppression through multiple pathways (Bai et al., 2022). ROS can directly damage immune cells and inhibit their function (Kennel and Greten, 2021). Oxidative stress can also interfere with intracellular signal transduction pathways, such as mitogen-activated protein kinase, NF-κB, and other immune-related signaling pathways (Kennel and Greten, 2021; Yao et al., 2022).
In the immunosuppressive state, the body’s resistance to infection is reduced, and infection may further aggravate the level of oxidative stress in the whole body. The inflammatory response caused by infection will produce a large amount of ROS, exacerbating oxidative stress (Wen et al., 2024). At the same time, the related injuries further activate the SNS, which interacts with inflammation and oxidative stress to form a cross-connected grid-like injury pathway (Faura et al., 2021; Santos Samary et al., 2016; Weber et al., 2022).
These processes not only cause damage at the site of brain injury, but also transmit related damaging factors through the blood circulation, further causing damage to remote organs (Balch et al., 2020; Wang et al., 2022a).
Molecular regulatory networks of remote organ damage
The molecular regulatory network of remote organ damage after stroke involves multiple signaling pathways and molecular factors, as shown in Figure 5, including inflammatory factors, oxidative stress products, apoptosis signals, cytokines, and miRNAs.
Due to the destruction of the BBB, the products of CNS damage, such as inflammatory factors, ROS, and DAMPs, will directly affect the normal physiological functions of remote organs, causing damage to remote organs (Chang et al., 2017; Hernandes et al., 2022; Lochhead et al., 2024). Newly generated damaging substances will also mediate brain damage, thereby increasing the burden on the body. After a stroke occurs, inflammatory mediators such as TNF-α, IL-1β, and IL-6 may affect the degree of damage to remote organs by promoting inflammatory responses and increasing vascular permeability. In addition, inflammation may lead to changes in the metabolism of cells in distant organs, dysregulation of tissue-resident leukocyte populations, and further regulate immune cells, promoting the continuous infiltration of spleen-mediated macrophages (Scheitz et al., 2022). Oxidative stress is considered to be an important mechanism leading to damage to remote organs. Key oxidative stress pathways include the nuclear factor erythroid 2-related factor 2/antioxidant response element pathway, ROS production pathway, antioxidant enzyme system, and more, which regulate the intracellular redox balance and affect the cell’s ability to cope with oxidative stress (Deng et al., 2023; Farina et al., 2021). The degree of cell apoptosis in remote organs may increase. Inflammatory factors and ROS activate ECs to destroy the BBB and promote apoptosis of cells in remote organs (Wang et al., 2022a). Apoptotic signaling pathways include mitochondrial pathways, death receptor pathways, endoplasmic reticulum stress (ERS), and more, which regulate the balance between cell survival and death and affect the tissue structure and function of remote organs (Viereck et al., 2014). Abnormal expression of cytokines and growth factors may affect remote organs. These molecules, such as angiogenin, fibroblast growth factor, and growth factors such as insulin-like growth factor (IGF), affect the repair and regeneration of remote organs by regulating processes such as cell proliferation, differentiation, and migration (Dordoe et al., 2023; Yang et al., 2024). After a stroke, miRNA may also be involved in regulating the damage and repair process of remote organs. miRNA regulates processes such as cell proliferation, apoptosis, and inflammatory response by inhibiting the expression of target genes, thereby affecting the function and structure of remote organs (Viereck et al., 2014; Wang et al., 2018).
Heart
Stroke causes disorders of the SNS and endocrine system, generally acting through the HPA axis, which is the main pathway for the human body to regulate physiological and emotional hormones. Through changes in neurotransmitters, the ANS is further activated, and the heart is directly or indirectly affected, leading to abnormal myocardial function, which, in turn, induces the occurrence of SHS (Wang et al., 2024a). Studies have shown that SHS may originate from structural or functional changes in the central autonomic network after stroke, which is specifically manifested in the activation of sympathetic nerves and inhibition of parasympathetic nerves, thereby affecting the rhythm of the myocardium. At the molecular level, the massive release of cortisol and increased levels of catecholamines are believed to be related to heart damage (Barugh et al., 2014). Cortisol and catecholamines have been shown to promote the progression of inflammation in related studies, as shown above.
Recent experimental studies support the important role of inflammatory response in the occurrence of SHS (Vornholz et al., 2021). In some previous animal models, stroke-induced cardiac dysfunction was accompanied by systemic inflammatory response, upregulation of proinflammatory factors in the myocardium, and infiltration of macrophages in the heart. Sympathetic nerve activation promotes inflammatory recruitment of the bone marrow and spleen (Vasamsetti et al., 2018). However, after splenectomy, stroke proinflammatory factors decreased and macrophage recruitment was reduced, indicating that spleen-mediated inflammatory pathways may be the key to myocardial injury (Singh et al., 2016a).
In the early stage of brain injury, localized inflammation occurs due to the activation of microglia. With the recruitment and infiltration of macrophages, ROS accumulate due to the stimulation of injury and inflammation, which promotes the occurrence of oxidative stress and further promotes the destruction of vascular ECs. The brain releases DAMPs, which stimulate pathogen recognition receptors and toll-like receptors (TLRs), such as TLR-4, and increase the production of proinflammatory mediators IL-6, IL-1β, TNF-α, chemokines, and their receptors. In this process, injury, inflammation, and oxidation signals cross the BBB and act on the heart (Santos Samary et al., 2016; Wang et al., 2022a). A previous study demonstrated that NOX-2 expression increases in cardiac tissue after stroke and, as a source of ROS, can regulate myocardial function and immune response by inducing neutrophil extracellular traps (NETs) (Li et al., 2020).
Systemic inflammation and sympathetic overactivity can both lead to coagulation activation, platelet hyperactivity, and endothelial dysfunction (Gupta et al., 2020). Many previous studies have shown that inflammatory factors are related to cardiovascular disease. It may be that the inflammatory factors are significantly activated and accumulated in the lower layer of ECs, inducing the rupture of atherosclerotic plaques and promoting the occurrence of acute coronary disease. The inflammatory response can also further aggravate cardiac oxidative stress by inducing cardiomyocyte apoptosis. This process mainly involves the shedding of ECs and astrocytes produced by inflammation into extracellular vesicles (EVs), which quickly cross the BBB and enter the blood, thereby damaging myocardial cells (Wang and Peng, 2022). Due to the damage to the antioxidant system, the reduction of antioxidant enzymes and nonenzyme antioxidant substances makes the heart less resistant to oxidative stress and more susceptible to oxidative stress damage.
As a small regulatory RNA, miRNA is also closely related to the occurrence of SHS. miRNA can regulate cardiac structural remodeling, inflammation, and fibrosis (Park and Kho, 2021). For example, miRNA-21 regulates the expression of apoptotic and inflammatory genes such as B cell lymphoma-2 (Bcl-2)-associated X protein and TNFα, miRNA-146a is upregulated after ischemic injury and can regulate the expression of inflammatory and antiapoptotic genes, such as NF-κB and Bcl-2, and some miRNAs, such as miRNA-210 and miRNA-155, are involved in regulating angiogenesis-related signaling pathways and genes, such as vascular endothelial growth factor and NOS (Boxhammer et al., 2024).
Lung
The occurrence of SAP is usually affected by multiple factors, including imbalance of the neuroendocrine system, suppression of immune function, weakened swallowing and cough reflexes, and postural retention of pulmonary secretions. In stroke mice, gut barrier dysfunction and immune disturbances after ICH promote intestinal bacterial migration and increase the risk of poststroke pneumonia. Sympathetic nerve activation may lead to suppression of immune function, including affecting the function of macrophages and other immune cells (Zhang et al., 2021). A study showed that tissue-resident macrophages play a key role in lung injury after trauma or stroke through inflammatory response, which is manifested by the release of circulating alarmins, which can bind to the receptor for advanced glycation end-products on the membrane and further activate the epidermal growth factor receptor (EGFR), leading to Rab5-mediated RAGE internalization and EGFR phosphorylation, followed by the recruitment and activation of P38, thereby exacerbating inflammation (Zhong et al., 2024). The possible mechanism of poststroke pneumonia is the transformation of lymphocytes from Th1 phenotype to Th2 phenotype, the decrease of lymphocytes and NK cells in the blood and spleen, and the impairment of monocyte and neutrophil defense mechanisms (Faura et al., 2021). One relative study showed that adoptive transfer of interferon-γ (IFN-γ) produced by T lymphocytes or early treatment with recombinant IFN-γ can prevent the occurrence of SAP (Wang et al., 2023b).
Stroke itself can cause immune system disorders, and sympathetic nerve activation may further aggravate this disorder, leading to immune deficiency and making patients more susceptible to infection, including lung infection. On the contrary, sympathetic nerve activation also leads to increased release of inflammatory mediators. Related studies have shown that as a systemic inflammatory factor, HMGB1 is associated with an increased risk of poststroke depression and stroke-related pneumonia (Li et al., 2024a). In addition, the specific enhancement of alveolar angiotensin-converting enzyme 2 (ACE-2) levels and inflammation in the lungs of mice after experimental stroke also confirms the reason for susceptibility to coronavirus disease 2019 after stroke (Singh et al., 2021).
NPE after acute stroke is a type of acute respiratory distress syndrome in which the pathological process focuses on sympathetic nerve stimulation and explosive release of catecholamines (Zhao et al., 2020). Previous studies have shown the connection between SAH and NPE, and melatonin can downregulate the expression/activation levels of mature IL-1β, myeloperoxidase, and matrix metallopeptidase-9 (MMP-9), thereby alleviating NPE caused by inflammatory damage (Chen et al., 2015; Zeng et al., 2022). In addition, the knockout of S100 calcium-binding protein A9 also demonstrated the anti-inflammatory effect on the occurrence of NPE (Wang et al., 2023a).
Liver
The liver is an important organ for metabolism and detoxification, but its rich blood supply and high metabolic activity make it extremely vulnerable to damage under conditions of systemic inflammation and oxidative stress. The liver contributes not only to poststroke immunosuppression but also to stress-induced hyperglycemia (Inderhees and Schwaninger, 2024). Sympathetic overactivation and hepatic inflammation have been reported to induce hepatic insulin resistance, glycogenolysis, and gluconeogenesis in experimental stroke (Inderhees and Schwaninger, 2024). Catecholamines may cause hepatic ERS and insulin resistance and contribute to poststroke hyperglycemia, possibly through higher levels of circulating catecholamines at hepatic adrenergic receptors (ARs), activating NF-κB and stimulating the expression of TNF-α and other proinflammatory cytokines.
The systemic inflammatory response induced by stroke can spread to the liver through the bloodstream, activate Kupffer cells (hepatic macrophages) in the liver, trigger local inflammation and oxidative stress responses, and lead to liver cell damage. As an antioxidant substance, glutathione (GSH) is produced in the liver and transported into the blood. Intracellular GSH acts mainly through GPX. Under severe oxidative stress, peripheral GSH can enhance antioxidant activity in the ischemic brain (Chen et al., 2020). Studies have shown that lipid metabolism levels can regulate glucose and GSH homeostasis, and at the same time, increased hepatic very low-density lipoprotein secretion after stroke may increase the utilization of phospholipids, PUFAs, and GSH (Inderhees and Schwaninger, 2024).
Immune response also plays an important role in liver damage after stroke. Related studies have shown that the number of ionized calcium binding adaptor molecule 1 (Iba1)+ and CD68+ macrophages in the spleen, thymus, and mesenteric lymph nodes decreased, while the number of Iba1+ and CD68+ macrophages in the brain and liver increased, indicating that immune cells were recruited into hepatocytes after stroke, accompanied by inflammation and apoptosis (Brea, 2023; Tan et al., 2021). Some studies have shown that cerebral IS can aggravate the liver damage caused by Hodgkin lymphoma. This effect may be due to the enhanced induction of cytochrome P450 2E1 (CYP2E1), which further promotes oxidative damage, inflammation, and hepatocyte apoptosis(Gong et al., 2012). Another study showed that the liver after stroke also showed histological changes indicative of liver damage, and a decrease in alanine transaminase was observed (Haley et al., 2020).
Digestive tract
GIB after acute ischemia is a relatively uncommon stroke complication but is associated with increased odds of death and severe dependency (O’Donnell et al., 2008). Stress ulcers caused by acute brain injury, increased gastric acid secretion, or mucosal ischemia caused by vagus nerve overactivity may be one of the causes of GIB (Wang et al., 2022a). In addition to stress ulcers and antiplatelet effects, systemic inflammation and oxidative stress are more likely pathophysiological causes of gastrointestinal mucosal damage after stroke (Camara-Lemarroy et al., 2014).
The intestinal microbiota plays an important role in the immune system. Intestinal dysbiosis can affect the intestinal lymphocyte population, which, due to its role in immune response and systemic inflammation, promotes the occurrence of immune dysbiosis after stroke (Balch et al., 2020). In a recent study, gram-negative bacteria release endotoxins, which trigger inflammatory responses by mediating the TLR4/myeloid differentiation primary response protein 88 signaling pathway (Zhang et al., 2024). The cytotoxic effects of inflammatory substances damage intestinal microvilli, affect the expression level of intestinal tight junction proteins, and aggravate the damage to intestinal barrier function. Endotoxemia and bacterial translocation can aggravate gastrointestinal complications such as intestinal bleeding, dysmotility, and intestinal paralysis. Microbes release short-chain fatty acids (SCFAs) that modulate G protein-coupled receptors to mediate hormone release, neurotransmitter release (i.e., serotonin, dopamine, NE, gamma-aminobutyric acid, acetylcholine, and histamine) and regulate inflammation and mood (Panther et al., 2022). One study showed that SCFAs play a protective role by inhibiting the NLRP3 inflammasome and promoting the development of the intestinal barrier. Some studies have shown that stroke can lead to dysbiosis of intestinal flora and epithelial barrier integrity, which is considered a factor in systemic infection (Peh et al., 2022). However, other studies have shown that stroke patients are at increased risk of depression due to an imbalance in their gut microbiota (GM) (Jiang et al., 2023). Some studies have shown that dysbiosis directly triggers inflammatory γδ T cells in the gut, which then traffic to the brain and exacerbate stroke development (Benakis et al., 2016). Following a shift in immune suppression after stroke, mesenteric lymph node dendritic cells promote the migration of protective regulatory T cells (Tregs) to the gut, thereby reducing the movement of γδ T cells to the brain (Arya and Hu, 2018; Pluta et al., 2021).
A Mendelian randomization study investigating the causal relationship between GM, stroke, and potential metabolic mediators showed that 10 GM taxa were causally associated with stroke; stroke affected the relative abundance of 27 taxa. The results confirmed that different GM have potential metabolite changes after stroke and may affect immune responses through metabolic functions (Wang et al., 2023c).
Spleen
The spleen is an important immune organ that filters blood and initiates immune responses (Han et al., 2021; Yu et al., 2021). Activation of the ANS, release of CNS antigens, and interactions between chemokines/chemokine receptors have been shown to be critical for effective brain–spleen cross talk after stroke (Wang et al., 2019). In mice, stroke modulated the macrophage subtypes of the spleen, and genes associated with the ability of macrophages to communicate with other immune cells, such as the production of costimulatory molecules and inflammatory cytokines, were also downregulated in the spleen after stroke (McCulloch et al., 2018).
During acute stroke, the spleen shrinks, and the number of spleen-derived immune cells increases first in the blood and then in the brain, suggesting that peripheral immune cells may migrate in the CNS (Seifert et al., 2012). First, activated splenocytes produce significantly higher levels of proinflammatory cytokines, including TNF-α, IFN-γ, IL-2, IL-6, and C-C motif chemokine ligand 2 (CCL2) (Offner et al., 2006a). Activated immune cells are recruited by chemokines CC and CXC after stroke and enter the CNS from the spleen mainly through the CCL2-C-C motif chemokine receptor 2 signaling axis (Xie et al., 2024). As time progresses, proinflammation weakens, immunosuppression strengthens, the number of cells in the spleen and thymus decreases dramatically, the response of spleen-derived T lymphocytes to mitogens weakens. Splenocytes express more anti-inflammatory cytokine IL-10 and less TNF-α, IFN-γ, and IL-6 (Offner et al., 2006a; Offner et al., 2006b).
In related studies, splenectomy can significantly reduce neurodegeneration after ischemic injury and also lead to a decrease in the number of activated microglia, macrophages, and neutrophils in brain tissue (Ajmo et al., 2008). Subsequently, in another animal experiment, it was shown that splenectomy did not alter the immune response to brain antigens or improve outcomes after stroke (Zierath et al., 2017). However, a recent meta-analysis analyzing the effects of splenectomy on stroke in mice indicated that the spleen and its functional cell populations are promising targets for therapeutic modulation of inflammation after stroke (Sternak et al., 2022).
In addition, the activation of the sympathetic nerves is accompanied by a large release of catecholamines. Since catecholamines bind to α and β ARs in the spleen, the activation of α1-ARs on the splenic smooth muscle capsule leads to contraction of the splenic capsule, thus causing a decrease in spleen volume after a stroke (Aboud et al., 1993). Another study also showed that circulating catecholamines can regulate the spleen’s response to stroke by activating α- and β- ARs (Ajmo et al., 2009).
Kidney
There is a strong bidirectional relationship between stroke and kidney disease, which may be related to their similarities in anatomy, hemodynamics, and vascular regulation (Shah et al., 2021). On the one hand, activation of the HPA axis promotes the release of glucocorticoids, which in turn directly or indirectly affects the integrity of the glomeruli and tubules (Lau et al., 2017). On the other hand, through the continuous excitement of the SNS, catecholamines and angiotensin II bind to renal artery receptors, causing renal artery constriction and renal ischemia, thereby causing the occurrence of AKI (Fujii et al., 2003). The renin–angiotensin–aldosterone system plays an important role in the occurrence of AKI after stroke. It damages the renal blood vessels through increased blood pressure and can also directly damage the kidneys through inflammation (Mogi et al., 2014). Angiotensin II can stimulate macrophages to aggregate in glomerular and tubular cells, increase the production of cytokines such as IL-1, TNF-α, and monocyte chemoattractant protein-1, and also increase the expression of glomerular cytokines and inflammatory and fibrotic factors, leading to renal damage and fibrosis (Imig and Ryan, 2013).
After stroke, macrophage-derived cytokines and inflammatory factors such as C-reactive protein, IL-6, IL-1β, TNF-α, and MMP-9 are associated with kidney injury (Clausen et al., 2005). In addition, a significant increase in the presence of various peripheral immune cells (including lymphocytes, monocytes, and neutrophils) was seen in the circulation, emphasizing the systemic nature of the immune activation, which also led to increased renal inflammation and oxidative stress damage (Chen et al., 2024). In the kidney, ROS mainly degrade the glomerular basement membrane and change the function of glomerular and tubular cells (Manning et al., 2005).
In the kidney, EVs can originate from blood cells, ECs, podocytes, or tubular epithelial cells and are associated with inflammation, thrombosis, and immunosuppression (Karpman et al., 2017). After stroke, miRNA expression levels change. While the specific mechanism is still unclear, some studies suggest that it may be associated with protection against renal fibrosis, while others focus on the role of miRNAs in target organ damage during arterial hypertension, by highlighting the involvement of miRNAs in pathological end-organ remodeling processes, and the potential of using circulating miRNAs as noninvasive biomarkers for early detection of hypertension-related stroke (Heggermont and Heymans, 2012; Li et al., 2014; Wang et al., 2018).
Comprehensive interventions
Inflammation plays an important role in the damage to remote organs mediated by stroke, and the most important therapeutic intervention strategy is to block or reduce the occurrence of inflammation. In addition, antioxidant therapy can also play an important role, as inflammation and oxidative stress are closely linked. Patients may also benefit from treatments targeting immunosuppression and sympathetic activation. Individualized treatment of the corresponding organs is also extremely important (Fig. 6). The treatment of remote organ damage requires comprehensive evaluation by professionals in consideration of the patient’s condition.

Treatment of molecular mechanisms
Controlling inflammation is considered an effective treatment option for stroke, and some studies have focused on the anti-inflammatory properties of microglia and macrophages (Wang et al., 2020). The latest research shows the relationship between the polarization dynamics of three reactive cell populations (microglia, astrocytes, and neutrophils) and inflammatory response in the context of stroke (Li et al., 2025). Granulocyte colony-stimulating factor promotes axonal growth and polarization of microglia toward an M2 phenotype and induces stem cell migration to the injury site (Duan et al., 2024). Fingolimod and minocycline can adjust the infiltration and number of immune cells in the CNS, thereby reducing inflammation and edema. Rosiglitazone, as an agonist of peroxisome proliferator-activated receptor-γ, can upregulate the expression of CD36, which has been shown to regulate the phagocytic activity of microglia and phagocytes in ICH, indicating that rosiglitazone can promote the clearance of hematomas after ICH (Liu and Wang, 2023). Rapamycin inhibits the action of mammalian target of rapamycin and increases the number of Treg cells in brain tissue and increases IL-10 expression (Duan et al., 2024; Liu and Wang, 2023). Other clinical drug trials are also evaluating and summarizing inflammatory biomarkers, neuroinflammation, and neuro-immunotherapy regimens to provide a theoretical basis and new prospects for clinical treatment (Cao et al., 2023; Safouris et al., 2021). At the same time, some studies have shown that inducing hibernation can promote neuroprotection and anti-inflammatory therapy, possibly by inhibiting neuroinflammation and NLRP3 inflammasome activation (Guo et al., 2021).
The use of antioxidants can help remove oxygen free radicals and other harmful oxidants in the body. The current mainstream antioxidants include vitamin C, vitamin E, and GSH, which mainly function by binding to free radicals and promoting the antioxidant system (Maggio et al., 2024). In the latest research, some new antioxidants such as α-phenyl-N-tert-butylnitrone and polyphenols were used after stroke (Marco-Contelles, 2024), and the application characteristics and research progress of nanomaterials in antioxidant treatment of stroke were updated (Luo et al., 2024). Since inflammation is closely related to oxidative stress, conventional anti-inflammatory treatments can also help reduce oxidative stress damage to remote organs after stroke. Conventional anti-inflammatory drugs include nonsteroidal anti-inflammatory drugs and glucocorticoids, but some studies have shown that the above drugs have limitations, including bleeding and induction of immunosuppression (Chuang et al., 2015; Courties et al., 2019).
Immunotherapy is another important treatment method. For immunosuppression after systemic immune activation, targeted transport and controlled release of immunopotentiators are particularly crucial. Nanomaterials such as liposomes, micelles, polymer nanoparticles, and dendrimers play a critical role (Wang et al., 2024b). A recent study reported a highly efficient neutrophil hijacking nanoplatform for targeted A151 (telomerase repeat) delivery to microglia without generating NETs (Yin et al., 2024). Another study reported a nanocarrier composed of sulfated chitosan polymers that promoted microglial polarization by releasing encapsulated rapamycin in response to high ROS after stroke (Cao et al., 2024). Stem cell therapy is a new treatment option. The differentiation of neural stem cells (NSCs) is related to the protection against inflammation. Maintaining a low concentration of hypochlorous acid can promote proliferation and differentiation into neurons and astrocytes and play a key role in the recovery of neural function (Huang et al., 2025; Wang et al., 2025). However, autologous NSCs after stroke are often limited in differentiation efficiency and transfer pathways. A recent study reported that induced pluripotent stem cell transplantation has neurorepair, angiogenesis, and anti-inflammatory effects after stroke (Darban et al., 2024). At the same time, how exogenous cell transplants implement immune evasion has become another important issue (Rust et al., 2024). A study reported the application of stem cells in immune evasion after stroke. The immune system recognizes foreign cells through the human leukocyte antigen (HLA) system. HLA genetic engineering in pluripotent stem cells and derived cells reduces allogeneic cell lineages, thereby evading the host immune system (Achón Buil et al., 2024).
Treatment of sympathetic activation is discussed in the treatment of remote organ damage. It is worth mentioning that VNS has been approved by the US Food and Drug Administration as an adjunct to intensive rehabilitation therapy for the treatment of chronic upper limb deficits after IS (Dawson et al., 2024). A recent article analyzed the efficacy of transcutaneous auricular VNS and implantable VNS for stroke, and the results showed no significant difference, which still needs further exploration (Malakouti et al., 2024).
Treatment of remote organ damage
For cardiac protection, beta-blockers can reduce the overactivation of the SNS and protect the heart. At the same time, it also has the effect of preventing cardiac remodeling and alleviating arrhythmias. Propranolol has been shown to exert neuroprotective effects by blocking the upregulation of IL-6, thereby improving outcomes after brain injury. The ACE inhibitor/angiotensin II receptor blocker can lower blood pressure, reduce myocardial burden, and have certain anti-inflammatory and antioxidant effects (Powers et al., 2019). Remote ischemic conditioning has become a safe and promising treatment for cardiovascular and cerebrovascular diseases, which can improve the recurrence of cardiovascular events and long-term neurological function in stroke patients (Li et al., 2024b). In general, the heart and brain are closely connected, and future research will focus on signal transduction and mediators between the heart and brain.
For patients with poststroke pneumonia, oxygen therapy or mechanical ventilation should be selected according to the patient’s blood oxygen concentration and other monitoring indicators. Patients diagnosed with pneumonia or suspected of pneumonia should also choose antibiotics to fight infection in the early stage. The most common bacterial infections in SAP are Pseudomonas aeruginosa and Staphylococcus aureus. Studies have shown that ampicillin + sulbactam is a good choice (Wang et al., 2022a). Regular suction of the patient’s sputum keeps breathing patency, and the use of postural drainage helps clear lung secretions. Expectorant drugs such as ambroxol can be used selectively, and bronchodilator drugs such as salbutamol can be used if bronchospasm occurs (Finlayson et al., 2011; Ge et al., 2020).
For gastrointestinal damage caused by stroke, appropriate nutritional support and gastrointestinal protection measures should be taken, such as the use of proton pump inhibitors (PPIs) and gastric mucosal protectants, to prevent and treat gastric mucosal damage. Study shows that antiplatelet drugs combined with PPIs or misoprostol can prevent GIB (Wang et al., 2022a). For patients with dysphagia after stroke, relevant rehabilitation training should be carried out to improve swallowing and eating functions, which will help prevent the occurrence of gastrointestinal damage (Camara-Lemarroy et al., 2014; Ye et al., 2021).
For patients with kidney injury, kidney support therapy should be performed, including appropriate fluid management and electrolyte balance, and avoidance of nephrotoxic drugs. For patients with severe AKI, blood purification therapy such as hemodialysis or hemofiltration can be considered (Gallacher et al., 2024). Studies have shown that for stroke patients with kidney damage, thrombolytic therapy and other treatments will increase the glomerular filtration rate and the risk of bleeding. Aspirin treatment for patients with CKD reduces the risk to a greater extent than the risk of bleeding (Chou et al., 2023). Some anticoagulants and thrombolytic drugs are very likely to aggravate the degree of renal damage. Further exploration is needed for the treatment of renal damage after stroke.
Some previous studies have confirmed the protective effect of splenectomy on stroke. However, as the spleen is an immune organ, long-term spleen loss will undoubtedly lead to immunosuppression. At present, some studies mainly support the spleen as a key target of immune regulation and suggest that induced stem cell transplantation replaces exogenous administration (Wang et al., 2022a). A recent study confirmed that multipotential adult progenitor cell treatment can increase the number of Treg cells in the spleen, upregulate serum IL-10 levels, reduce the release of IL-1β and IL-6 by splenocytes, and restore stroke-induced spleen mass reduction (Yang et al., 2017).
For patients with liver dysfunction after stroke, avoiding drugs that may be toxic to the liver, especially when liver function is already impaired should be necessary. Liver protection drugs, such as GSH, can also be used to help protect liver cells (Inderhees and Schwaninger, 2024). Currently, there are few studies on the treatment of liver damage after stroke. In the future, the impact of liver metabolism on stroke should become a major research direction.
Conclusion
Stroke is not only accompanied by damage to the nervous system but also affects remote organs through various pathways. Inflammation, as the initiator and promoter of this pathway, plays a crucial role in the damage of remote organs. Oxidative stress and sympathetic nerve activation could also drive remote organ damage. Immune activation is followed by immunosuppression, which aggravates the damage to the nervous system and remote organs. The treatment of stroke includes not only simple brain damage treatment but also systemic intervention.
Clarifying the damage association and mechanism between the CNS and peripheral remote organs will help us better understand stroke pathophysiology and provide more precise treatment.
Footnotes
Acknowledgment
All figures were created with BioRender.com.
Authors’ Contributions
J.W. wrote the article; S.G. edited the figures; Y.C. reviewed the language; X.-Z.L. and X.-X.C. made suggestions for some of the work; and C.-H.H. and W.L. revised the work. All authors contributed to the article and approved the submitted version.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This study was funded by the National Natural Science Foundation of China (No.82130037 for C.-H.H., No.82171323 and No.82371344 for W.L.) and the Key Project of Medical Science and Technology Development Foundation, Nanjing Department of Health (No.ZKX23025 for W.L.).