
Abstract
Significance:
Glucose-induced lipid metabolism is essential for preserving functional β-cells, and its disruption is linked to type 2 diabetes (T2D) development. Lipids are an integral part of the cells playing an indispensable role as structural components, energy storage molecules, and signals.
Recent Advances:
Glucose presence significantly impacts lipid metabolism in β-cells, where fatty acids are primarily synthesized de novo and/or are transported from the bloodstream. This process is regulated by the glycerolipid/free fatty acid cycle, which includes lipogenic and lipolytic reactions producing metabolic coupling factors crucial for insulin secretion. Disrupted lipid metabolism involving oxidative stress and inflammation is a hallmark of T2D.
Critical Issues:
Lipid metabolism in β-cells is complex involving multiple simultaneous processes. Exact compartmentalization and quantification of lipid metabolism and its intermediates, especially in response to glucose or chronic hyperglycemia, are essential. Current research often uses non-physiological conditions, which may not accurately reflect in vivo situations.
Future Directions:
Identifying and quantifying individual steps and their signaling, including redox, within the complex fatty acid and lipid metabolic pathways as well as the metabolites formed during acute versus chronic glucose stimulation, will uncover the detailed mechanisms of glucose-stimulated insulin secretion. This knowledge is crucial for understanding T2D pathogenesis and identifying pharmacological targets to prevent this disease. Antioxid. Redox Signal. 41, 865–889.
Introduction
The activity of pancreatic β-cells is crucial for maintaining glucose homeostasis in the body. These cells primarily sense glucose, whose metabolism is vital for insulin secretion. However, complex diets and specific metabolism-related chronic conditions leading to pathologies, including type 2 diabetes (T2D), expose β-cells to other nutritional stimuli, such as fatty acids (FAs) in their various quantities and qualities. Prolonged fasting, when glycogen stores are depleted, and chronic overnutrition, leading to ectopic fat deposition, are particularly associated with FA sensing by β-cells. These are rather extreme conditions; however, FAs significantly enhance insulin secretion during the postprandial period under normal physiological conditions. The metabolism of FAs within β-cells is essential for insulin secretion and overall cellular function. β-cells synthesize FAs, a process tightly linked to glucose metabolism or transport them from the bloodstream using various transporters. FAs also play a significant signaling role in β-cells, as indicated by the presence of numerous receptors on their membranes. Depending on the metabolic status of the β-cells and systemic nutritional conditions, β-cells utilize the glycerolipid/free FA cycle (GL/FFA cycle) and/or FA oxidation. FAs can be stored in lipid droplets (LDs), and used for signaling, both under the regulation of glucose or can be partially oxidized to provide cellular energy under specific conditions. Glucose metabolism and FA oxidation in peroxisomes and mitochondria alter cellular redox status further regulating insulin secretion and potentially local inflammation. Moreover, FAs contribute to the formation of complex lipids. These compounds are crucial for pancreatic β-cell physiology, impacting membrane structure, signal transduction, insulin granules trafficking, and insulin secretion. Dysregulation of these lipids, often accompanied by altered redox cellular status can lead to β-cell apoptosis and dysfunction, contributing to the development of non-physiological states. This review explores the detailed roles of FA and lipid signaling in β-cells, focusing on their involvement in glucose-stimulated insulin secretion (GSIS) and β-cell function under physiological and pathological conditions. We examine how imbalances in these processes along with prooxidative cellular status lead to toxicity, altered signaling, and the development of T2D.
Tracing the Source: Fatty Acids in Pancreatic β-Cells
Biogenesis of fatty acids
FAs constitute a major part of the lipid species in pancreatic β-cells. They can either be synthesized de novo from other metabolic precursors or taken up from the blood through various translocators located at the plasma membrane (see below). FAs are synthesized de novo in the cytoplasm from malonyl-CoA with the help of the fatty acid synthase complex (FAS) (Fig. 1). The main product, 16C-palmitate, can be further elongated in the endoplasmic reticulum (ER) by elongases, while double bonds are introduced by desaturases to form unsaturated FAs. There are two pathways of anaplerosis of short-chain fatty acyl-CoAs (SC-FA-CoAs) that precede FA synthesis in β-cells: i) the citrate pathway and ii) the acetoacetate pathway (El Azzouny et al., 2016; MacDonald et al., 2008). The provision of malonyl-CoA for FA synthesis is essential for proper insulin secretion through nascent metabolic coupling factors (MCFs). It has been suggested that both anaplerotic metabolic pathways are simultaneously active at rest, while the acetoacetate pathway predominates upon stimulation by glucose (El Azzouny et al., 2016).

In β-cells, acetyl-CoA carboxylase (ACC) plays a crucial role in malonyl-CoA production, which inhibits the transport of FA-CoAs into mitochondria and their subsequent β-oxidation. Instead, these activated FAs in the form of FA-CoAs serve as MCFs for insulin secretion (Zhang and Kim, 1998). Glucose-derived carbons are rapidly incorporated into lipids in β-cells highlighting the lipogenic nature of β-cells rather than their role in catabolizing lipids as fuel (MacDonald et al., 2008). De novo lipid synthesis correlates well with insulin secretion (MacDonald et al., 2008). However, the extent to which β-cells synthesize FAs de novo versus taking up FAs from the circulation likely varies with the nutritional state and remains to be fully elucidated.
Lipoproteins/FAs translocators and receptors in β-cells
Lipids and FAs are supplied to the β-cells via the bloodstream mainly in the form of lipoprotein particles (based on their size and density they are divided into Very Low-Density Lipoprotein/VLDL, Low-Density Lipoprotein/LDL, High-Density Lipoprotein/HDL) or free FAs associated with serum albumin (Fig. 2). It is assumed that due to the narrow diameter of the vessels within the rich vasculature of the islets, only remnants of chylomicrons which also contain triacylglycerols (TAGs) can reach the β-cells. Chylomicrons are lipoprotein particles with dietary lipids produced in intestinal enterocytes and released through the lymphatic system to the blood. The TAGs in the chylomicrons are hydrolyzed to a high degree by endothelial lipases when in circulation.

Lipoprotein particles carrying cholesterol enter the β-cells via a receptor system that specifically recognizes and binds LDL, i.e., the LDL receptor (LDLR), HDL, i.e., the scavenge B1 receptor (SR-B1), VLDL i.e., the VLDL receptor and oxidized LDL, i.e., CD36 (Fryirs et al., 2009; Grupping et al., 1997) (Fig. 2A). In addition, LDLR requires its co-receptors such as LPR5, whose knockout shows impaired insulin secretion and glucose tolerance (Fujino et al., 2003). The effects of cholesterol uptake from plasma lipoproteins on insulin secretion have been studied mainly on LDLR, as it is highly expressed in pancreatic β-cells. Its expression is regulated by sterol regulatory element binding protein 2 (SREBP2) and ultimately by post-translational modifications in β-cells (Ishikawa et al., 2008). Interestingly, apart from being the main regulator of cholesterol homeostasis, it was recently found that loss of Srebf2 (coding for SREBP2) inhibits the production of pro-inflammatory chemokines but amplifies type I interferon response genes in response to inflammatory stimulus in endothelial cells (Fowler et al., 2023). In addition, recent findings have revealed the presence of the protein PCSK9, which controls the abundance of LDLR in the plasma membrane of β-cells (Da Dalt et al., 2019, Marku et al., 2022). PCSK9 knockout mice, which allow increased expression of VLDL, have lower plasma cholesterol levels but increased cholesterol content in the islets. They show glucose intolerance as a result of impaired insulin release, probably due to redistribution of lipid rafts and decreased expression of SNAP25 and VAMP2 (Marku et al., 2022). However, the embryonic-derived human cell line EndoC-βH1 in which PCSK9 was inhibited, showed no GSIS regression, only upon exposure to LDL (Ramin-Mangata et al., 2021). Interestingly, other endocrine cells in islets, namely the δ-cells, also express PCSK9. Recently, PCSK9 was found to play the role independent of cholesterol levels, in pro-inflammatory cytokine production affecting oxidative modifications within atherosclerotic lesions, demonstrating its pleiotropic effect (more in Punch et al., 2022).
In contrast to LDL, HDL, which contains more than 80 bioactive proteins and even more lipid species, has been shown to have a protective effect against stress conditions induced by cytokines, high glucose, but also oxidized/LDL and VLDL. It protects β-cells from apoptosis and ER stress (Petremand et al., 2012). It slows the oxidation of LDL and removes the products of lipid oxidation (Marin et al., 2015). Protective HDL function depends on its lipidome composition. The increased lipid peroxidation of HDL caused by circulating myeloperoxidase restricts its protective function. HDL with peroxidized lipids is increased along with higher oxidative stress and inflammation in patients with diabetes (Morgantini et al., 2014). Interestingly, apolipoprotein A1 (APOA-1) and sphingosine- 1- phosphate carried by HDL are protective against apoptosis triggered by glucose and IL1β when accompanied by oxidative stress (Abderrahmani et al., 2007; Petremand et al., 2012; Rutti et al., 2009). Similarly, a positive effect on insulin secretion by lipid-free recombinant of APOA-1, APOA-2, or discoidal reconstituted HDLs has been reported (Li et al., 2004). However, β-cells can synthesize cholesterol via the mevalonate pathway in the ER, so the uptake of cholesterol from blood circulation mainly reflects nutritional status. Cholesterol is part of membrane domains, most of which are located in the plasma membrane and insulin granules (60–90%) (Lange et al., 1989). It has also been shown that excessive cholesterol in the ER inhibits the activity of the calcium pump SERCA2b (Lange et al., 1989; Li et al., 2004). Its presence influences cellular permeability, signaling activity, vesicular transport, and organelle architecture. Its dysregulation leading to intracellular accumulation can cause toxicity through its oxidation, resulting in the formation of harmful cholesterol peroxides and resulting oxidative stress in β-cells. Oxidized LDL, but also VLDL, induces the JNK signaling pathway leading to ER stress and apoptosis (Bogan et al., 2012; Plaisance et al., 2016). Moreover, more small dense LDL particles are formed in adults with T2D which are then prone to oxidation by myeloperoxidase (Tribble et al., 1992). Since cholesterol is a part of membranes, its excess has been shown to lead to enlargement of insulin granules, altered membrane curvature, and accumulation of immature insulin granules, which limits the ability to properly keep glucose homeostasis (Bogan et al., 2012). Another negative effect of LDL accumulation has been shown on the activity of NOS which by S-nitrosylation of glucokinase impacts its translocation from insulin granules to cytoplasm thus inhibiting insulin secretion (Hao et al., 2007). On the other hand, its depletion leads to the re-localization of syntaxin1 and SNAP25 from lipid rafts, reducing the number of docked granules (Vikman et al., 2009). Its comprehensive function therefore requires strict regulation in the cells largely dependent on nutritional status. (Bogan et al., 2012).
To avoid an excess of sterols in the cells, which are highly toxic and contribute to ER and oxidative stress, the cells convert the sterols into cholesteryl esters by the enzyme ACAT1. Cholesteryl esters are then stored in LDs (Sugii et al., 2003). It was reported that aged and T2D islets are known to be highly enriched in LDs. In case of LDs overload and cholesterol abundance, β-cells possess an efflux machinery. These are represented in the β-cells by SR-B1 (also mediates HDL uptake), ABCA1 (specific for HDL), ABCG1 (non-specific acceptors such as LDL, cyclodextrin) (Neumann et al., 2017) (Fig. 2A). The homeostasis of cholesterol in the cells is mainly maintained by the ER as the site of its synthesis and by the lysosomes. The ER sensor for cholesterol is caveolin1 (CAV1). It is directly related to insulin secretion as CAV1 forms the complex with insulin granule proteins VAMP2, CDC42, and βPIX and its ablation has been associated with hyperinsulinemia under physiological glucose and lipid levels (Cohen et al., 2003; Nevins and Thurmond, 2006). β-cell specific deletion of Abca1 displayed impaired glucose tolerance (Kruit et al., 2011), which was worsened in mice with additional deletion of Abcg1 causing substantial islet inflammation and diabetes (Kruit et al., 2012). Lysosomes engulf LDL via clathrin-coated vesicles and further metabolize it by acid lipases. However, this transport has not been fully elucidated, as some studies in other cells have shown that 30% of the cholesterol in the lysosomes enters the ER where it joins the pool of newly synthesized cholesterol (Vance, 2022). In addition, the excess cholesterol from the lipid rafts can be channeled back to the ER via retrograde transport mediated by STARD4/ORP1/ORP2-dependent pathways (Trinh et al., 2020, Vance, 2022). Thus, the level of various types of cholesterol needs to be strictly regulated in β-cells.
Besides being released from TAGs of lipoproteins by extra/cellular lipases or phospholipase A2 (PLA2) in membranes, FAs can be delivered as circulating non-esterified FAs, which are mainly transported with serum albumin. The optimal ratio of FA:albumin in healthy humans is 1:1 to 3:1, while the amount of FAs increases in pathological conditions of obesity and T2D (Kleinfeld et al., 1996). Transport rates by albumin depend on the size of FAs and the degree of unsaturation as well as the composition of membrane lipids and proteins. The uptake of FAs into cells occurs by membrane diffusion or is facilitated by transport membrane proteins (Fig. 2B). Importantly, FA composition and load is one of the critical factors for the development of lipotoxicity in β-cells during the development of T2D which is accompanied by oxidative stress. LC-FAs dissociate at physiological pH in solution, i.e., they release a proton and thus become negatively charged. They must first be deionized to lose the charge so that they can flip-flop across the cell membrane. However, this process is very slow, which is why the cells require protein facilitators for efficient influx.
The best-described FA translocator in β-cells is CD36, an N-linked glycosylated transmembrane protein. After transfer into the cytoplasm, FAs become activated through the addition of acyl-CoA by LC-FA-CoA synthetase/ligase (ACSL). They can then enter the GL/FFA cycle or undergo β-oxidation in peroxisomes or mitochondria or be incorporated into the membrane phospholipids or participate in sphingolipid synthesis, producing molecules such as ceramide or sphingosine-1-phosphate (Fig. 2B). The choice of a specific pathway depends on the metabolic status of the body, the metabolic needs of the β-cells, the amount and type of FAs, and mainly the blood glucose level. CD36 can bind lipoproteins (mainly in oxidized form), microbial lipids, and non-lipids (e.g., collagen), and thus has a rather pleiotropic cellular effect (Silverstein and Febbraio, 2009). Its activity is regulated by numerous post-translational modifications such as palmitoylation, ubiquitination, and glycosylation, which are important for folding and trafficking to the plasma membrane. CD36 localizes to lipid rafts or cellular caveolae (supported by CAV1), and it has been suggested that its function in FA transport is dependent on this localization (Ring et al., 2006). Chronic elevation of glucose and oxidized lipoproteins were shown to induce CD36 leading to activation of Rac1 signaling towards increased activity of NADPH oxidase and downregulation of Nrf2 (Guichard et al., 2008; Li et al., 2010). Downstream signaling pathways stimulate tyrosine phosphorylation of downstream proteins such as Scr family kinases (e.g., Fyn). CD36 thus mediates enhancement of oxidative stress under sustained nutritional pressure contributing to pancreatic β-cell dysfunction. When overloaded with FAs, these signaling pathways can trigger proinflammation (through NFκB activation) and apoptosis (Silverstein et al., 2010). CD36 activation by oxidized LDL were shown to signal towards lipid oxidation and play a key role in immune dysfunction within tumor microenvironment (Xu et al., 2021). CD36 also activates AMPK and suppresses insulin receptor signaling in peripheral tissues (Samovski et al., 2015). In β-cells, CD36 has been found not only in the plasma membrane but also in the insulin granules. It mediates both the stimulatory and inhibitory effects of palmitate on insulin release depending on the length of treatment (Noushmehr et al., 2005). Further research is needed to clarify its role in insulin granules (Noushmehr et al., 2005).
Another family of FA transporters are the fatty acid binding proteins (FABP) bound to the plasma membrane. They express several isoforms (up to 7 isoforms in mouse islets, our observation) (Fig. 2B). There is also a group of cytosolic FABPs that serve as metabolic acceptors of FAs allowing transportation to mitochondria/peroxisomes for their oxidation. Depending on the isoform, they bind various FAs and their derivatives such as eicosanoids and lipids (Veerkamp et al., 1999). It has been hypothesized that in addition to their translocation function, they also scavenge cytotoxic reactive lipids by covalent modification of Cys120 in epithelial FABP (Bennaars-Eiden et al., 2002). Although the expression of some isoforms in mouse islets is relatively high compared to CD36 (our observation), knowledge about their role in islet cells is limited. Specifically, FABP3 and FABP5 have been suggested to be regulated by FAs and glucose in β-cells further impacting insulin secretion (Hyder et al., 2010). Indirect effect of circulating FABP4 was observed to induce inflammation in patients with T1D. FABP4 activated innate responses in islets by enhancing the infiltration and polarization of macrophages to the pro-inflammatory subtype, thus creating an inflammatory milieu required for activation of other immune cells (Xiao et al., 2021).
Another known FA transporter in β-cells is the fatty acid transport protein (FATP) from the SLC27 family. (Fig. 2B). It is primarily an intracellular protein located at ER and mitochondria, where it controls the uptake of FAs (Ibrahim et al., 2020). FATP2 isoform working as an FA transporter and acyl-CoA synthetase is upregulated in human islets upon glucose stimulation (Schrimpe-Rutledge et al., 2012). However, other isoforms are expressed in rodent β-cells (our observation) and further research needs to uncover their role.
The plasma membrane of β-cells contains receptors of the G-protein coupled receptor (GPCR) family for binding of FA, called FFAR, which play an important role in significantly enhancing insulin secretion. Human and rodent β-cells express four types: FFAR1 (GPR40), FFAR2 (GPR43), FFAR3 (GPR41), and FFAR4 (GPR120), which differ in specificity for FAs based on chain length and degree of saturation (Fig. 2B). FFAR1 and FFAR4 are activated by long- and medium-chain (LC/MC)-FAs, whereas FFAR2 and FFAR3 are activated by short- chain (SC)-FAs. FFAR1 has been shown to enhance GSIS in pancreatic β-cells by activating PLCβ/PIP2/IP3/DAG signaling pathway further directly modulating insulin granule exocytosis and increasing intracellular calcium through activation of ER stores and also influx via L-type calcium channels (Usui et al., 2019). Interestingly, peroxisome proliferator-activated receptor γ (PPAR-γ) activation positively regulates FFAR1 towards insulin secretion through the mobilization of calcium (Kim et al., 2013; Usui et al., 2019). Note that glucose inhibits the expression of PPAR-γ through activation of protein phosphatase 2A and subsequent inactivation of AMPK, however, FAs activate further signaling. (Nagasumi et al., 2009; Ravnskjaer et al., 2006). Ablation of FFAR1 protects mice from obesity-induced hyperinsulinemia, hyperglycemia, and glucose intolerance, while its overexpression leads to impaired β-cell function and diabetes, but increases insulin sensitivity in obese mice (induced by high-fat diet [HFD]) (Nagasumi et al., 2009; Steneberg et al., 2005). The key regulation might involve NADPH oxidase and derived redox signaling (Nunes Marsiglio-Librais et al., 2020). Moreover, recently FFAR1 was found to be essential for GSIS for neonatal β-cell function and their adaptation to maternal high-fat feeding. The strongest stimulators of FFAR1 are unsaturated FAs with the polyunsaturated docosahexaenoic FA, but palmitate and palmitoleic acid have also been recognized as ligands. In general, FFAR1 recognizes FAs with the chain length C8-C22. Since FFAR1 is also expressed in L- and K- intestinal cells, which secrete incretins (GLP-1 and GIP) via adenylate cyclase/cAMP signaling, its effect on the induction of insulin secretion is also indirect via these hormones. It has been suggested that FFAR1 signaling is responsible for almost 50% of FA-induced insulin secretion (Latour et al., 2007). FFAR1 activation was found to attenuate chronic inflammation-induced impact through inflammation-mediated oxidative and ER stresses in pancreatic β-cells (Verma et al., 2014). FFAR4 is a sensor for unsaturated (ω−3, ω−6, ω−9, C16-C22) and saturated (C14-C18) LC-FAs, with docosahexaenoic acid and α-linoleic acid (both ω−3 FAs) being the most potent (Hirasawa et al., 2005) (Fig. 2B). These FAs enhance insulin secretion via incretin secretion from intestinal cells and show an anti-apoptotic effect via ERK, and PI3K pathway (Hirasawa et al., 2005; Katsuma et al., 2005). FFAR4-induced signaling diminishes inflammatory outbreak by inhibiting NFκB, c-JNK1/2 and p38/MAPK signaling pathways (Wang et al., 2019). Its anti-inflammatory potential was observed in other cell types and tissues, namely adipose tissue, macrophages, Kupfer cells, hypothalamus etc. (Pærregaard et al., 2016). Knockout of FFAR4 in mice leads to glucose intolerance and hyperglycemia (Suckow et al., 2014). Its expression in mouse pancreatic islets is very low (our observation) and its signaling directly in β-cells has not been described, however, the receptor was found to be rather expressed by δ-cells, thus regulating somatostatin production (Stone et al., 2014). FFAR2 and FFAR3 are receptors for microbe-derived SC-FAs such as propionate, butyrate, and valerate (Fig. 2B). Their activation has been shown to rather inhibit insulin secretion by coupling to G-proteins, as their deletion increases insulin secretion (Tang et al., 2015). However, it has been demonstrated that the amount of circulating SC-FAs is insufficient for their activation, so they might act in a cell-autonomous manner in insulin secretion. SC-FAs can contribute also to the modulation of inflammatory responses through inflammatory gene expression networks; however, it remains unclear whether it has pro- or anti-inflammatory consequences (Ratajczak et al., 2019; Schlatterer et al., 2021). Interestingly, FFAR2 has also been found to regulate β-cell mass by stimulating β-cell growth and proliferation (Villa et al., 2016). Another G-protein-coupled receptor, GPCR119, is quite specific for metabolites of FAs such as lysophosphatidylcholine, oleoylethanolamine, and 2-monoacylglycerol, and is another poorly studied insulin secretagogue (Villa et al., 2016) (Fig. 2B). The GPCR119 agonist was able to increase insulin secretion in rodent islets (Chu et al., 2007), but it was also detected in enteroendocrine cells, thus indirectly amplifying the effect on insulin secretion.
Nevertheless, we still do not know much about the mutual interaction of FFARs with FA translocators in subsequent FA metabolism and signaling. Samovski et al. suggested that FFARs do not modulate FA metabolism, but they may play a role in mitigating the negative effects of abnormal FA metabolism leading to impaired insulin secretion due to dysfunctional CD36 (Samovski et al., 2023). The potential synergistic effect of FFAR1 and CD36 or other FA translocators remains to be tested (Samovski et al., 2023). Similarly, isoform specificities in these FA translocators and receptors between species will dictate their impact on β-cell function.
Metabolic Fate of Fatty Acids in Pancreatic β-Cells: Storage, Turnover, and Oxidation
Glycerolipid/free fatty acid cycle
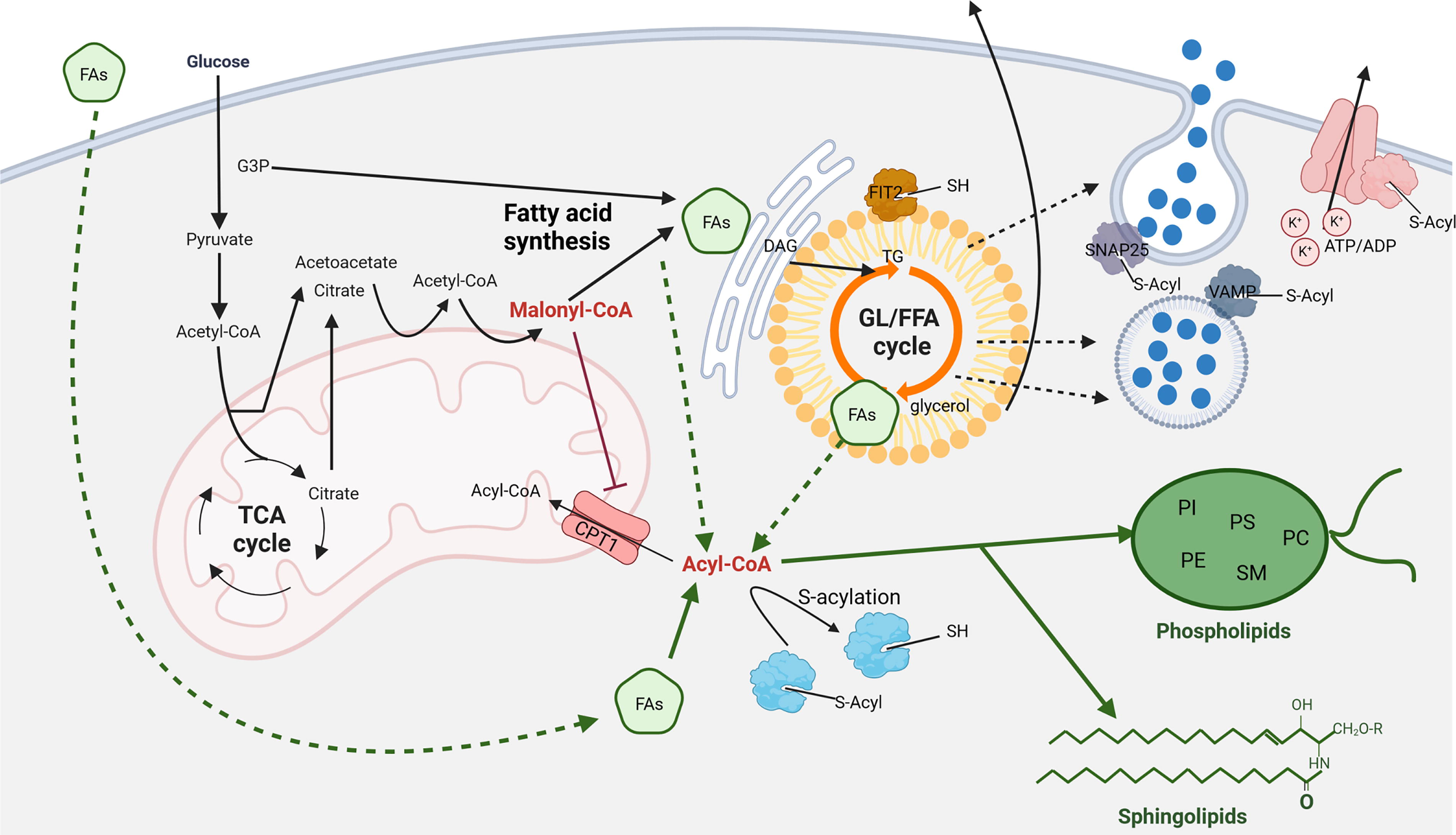
In general, FAs must undergo activation through the addition of CoA by ACSL to enter β-oxidation or lipogenesis. The reverse reaction, which involves its hydrolysis is catalyzed by acyl-CoA thioesterase 7 (ACOT7). Notably, ACOT7 expression is relatively low in β-cells (Martinez-Sanchez et al., 2016) but is upregulated in patients with T2D (Marselli et al., 2010). Storage of FAs induces GL/FFA cycle. The GL/FFA cycle consists of lipogenic and lipolytic arms (Prentki and Madiraju, 2012) (Fig. 3). Lipogenic pathway gives rise to triglyceride (TAG) from glycolysis-derived glycerol-3-phosphate and FA-CoA while the lipolytic part hydrolyzes TAGs back to FFAs by several lipases (Fig. 3).

In β-cells, the GL/FFA cycle provides MCFs, such as 1,2-DAG and 1-MAG, which amplify insulin secretion in the presence of glucose (Gembal et al., 1993; Prentki et al., 2020). Both 1-MAG and 1,2-DAG induce insulin secretion by facilitating exocytosis via the protein Munc13-1, with 1,2-DAG additionally regulating insulin secretion through protein kinase C (PKC) (Rhee et al., 2002; Sheu et al., 2003). The GL/FFA cycle also functions in β-cells as a detoxification pathway during nutrient excess by storing lipids and releasing glycerol and FAs (Mugabo et al., 2016b; Oberhauser and Maechler, 2021).
Free fatty acids and lipid storage
As described above, activated FAs are stored in LDs. However, LDs are not just a lipid depot when there is an excess of FAs, but rather dynamic particles involved in lipid metabolism that enable the flexible release of FAs and subsequent signaling. The LDs in pancreatic β-cells are composed of phospholipid membrane monolayer and a neutral lipid core consisting of TAGs and cholesterol esters. Human β-cells contain rather small LDs, most being smaller than 1.2 μm (Liu et al., 2020). The membrane harbors several proteins that possess a short N-terminal hydrophobic domain (e.g., AAM-B, UBXD-8 or ALDI) (Zehmer et al., 2008) and proteins of the perilipin family (PLIN) serving as markers which are bound to the membrane via the N- and C-terminal region (Subramanian et al., 2004) (Fig. 3). PLINs are actively involved in the composition of the lipid cargo, the size of LDs, and, most importantly, in the interaction with certain proteins such as lipases, colipases, transcription factors, chaperones, and other proteins involved in LDs transport and interorganelle communication. There are five isoforms of the PLIN family (PLIN1-5), which are differently represented in different tissues. LD formation is initiated by the final step of TAGs accumulation by DGAT1 in the seipin-rich part of the ER into the lens structure. Other proteins such as FIT2, PLIN3, expression-enhancing protein 5 or cytoskeletal septin-7 also contribute to initiating the formation of the cytoplasmic LDs (Chen et al., 2021). Interestingly, FIT2 was recently found to be regulated by FA signaling, i.e., it is removed from the ER by direct S-acylation at cysteine residues in β-cells (Zheng et al., 2022). Further accumulation of TAGs in the lens structure of the ER with the help of LDAF1 leads to the formation of the lipid core of LDs after being covered by a phospholipid monolayer. Further enlargement of LDs using CIDE protein family and the recently discovered STX18-SNAP23-SEC22B complex enables the subsequent activity of LD-associated enzymes such as DGAT2 (Fu et al., 2023). Similarly, ACAT1/SOAT1 and ACAT2/SOAT2 synthesize sterol esters to be stored in LDs. This process is very efficient and is greatly accelerated by glucose, which inhibits FA oxidation. In parallel, glucose also induces lipolysis. PLIN proteins are incorporated into the nascent LDs shortly after they emerge from the ER and contribute to the regulation of lipid metabolism, primarily by recruiting neutral lipases to initiate lipolysis of stored TAGs (except PLIN2) but also through increasing the stability of LDs (PLIN2). The major lipase associated with glucose-induced LDs is adipose triglyceride lipase (ATGL) in human β-cells. Its downregulation leads to a decrease in syntaxin-1 vesicle docking protein, likely through decreased palmitoylation which accelerates its degradation (Zhao et al., 2014a). In other tissues, oxidative stress resulting from the overproduction of reactive oxygen species (ROS) often correlates with the increased biogenesis of LDs. The causal mechanisms need still to be clarified, but potential one might be lipogenesis activation by SREBP signaling as well as phospholipid turnover (Bailey et al., 2015; Liu et al., 2015). In line with the key role of LDs in a cell, a reduction in a cell’s ability to form LDs may result in severe oxidative stress leading to apoptosis (Cheng et al., 2020). The biogenesis of LDs requires increased expression of tissue-specific PLINs, which were suggested to be regulated by ROS on transcriptional (through PPAR signaling) and translational (redox modification of cysteines residues) levels (Ding et al., 2020; Poulsen et al., 2012). Moreover, LDs were found to physically associate with mitochondria, enabling them to reflect their mutual metabolic and redox status. LDs have also been found to modulate immune cell function thus driving inflammation. They were suggested to provide structural components and energy for the production of inflammatory mediators such as leukotrienes, cytokines, and prostaglandins (more in Monson et al., 2021). Interestingly, LD accumulation signals toward formation of inflammasome and vice versa (Zhang et al., 2023). Anyway, the redox regulatory role in LD formation and its contribution to GSIS in pancreatic β-cells remains to be clarified.
PLIN2 is the most abundant isoform in pancreatic β-cells. It promotes the enlargement of the LD pool by preventing lipolysis and lipophagy (Sztalryd and Brasaemle, 2017) and therefore correlates well with LD accumulation in β-cells. Its deficiency also likely impairs GSIS through downregulation of specific OXPHOS proteins, leading to mitochondrial dysfunction as described in rat β-cells and human pseudoislets (Mishra et al., 2021). In HepG2 cells, ROS promote LD formation by increasing PLIN2 expression (Jin et al., 2018). PLIN5 is moderately expressed in mouse islets, with higher expression levels in humans (Trevino et al., 2015). However, when overexpressed in mouse MIN6 cells, it induces lipolysis thus improving GSIS. PLIN5 has also been implicated during mice fasting. Its upregulation allows β-cells to store TAGs that can be activated by lipolysis upon resumption of food intake to facilitate insulin secretion (Trevino et al., 2015). Interestingly, PLIN5 related to mitochondria-LD contacts reduces ROS levels and improves mitochondrial function in HepG2 cells in response to stress (Tan et al., 2019).
PLIN1 has also been shown to play a role in protecting islets chronically exposed to palmitate and HFD by activating the PI3K/ERK-mediated Nrf2-ARE signaling pathway, resulting in antioxidant protection (Zhu et al., 2020). The role of PLIN1, PLIN3, and PLIN4 in β-cell function is unknown. Moreover, PLIN1 and PLIN4 have weaker expression in human islets compared to PLIN2, PLIN3, and PLIN5 (Benner et al., 2014). We observe a similar expression pattern in mouse islets (our unpublished data). LDs dynamically interact with mitochondria, ER, lysosomes, peroxisomes, oleate, and nuclei to coordinate lipid metabolism in the cell in reflection of subcellular redox status. For example, PLIN5 and a mitofusin2-PLIN1 complex are involved in the formation of contacts between LDs and mitochondria in brown adipose tissue (Benador et al., 2018). LDs also bind to peroxisomes through spastin, which physically interacts with the peroxisomal FA transporter ATP binding cassette subfamily D member 1 enabling entry of FAs to β-oxidation in peroxisomes (Chang et al., 2019) (Fig. 4). This could have consequences for the cellular redox status, as a weaker expression of catalase in peroxisomes of β-cells could lead to production of ROS, which serve as signals at physiological conditions or cause oxidative stress during pathological gluco/lipotoxic conditions.

The formation of LDs is regulated by diet, as HFD and fasting in vivo increase the content of TAGs together with the expression PLIN2 and PLIN5 (Hsieh et al., 2012; Trevino et al., 2015). Thus, LDs not only accumulate when islets are exposed to FAs and glucose to avoid their toxicity in their free state but also increase under nutritional stress, oxidative stress, and dysfunctional human β-cells in vivo (Dai et al., 2016). β-cell maturity and aging are other factors associated with the accumulation of LDs in human β-cells (Tong et al., 2020). They are absent in less mature, poorly glucose-responsive β-cells at an earlier stage of development (Tong et al., 2020). Active LDs are therefore a key component of healthy β-cells. However, abnormal lipid metabolism leading to LD accumulation and altered lipid management often accompanied by oxidative stress may contribute to the development of T2D. Interestingly, no visible LDs were detected in mouse islets despite the increased expression of PLIN isoforms during HFD feeding or fasting probably due to a different nutritional regulation (Faleck et al., 2010, Tong et al., 2022). Mouse islets also fail to induce lipolysis in response to glucose.
LD metabolism, particularly lipolysis, allows insulin secretion to be supported by selected pathways that are regulated by the nutritional status of the cells: (i) the release of FAs can activate FFAR1 on the cell surface to potentiate GSIS, (ii) the release of lipid metabolites such as MAG can directly activate insulin granule exocytosis via Munc13 in the SNARE complex, (iii) the modification of FAs proteins by S-acylation, which allows their proper transport into the membrane and subsequently their proper functioning. However, LDs are also accumulated in mammalian cells under ER stress. It activates MAPKs, which can phosphorylate and activate phospholipases such as cPLA2α to promote LD biogenesis (Gubern et al., 2008). Oxidative stress is also associated with increased LD biogenesis. Under oxidative stress, phospholipases release polyunsaturated FAs (PUFAs) from membranes to be stored in LDs to protect them from peroxidation (Bailey et al., 2015; Welte and Gould, 2017). Thus, LDs limit cell damage, prevent cell death, and reduce the inflammatory response by sequestering PUFAs and reducing their availability for oxygen delivery. The redirectioning of saturated FAs into LDs also protects β-cells from synthesizing ceramides, which can trigger cell dysfunction and inflammatory signaling. The type of FAs in LDs is thus crucial for cell function. It has been found that TAGs in LDs are enriched in unsaturated FAs and can be converted into phospholipid pool under stress conditions which prevents cellular stress by avoiding the use of fully saturated lipids that can be potentially toxic (Ackerman et al., 2018). There is still the question of how efficient the activity of LDs is in terms of internal FA synthesis versus external influx of FAs. The de novo synthesis of FAs in β-cells is not as efficient and the incorporation of FAs into TAGs in LDs is highly dependent on the immediate nutrient supply of the individual and potential stress conditions. It is known that PPARα is a key factor in mediating oxidative lipid metabolism in many tissues including the heart, liver, etc. It is directly activated by FAs and their metabolites. The turnover of LDs appears to be necessary for the activation of PPAR signaling by exogenously derived FAs in cardiac cells (Haemmerle et al., 2011). Whether exogenous FAs must first enter GL/FFA cycle in LDs in β-cells to exert the signaling effect remains to be clarified. It has been suggested that the activity of the cycle is useful to precisely match the FA supply from the bloodstream with the mitochondrial capacity for OXPHOS and peroxisome activity to avoid cytotoxic cell damage potential induced by the generation of ROS (Jarc and Petan, 2020).
Neutral lipids that do not contain PLIN protein represent lysosome-derived lipofuscins. Lipofuscin is of pathophysiological relevance as it generates ROS due to its ability to incorporate redox-active metals such as iron, thus promoting Fenton reaction (Höhn et al., 2010). Human β-cells can contain about 40% of neutral lipids (Tong et al., 2020). As it is often detected together with lysosomal markers, it is questionable whether LDs delivered to lysosomes by lipophagy do not interfere (Zechner et al., 2017). LDs can be directed into autophagy as an alternative pathway to mobilize lipid stores (Wang, 2016). After LDs are channeled into autophagosomes, the contents are degraded by lysosomal acid lipases or by chaperone-mediated autophagy, lysosomal proteolysis carried by HSP70 and lysosome-associated membrane protein-2 (LAMP-2) (Cuervo and Dice, 1996; Chiang et al., 1989, Tong et al., 2020). Moreover, chronic inhibition of lipophagy (over 48 hrs) in rat and human islets significantly blunts GSIS. The interplay between LDs lipolysis and lipophagy is still unknown; however, it is suggested that lipophagy is an alternative way of prompt lipid degradation (Schmucker and Sachs, 2002).
β-oxidation in pancreatic β-cells
FAs can be also metabolized by degradation in the process of β-oxidation. They can serve as a major energy source during periods of low substrate supply or high energy demand states such as exercise, but it is becoming clear that β-oxidation participates in FAs turnover and regulation of overall lipid homeostasis in the cells. In general, there are two major places where β-oxidation occurs: mitochondria and peroxisomes (Fig. 4). Both organelles are equipped with enzymes that are able to carry out all the reaction steps during FA degradation; however, there are certain differences that have important functional implications. First of all, the enzymes are coded by distinct genes resulting in the expression of different proteins (Wanders et al., 2015) (Fig. 4). The reactions involve four sequential steps including dehydrogenation, hydration, dehydrogenation again, and thiolytic cleavage. The mitochondrial enzymes catalyzing the first step are FAD-dependent dehydrogenases, which provide the electrons to the respiratory chain via the Electron-Transfer-Flavoprotein cycle. On the contrary, the peroxisomal counterparts are FAD-dependent acyl-CoA oxidases donating their electrons directly to molecular oxygen (Wanders et al., 2015). Another difference is how the FAs are transported to these organelles. In the case of peroxisomes, the FAs enter either in their free form or as activated CoA esters. In contrast, transport to mitochondria involves the binding of FAs to carnitine and the subsequent so-called carnitine shuttle via the concerted action of carnitine palmitoyl transferase 1 (CPT1), carnitine acylcarnitine translocase (CACT), and carnitine palmitoyl transferase 2 (CPT2) (Gehrmann et al., 2010). Both organelles can not only metabolize saturated SC-, medium- and LC-FAs but also catalyze the oxidation of certain monounsaturated fatty acid (MUFAs) and polyunsaturated fatty acids (PUFAs) as well as 2-(R)-methyl branched-chain FAs and 2-hydroxy-FAs (Wanders et al., 2015). On top of that very LC-FAs (C24-26) are almost solely metabolized in peroxisomes (Wanders, 2014).
The activity of β-oxidation is highly dependent on the cellular metabolic status. In this respect, the interplay between peroxisomes and mitochondria is especially important involving the end-products of β-oxidation such as NADH, acetyl-CoA, propionyl-CoA, and a variety of acyl-CoAs shortened in peroxisomes. (Wanders et al., 2015). Peroxisomes depend on the provision of re-oxidized NAD+ to continue the β-oxidation, which can only be achieved in mitochondria. However, a carrier system able to mediate the exchange of NADH and NAD+ on the peroxisomal membrane appears to be lacking in higher eukaryotes including humans. The existence of metabolite-based redox shuttle has been proposed analogous to the well-known shuttles providing such cycling between mitochondria and cytoplasm. The evidence in favor of the existence of a lactate/pyruvate-based redox shuttle has been provided in hepatocytes peroxisomes (Baumgart et al., 1996). As mentioned above the products of peroxisomal β-oxidation need to be transferred to mitochondria for final oxidation to CO2 and H2O and ATP production. Transport of different acyl-CoAs is mediated either by carnitine shuttling or as free FAs. The first pathway involves the conversion of acyl-CoAs to acyl-carnitines in peroxisomes via carnitine-acetyltransferases and carnitine-octanoyl transferases reactive with short or medium-length acyl-CoAs respectively (Wanders et al., 2015). The alternative, free FAs route involves cleavage of the acyl-CoA esters by one of the different thioesterases found in peroxisomes (Hunt et al., 2012). The free FAs leave the organelle probably through a porine (PXMP2) (Rokka et al., 2009) and enter mitochondria most likely in their protonated form (Baumgart et al., 1996; Wanders et al., 2015).
In heart and kidney mitochondria, over 95% of ATP is produced via β-oxidation of LC-FAs. The enzymes involved in β-oxidation are physically linked to respiratory supercomplexes (respirasomes), which generate high levels of ubiquinol (QH2) and reverse electron flow through complex II, reducing fumarate to succinate. This process is enhanced by the presence of succinate. The acyl-CoA dehydrogenase complex reduces ubiquinone to QH2, which is quickly oxidized by the supercomplexes, energizing mitochondria and reversing electron flow through complexes II and I, raising the NADH/NAD+ ratio (Schönfeld et al., 2010). Nicotinamide nucleotide transhydrogenase (NNT) transfers hydride across the mitochondrial membrane, reducing NADP+ to NADPH, supplying ATP and reducing equivalents for the heart. In well-energized mitochondria, excess electrons reduce components of complex I, accelerating superoxide radical production (Panov and Orynbayeva, 2018). However, NNT can mitigate this by transferring energy to the cytoplasm, reducing superoxide formation. While reverse electron transfer from succinate generates ROS, fatty acid oxidation does not lead to high ROS levels despite being a strong source of FADH2 (Panov and Orynbayeva, 2018). Nevertheless, these findings cannot be applied very well to pancreatic β-cells, as they prefer glucose over lipids for ATP generation (Oberhauser and Maechler, 2021).
In β-cells, β-oxidation is primarily recognized for its role in two scenarios: (i) during low substrate conditions (starvation) and (ii) under pathological conditions with elevated free FA levels (lipotoxicity). In the first scenario, mitochondrial β-oxidation of stored FAs and free FAs, which are increased in the blood during starvation, is believed to be a primary source of ATP production (Imai et al., 2020). However, β-cells typically favor other carbon sources over FAs for respiratory chain substrates, suggesting that β-oxidation plays a minor role in substrate delivery. In the second scenario, under lipotoxic conditions, FAs are either broken down in peroxisomes or stored in complex lipids (e.g
Functional Aspects of Complex Lipids in Pancreatic β-Cells
Role of phospholipids in β-cell function
Phospholipids are a unique class of complex lipids composed of phosphatidylinositol with a myoinositol ring that can be phosphorylated at the D3, D4, and/or D5 position, giving rise to seven distinct derivates—the phosphoinositides (Wuttke, 2015). The distribution of the phosphoinositides within the cellular membranes differs greatly and together with the rapid turnover is precisely regulated by at least 19 mammalian phosphoinositide kinases and 28 phosphoinositide phosphatases, of which some have multiple isozymes (Sasaki et al., 2009). Their role in β-cells is defined by (i) their presence and composition of the cellular membranes affecting various processes including membrane fluidity, excitability, and ability to fuse with insulin granules (which also contain phospholipids in their membranes) and ii) by their cleavage to provide second messengers important for regulation of insulin release.
β-cells were shown to have significant differences in the distribution of phosphatidylserine (PS), phosphoinositol (PI), phosphatidylethanolamine (PE), phosphatidylcholine (PC), and sphingomyelin (SM) among insulin granules, mitochondria, ER and plasma membranes (MacDonald et al., 2015) (Fig. 5). Additionally, the abundance of various SFAs, MUFAs, and PUFAs in these phospholipids differs across these compartments. Interestingly, glucose stimulation of β-cells leads to reversible changes in the FA composition of phospholipids within insulin granules. These modifications and the resulting specific phospholipid signatures may alter their biophysical properties. Consequently, this can affect the interaction of granules with soluble proteins (e.g., SNAP receptors, SNAREs), target membrane proteins within the granules (e.g., VAMP), or plasma membrane docking proteins (e.g., syntaxins), ultimately impacting insulin secretion (Maulucci et al., 2019). Beyond the inherent composition and turnover of phospholipids in subcellular organelles of β-cells, it is crucial to highlight the significant impact of increased availability of dietary-free FAs, both essential and non-essential, and their incorporation into phospholipids. This becomes particularly consequential when β-cells are exposed to high levels of SFAs (e.g., palmitate), either alone or in combination with high glucose levels leading to (gluco)lipotoxic effects that often contribute to the decline in β-cell mass and function (Imai et al., 2020). The FA composition in membrane phospholipids is continually remodeled due to the availability of free FAs, the enzymatic activity of phospholipases, and various stressful conditions (e.g., nutritional deficiencies or overloads) or metabolic diseases. This remodeling is a dynamic and rapid process that shifts the balance between the hydrolysis of FAs from phospholipids by PLA2 and their reattachment to the phospholipid backbone by lysophospholipid acyltransferase (LPAT) (Wang and Tontonoz, 2019) (Fig. 5). Preferentially, PUFAs are released from the phospholipids in β-cells (Maulucci et al., 2019). They can enter GL/FFA cycle or are converted to bioactive metabolites. These metabolites subsequently regulate various cellular functions in autocrine and/or paracrine fashions. It has been shown that endogenous PUFA metabolites, such as 20-hydroperoxyeicosatetraenoic acid (20-HETE), prostaglandin E1, E3, J2, and I2, or endocannabinoids impact β-cell functions (Jourdan et al., 2016; Luo and Wang, 2011; Tunaru et al., 2018). Interestingly, the exposure of β-cells to glucose and palmitate leads to significant changes in SFAs, MUFAs, and PUFAs in membrane phospholipids. Under high glucose incubations, the abundance of PUFAs significantly decreased, while MUFAs increased, and SFA levels remained constant. Notably, the total FA content did not change under these conditions. This study also revealed that the released PUFAs (e.g., arachidonic and linoleic acids) were rapidly peroxidized to 4-hydroxynonenal (4-HNE). This compound subsequently activated PPARδ, further enhancing GSIS (Cohen et al., 2011). In a follow-up study, authors challenged pancreatic β-cells with high glucose/palmitate levels, which caused incorporation and exchange of palmitate for SFAs, MUFAs, and PUFAs in membrane phospholipids, generation of arachidonic and linoleic acid and their peroxidation, creation of 4-HNE, but without the increase in insulin secretion. The increased synthesis of these compounds leads to ER stress induction, apoptosis, and cell death emphasizing the importance of cell adaptation mechanisms during different metabolic statuses under physiological and pathological conditions (Cohen et al., 2015).

In addition to affecting phospholipid structure and composition, cellular redox status influences enzymes involved in phospholipid turnover. Group VIA phospholipase A2, crucial for transcription, proliferation, apoptosis, and secretion, is expressed in β-cells and regulated by redox-dependent thiol modifications (Song et al., 2006). Other proteins involved in phospholipid processing are likely regulated by redox modifications, though no experimental evidence has been shown yet. One of the most studied mechanisms in phospholipid biology is the hydrolysis of PI(4,5)P2 by PLC generating two important second messengers inositol-3-phosphate (IP3) and DAG. IP3 is released to the cytoplasm, mobilizing calcium from intracellular stores (Idevall-Hagren and Tengholm, 2020) (Fig. 5). DAG remains in the plasma membrane, interacting with several DAG-interacting proteins including PKC (Kaneko and Ishikawa, 2015). In β-cells, PLC is activated after the stimulation of muscarinic receptors by acetylcholine released within islets after food intake (Gilon and Henquin, 2001). The nerve endings are the major source of acetylcholine in rodent islets, but it was also suggested that α-cells could be the source of acetylcholine in human islets (Rodriguez-Diaz et al., 2011). Islet PLCs are also activated by glucose and depolarizing stimuli, mainly mediated by the influx of calcium (Thore et al., 2004). Oscillations of calcium lead to oscillatory PLC activity and asynchronous oscillations of the plasma membrane PIP2 concentration (Thore et al., 2004).
The PLC pathway is also activated by ATP, which is released from the nerve endings and insulin secretory vesicles (Wuttke, 2015) (Fig. 5). It acts through the binding to purinergic receptors—ligand-gated cation channels P2X receptors and Gq protein-coupled P2Y receptors (Petit et al., 2009). Extracellular ATP increases intracellular calcium levels leading to the enhancement of insulin secretion and the synchronization of cytosolic calcium oscillations and pulsatile insulin secretion (Petit et al., 2009).
Experimental evidence further suggests that phosphoinositides can act also independently of the PLC pathway and several mechanisms were suggested for how they affect insulin secretion (Wuttke, 2015). PI(4,5)P2, PI(4)P, and likely PI(3,4,5)P3 bind to the Kir6.2 subunit of KATP channels. The negative charges of these lipids counteract the inhibitory effect of ATP, stabilizing the open state of the channel (Baukrowitz et al., 1998; Shyng and Nichols, 1998). This is especially significant in β-cells, where KATP channels exhibit paradoxically high sensitivity to ATP in excised membrane patches, suggesting they would be constantly closed at physiological ATP concentrations. ADP is proposed to modulate ATP sensitivity in vivo (Tarasov et al., 2006), but the interaction between KATP channels and membrane phospholipids is also crucial for proper membrane excitability and insulin secretion (Lin et al., 2005).
Calcium-triggered exocytosis is preceded by the transport of secretory vesicles to the plasma membrane and the assembly of the exocytotic machinery, a process known as priming. Priming depends on ATP and phosphoinositide synthesis (Eberhard et al., 1990; Hay et al., 1995). The calcium-dependent activator protein for secretion is a phosphoinositide-binding priming factor that aids in fusion complex assembly (James et al., 2010), and several studies underscore its importance in insulin secretion (Gromada et al., 2005; Olsen et al., 2003; Waselle et al., 2005). Another potential link between phosphoinositides and exocytosis is Munc18-interacting proteins (MINTs) (Okamoto and Südhof, 1997), of which some isoforms are present in insulin-secreting β-cells (Zhang et al., 2004b), and silencing of Mint-1 was found to reduce exocytosis in INS-1E cells (Waselle et al., 2005).
DAG is one of the most important lipid signal messengers in β-cells (Fig. 5). DAG is metabolized through three major pathways: i) phosphorylation by diacylglycerol kinase (DGK) to produce phosphatidic acid, ii) hydrolysis of fatty acyl chain by diacylglycerol lipase to generate a monoacylglycerol and FAs, and iii) formation of TAG by diacylglycerol acyltransferase (DGAT). Since the expression of DGK, diacylglycerol lipase, and DGAT is detected in isolated islets (Kaneko and Ishikawa, 2015), DAG level and DAG signaling in β-cells might be strictly and intricately regulated by these enzymes. In most cases, however, the conversion to PA catalyzed by DGK seems to be the primary pathway for DAG metabolism in β-cells (Baldanzi, 2014). It is widely acknowledged that DAG produced through the activation of PLC, following the stimulation of muscarinic receptors or exposure to high glucose concentrations, is essential for the normal functioning of β-cells (Peter-Riesch et al., 1988; Wolf et al., 1990). The main role of DAG is to activate PKC. However, the involvement of PKC in insulin secretion is still debated. Some studies indicate that conventional PKC isoforms enhance insulin secretion (Tian et al., 1996; Zawalich and Zawalich, 2001; Zhang et al., 2004a), while others suggest these isoforms are not involved (Carpenter et al., 2004; Zawalich and Zawalich, 2001, Zhang et al., 2004a). Additionally, the impact of novel PKC isoforms on insulin secretion is also disputed, indicating that the role of PKC in β-cell function is likely complex (Cantley et al., 2009, Schmitz-Peiffer et al., 2007). These varying findings might be partly due to species differences between rats and mice (Zawalich and Zawalich, 2001). Besides the DAG/PKC pathway, DAG has been shown to impact other pathways in β-cells, such as those involving Ras guanine nucleotide-releasing protein (Ras-GRP), protein kinase D, and transient receptor potential (TRP) channels (Kaneko and Ishikawa, 2015). Furthermore, DAG regulates insulin secretion by affecting the pathway dependent on Munc13, a synaptic protein that controls vesicle release independently of PKC (Rhee et al., 2002)(Fig. 5). Munc13-1 heterozygous mice exhibit impaired GSIS both in vivo and in isolated islets. Thus, Munc13 may function as an effector molecule in β-cell lipid signaling (Kwan et al., 2006).
Role of sphingolipids in β-cell function
Sphingolipids (SLs) comprise a class of complex lipids derived from the amino alcohol sphingosine and include sphingomyelins (SM), ceramides, and sphingosine-1-phosphate (S1P). SLs are synthesized through the condensation of palmitate with serine, forming a sphingoid backbone. Six different ceramide synthases (CerS1-6) then attach a second fatty acid of specific chain length to this backbone, producing ceramides (Cer). This second FA can range from 14 to over 30 carbon atoms. Cer can be further modified at the C1 hydroxyl residue on the sphingosine backbone by the addition of sugar moieties or phosphatidylcholine, resulting in the formation of hexosylceramide (HexCer) and SM species. Consequently, this process generates hundreds of SLs with potentially diverse biological functions (Griess et al., 2023). SLs, such as Cer, have become recognized as significant factors in metabolic diseases (Summers et al., 2019). While the accumulation of ceramides is generally viewed as harmful, contributing to glucolipotoxicity-induced apoptosis and β-cell dysfunction (Manukyan et al., 2015; Véret et al., 2011; Yano et al., 2011), recent research suggests a more nuanced role. Cer with different acyl chain lengths have distinct effects on glucose metabolism. An increase in long-chain Cer (C16–C22) is associated with insulin resistance, whereas an increase in very-long-chain (VLC) Cer (C >22) enhances insulin signaling and protects mice from obesity and glucose intolerance induced by a HFD (Montgomery et al., 2016; Turpin et al., 2014). Synthesis of C24:1 SLs is associated with oleate-induced β-cell proliferation in rat islets as an adaptive β-cell mass expansion in response to nutrients (Castell et al., 2022). Recently, imbalance in VLC-SLs and LC-SLs has been shown to contribute to T2D-associated β-cell failure. As the specific ablation of Cer synthase, isoform 2, which is necessary for the generation of VLC-SLs selectively reduces insulin content, impairs insulin secretion, and disturbs systemic glucose tolerance. CerS2 ablation affects SL binding to several ER–Golgi transport proteins, including Tmed2, which has been defined as an endogenous regulator of the essential proinsulin processing enzyme Pcsk1 (Griess et al., 2023).
The Importance of Glucose-Induced Lipid Metabolism in β-Cell Insulin Secretion
Experiments show that β-cells deprived of lipids cannot secrete insulin upon glucose stimulus, highlighting the importance of intracellular lipid metabolism in GSIS (Stein et al., 1996). Glucose increases malonyl-CoA, which inhibits CPT1 and mitochondrial FAO, triggering lipogenesis in LDs and the synthesis of TAGs, cholesteryl esters, phospholipids, sphingolipids, and other specialized lipids in concert with ER. This process is driven by increased expression of lipogenesis enzymes (FASN, ACLY, and DGAT2) via the ChREBP transcription factor (Sae-Lee et al., 2016) (Fig. 6). Glucose metabolism produces mild prooxidative redox status, which initiates redox signaling leading to effective insulin secretion (Plecita-Hlavata et al., 2020). How lipid metabolism is linked to redox intracellular status needs to be resolved, although we observed redox modification of enzymes participating in lipid metabolism of β-cells upon glucose stimulation (data not shown) (Holendová et al., 2024b). Many of the enzymes within lipogenesis were shown to be redox modulated mainly by H2O2 in adipocytes. H2O2 produced by NOXs increases lipogenesis by enhancing substrate transport and NADPH along with stimulating pyruvate dehydrogenase (Abou-Rjeileh and Contreras, 2021). SREBP is also involved in this process through ROS activation of mTORC1 (Crewe et al., 2019). Elevated glucose also induces lipolysis in LDs, producing metabolites (DAG and MAG) that enhance insulin secretion and increase LC-FA-CoAs for cell signaling through PKC, mTORC, PPARγ, and FFAR receptors (Ardestani et al., 2018; Yaney et al., 2000). LC-FA-CoAs are crucial for posttranslational modifications, particularly S-acylation, affecting ion channels, vesicle trafficking, and protein sorting (Prentki et al., 2020; Roduit et al., 2004). Note, S-acylation is a posttranslational modification of cysteine residues within target proteins upon glucose stimulation. At the same time, enhanced glucose metabolism modifies cysteines of specific proteins in redox relay signaling (Holendova and Plecita-Hlavata, 2023, Holendová et al., 2024b). How and if these types of glucose-induced signaling modifications are linked and their relevance to lipid metabolism remains to be resolved. The glucose level thus regulates the amount of cytosolic LC-FA-CoA together with the GL/FFA cycle in LDs (Corkey et al., 2021). LC-FAs can also enter the β-cells from the bloodstream (see above); however, it is not known how efficient the contribution of LC-FAs influx to the cell is when glucose is present. When cells are starved, they accumulate FAs from the media rather than synthesize them, as glycolysis is downregulated. LC-FA-CoA-induced S-acylation modulates KATP channel activity and the function of SNARE complex proteins involved in vesicle fusion, indicating the importance of the compartmentalization of lipid metabolites within the β-cell (Cheng et al., 2003, Prescott et al., 2009). Destabilization of FIT2 by S-acylation (a critical LD protein) in β-cells induced by saturated FAs leads to ER stress, lower ATP levels, reduced calcium signaling, and attenuated insulin vesicle exocytosis (Zheng et al., 2022). Interestingly, this effect was not observed with unsaturated FAs. The important role of S-acylation, specifically palmitoylation, is shown in the work of Dong. et al., where the deletion of APT1, an acyl-protein thioesterase that reverses palmitoylation, leads to hypersecretion of insulin and thus to β-cell failure (Dong et al., 2023). Hence, the metabolic state of β-cells, reflected in LC-FA-CoA levels, impacts insulin secretion.
Toxicity of Free Fatty Acids/Lipids
Plasma FAs are strictly regulated and reach a concentration 100–1000 μM (Huber and Kleinfeld, 2017). Their profile consists of more than 30 different species, with 78% of all FAs in the bloodstream consisting of palmitic, stearic, and oleic acids (Quehenberger et al., 2010). The increased concentration of even-carbon chain LC-FAs was associated with an increased risk of developing T2D, while odd-carbon chain LC-FAs reduced the development of T2D (Forouhi et al., 2014). Reducing the carbon chain length of even-chain FAs such as C14:0 or C12:0 makes them less toxic. Unsaturated FAs (both MUFAs and PUFAs) do not trigger apoptosis of β-cells, and the effect is independent of chain length (Newsholme et al., 2007). Ericson et al. came up with the explanation that even-chain LC-FAs in T2D are predominantly a result of lipolysis and lipogenesis from excess carbohydrates, while shorter FAs (C4-C10) and odd-carbon chain LC-FAs (C15:0 and C17:0 and their phosphoryl esters) are mainly derived from dairy products and correlate with a lower risk of T2D (Ericson et al., 2015). There is also an effect of the degree of desaturation. The recent meta-analysis of randomized controlled trials suggests that a diet high in MUFAs leads to lower fasting blood glucose than a diet high in carbohydrates or PUFAs (Qian et al., 2016). Different FAs can therefore be converted into different signals, e.g., proinflammatory and anti-inflammatory signaling molecules. Although studies have examined the quantification and impact of FAs in the circulation in the context of T2D, there is no consensus on their role. Anyway, an increase in circulating FAs along with altered lipid metabolism and ectopic fat accumulation is associated with the development of T2D early before T2D is diagnosed. However, hypertriglyceridemia in individuals with lipoprotein lipase deficiency improves GSIS and lowers blood glucose levels, suggesting that hypertriglyceridemia and high FAs alone are not deleterious to β-cells (Tamasawa et al., 2006). Differences between the sexes and races play also an important role. Compared to men, women have about 15% higher levels of circulating FAs and release about 40% more FAs relative to energy expenditure at rest than men (Karastergiou et al., 2012). South Asian populations also develop T2D at a lower BMI and show differential ectopic accumulation of fat depots. (Anand et al., 2011).
The direct deleterious effect of chronic action of FAs (especially palmitate) on β-cells involves transcriptional changes associated with inflammation, ER stress, oxidative metabolism, autophagy, and apoptosis pathways. We showed that chronic overnutrition of mice induced by HFD activates NOX4 leading to the formation of inflammasome and the production of proinflammatory IL1β that differentiates macrophages/monocytes toward local inflammation (Holendová et al., 2024a). Chronic treatment of β-cells with palmitate activates ER stress via the PERK, IRE1, and ATF6 pathways, impairing calcium stores and triggering ER stress-induced apoptosis. The palmitate-impaired mitochondrial function involves the opening of the mitochondrial permeability transition pore through altered calcium signaling from the ER, causing the swelling of mitochondria and apoptosis. However, chronic palmitate excess also generates H2O2 by peroxisomal β-oxidation (Elsner et al., 2011). Given that β-cells show weak expression of the H2O2-inactivating enzyme catalase in peroxisomes, peroxisomal H2O2 formation contributes to palmitate-induced oxidative stress. Mitochondrial stress contributing to prooxidative status leads to their fragmentation and impaired network dynamics where any shift toward the fusion of the mitochondrial network prevents lipotoxicity-induced apoptosis (Molina et al., 2009). Sustained overload of FAs induces also H2O2 through NOX2 activity that can be also induced upon stress conditions by UPR response of the ER (Vilas-Boas et al., 2021a; Vilas-Boas et al., 2021b). Lipotoxicity- induced oxidative stress and ER stress then activate inflammatory signaling. The ER stress activates NFκB via both IRE1 and PERK branches; however, it can also induce inflammation by NFκB-independent mechanisms (Goodall et al., 2010; Nakamura et al., 2011). FA excess also alters cardiolipin function, leading to dissociation of cytochrome c, which in turn triggers apoptosis (Koshkin et al., 2008). Palmitate together with glucose causes changes in the composition of membrane phospholipids and their peroxidation, which in the short-term augments insulin secretion, but in the long term causes ER stress,prooxidative metabolism and apoptosis (Cohen et al., 2015). Glucolipotoxicity is associated with increased deposition of SLs, apoptosis, and β-cell failure (Manukyan et al., 2015; Véret et al., 2011; Yano et al., 2011); however, current research shows that VLC-Cers can have beneficial effect enhancing proinsulin processing, insulin signaling and protecting mice from obesity and glucose intolerance induced by a HFD (see above and more in Griess et al., 2023, Montgomery et al., 2016; Turpin et al., 2014). Chronic exposure to FAs and glucose in in vivo studies increases UCP2 expression, which is associated with impaired insulin secretion, hyperglycemia, and T2D (Joseph et al., 2002). In in vitro experiments in the absence of glucose, FAs are degraded in excess by peroxisomes/mitochondria to generate H2O2 establishing oxidative stress upon increased nutritional pressure. Note, β-cells are not able to cope with prooxidative treatment in chronic period (Benáková et al., 2021; Holendova and Plecita-Hlavata, 2023). Palmitate-induced proinflammatory signaling includes activation of NF-κB, JNK, ds RNA-dependent protein kinase, and NLRP inflammasome (Salvado et al., 2015). Moreover, β-cells also produce chemokines via a TLR4 receptor that recruits proinflammatory M1-type monocytes (Inoue et al., 2018). Activation of TLR4 signaling is crucial for maturation of M1 proinflammatory macrophages, leading to secretion of TNFα, IL1, IL6 IL12, activation of inducible NOX and expression of chemoattractants such as MCP1m RANTES (Winer and Winer, 2012). In contrast, oleate, n-3, and n-6 FAs inhibit NLRP inflammasome. There is an interaction between the stress pathways, i.e., the proinflammatory stress response triggered by ER stress via NF-κB and JNK and the JNK pathways stimulating autophagy. Chronic palmitate treatment also significantly downregulates genes related to β-cell identity and insulin expression (Brun et al., 2015; Cnop et al., 2014). However, the studies are often performed with selected FAs (palmitate) in different FAs/albumin ratios and qualities. Contrariwise, oleate does not have as deleterious effect as palmitate when reducing H2O2 formation and stimulating autophagic flux and clearance of autophagosomes compared to palmitate (Gehrmann et al., 2015; Chu et al., 2019). The toxicity of FAs is also dependent on the experimental species. Human cell lines have been shown to express a higher amount of stearoyl-CoA desaturase, which efficiently reduces the chronic toxicity of saturated FAs (Oshima et al., 2020). In addition, they convert saturated LC-FAs into LDs, in contrast to rodent β-cells, for which LC-FAs then become more toxic. Thus, we are still missing many details about the effect of chronically elevated FAs on β-cell function and glucose homeostasis.
The Development of T2D and β-Cell Lipid Metabolism
Disturbances in lipid metabolism, particularly lipogenesis and lipolysis within the GL/FFA cycle in β-cells, alter insulin secretion and contribute to type 2 diabetes. Obesity coupled with decreased sensitivity of peripheral and hepatic tissue to the actions of insulin leads to ectopic fat accumulation and increased circulating FAs, stressing β-cells and heightening diabetes risk if chronic. This is also associated with decreased production of anti-inflammatory mediators (e.g., adiponectin) and an increase in the secretion of proinflammatory signaling molecules by adipocytes and immune cells residing in adipose tissue. Although in high quantity, not all FAs are toxic to β-cells (see above). However, the activity of lipogenesis versus lipolysis within the GL/FFA cycle in GSIS, especially under T2D conditions, is not well understood. It is suggested that increased cycling activity serves as an adaptive response, maintaining GSIS under glucolipotoxic conditions and detoxifying excess fuels (Mugabo et al., 2016a). Excess cytoplasmic FAs are processed into LDs as TAGs through mTOR pathway activation, but chronic FA overload improperly activates mTORC1, impairing insulin secretion (more in (Ardestani et al., 2018, Yuan et al., 2017). Malonyl-CoA’s direct binding to mTORC1 has been recently reported (Nicastro et al., 2023). Elevated intracellular LC-FA-CoA reduces β-cell excitability and insulin secretion via KATP channel activation (Webster et al., 2008). Most studies focus on the toxicity of circulating FAs on β-cell physiology, often ignoring the GL/FFA cycle and intracellular lipid management. Moreover, in vitro experiments typically use nonphysiological FA and glucose concentrations. Furthermore, processing of FAs results in proinflammatory lipid mediators, another potentially significant source of inflammation in islets such as eicosanoids including leukotriens, lipoxins eoxins, and resolvins. These can activate Jun/MAPK pathway and produce cytokine IL12 that helps to differentiate Th1 cells (Chen et al., 2005, Middleton et al., 2006). While lipid metabolites, both intracellular and extracellular, enhance GSIS, they can become toxic in large amounts or with prolonged exposure, increasing ER stress and excessive ROS production, potentially triggering inflammation—a hallmark of diabetes.
Conclusion
Proper FA and lipid metabolism is essential for maintaining healthy β-cells, while its disruption is closely linked to T2D. Despite significant research on the individual steps of this metabolic process, a comprehensive understanding under normal and pathological glucose signaling remains elusive. The interplay between glucose signaling and derived redox signaling with lipid metabolism is complex and presents technical challenges, such as the simultaneous activation of lipolytic and lipogenic pathways in the GL/FFA cycle and dynamic redox homeostasis. The precise roles of external FAs and the regulation of FA/lipid metabolism by glucose are still not fully understood, with many studies being conducted under nonphysiological conditions.
It is now recognized that lipid metabolism and LD accumulation are crucial for cellular signaling beyond mere fat storage. Lipid metabolism was suggested to be dependent on glucose metabolism and derived redox status; nevertheless, no clear evidence exists. Lipid metabolites are important for signaling and protein modification in β-cells, influencing the composition of lipid rafts. However, current analytical techniques often fall short in detailing the fluxes of lipid metabolism due to their reliance on bulk FA/lipid content analysis, which lacks specificity in subcellular localization, similarly as redox status. Advanced tools that target individual cellular compartments are necessary for a more detailed understanding.
Moreover, differences between rodent and human islets require further investigation to ensure findings are translatable. Despite these challenges, glucose-induced lipid metabolism remains fundamental for β-cell homeostasis, and its dysregulation is strongly associated with T2D accompanied by oxidative stress and metainflammation, particularly in the context of obesity and metabolic syndrome.
Footnotes
Acknowledgement
The authors thank BioRender.com where the figures were produced.
Authors’ Contributions
B.H.: Conceptualization; article writing and editing; L.S: article writing; L.P.H.: Conceptualization; article writing and editing, funding acquisition, guarantor of this work.
Author Disclosure Statement
The authors have declared no competing interests and no personal financial interests. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding Information
This work was supported by the Grant Agency of the Czech Republic, grant No. 22-11439S to L.P.H. and by the project National Institute for Research of Metabolic and Cardiovascular Diseases (Program EXCELES, ID Project No.