
Abstract
Background:
Inflammation is one of the most important pathways in innate immunity and its relationship with redox biology is becoming increasingly clear in the last decades. However, the specific redox modes and pathways by which inflammation is produced are not yet well defined.
Significance:
In this review, we provide a general explanation of the reactive oxygen species (ROS) production and quenching modes occurring in mammalian mitochondria, as well as a summary of the most recent advances in mitochondrial redox biology and bioenergetics regarding sodium (Na+) homeostasis. In addition, we provide a collection of examples in which several inflammatory pathways have been associated with specific modes of either mitochondrial ROS production or quenching.
Innovation:
The role of Na+ in mitochondrial biology is being developed. Since its discovery as a second messenger, the research of its role in the immune system has emerged. Now, the role of Na+ in mitochondrial bioenergetics has recently been identified, which owns unprecedented applications. The potential implication of Na+ in inflammatory mechanisms grows as its role does not only cover ROS production and respiration but also the control through the management of mitochondrial membrane potential.
Future directions:
Na+ is becoming relevant for mitochondrial biology. Thus, processes regarding mitochondrial bioenergetics, redox state, or metabolism may probably need to include the study of Na+ in their road map. Some of these pathways are involved in inflammation and more are possibly to come. This review is expected to serve as a bridge between both fields. Antioxid. Redox Signal. 42, 868–884.
Introduction
In the last decades, the importance of mitochondria and, particularly, their redox reactions have caught up the attention of immunologists. Among the most conserved responses in innate immunity, inflammation is perhaps one of the most studied ones, which is aimed to combat cell injury or infection. In this process, mitochondria play important and specific roles (Marchi et al., 2023) that require a fine-tuned coordination among structure and function. In particular, the role of sodium (Na+) in core mitochondrial functions has recently reshaped our understanding of this organelle in (patho)physiology. Therefore, it is crucial to revise our perspective on how the regulation of this cation, in relation to mitochondria, could influence critical homeostatic processes, such as inflammation.
Mitochondria are required for energy transduction, in which the redox power of reducing equivalents is transformed into adenosine trinucleotide phosphate (ATP) through a phosphorylation reaction, a process called oxidative phosphorylation (OXPHOS). Reducing equivalents are nicotinamide adenine dinucleotide hydrogen (NADH) or succinate, which reduces either complex I (CI) or complex II (CII) which subsequently transfer their electrons to ubiquinone (CoQ), forming ubiquinol (CoQH2). CoQH2 is oxidized by complex III (CIII), which, in turn, reduces cytochrome c (cyt c). Finally, cyt c transports these electrons to complex IV (CIV), which transfers them to oxygen (O2), producing H2O. All these complexes form what is called the mitochondrial electron transport chain (mETC). As CI, CIII, and CIV transfer electrons, the energy released by their redox reactions is used to pump H+ from the matrix to the intermembrane space, creating a pH difference (proton gradient, ΔpH) across the inner mitochondrial membrane (IMM). This difference in charges also generates an electric potential, called mitochondrial membrane potential (ΔΨmt). The sum of ΔΨmt and ΔpH components results in the so-called H+-motive force (Δp), being ΔΨmt around 80%–90% of Δp (Nicholls, 1974). The buildup of Δp is essential for the activity of complex V (CV) as it moves H+ back to the matrix, using the energy released to phosphorylate adenosine dinucleotide phosphate (ADP) into ATP. OXPHOS can be organized in a panoply of plastic quaternary assemblies called supercomplexes that optimize substrate oxidation and metabolic wiring (Hernansanz-Agustín and Enríquez, 2022).
Most of the processes in mitochondria are governed by ΔΨmt, one of them being the movement of molecules and ions across the IMM (Fig. 1). Ca2+ enters the mitochondrial matrix through the mitochondrial calcium uniporter (MCU), which is powered by the ΔΨmt. Once in the matrix, Ca2+ can activate multiple enzymes and processes, such as the enzymes isocitrate dehydrogenase (IDH) or the 2-oxoglutarate dehydrogenase (2OGDH) in the tricarboxylic acid cycle (TCA cycle) (Hansford, 1991), which increase the production of NADH and CI-linked respiration, ΔΨmt, and ATP production (Fig. 1). Interestingly, Ca2+ can be retained in the matrix by reacting with phosphate to form calcium phosphate-like precipitates, which are increasingly considered one of the largest stores of Ca2+ within the cell (Chinopoulos and Adam‐Vizi, 2010; Wolf et al., 2017). Ca2+ can leave the mitochondria through two different pathways. One is the Na+-dependent pathway, which is triggered by the mitochondrial Na+/Ca2+ exchanger (NCLX). This pathway is known to be a major mitochondrial Ca2+ exit pathway (Samanta et al., 2018), which becomes inhibited with decreasing mitochondrial pH and ΔpH (Kostic et al., 2018). In contrast, a Na+-independent pathway, which is exerted by the transmembrane BAX inhibitor motif containing protein 5 (TMBIM5), interchanges Ca2+ by H+, directly linking Ca2+ exit with the H+ gradient generated by the mETC (Austin et al., 2017; Patron et al., 2022). TMBIM5-dependent mitochondrial Ca2+ efflux is sizable at low mitochondrial pH and ΔpH, whereas it becomes inhibited at higher, rather physiological mitochondrial pH (Austin et al., 2017), contrasting with NCLX activity. This may reflect the possibility that both pathways function alternatively in mitochondria working under different conditions and circumstances. The mitochondrial ΔpH generated by the mETC and the activity of NCLX are linked through the function of the mitochondrial Na+-specific, Na+/H+ exchanger (NHE), which we have recently identified as CI (see The Novel Roles of Na+ in Mitochondrial Biology; Hernansanz-Agustín et al., 2024a). In contrast, there is a little activity linking other cations with the H+ gradient under normal bioenergetic conditions. This activity is exerted by the Na+-unspecific NHE, most probably driven by the leucine zipper-EF-hand 1 (Fig. 1). Very interestingly, the entry/exit pathway of mitochondrial Mg2+ is exerted by a protein axis similar to the mitochondrial Ca2+ machinery as follows: a uniporter entering Mg2+, called mitochondrial RNA splicing 2 (Daw et al., 2020), and a Na+-dependent Mg2+/Na+ exchanger, the solute carrier 43A1 (Mastrototaro et al., 2016) (Fig. 1). This may be of central importance for physiology since Mg2+ is required for adenine nucleotides usage and strikes on the potential importance of mitochondrial Na+ in bioenergetics, physiology, and metabolism.
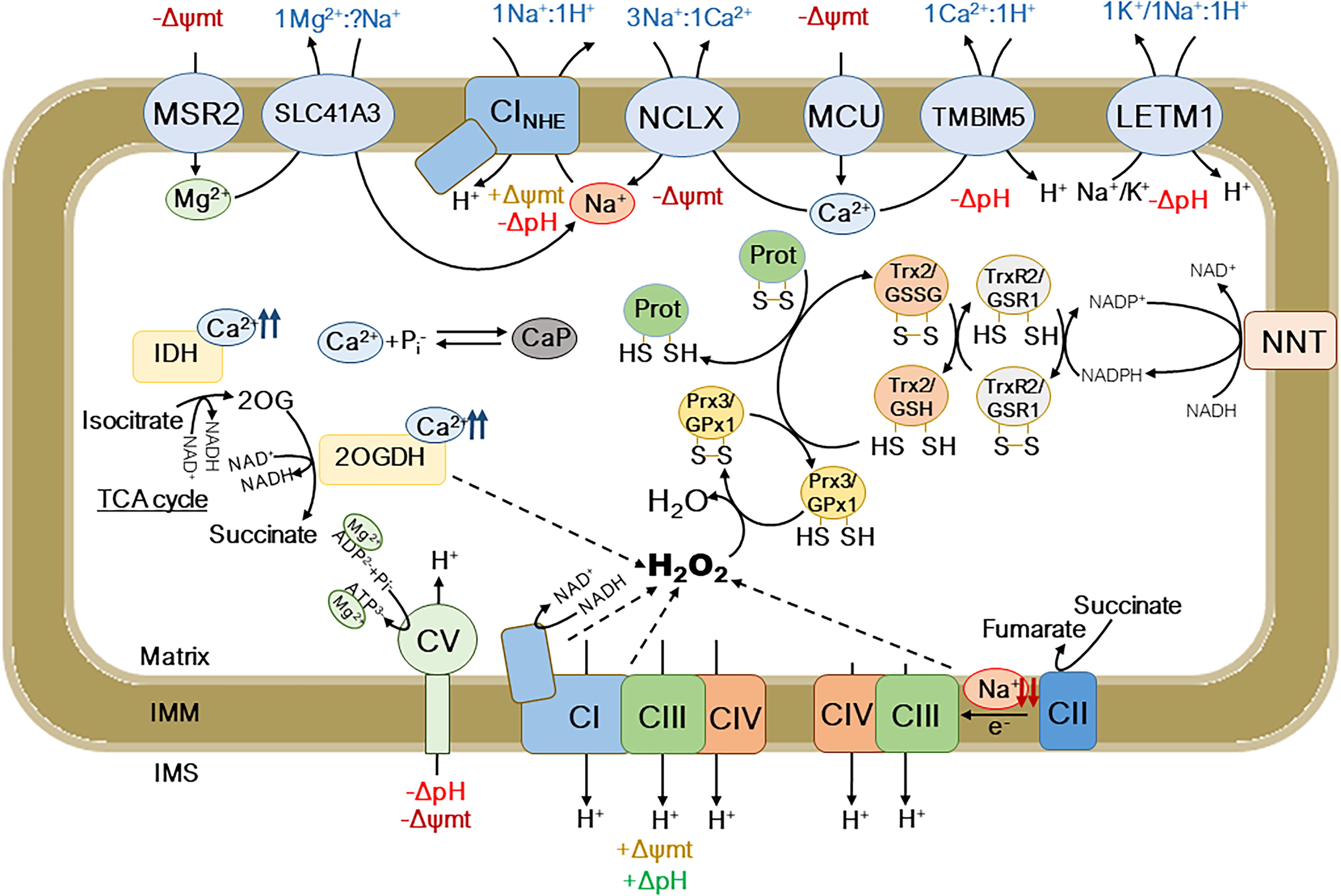
How Mitochondria Elevate Reactive Oxygen Species Under (Patho)Physiological Conditions?
Reactive oxygen species (ROS) are by-products of O2 metabolism, which are formed by its one electron reduction. These reactions can either be spontaneous or catalyzed by enzymes in the antioxidant system. Donation of one electron to O2 yields superoxide anion (O2 ●−), which occurs at the level of the mETC or TCA cycle enzymes. Superoxide dismutase (SOD) carries out dismutation of this radical into hydrogen peroxide (H2O2), a reaction that requires the donation of another electron. In the mitochondrial matrix, this is achieved by the MnSOD or SOD2. Then, H2O2 can have multiple roles, as it is a second messenger; however, it can also be quenched by several enzymes in the cell. H2O2 reacts with thiols in the structure of proteins within the cell, which not only allows the modification of their function, location, or stability but also is part of the antioxidant system, permitting the correct quench of this type of ROS. Within the mitochondria, the thiols in peroxiredoxin 3 (PRX3) or glutathione peroxidase 1 (GPX1) react with H2O2, forming water and becoming oxidized. PRX3 and GPX1, then, become re-reduced by thioredoxin 2 (TRX2)-thioredoxin 2 reductase (TRXR2) or glutathione/glutathione reductase (GR) systems, respectively, restoring their basal state. To note, other proteins with their thiols oxidized become re-reduced by either system, as well as by glutaredoxin 2 (GRX2). Finally, TRXR2 becomes reduced again by the pool of nicotinamide adenine dinucleotide phosphate (NADPH) within the mitochondria, which ultimately depends on the action of several enzymes, among which the nicotinamide nucleotide transhydrogenase (NNT) stands out (Fig. 1) (Ronchi et al., 2013).
Mitochondrial ROS levels depend on both the production and the quenching of all their different types. Changes in the physiological state and defects in any enzyme or substrate in the antioxidant system will evoke a rise in the level of the corresponding oxidant species. A particular case is that of SOD and similar O2
●− dismutating agents, in which raises in their activity will not only produce a decrease in O2
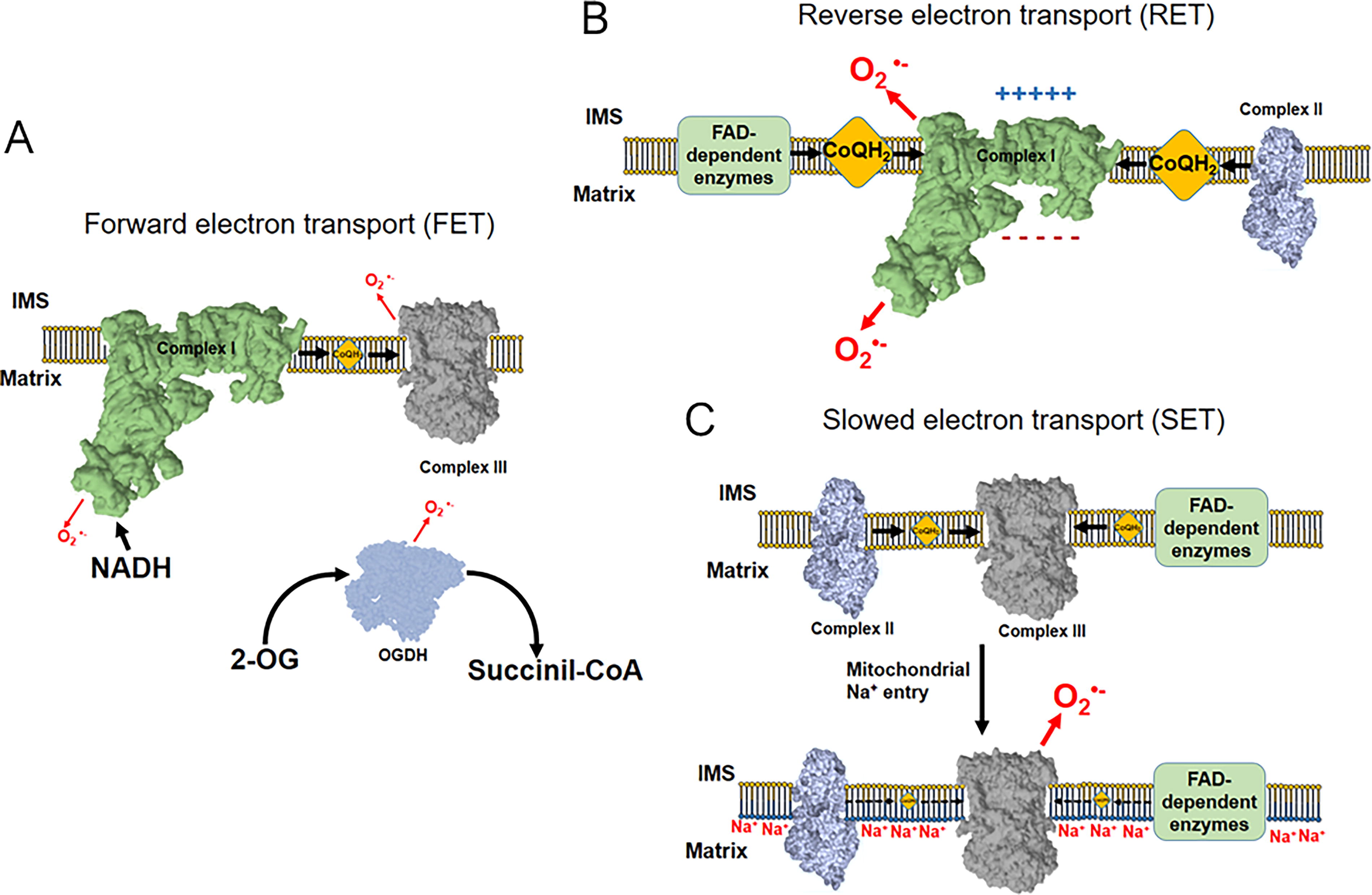
●− levels but also increase in the levels of H2O2 (Hernansanz-Agustín et al., 2014). In addition, increased production of ROS by enzymes will elicit raises in their steady-state levels. Thus, there are three main ways by which mitochondria produce ROS under physiological conditions (i.e., proven in whole cells or tissues) (Hernansanz-Agustín and Enríquez, 2021) as follows: Within the mitochondria, the primary ROS produced is O2
●− as it is a by-product of the activity in the respiratory chain. In contrast, other enzymes in the mitochondria also produce O2
●−, such as those in the TCA cycle (for further discussion on the molecular mechanisms, see Hernansanz-Agustín and Enríquez, 2021; Fig. 1). In the normal physiological function of the mitochondria, the mETC and TCA cycle enzymes produce O2
●− as a consequence of the nonperfect coupling of the electron flow in the bioenergetic system. The enzymes mostly contributing to it are CI, CIII, and 2OGDH. This type of ROS production is called forward electron transfer (FET) (Quinlan et al., 2014, 2013; Wong et al., 2017) and occurs under normal circumstances (Fig. 2A). A particularly interesting case is that of supercomplexes since the absence of the supercomplex assembly factor 1 promotes larger ROS production by the N-respirasome (i.e., supercomplex I+III2+IV) in FET (Calvo et al., 2020). When succinate or other substrate of any other mitochondrial flavin adenine dinucleotide (FAD) enzyme accumulates in the mitochondria, there is a possibility that CI reverses its reaction and oxidizes CoQH2, reducing NAD+, and bringing back four H+ into the mitochondrial matrix. This reaction, as CI consumes large amounts of ΔΨmt and requires CoQH2, can be inhibited by either CI Q-site inhibitors, such as rotenone or piericidin A, or by OXPHOS uncouplers, such as carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP). In addition, it is inhibited by ADP, as it activates forward CV activity and partially dissipates Δp. This is the so-called CI reverse electron transfer (RET) and it is one of the largest O2
●− producers in the mitochondria. The precise location of O2
●− production within CI is not yet clear (Fig. 2B), as there is a debate on whether Q module or N module is responsible for the actual electron transfer to O2 (Brand et al., 2016; Genova et al., 2001; Komlódi et al., 2018; Pryde and Hirst, 2011). This simple yet quite productive mode of ROS generation has been observed in inflammation (see Different Modes of ROS Production in Inflammation). In the last few years, there has been an advance in the relationship between one mode of mitochondrial ROS production and inflammation. This mode is that occurring under hypoxic conditions which, given that it is also present under other physiological conditions and not only under hypoxia, it is preferable to name it slowed electron transfer (SET). This type of Na+ dependent ROS production by mitochondria was first described to occur under situations of acute hypoxia and in nonspecialized cells (Hernansanz-Agustín et al., 2020). In summary, CI undergoes a conformational change under acute hypoxia by which it loses its catalytic activity and H+ pumping capacities (Hernansanz-Agustín et al., 2017). This makes the mitochondrial matrix to acidify, which, in turn, partially dissolves the matrix calcium phosphate precipitates, releasing soluble Ca2+. This activates NCLX which introduces Na+. This monovalent cation interacts with phospholipids in the inner side of the IMM, reducing its fluidity and slowing down CoQH2 transfer from CII or other FAD-containing enzymes to CIII, promoting the production of O2
●−. Importantly, this does not occur between CI and CIII, since most CI is attached to CIII into supercomplexes, maximizing their proximity and disabling the effects of IMM fluidity in CoQ-dependent electron transfer. The basis of this mode of ROS production lies on the fact that IMM fluidity becomes altered due to mitochondrial Na+ entry into the matrix and interaction with its phospholipids (Fig. 2C). The major Na+ entry pathway in the mitochondrial matrix is NCLX, though there may be other possible routes, depending on the cell type and metabolic condition (Hernansanz-Agustín and Enríquez, 2022). In this mode of ROS production, there is not only an increase in the production of oxidant species but also a decline in respiration, particularly that of CII or FAD-linked enzymes. SET has been observed to activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) inflammatory pathway (see Different Modes of ROS Production in Inflammation).
In addition, there are several cases on how lowering enzymes from the antioxidant system can impact on the levels of ROS by mitochondria in vivo: In line with the pro-oxidant role of Na+ in mitochondrial redox biology, increasing cytosolic Na+ levels promote a decrease in the steady state of mitochondrial Ca2+. This occurs in failing cardiac myocytes. Lowering mitochondrial Ca2+ levels reduce the activity of the TCA cycle dehydrogenases, impacting the regeneration of NADH (Fig. 1) and, thus, of NADPH, one of the main antioxidant buffers in the cell. This translates into an increase in the levels of H2O2. As in the previous mode of ROS production, this pathway reflects the intimate relationship between cation homeostasis and metabolic control of redox processes (Kohlhaas et al., 2010). This type may also reflect a decrease in CI-dependent respiration. Another example of how ROS levels can vary as a function of the action of antioxidant enzymes is the case of NNT. This protein reversibly converts NADH in NADPH, acting in this way as a source of NADPH in the mitochondria under normal conditions. Pathological metabolic demand reverses the action of NNT, now consuming NADPH to fuel NADH pool and ATP production. This occurs in heart failure, in which excessive NADPH expenditure increases the levels of H2O2 and causes oxidative stress and necrosis. Animals lacking functional NNT show ameliorated pressure overload-induced heart failure. This is due to the increased levels of NADPH, which cannot be converted into NADH, and that can better quench the strong rise in H2O2 during this pathological situation (Nickel et al., 2015). In contrast, in liver, NNT functions using NADH to replenish NADPH stores. Thus, the lack of NNT promotes NADPH deprivation, large H2O2 production, and higher susceptibility to Ca2+-induced permeability transition (Ronchi et al., 2013). This again highlights the intricate relationship between the mETC function with the control of redox homeostasis at the molecular level in different tissues.
The Novel Roles of Na+ in Mitochondrial Biology
In the last few years, there have been important advances on the role of Na+ in mitochondrial biology. Until recently, Na+ was considered as a partially innocuous cation with a collateral function in the maintenance of plasma membrane potential, thanks to the role of the Na+/K+ ATPase.
Old functions in a world under construction
Two of the first investigators interested in the role of Na+ in mitochondrial biology were Peter Mitchell, who received the Nobel prize for the chemiosmotic theory in 1978, and Albert Lehninger. These researchers discovered that Na+, in contrast to other monovalent or divalent cations, was able to reversibly (i) enter mammalian mitochondria, (ii) in an exchange for H+, and (iii) electroneutrally (Gear and Lehninger, 1968; Mitchell and Moyle, 1969). In addition, Mitchell observed that Na+/H+ antiport was faster than any other channel-mediated exchange (Mitchell and Moyle, 1967) while others that there was a Na+-specific and a Na+-unspecific NHE (Nakashima and Garlid, 1982). All this work was necessary to functionally define each of their features as follows: the Na+-specific NHE is fast-acting antiporter, becomes inhibited by alkaline pH, low amounts of Li+, and high amounts of Mg2+, whereas the Na+-unspecific NHE can transport other alkali metals, such as K+ or Li+, its kinetics are slow under physiological conditions, it becomes activated under alkaline pH, and is inhibited by low/ physiological amounts of Mg2+ (Brierley et al., 1988, 1978; Douglas and Cockrell, 1974; Nakashima and Garlid, 1982). All the results point to the fact that one NHE activity is needed under basal, physiological conditions, whereas the other would be only required under certain specific scenarios. These data also helped to set the conditions necessary for the discovery of the pathway responsible for mitochondrial Ca2+ exit, which was observed to be facilitated by Na+. Interestingly, Na+-dependent mitochondrial Ca2+ exit was also triggered by Li+ (Carafoli et al., 1974). After these discoveries, the field stopped their interest in the roles of Na+ on mitochondria, but for some publications during the 80–90s (Baysal et al., 1994, 1991; Jung et al., 1995, 1992). During the posterior two decades, the role of Na+ in mitochondrial biology was neglected. These seminal discoveries were part of the basis for the new world that was opening, which we all know as bioenergetics.
New players and unexpected roles of Na+ in signaling and bioenergetics
The functions mentioned above have not been assigned molecularly to players until very recently. In 2010, a seminal article by Israel Sekler’s group identified NCLX for the first time (Palty et al., 2010), for which its sensitivity to Li+ ions became a critical feature (Palty and Sekler, 2012). This discovery served as a platform to study the involvement of NCLX in hypoxic adaptation.
In 2020, we discovered SET that occurred during the first minutes of hypoxia and that was initiated by a conformational change, the so-called active–deactive transition, of CI. Interestingly, SET involves the action of a new second messenger system, being constituted by a primary effector, CI, a second messenger, Na+, and a secondary effector, the IMM (Hernansanz-Agustín and Enríquez, 2022). However, the nature of the signal transducer, which is involved in O2 sensing during acute hypoxia, is not yet known in nonspecialized cells.
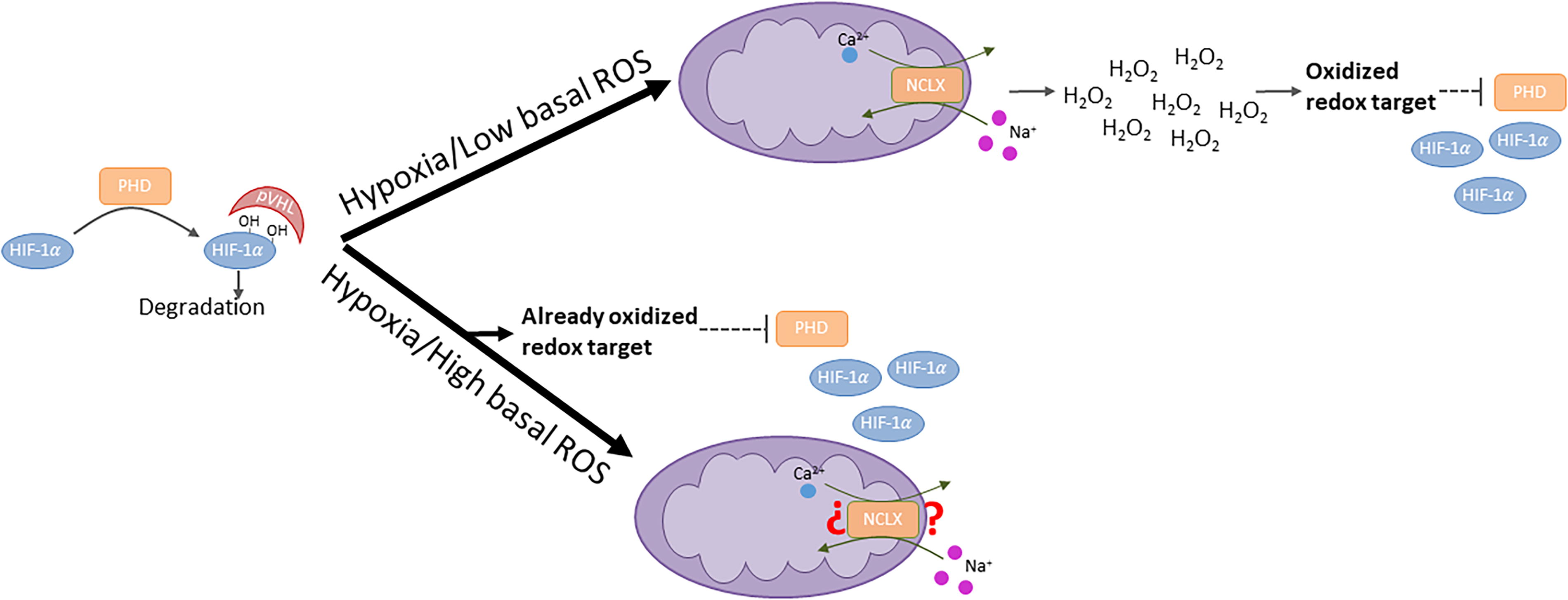
Downstream the action of Na+ is SET-dependent ROS production by CIII, which controls adaptive processes, such as hypoxic pulmonary vasoconstriction (Hernansanz-Agustín et al., 2020) and hypoxia-inducible factor (HIF)-α stabilization (Choya-Foces et al., 2024). We have recently observed that pharmacological or small interfering RNA inhibition of NCLX was able to decrease the hypoxic stabilization and activity of HIF-α in culture and ex vivo. Strikingly, we also observed that mouse embryonic fibroblasts (MEFs) chronically lacking NCLX were able to stabilize it during hypoxia up to control levels. This apparent contradiction could be solved by considering that chronically NCLX-depleted MEFs displayed higher levels of basal ROS than acutely inhibited cells (Hernansanz-Agustín et al., 2020) and that this could mediate the observed stabilization. Indeed, we incubated NCLX-depleted MEFs with either N-acetyl cysteine or l-cysteine only during the 2 h of normoxia before hypoxia, removing them upon hypoxia, and both consistently lowered the hypoxic stabilization of HIF-α, whereas wild-type controls remained unchanged. This meant that lowering normoxic, basal ROS levels in cells with altered redox homeostasis impacted HIF-α during hypoxia (Fig. 3). In addition, we observed that this did not occur in isogenic mouse adult fibroblasts (MAFs) without active NCLX, pointing to the specific metabolic features of MEFs (i.e., embrionic cells) as the causatuve difference in HIF-α stabilization (Choya-Foces et al., 2024). These results have extensive implications as they call for caution when assessing HIF-α stabilization since redox homeostasis can be different in diverse cell types, environments, and upon different genetic transformations (e.g., chronically vs acute NCLX-depletion vs. wild type; MEFs vs. MAFs; etc). Thus, given this diversity in redox responses and cell types, conclusions should be made with extreme caution.
As mentioned above, the historical functional characterization has been essential for the molecular identification of NCLX. Now, we have molecularly characterized the identity of the Na+-specific NHE based on the following: (i) the functional characterization in the previous 60 years (Hernansanz-Agustín et al., 2024b) and (ii) a collection of genetic models (Hernansanz-Agustín et al., 2024a). CI is a modular complex, composed by 44 different subunits and evolutionarily related to both bacterial NiFe hydrogenases and Na+/H+ antiporters (Kampjut and Sazanov, 2020): The N module accepts electrons from NADH and sends them, through a series of iron-sulfur clusters, to the Q module, which, in turn, reduces CoQ. The Q module transmits this energy to the P module, which pumps four H+. As noted before, the P module is evolutionarily related to Na+/H+ antiporters, a feature that called the attention of other researchers, which investigated this potential function in bacteria, yeast and beef in vitro to find that pure CI in proteoliposomes were able to perform NHE (Lin et al., 2008; Roberts and Hirst, 2012; Stolpe and Friedrich, 2004). However, as mentioned above, the complexity of the NHE function in mitochondria is enormous, and further characterization was required. To do it, we approached the problem from four different angles: First, we took advantage of the historical features present in the literature and observed that, between both types of NHE, CI could be the Na+-specific NHE, since it was fast acting, Na+-specific, inhibited at high pH, also by low Li+ and high Mg2+ (Hernansanz-Agustín et al., 2024a). In addition, we used genetic models deleting different subunits in CI structure. This was possible since removing some of its subunits does not always promote whole CI disassembly. Indeed, eliminating subunits from the N module usually let the rest of the complex to stay assembled (Stroud et al., 2016). Thus, we deleted ND6, ND4, and NDUFB11, all in the CI P module, and observed a clear and profound loss in mitochondrial Na+-specific NHE function as the whole CI disappeared (Hernansanz-Agustín et al., 2024a). Importantly, using the G11778A mutant of the mitochondrial DNA (mtDNA), causing Leber’s hereditary optic neuropathy (LHON), which provokes a point mutation in ND4/R340H and does not affect assembly or any other activity of CI, we observed that CI NHE function was specifically altered. However, when the N module subunit NDUFS4 was deleted, we observed an increase in Na+-specific NHE activity, pointing to a role of the CI P module in it. We used pharmacological approaches to inhibit different modules of CI and observed that only when the inhibition took place at the P module, a decrease in Na+-specific NHE function was observed. All the above approaches were also performed in pure CI reconstituted into proteoliposomes.
But most importantly, we observed that the Na+-specific NHE activity of CI was necessary to establish full ΔΨmt and to set normal levels of respiration in mammalian mitochondria. We observed that the physiological function of CI NHE was to partially dissipate the mitochondrial ΔpH, increasing respiration and setting a parallel Na+ gradient, which contributed up to half of the ΔΨmt. This unprecedented role of Na+ in mitochondrial biology has extensive implications: (i) the Na+ gradient serves as a reserve of ΔΨmt in respiring mitochondria in healthy cells, as it cannot be used by CV; (ii) deregulation of CI NHE activity can depolarize mitochondria, lower their respiration, and lead to cell death, as occurring in LHON; and (iii) diminishing CI NHE may increase intramitochondrial Na+ and increase ROS production by SET. All these features are of critical importance as they link mitochondrial bioenergetics, physiology, and metabolism, all through CI (Fig. 4) (Kowaltowski and Abdulkader, 2025).
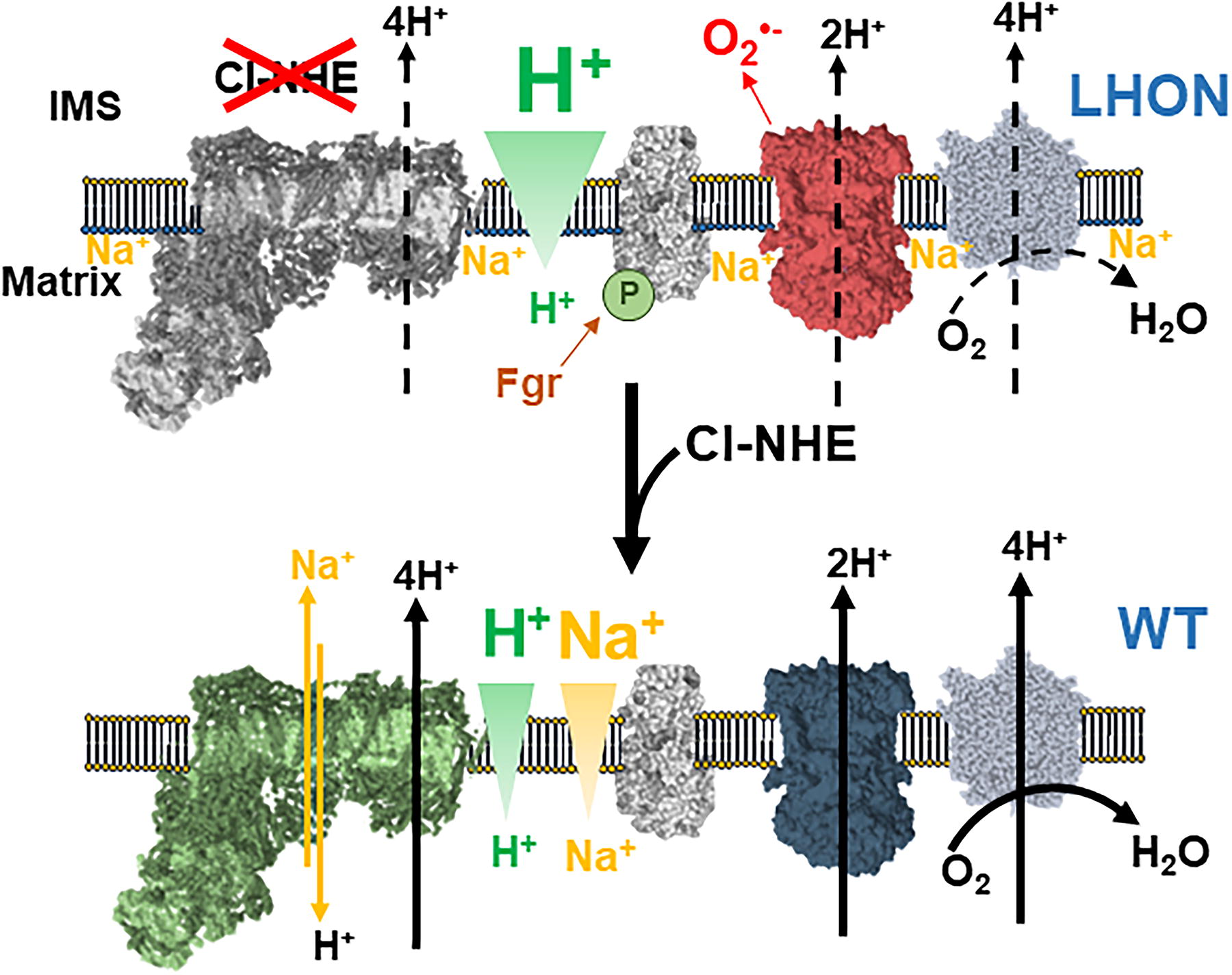
It is to note that CI, after undergoing the active/deactive transition, increases its NHE activity (i.e., mitochondrial Na+ exit pathway) (Hernansanz-Agustín et al., 2024a; Roberts and Hirst, 2012), but, in parallel, the hypoxic intramitochondrial Na+ content and SET increase. This apparent contradiction can be easily solved by considering that the Na+ gradient can only parallel the ΔpH. Thus, deactivation of CI lowers its H+ pumping activity and, in turn, the ΔpH, lowering the Na+ gradient and letting Na+ in the mitochondrial matrix.
Inflammation and Mitochondrial ROS
Inflammation is characterized as the reaction initiated by immune cells in response to infection or tissue damage, playing a role in both physiological and pathophysiological processes (Medzhitov, 2008). A well-regulated inflammatory response is typically beneficial, as it contributes to healing, tissue repair, and the prevention of additional tissue or cellular damage. However, when this response becomes uncontrolled, it can have harmful consequences (Medzhitov, 2008). Numerous diseases are associated with chronic unregulated inflammation, including autoimmune conditions such as multiple sclerosis (Filippi et al., 2018).
The relationship between the redox responses by mitochondria and the molecular inflammatory pathways has been a matter of interest since long ago. This is particularly important in the context of immune cells, where the cytosolic volume occupied by mitochondria is especially small in comparison with other cell types. In this way, the sensitivity of the redox pathways is higher as the distance traveled by oxidant and reducing molecules is shorter from their source to their target. Mitochondrial Ca2+ elevations activate the production of tumor necrosis factor-α, interleukin (IL)-1β, and IL-6 in cardiomyocytes, which drives cardiac inflammation and dysfunction after a physical insult (Maass et al., 2005). This could be related to mitochondrial ROS production since blockade of MCU decreased their levels, concomitantly with pro-inflammatory mediators (López-Armada et al., 2013). However, although the precise mechanism is still unknown, the evidence suggests a strong relationship between mitochondrial Ca2+ management, ROS production, and the trigger of inflammatory pathways. In this way, it would be interesting to study whether SET or the activation of TCA cycle dehydrogenases (Fig. 1) could be behind this scenario.
Inflammatory signaling is induced by ROS production, such as NF-κB or NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome. NF-κB becomes activated and translocates to the nucleus upon elevations in oxidant species (Canty et al., 1999), and the ROS-dependent NLRP3 inflammasome activation depends on several mechanisms. A recent communication studied the relationship of mitochondrial ROS production with activation of NF-κB tissues. In particular, SET-dependent generation of oxidant species is the mechanism responsible for amplifying NF-κB signaling, which in turn promotes the neuroimmune cascade associated with neurodegeneration: astrocytic mitochondria, with high NCLX expression, are primed to produce elevated CIII-derived ROS in pathological conditions. This study highlights that the SET pathway may also be involved in other cellular responses, such as inflammation, ensuring further research in the field (Barnett et al., 2024).
An inflammasome is a multi-protein complex that, when activated, triggers the stimulation of caspase-1. This, in turn, promotes the upregulation of various inflammatory cytokines, such as IL-1β and IL-18, ultimately driving the initiation of an inflammatory response (Strowig et al., 2012). Mitochondrial ROS have the ability to activate the NLRP3 inflammasome (Bulua et al., 2011; Cruz et al., 2007; Zhou et al., 2011). In general terms, ROS produced by CI and CIII are necessary for the formation of the inflammasome complex (Billingham et al., 2022). When oxidant species are not effectively quenched by the scavenging mechanisms, they can lead to an uncontrolled inflammatory response in certain pathological conditions (López-Armada et al., 2013).
There are three general mechanisms by which mitochondrial ROS can induce activation of the NLRP3 inflammasome. The first depends on TRX-interacting protein. Inflammasome activators promote the ROS-dependent dissociation of TRX-interacting protein from TRX, enabling its binding to NLRP3. The absence of this interactor protein disrupts the activation of the NLRP3 inflammasome and the subsequent release of IL-1β. Concomitantly, animals with absent TRX-interacting protein show improved glucose tolerance and insulin sensitivity (Zhou et al., 2010).
The other two mechanisms are based on the binding of mtDNA to NLRP3. In one of them, excessive mitochondrial ROS production damages mtDNA, causing strand breaks and the formation of oxidized bases (Maynard et al., 2009). As a result, the mtDNA is released into the cytoplasm, where it binds to NLRP3, promoting its activation (Nakahira et al., 2011). Under inflammatory conditions, the damage cannot be fully repaired, leading to the accumulation of breaks in the mtDNA and, consequently, the sustained activation of the NLRP3 inflammasome (Zhao et al., 2020).
In the third mechanism, increased mitochondrial ROS production induces an oxidative form of mtDNA, which can bind to NLRP3 and produce its activation. It has also been suggested that a priming signal, such as the involvement of lipopolysaccharide (LPS)/Toll-like receptor 4, is required to activate NLRP3 (Shimada et al., 2012). Activation of the mtDNA/NLRP3 pathway leads to the release of IL-1β and consequently to increased vascular permeability (Wu et al., 2019). This, coupled with ATP depletion due to ROS-induced mitochondrial damage, compromises immune function and aggravates tissue hypoxia, exacerbating the immunological disorder characteristic of sepsis (Zhao et al., 2020).
These pathways highlight the importance of mitochondrial ROS production in the activation of inflammatory pathways at the molecular level. However, a central question is whether there are different modes of oxidant species production by mitochondria that signal toward inflammation and in what contexts these become active.
Different Modes of ROS Production in Inflammation
CI-dependent ROS in inflammation
An effort has been lately made to pinpoint the mechanisms leading to inflammation that are related to specific modes of ROS production by mitochondria. Macrophage pro-inflammatory phenotype, which is driven by their activation (e.g., by LPS) and metabolic reprogramming, makes them to shift from ATP production by OXPHOS toward a mitochondria-producing ROS by RET. Succinate-driven ROS production by mitochondria is able to promote the production of IL-1β through the enhancement of HIF-1α stabilization (Mills et al., 2016) (Fig. 5A). These interesting findings correlate well with recent results on neuroinflammation, in which CI is involved in neurotoxic damage through RET. These studies have shown that CI activity induced a state of constant activation in myeloid cells (Fig. 5A). This is favored by the production of ROS generated during RET. Inhibition of CI by small molecules or mutant mouse models disrupts oxidant species production by RET, providing protection against ROS-induced neuroinflammation (Peruzzotti-Jametti et al., 2024). In this context, Sanmarco et al., 2023 have identified a molecule capable of limiting oxidant species production by mitochondria. Cytochrome c oxidase subunit (NDUFA4)-like 2 subunit, a homolog of the CIV subunit NDUFA4 (Balsa et al., 2012), is induced by HIF-1α (Tello et al., 2011), for which stabilization under inflammatory conditions is mediated by lactate production in activated dendritic cells. This mechanism reduces CI activity and consequently regulates ROS production, which is involved in the control of pathogenic autoimmune T cells. This supports the idea that modulation of CI may offer protection against oxidant-mediated neurotoxicity (Sanmarco et al., 2023). These consequences in inflammation, after CI inhibition, highlight that CI-dependent ROS production is critical for the modulation of inflammatory responses and that one of the consequences of CI dysfunction is the dysregulation of the molecular mechanisms leading to inflammation. In this way, it is to note that one of the drivers in Leigh syndrome (CI disorder) pathogenesis is the activation of microglia and neuroinflammation (Aguilar et al., 2022; Daneshgar et al., 2022), which opens new and interesting venues for research. A detail that may pass unnoticed is that CI deficiency activates the tyrosine kinase Gardner-Rasheed feline sarcoma viral (v-fgr) oncogene homolog or FGR kinase to phosphorylate CII and increase its activity to compensate a lower CI electron transfer to O2 (Acín-Pérez et al., 2014). This mechanism may not only be sufficient to metabolically compensate TCA cycle intermediates but also to promote ROS production by CIII, supporting the role of CI in the activation of the inflammatory response (Zmijewski et al., 2009).

CII in mitochondrial ROS production during inflammation
The increase in CII activity is also required for pro-inflammatory macrophage-dependent host defense (Garaude et al., 2016) and diet-induced obesity (Acín-Pérez et al., 2020). This activation occurs through the action of the ROS-dependent FGR kinase, which phosphorylates succinate dehydrogenase A and activates CII activity, pushing electron transfer to CI and CIII (Fig. 5). The involvement of this kinase in inflammation through CII also correlates well with a recent finding linking CII subunits with the control of pro-inflammatory pathways in activated macrophages (Gobelli et al., 2023). It is worth mentioning that succinate can also promote inflammation through the activation of its cognate receptor succinate receptor 1, a G-protein-coupled receptor activated by succinate, able to mediate the production of pro-inflammatory cytokines, such as IL-1β (Mills et al., 2021).

SET-dependent ROS in inflammation
As mentioned above, SET is arising as a mechanism of action in inflammation. Seminal studies pointed that Na+ was able to promote inflammatory responses in mononuclear cells (Shapiro and Dinarello, 1995). Later, the role of Na+ in activating pro-inflammatory macrophages and T helper 17 (Th17) has been clarified (Fig. 5B) as follows: Na+ elicits profound metabolic changes, which include the stabilization of HIF-1α and nuclear factor of activated T cells 5 (NFAT5), among others (Jobin et al., 2021; Müller et al., 2019). Interestingly, Th17 response also elicits gut-initiated neurovascular and cognitive dysfunction (Faraco et al., 2018), as well as dysfunctional regulatory T cells (Tregs) (Safa et al., 2015) (Fig. 5B). All these events are mechanistically related to a metabolic switch, which has been proposed to occur through the alteration in mitochondria (Miyauchi et al., 2024). As mentioned above, the main mitochondrial Na+ entry pathway is through NCLX, which promotes SET under hypoxic conditions. However, mitochondrial Na+ also inhibits joint CII+CIII activity in macrophages (Geisberger et al., 2021) and Tregs (Côrte-Real et al., 2023), the latter mimicking the metabolic and gene expression profiles of Tregs after deletion of CIII (Weinberg et al., 2019), both in humans and mice (Fig. 5B). Interestingly, the inhibition of CII+CIII is accompanied by an accumulation of succinate (Côrte-Real et al., 2023). All these point to SET as a driving mechanism, leading to inflammation through the production of mitochondrial ROS.
Indeed, the SET-dependent pathway was identified as a mechanism able to activate NF-κB signaling, thereby driving the neuroimmune cascade implicated in neurodegeneration. Because of their high NCLX expression, astrocytic mitochondria seem functionally primed to generate elevated levels of CIII-derived ROS generation in the pathological scenario under study. In this study, the authors also observed that astrocytic CI-dependent ROS production is active under basal conditions in culture, but does not increase further upon stimulation. This absence could potentially result from the deactivation of CI by certain stimuli, pointing to a (patho)physiological regulation of CI deactivation in a context different from hypoxia (Barnett et al., 2024). The implication of SET in other, non-hypoxic scenarios reflects that this pathway may be implied in many potential yet unexplored cellular responses. Specifically, it seems clear that Na+ is becoming a very important player in the activation of the pro-inflammatory phenotype, which will surely deserve further investigation.
To note, HIF-α is a master regulator of inflammatory responses and its stabilization depends on mitochondrial Na+ accumulation (Choya-Foces et al., 2024; Hernansanz-Agustín et al., 2017). Thus, it is tempting to speculate that acute inhibition of Na+ entry into the mitochondria or substitution of Na+ by molecules unable to interact with phospholipids may be a suitable approach for the treatment of pro-inflammatory diseases. However, it is also possible that another, yet unknown, Na+ sensor partially controls diseases in which inflammation is involved. In this regard, either direct or indirect activation of p38/mitogen-activated protein kinase, NFAT5, or serine/threonine-protein kinase 1 by high Na+ has been shown to induce pro-inflammatory cytokine production and autoimmune disease (Kleinewietfeld et al., 2013).
Other sources of mitochondrial ROS in inflammation
Monoamine oxidases (MAOs), a class of enzymes that catalyze the degradation of catecholamines and biogenic amines, produce large amounts of ROS when stimulated. Inhibition of MAOs effectively reversed oxidative stress and inflammation in airway epithelial cells (Cui et al., 2017), in adipose tissue (Nagy et al., 2018) and cardiac cells (Deshwal et al., 2018), highlighting the role of these enzymes in the modulation of the activation of inflammatory pathways. Interestingly, as occurring with other modes of ROS production, MAOs are related to the formation of mitochondrial damage-associated molecular patterns, which are molecules released from mitochondria into the extracellular space during cell death and that include oxidized proteins, mtDNA, and lipids (Beucher et al., 2024).
Inflammation in the Context of a Mitochondrial Na+-Related Neuropathology
Recently, we have pinpointed the functional defect by which mutations in CI genes produce LHON. LHON is a mitochondrial disorder characterized by the sudden loss of central vision, primarily affecting young males with mutations in their mtDNA. There are three main mutations as follows: G11778A, T14484C, and G3460A, all of which affect different CI P-module subunits. G11778A mutants showed an intact CI NADH:CoQ oxidoreductase activity with a substantially lowered CI substrate-dependent respiration (Hofhaus et al., 1996). This is accompanied by an increase in mitochondrial ROS production (Howell, 2003). This paradoxical feature did not fit well with a chemiosmotic model only reliant on ΔpH as a driving force for the buildup of ΔΨmt. In this mutant, the CI NHE activity is decreased (Hernansanz-Agustín et al., 2024a), lowering their capacity to dissipate the mitochondrial ΔpH and to form a ΔNa+. Therefore, they lack the capacity to create a ΔΨmt deep enough to keep up standard energy requirements and respiration, and consequently, they cannot cater to the bioenergetic needs of the cell.
Vision loss in LHON is due to the degeneration of retinal ganglion cells (RGCs), ultimately causing optic nerve atrophy. RGCs death could be caused by bioenergetic failure and subsequent apoptosis, but this does not fully explain why these cells are affected while the rest remain healthy, neither elucidate why there are cases with spontaneous reversion of the disease. Thus, CI NHE most probably relates to nerve activity rather than with its survival, reflecting the involvement of mitochondria in neuron function and fitness.
In contrast, a lower CI NHE activity increases intramitochondrial Na+, triggering oxidant species production by SET and HIF activation. Given that ROS and HIF activity are linked to inflammation, it is possible that the autoimmune cases observed in LHON patients are related to overactivation of pro-inflammatory pathways due to this mechanism (Gofas et al., 2023; Govan et al., 1994; Kovacs, 2004; Zainal et al., 2002). In this regard, the potential relationship between mtDNA mutations and inflammatory responses awaits further interesting investigation.
Different Modes of Lowering Mitochondrial ROS Quenching-Producing Inflammation
Contrary to previous sections, here we will provide some specific examples on how decreasing the antioxidant capacity of mitochondria can produce inflammation. PRX3 is necessary for the quenching of H2O2 and oxidizes TRX2, which, in turn, oxidizes TRXR2 that relies on the NADPH pool.
Mitochondrial antioxidant enzymes involved in the control of inflammation
A decrease in PRX3 in mouse adipose tissue leads to increased oxidant species and protein carbonylation. This damage is intricately correlated with lower mitochondrial biogenesis, impaired glucose tolerance, inflammation, adipocyte hypertrophy, and increased fat mass. Thus, PRX3 is an essential component for the maintenance of normal adipocyte characteristics and redox state and function to avoid metabolic alterations (Huh et al., 2012). In addition, the absence of PRX3 exacerbated fibrosis, inflammation, and chronic kidney disease, coupled with mitochondrial oxidative stress, in both obstructed and diabetic kidneys, as well as in proximal tubular epithelial cells. This was due to the activation of macrophages, resulting in increased levels of pro-inflammatory cytokines, even under basal conditions in both tissues and cells (Fig. 6). This highlights the importance of the modulation of interactions between macrophages and tubular epithelial cells, particularly under a pro-inflammatory context (Hwang et al., 2019). In the same way, PRX3 knockdown aggravated acetaminophen-induced liver injury through the contribution to inflammation by increasing of NLRP3 inflammasome activity (Wang et al., 2021).
During pancreatitis, mice deficient in sulfiredoxin (SRX) exhibited a significant accumulation of p53 within mitochondria, along with increased levels of mitochondrial H2O2 and enhanced pancreatitis (Fig. 6). p53 contributes to ROS accumulation by inactivating PRX3 through hyperoxidation. The action of SRX is exerted by inactivation of p53-induced mitochondrial ROS production, modulating the entry into necrosis and, thereby, playing a protective role in maintaining cellular homeostasis (Rius-Pérez et al., 2022).
The upregulation of GRX2 increased the activation of nuclear factor erythroid 2-related factor 2 (NRF2) signaling, which was linked to elevated phosphorylation of Ras-related C3 botulinum toxin substrate 1 (RAC)-α serine/threonine-protein kinase and glycogen synthase kinase-3β (GSK-3β). The activation of this pathway protected against hypoxia/reoxygenation-induced apoptosis, oxidative stress, and inflammation (Li et al., 2021). In line with this, GRX2 plays a critical role in mitigating high-fat diet-induced brain injury, at the hippocampal level, by alleviating insulin resistance, inflammation, oxidative stress, and mitochondrial dysfunction, primarily through the regulation of GSK-3β signaling (Wohua and Weiming, 2019).
Increasing the amount and activity of TRXR2, the rate-limiting enzyme in the mitochondrial TRX system leads to increased resistance to metabolic dysfunction induced by high-fat diet and to raised glucose tolerance and decreased fat deposition. Interestingly, this phenomenon was partially due to enhancements in the TCA cycle enzymes citrate synthase, IDH, and malate dehydrogenase. In parallel, OXPHOS function also increased at the level of CI, CII, CIV, and CV. Given that the protein levels of these mitochondrial components were not varied, it is possible that TRXR2 overexpression performed its function by reducing thiols groups in all the mentioned enzymes, precluding overoxidation and inhibition (Chocron et al., 2022). The latter is known to occur in CI (Onukwufor et al., 2022), CII, CIII (Taylor et al., 2003), CIV (Puntel et al., 2013), CV (Cobley et al., 2020), and IDH (Yoshida and Hisabori, 2014). These findings suggest an interesting line of research studying how TRXR2 controls the metabolism and its plasticity upon (patho) physiological situations (Fig. 6).
Another example is that loss of TRX2 leads to an increase in mitochondrial ROS levels, dysregulation of the mitochondrial integrity, and release of mtDNA to the cytosol, activating aberrant inflammasome pathways in brown adipose tissue (Fig. 6). This leads to the impairment of adaptive thermogenesis by suppression of fatty acid oxidation (Huang et al., 2022).
Transcriptional modulators acting on the mitochondrial antioxidant system
All these pathways can be modulated by the transcription coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α). It plays a key role in regulating mitochondrial antioxidant defense in cells by enhancing the expression of various mitochondrial antioxidant enzymes, including MnSOD/SOD2, PRX3, uncoupling protein 2, TRXR2, and TRX2 (Olmos et al., 2009; Valle et al., 2005). This regulation helps protect cells from mitochondrial failure. Notably, the upregulation of antioxidant defenses by PGC-1α is essential for preventing cell death associated with mitochondrial failure (Valle et al., 2005). Conversely, PGC-1α deficiency is linked to greater susceptibility to oxidative stress in mice (Lin et al., 2004; Pérez et al., 2019; Sánchez-Ramos et al., 2017). Importantly, PGC-1α-driven upregulation of antioxidant target genes is not only crucial for preventing oxidative damage but also for reducing mitochondrial ROS levels and maintaining mitochondrial integrity during cell differentiation (Baldelli et al., 2014; Rius-Pérez et al., 2020). This central role of PGC-1α in the regulation of the mitochondrial antioxidant system translates can be seen in the context of inflammation. Mouse lacking this protein displays spontaneous subclinical kidney injury characterized by tubulointerstitial inflammation, leading to premature death (Fontecha‐Barriuso et al., 2019). In aged mouse, the levels of PGC-1α diminish in muscle, which has been related to systemic inflammation (Sczelecki et al., 2014).
Another critical regulator of the mitochondrial antioxidant defense is the NRF2. Under normal conditions, NRF2 is bound to its cytoplasmic repressor, Kelch-like ECH-associated protein 1 (KEAP1), which facilitates the ubiquitination of NRF2 and subsequent proteasomal degradation through its association with the cullin 3-based E3 ubiquitin ligase complex. After the oxidation of highly reactive cysteine residues (sensor residues) on KEAP1, its ability to target NRF2 for degradation becomes impaired. As a result, NRF2 stabilizes, binds to the antioxidant response element in heterodimeric complexes with small MAF transcription factors, and activates the transcription of cytoprotective (phase 2) genes. The sensor residues on KEAP1 can be modified by ROS or small molecules, such as tert-butylhydroquinone, sulforaphane, fumarate, hydroxytyrosol, or curcumin (Feissner et al., 2009). NRF2 enhances the transcription of genes involved in the antioxidant control of the mitochondria. This is the case of the Parkinson’s disease (PD) protein 7, widely known as DJ-1, which maintains CI activity, precluding mitochondrial ROS production (Hayashi et al., 2009). NRF2 also controls the expression of genes, critical for mitochondrial metabolism, such as IDH (Mitsuishi et al., 2012; Wu et al., 2011) and aldehyde dehydrogenase (Ushida and Talalay, 2013), which are also enzymes controlling the NADPH/NADP+, a critical ratio essential for the maintenance of the redox balance within mitochondria. This variety of functions permits NRF2 to maintain hormesis (aka, the response in an adaptive manner to an external factor or agent that would be harmful at high doses) in this organelle after its activation. In this way, activation of NRF2 has been shown to protect models of PD through the maintenance of a hormetic-like environment. This may relate to mitochondrial metabolism through the regulation of neuronal nitric oxide (NO•) synthase (Sampath et al., 2022), in which production of NO• may exert their neuroprotective effects through the partial inhibition of CIV and decrease in ROS production (Calabrese et al., 2007).
Concluding Remarks
The type, location, and the functional state of the ROS-producing enzymes are all critical factors in physiology and disease. Thus, understanding the molecular mechanisms by which oxidant species control processes as important as inflammation is essential for the development of future tools to take over them. For the moment, there are two clearly defined modes of mitochondrial ROS production involved in inflammation, RET and SET, and it will be fundamental to investigate whether and which others, if any, are controlling redox processes of inflammation. In addition, the comprehension of the further role of the antioxidant system in redox signaling contributes for the expansion of the frontiers in the control of inflammatory pathways.
Footnotes
Acknowledgments
Authors’ Contributions
Conceptualization: P.H.-A. Writing: M.G.-H., L.G.-A., and P.H.-A. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This study was supported by MICIU IJC2020-042679-I and RYC2022-036516-I.