
Abstract
Aims:
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder among the elderly. Uric acid (UA), the end product of purine metabolism, functions as a potent free radical scavenger and helps mitigate oxidative stress. Several epidemiological studies revealed that serum UA levels are negatively correlated with the risk of AD; however, the molecular mechanisms remain unclear. Notably, β-amyloid (Aβ) deposition is implicated in the disruption of mitophagy, leading to neuronal apoptosis. In this study, we aim to elucidate the link between UA and AD and explore the underlying mechanisms.
Results:
We demonstrated that UA improved cognitive impairment in 5×FAD mice and reduced neuronal apoptosis both in vivo and in vitro. UA reversed the expression of phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1), p-ParkinS65, parkin, microtubule-associated protein 1 light chain 3 II/I, and p62 proteins inhibited by Aβ treatment, alleviated Aβ induced mitochondrial dysfunction, and disturbed dynamics. We found that UA activated nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1(HO-1) signaling both in vivo and in vitro. Furthermore, ML385, a Nrf2-specific inhibitor, reversed the increase in mitochondrial membrane potential and mitophagy promoted by UA and increased neuronal apoptosis in HT22 cells. The antiapoptotic effects of UA in HT22 cells were prevented by treatment with small interfering RNAs targeting PINK1.
Conclusions and Innovation:
These data suggest that UA stimulates PINK1/parkin-mediated mitophagy reducing Aβ-induced neuronal apoptosis through the Nrf2/HO-1 pathway, which plays a neuroprotective role in AD. Our findings confirmed that UA effectively reduces neuronal damage and cognitive impairment, highlighting its potential clinical applications in the treatment of AD. Antioxid. Redox Signal. 43, 381–399.
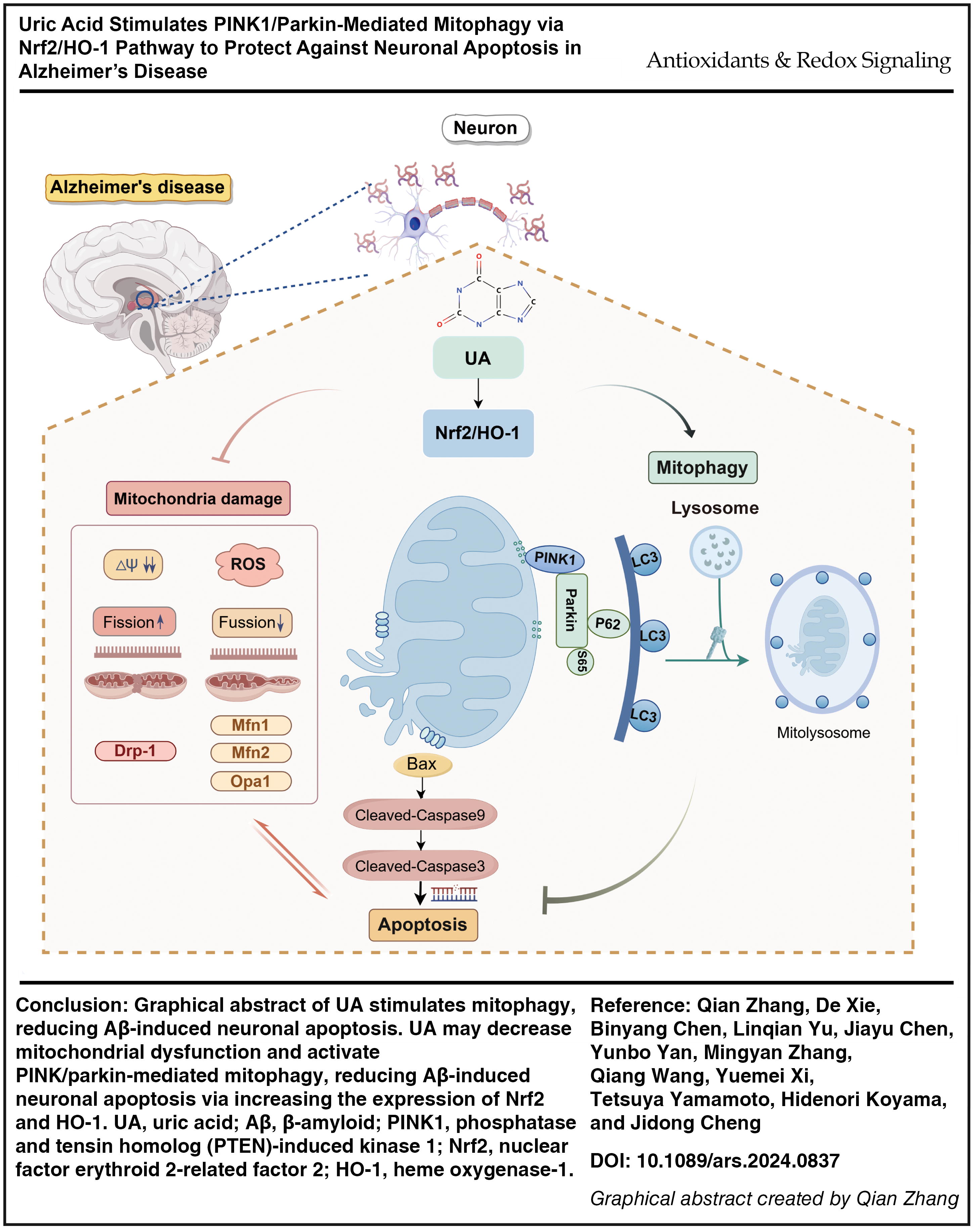
Introduction
Alzheimer’s disease (AD), the most prevalent kind of dementia (Citron, 2010), is a chronic, terminal neurological disorder characterized by progressive cognitive deficits. Multiple etiologies and pathogenic mechanisms are linked to AD, including β-amyloid (Aβ) aggregation and plaque development, excessive tau phosphorylation and neurofibrillary tangle formation, oxidative damage, and dysfunctional mitochondria (Zeng et al., 2022). Mitochondria serve as the major energy source by providing adenosine triphosphate (ATP) and play essential roles in calcium homeostasis and iron–sulfur biosynthesis in physiological neurons (Lin and Beal, 2006). During the prodromal phase of AD, mitochondrial shifts may occur, leading to bioenergetic dysfunction and neuronal dysfunction (Naia et al., 2023). Notably, the release of cytochrome c from Aβ-damaged mitochondria can trigger intrinsic neuronal apoptosis via Bax-regulated caspase pathway (Kerr et al., 2017), which exacerbated AD progression. Thus, maintaining mitochondrial homeostasis and inhibiting neuronal apoptosis emerge as promising therapeutic targets for AD.
Uric acid (UA) is the ultimate product of purine metabolism in humans. As a potent antioxidant, UA has been shown to play protective roles in Parkinson’s disease (Bao et al., 2018), ischemic stroke (Ya et al., 2018), and multiple sclerosis (Squadrito et al., 2000). In addition, several epidemiological studies have revealed a negative correlation between serum UA levels and the risk of AD (Du et al., 2016; Lu et al., 2016). A prospective cohort study of 2767 American individuals aged ≥60 years indicated that those with higher plasma UA levels have superior cognitive function (Geng et al., 2022). The majority of current research on UA and AD is epidemiological, whereas data from cellular and animal experiments are not widely accessible, which require further investigation.
Mitophagy, a selective process for removing defective mitochondria, is vital for neuronal function and survival since it maintains a healthy mitochondrial pool and prevents neuron from death and apoptosis (Kubli and Gustafsson, 2012; Palikaras et al., 2018). Among the various mechanistic pathways, the phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1)/parkin-mediated mitophagy has been extensively studied in neurodegenerative diseases, including AD (Goudarzi et al., 2021). In malfunctioning mitochondria, a decrease in mitochondrial membrane potential (MMP) leads to accumulation of PINK1, which subsequently recruits the E3 ubiquitin ligase parkin to mitochondria. Then, parkin ubiquitination promotes subsequent microtubule-associated protein 1 light chain 3 (LC3)-mediated autophagic degradation (Geisler et al., 2010; Gkikas et al., 2018; Wang et al., 2023; Zhang et al., 2015). Impaired mitophagy has been observed in AD-induced pluripotent stem cell-derived neurons, transgenic AD mouse models, and postmortem human AD brain tissues (Martin-Maestro et al., 2019). In addition, genetic and pharmacological strategies targeting mitophagy resulted in a rescue of several AD stigmata (Fang et al., 2019; Mary et al., 2023). Collectively, restoring mitophagy levels represents a promising therapeutic approach in AD.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is crucial for maintaining cellular homeostasis, as it hampers cell damage caused by external stimuli (Zgorzynska et al., 2021). Nrf2 sustains mitochondrial integrity, preserves cellular homeostasis, and promotes cell survival by upregulating key mitophagy and autophagy proteins and promoting continuous mitochondrial biogenesis (Dinkova-Kostova and Abramov, 2015; Gureev et al., 2019). One notable target of Nrf2 is heme oxygenase-1 (HO-1), which is renowned for its roles in combating oxidative stress (Wu et al., 2021) and regulating mitochondrial function (Bansal et al., 2014). Upregulation of Nrf2/HO-1 reduces oxidative stress, protects mitochondrial integrity, and may shield against mitochondrial dysfunction (Yang et al., 2024). Interestingly, UA treatment has been shown to activate the Nrf2 pathway and modulate neurotrophic factors, thereby providing neuroprotection in focal cerebral ischemia/reperfusion mouse model (Ya et al., 2018).
Nevertheless, the neuroprotective effect of UA to AD has not been thoroughly understood. In this study, we investigated whether UA can inhibit Aβ-mediated neuronal apoptosis and its underlying mechanism. We hypothesize that UA activates the Nrf2/HO-1/PINK1/parkin signaling pathway, promoting mitophagy and suppressing neuronal death in AD. We mainly study the effects of UA on mitophagy and its regulation of neuronal apoptosis in Aβ-induced AD models, which may provide a viable therapeutic strategy for AD (graphical abstract).
Results
UA improves cognitive impairment in 5×FAD mice and regulates transcriptome of Aβ-induced primary neurons
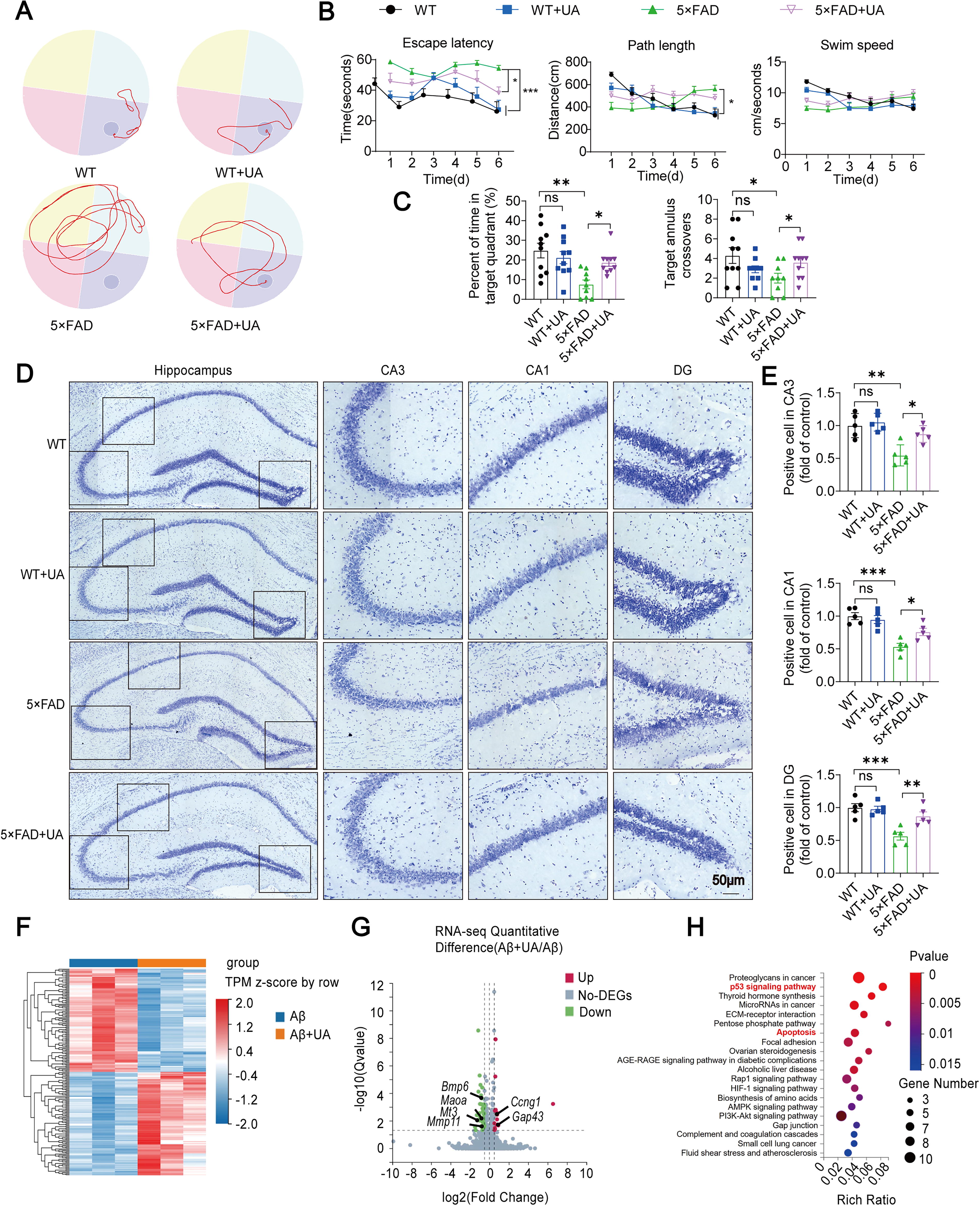
After 2-month treatment, Morris water maze (MWM) test was performed to evaluate the therapeutic effect of UA. UA improved the escape latency of transgenic mice with five familial Alzheimer's disease (5×FAD) mice in the MWM test, with no effect on swimming distance and speed (Fig. 1A, B). On the testing day, UA-treated AD mice spent more time in the target quadrant and appeared to prefer the target platform location (nonsignificant) (Fig. 1C). UA showed no influence on wild-type (WT) mice in the aforementioned tests. In addition, the level of injury in the hippocampal neurons was evaluated by Nissl staining. Compared with WT mice, 5×FAD mice exhibited neuronal damage, disordered and sparse neuron arrangement, and blurred or absent Nissl body shapes (Fig. 1D). UA reduced the amount of Nissl staining-identified degenerative cells within multiple regions of the hippocampus (CA3, CA1, and DG) (Fig. 1E). Compared with the 5×FAD group, neurons in 5×FAD+UA group were tightly arranged, and the number of Nissl bodies was increased. In addition, our data revealed a significant reduction in Aβ plaque deposition and soluble Aβ42 oligomer levels in the hippocampus of 5×FAD+UA mice (Supplementary Fig. S1). Then, the transcriptome of cultured primary neurons was analyzed using RNA sequencing (RNA-seq) after being stimulated by Aβ1–42 with or without UA. Compared with the Aβ1–42-treated group, Aβ1–42+UA neurons displayed 200 differentially expressed genes (DEGs), including 100 upregulated and 100 downregulated genes (Fig. 1F). Volcano plot showed the downregulation of Mmp11, Mt3, Maoa, and Bmp6, along with the upregulation of Ccng1 and Gap43 in Aβ1–42+UA group (Fig. 1G). Transcriptome-based Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis indicated that the apoptosis pathway was enriched (Fig. 1H). Based on the transcriptome analysis, we further explored the relationship between UA and apoptosis signaling pathway.
UA inhibits neuronal apoptosis in 5×FAD mice, primary neurons, and HT22 cells
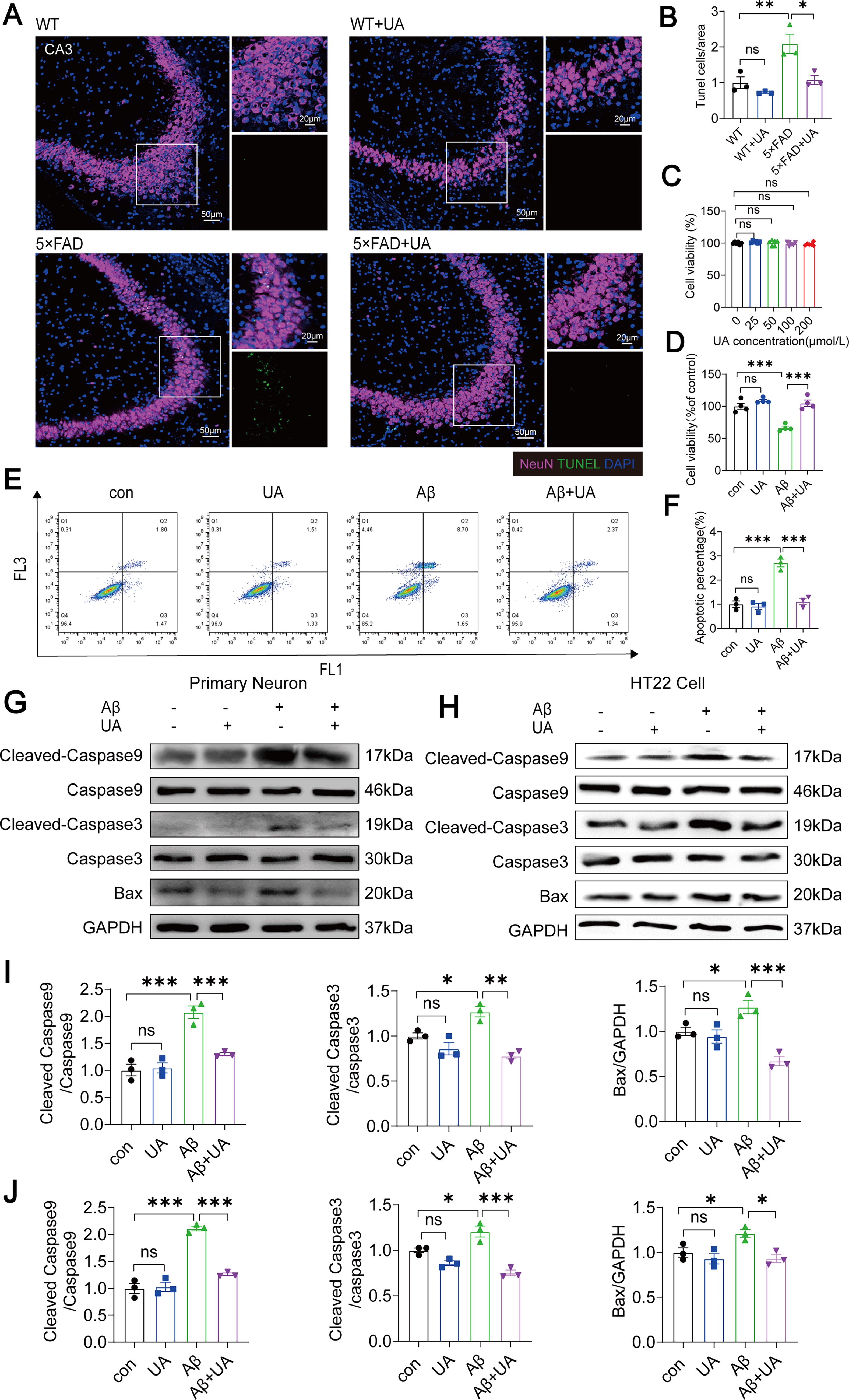
To assess the effect of UA on neuronal apoptosis, TdT-mediated dUTP nick-end labeling (TUNEL) and neuronal nuclei (NeuN) double immunofluorescence (IF) labeling were used to identify neuronal death in mice hippocampus. Compared with WT mice, the number of apoptotic neurons in hippocampal CA3 area was higher; notably, UA treatment reduced apoptotic neurons in 5×FAD mice (Fig. 2A, B). To further study the effect of UA on Aβ1–42–induced neuron apoptosis in vitro, we used Cell Counting Kit-8 (CCK8) assay to identify the impact of UA on HT22 cell viability. The data showed that 100 µM UA significantly attenuated cytotoxicity of hydrogen peroxide (H2O2) (Supplementary Fig. S2) and had no significant effect on the viability of HT22 cells (Fig. 2C). CCK8 assays revealed that cell viability decreased after 20 µM Aβ1–42 treatment, and UA offsets the decreased viability (Fig. 2D). Then, apoptotic HT22 cells were stained with fluorescently labeled annexin V (annexin V-FITC)/propidium iodide (PI) and recorded with flow cytometry. The number of apoptotic cells (both early and late) was increased in the Aβ1–42 group compared with the control group, and UA protected cells from Aβ1–42-induced apoptosis (Fig. 2E, F). Furthermore, we assessed the expression levels of apoptosis-related proteins. UA treatment inhibited Aβ1–42-induced expression of Cleaved-Caspase9, Caspase9, Cleaved-Caspase3, Caspase3, and Bax in primary neurons and HT22 cells (Fig. 2G–J).
UA alleviates mitochondrial dysfunction in 5×FAD mice and HT22 cells
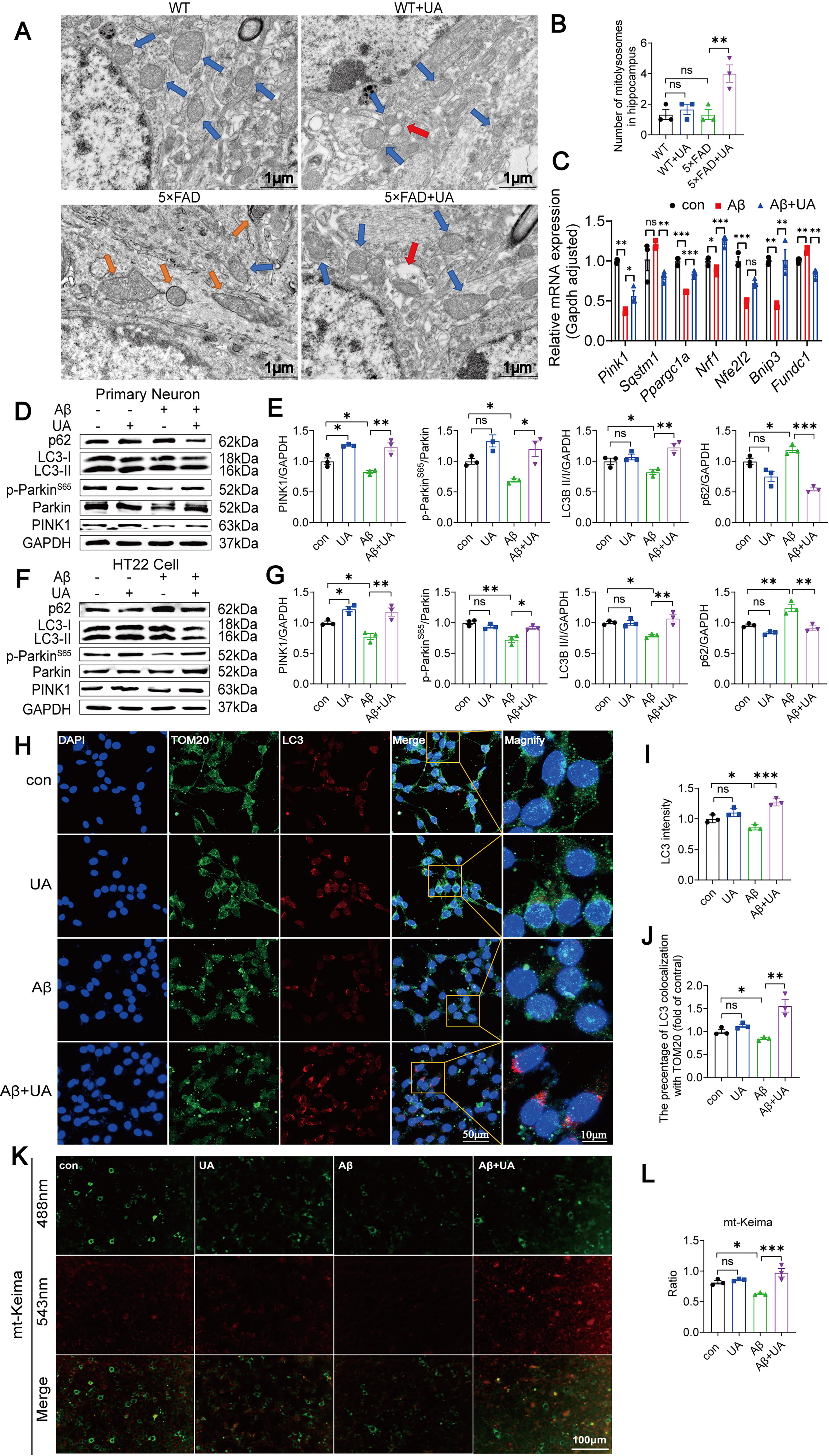
Damaged mitochondria may bring about an increase in pro-apoptotic signaling, impairing neuronal structure and function related to cognitive ability (Bao et al., 2021; Guo et al., 2023). To evaluate the morphology and integrity of mitochondria in hippocampal neurons, we used transmission electron microscopy (TEM) analysis and found aberrant mitochondrial morphology in 5×FAD mice, including significant destruction of the cristae, swollen and vacuolated mitochondria, and reduced electron density in the matrix (Fig. 3A). UA suppressed these morphological damages and decreased the buildup of defective mitochondria in the hippocampus of 5×FAD mice (Fig. 3B). To analyze mitochondrial dynamics, we detected the expression of the mitochondrial fusion genes mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy 1 (Opa1) and the mitochondrial fission genes dynamin-related protein 1 (Dnm1). We found that the mitochondrial fusion genes were upregulated (Mfn1, Mfn2, and Opa1), whereas the mitochondrial fission gene (Dnm1) was downregulated in UA-pretreated HT22 cells, which antagonized the effects induced by Aβ1–42 (Fig. 3C–G). Furthermore, we found that UA inhibited Aβ1–42-induced reactive oxygen species (ROS) and mitochondrial reactive oxygen species (MtROS) production in HT22 cells by 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent staining and fluorogenic dye MitoSOX (Fig. 3H–J). The percentage of cells generating JC-1 aggregates and JC-1 monomers was determined by a fluorescence microscope (Fig. 3K) and flow cytometry (Fig. 3M). Consistent with previous study (Bao et al., 2021), Aβ1–42 exposure resulted in a higher green fluorescence intensity and a lower red/green ratio compared with the control group. Conversely, UA significantly reduced the Aβ1–42-induced rise in green fluorescence, revealing that UA treatment conserved MMP after Aβ1–42 exposure (Fig. 3L, N). Given the pivotal role of mitophagy in mitochondrial function, we assessed ATP production in HT22 cells using chemiluminescence assays. The results showed that Aβ1–42 decreased ATP production in HT22 cells, which was reversed by UA treatment (Fig. 3O).

UA activates mitophagy in 5×FAD mice, primary neurons, and HT22 cells
Mitophagy is a selective autophagy process that operates as a self-protective mechanism for the cell (Pickles et al., 2018). Anomalies in mitophagy affect mitochondrial clearance, resulting in the accumulation of broken mitochondria and neuronal death, and play an essential position in AD pathogenesis (Fang et al., 2019). We performed TEM analysis to observe whether UA induces mitophagy in 5×FAD mice hippocampal neurons. We found this mitochondrial structure enveloped by lysosomes, which appears as a single-membrane structure that engulfs and encapsulates damaged mitochondria (red arrow, Fig. 4A). And the presence of mitochondrial engulfment into lysosomes increased in 5×FAD+UA group (Fig. 4B). We measured the mRNA and protein levels of mitophagy signaling pathway in primary neurons and HT22 cells. UA suppresses the Aβ1–42-induced mRNA expression levels of p62 and increases the expression of PPARγ coactivator-1α (Ppargc1a), Nrf1, Nfe2l2, and Pink1 mRNA. Interestingly, we discovered that the UA increased BCL2 and adenovirus E1B 19-kDa-interacting protein 3 (Bnip3) mRNA expression while inhibiting FUN14 domain containing 1 (Fundc1) (Fig. 4C). Compared with the Aβ1–42 group, UA pretreatment enhanced the expression of PINK1, p-ParkinS65, parkin, and LC3II while reducing the expression of p62 in both primary neurons (Fig. 4D and E) and the HT22 cells (Fig. 4F and G), suggesting that UA restored the inhibition of PINK1/parkin-mediated mitophagy than Aβ1–42. To determine whether mitochondria are enwrapped by autophagosomes, we performed IF staining using antibodies against translocase of the outer membrane 20 (TOM20) and the autophagosome marker protein LC3. The Aβ1–42+UA group showed increased colocalization of TOM20 and LC3, as shown by double IF labeling, indicating enhanced mitophagy (Fig. 4H–J). In addition, mitophagy flux was determined by mt-Keima. Compared with the Aβ1–42 group, mt-Keima displayed a higher fluorescence ratio (543 nm/458 nm) in the Aβ1–42+UA groups, indicating that the level of mitophagy flux was increased (Fig. 4K, L).
UA enhances PINK1/parkin-mediated mitophagy by activating Nrf2/HO-1 pathway
Nrf2 expression strongly improves mitochondrial integrity and function by promoting the expression of nuclear-encoded mitochondrial proteins (Dinkova-Kostova and Abramov, 2015). To investigate how UA activates mitophagy, we examined changes in Nrf2 and HO-1 expression. UA treatment enhanced Nrf2 and HO-1 protein levels inhibited by Aβ1–42 in 5×FAD mice (Fig. 5A, B), primary neurons (Fig. 5C, D), and HT22 cells (Fig. 5E, F). The concentration of ML385, a specific Nrf2 inhibitor, was selected based on previous research studies (Chen et al., 2020; Qiu et al., 2020). To further verify whether UA-induced mitophagy is mediated by Nrf2 activation, HT22 cells were pretreated with UA or UA+ML385 for 12 h, followed by 48 h-Aβ1–42 oligomer stimulation. ML385 suppressed the protein levels of Nrf2 and HO-1, reflecting that it significantly hinders the Nrf2 signaling pathway (Fig. 5M, N). Next, we used JC-1 staining (Fig. 5G, H) and flow cytometry (Fig. 5I, J) to detect MMP changes in HT22 cells. ML385 reversed the ratio of JC-1 red/green fluorescence intensity and improved the green fluorescence intensity compared with the UA+Aβ1–42 group. Western blot (WB) results showed that PINK1, p-ParkinS65, parkin, and LC3II were decreased, while the expression of p62 was increased in the UA+Aβ1–42+ML385 group (Fig. 5K–N). Collectively, these results showed that the improvement of mitophagy by UA was reversed upon ML385 treatment, suggesting that UA enhances PINK1/parkin-mediated mitophagy by activating Nrf2/HO-1 pathway.
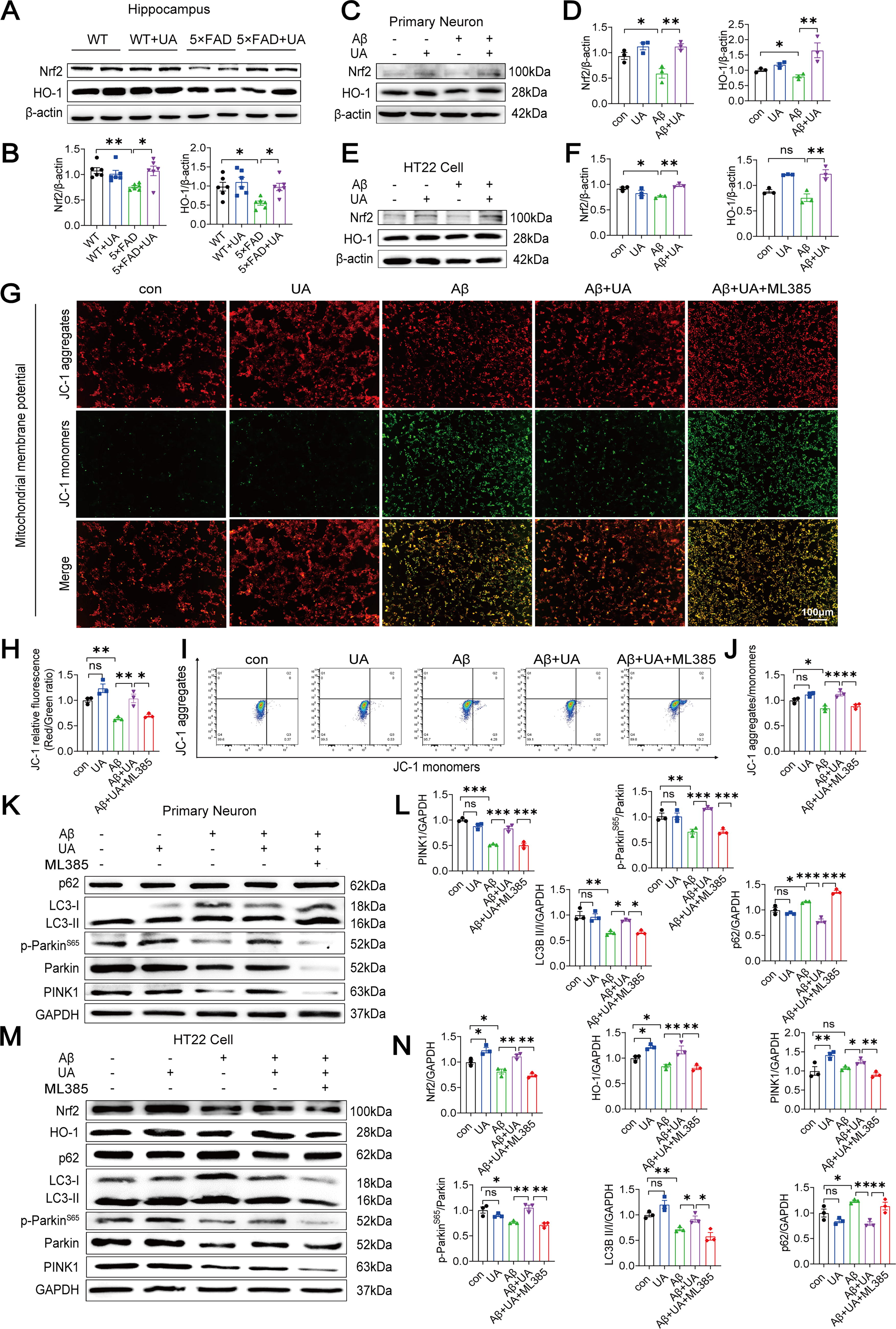
UA enhances PINK1/parkin-mediated mitophagy by activating the Nrf2/HO-1 pathway, which suppresses Aβ-induced neuronal apoptosis
To further explore the protective effect of UA-enhanced PINK1/parkin-mediated mitophagy against Aβ-induced apoptosis, small interfering RNA (siRNA) was used to knock down the PINK1 gene in HT22 cells. We used si-PINK1 (Supplementary Fig. S3) and then used annexin V-FITC/PI staining and flow cytometry to recognize apoptotic cells. The number of early and late apoptotic cells dramatically increased in PINK1-knockdown cells (Fig. 6A, B). This demonstrates that PINK1-mediated mitophagy is critical for the survival of HT22 cells. In addition, after 12 h of treatment with ML385, the ratio of early and late apoptotic cells (Fig. 6C, D) was significantly increased compared with UA+Aβ1–42 group. The results from the CCK8 also confirmed that ML385 reversed the increase in cell viability promoted by UA (Fig. 6E). In addition, the expression levels of Cleaved-Caspase9, Caspase9, Cleaved-Caspase3, Caspase3, and Bax were significantly increased compared with UA+Aβ1–42 group in primary neurons and HT22 cells (Fig. 6F–I). Therefore, we can conclude that UA enhances mitophagy by stimulating the Nrf2/HO-1/PINK1/parkin pathway, eventually declining neuronal death caused by Aβ1–42.
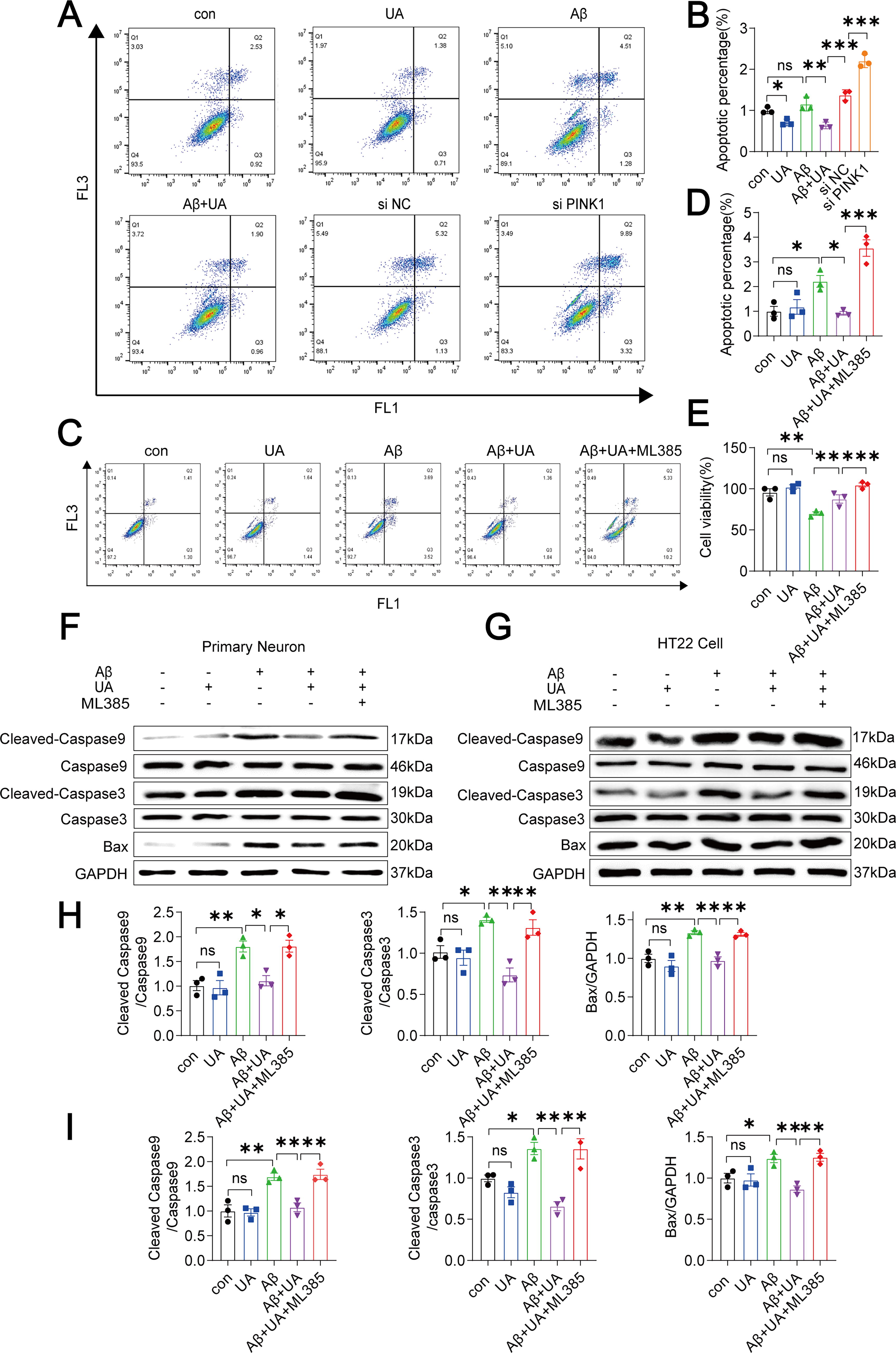
Discussion
AD, the leading cause of dementia, poses a growing global health challenge in an aging society. In our work, we investigated the therapeutic potential of UA in AD using HT22 cells, primary neurons, and 5×FAD mice. We found that UA improved cognitive impairment in 5×FAD mice and reduced neuronal apoptosis both in vivo and in vitro. In addition, UA treatment restored the depolarization of the MMP, ameliorated disrupted mitochondrial dynamics, and activated mitophagy in Aβ-treated neurons. Mechanistically, our data demonstrate that UA alleviates Aβ-induced apoptosis by activating PINK1/parkin-mediated mitophagy in hippocampal neurons through the Nrf2/HO-1 pathway. Collectively, these findings suggest that UA may present a novel approach for the prevention and treatment of AD.
According to several studies, UA may be associated with a lower risk of dementia (Cao et al., 2015; Shao et al., 2016; Wu et al., 2013). UA offers several advantages as a therapeutic agent for AD. As the end product of purine metabolism, UA is ubiquitously present in intracellular and extracellular fluids, providing a robust safety basis for its clinical application. Moreover, UA contributes approximately 50% of the total plasma antioxidant capacity (Wen et al., 2024), which may mitigate the extensive oxidative stress damage in AD. However, excessive peripheral supplementation of UA could lead to hyperuricemia. To avoid hyperuricemia-associated complications, the administration routes and doses of UA in our study were selected based on previous studies (Bao et al., 2018). Given the restrictive nature of the blood-brain barrier (BBB) (Bowman et al., 2010), future studies should explore targeted delivery strategies to effectively increase urate levels in the brain, such as nanomedicines (Fernandes et al., 2021; Hasan et al., 2024) or intranasal administration (Keller et al., 2022).
Mitochondria are vital for maintaining neuronal stability and physiological function, relying on the mitochondrial quality control systems, such as mitophagy and mitochondrial fission and fusion (Jiang et al., 2022; Schon and Przedborski, 2011). Specifically, AD neurons displayed a reduced number of mitochondria (Silva et al., 2012), a higher proportion of mitochondria with broken cristae (Wang et al., 2008), and reduced basal levels of mitophagy (Fang et al., 2019; Vaillant-Beuchot et al., 2021). Mechanistically, the AD cortex exhibited increased Drp1 and reduced levels of PINK1, parkin, Mfn1, Mfn2, and Opa1 (Manczak et al., 2011). Furthermore, Aβ peptides, whether as monomers or oligomers, have been shown to impair the mitophagy process (Guglielmotto et al., 2014; Mary et al., 2023; Wang et al., 2020). In our study, UA pretreatment effectively countered these mitochondrial alterations in Aβ-damaged neurons by reducing Drp1 expression while elevating the levels of Mfn1, Mfn2, Opa1, PINK, parkin, and LC3-II/I ratio. Moreover, UA treatment restored mitochondrial structure, enhanced ATP production, and reduced MMP depolarization in hippocampal neurons, suggesting UA’s protecting effect in mitophagy and mitochondrial dynamics against Aβ toxicity.
Oligomers and fibrillar Aβ peptides are toxic to neuronal cell membrane and mitochondria, leading to neuronal dysfunction and incident apoptosis (Hou et al., 2014; Kerr et al., 2017; Reiss et al., 2018). Mechanistically, the pro-apoptotic proteins Bak and Bax are central regulators of mitochondrial apoptosis by controlling mitochondrial membrane permeabilization (Kubli and Gustafsson, 2012) and activating of the downstream caspase cascade (Johri, 2021; Li et al., 2023). In our study, UA reduced the percentage of Aβ-induced apoptotic cells both in vivo and in vitro, accompanied by decreased activation of Bax, caspase9, and caspase3. Notably, siPINK1 reversed the antiapoptosis caused by UA. Previous research revealed that PINK1 deficiency results in severe neurodegeneration or postpartum death, highlighting its indispensability for neuronal survival (Han et al., 2023). Furthermore, UA promoted Bnip3 expression, which is suppressed by Aβ. BNIP3 and BNIP3L form homodimers and serve as regulatory targets of Parkin RBR E3 ubiquitin protein ligase (PRKN), suggesting potential cross talk between PINK1/parkin-dependent and -independent pathways (Yao et al., 2021). Further investigation is warranted to determine whether antiapoptosis properties of UA rely on PINK-independent mitophagy. Considering the protective effects of physiological apoptosis in neurons, further investigation should flesh whether antiapoptotic effects of UA are additionally associated with the endosomal–lysosomal pathway.
Nrf2 is a key transcription factor that regulates the expression of cellular protective proteins and has emerged as a promising therapeutic target for AD (George et al., 2022). Nrf2 is intimately interrelated with mitochondrial function and mitochondrial biogenesis by regulating the transcription of PGC1α, Mfn2, and Nuclear Respiratory Factor 1 (NResF1) (Baldelli et al., 2013; Dinkova-Kostova and Abramov, 2015; Kasai et al., 2020; Piantadosi et al., 2008). Particularly, Nrf2 activation promoted mitophagy to reduce oxidative stress injury by Nrf2/PINK1 signaling pathway (Peng et al., 2023; Xiao et al., 2017). Our study demonstrates that UA activates the Nrf2/HO-1 signaling pathway both in vivo and in vitro. Moreover, ML385, a specific Nrf2 inhibitor, suppressed UA-induced restoration of ΔΨm, PINK/parkin-related mitophagy, and the inhibition of neuronal apoptosis in the presence of Aβ. In addition, HO-1 catalyzes the oxidation of heme to produce biliverdin, which subsequently activates biliverdin reductase-A (BVR-A) (Mancuso, 2025). Previous studies have shown that BVR-A regulated the phosphorylation of insulin receptor substrate-1 and modulates the activation of PI3K-AKT-mTORC1 pathway, thereby preventing brain insulin resistance in AD and mild cognitive impairment (MCI) brains (Barone et al., 2016; Barone et al., 2019). Given that, UA treatment activated Nrf2/HO-1 pathway in neurons, which raises the question of whether neuroprotective effects of UA against apoptosis are independent of BVR-A activation.
The novelty of our research lies in the explicit demonstration of the ability of UA to promote mitophagy in neurons and in elucidating how UA attenuates Aβ-induced cell apoptosis by modulating mitochondrial function and homeostasis. However, several limitations should be considered in this study. First, our study focuses on the promotion of mitophagy via the Nrf2/HO-1/PINK1/parkin pathway. However, HO-1 is not exclusively regulated by Nrf2, and further verification is needed to delineate the role of HO-1 in UA-induced mitophagy. Second, although our study demonstrates that Nrf2 is a potential target of UA, further validation is required to determine whether UA interacts directly or indirectly with Nrf2. Third, despite the observed improving cognitive dysfunction, further research is needed to explore the additional molecular targets of UA and to establish the correlation between UA concentration ranges and its protective effects against AD in humans.
Conclusions
These data suggest that UA stimulates PINK1/parkin-mediated mitophagy reducing Aβ-induced neuronal apoptosis through the Nrf2/HO-1 pathway, which plays a neuroprotective role in AD.
Materials and Methods
Reagents and antibodies
The antibodies utilized in this investigation are summarized in Table 1. UA was supplied by Sigma (U2625, USA). ML385 came from MCE (USA). Aβ1–42 peptide was purchased from GL Biochem (054949) and diluted to 100 µM for storage purposes. H2O2 was purchased from Sinopharm Chemical Reagent Co., Ltd (China).
Primary and Secondary Antibodies Used in This Study
Drp1, dynamin-related protein 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HO-1, heme oxygenase-1; LC3, microtubule-associated protein 1 light chain 3; NeuN, neuronal nuclei; Mfn2, mitofusin 2; Nrf2, nuclear factor erythroid 2-related factor 2; PINK1, phosphatase and tensin homolog (PTEN)-induced kinase 1; SQSTM1/p62, protein sequestosome 1; TOM20, translocase of the outer membrane 20; HRP, horseradish peroxidase; WB, Western blot; IF, immunofluorescence.
Animals and drug treatment
Eight-week-old 5×FAD mice (an animal model of AD) were acquired from the Xiamen University Laboratory Animal Center (XMULAC), housed in standard conditions (unfettered access to food and water in temperature and humidity-controlled surroundings with a light-dark cycle of 12 h). 5×FAD mice with human APP variants (K670N/M671L, I716V, and V717I) and PSEN1 mutations (M146L and L286V) were bred on C57BL/6 genetic heritage. C57BL/6 or WT littermates of 5×FAD mice were used as WT controls. WT and 5×FAD mice (12 months old) received intraperitoneal injections of UA or vehicle at a dose of 200 mg/kg daily for 2 months (n = 9–10 per group). Each mouse underwent behavioral testing before being euthanized. The experimental mice included 6–7 male and 3–4 female mice in behavioral experiments. The mice were perfused with 0.9% saline, and their brains were separated for further experimentation. All animal experiments in the research were authorized by the Institutional Animal Care and Committee of Xiamen University, China (Animal Ethics No. XMULAC20240246).
Primary cell culture and treatments
Primary neural cells were collected from embryonic mice (C57BL/6) at 17.5 days of gestation. Brain tissue was extracted and processed briefly with 2 mg/mL papain for 30 min at 37°C. The pipette was used to triturate the digested tissue in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Glass bottom dishes were coated with poly-D-lysine, and then dissociated neurons were grown in neurobasal media with 2% B27 supplement, 0.5 mM L-glutamine, and 50 U/mL penicillin–streptomycin at a density of 1.5 × 105 cells per well. The medium was substituted after 4 h, with 50% replaced every 3 days. On day 7, neurons were exposed to Aβ1–42 (20 μM) for 48 h with or without UA (100 µM) for 12 h. The cells were gathered for WB.
Hippocampal HT22 cell culture
HT22 cells were grown in DMEM (10% FBS, Gibco, USA) in a 5% CO2 incubator at 37°C. Regarding drug treatments, HT22 cells were cultured with Aβ1–42 (20 µM) for 48 h with or without UA (100 µM) for 12 h with an equal amount of dimethyl sulfoxide as a control. Cell lysates were acquired through radio-immunoprecipitation assay (RIPA) lysis for the subsequent trials.
MWM test
The MWM test was applied to assess the remembering and learning capacities of mice and was conducted daily. Mice were trained to locate a concealed platform over 6 days, with 2 trials per day. Mice that were unable to identify the platform within 60 s were directed to it and held there for 10 s. On the seventh day of probe testing, the platform was removed. Mice were put in a testing pool opposite the chosen quadrant for 60 s of exploring. Trajectories of the mice were recorded using EthoVision XT 14.0 (Noldus), and their behavioral data (swim speed, traveled distance, latency, percentage of time in each quadrant, and target annulus crossings) were computed automatically.
Nissl staining
To evaluate neuronal vitality, 20-µm coronal cryosections of the brain were stained with Nissl staining solution (Beyotime, C0117, Shanghai, China) for 10 min at 37°C. All of the samples were subsequently cleaned with 95% and 100% ethyl alcohol for 1 min each. They were rinsed twice with xylene for 35 min. After being sealed with neutral balsam, the slides were examined under a light microscope at 200× magnification.
High-throughput RNA-seq and transcriptomic profiling
Primary neurons were treated with Aβ1–42 for 24 h with or without UA (100 μM) for 12 h, and then total RNA was isolated. The Beijing Genomics Institute (BGI, China) afterward carried out RNA-seq and data analysis (https://biosys.bgi.com/#/report/login). All the DEGs were used for heatmap analysis, volcano map analysis, and KEGG enrichment analyses.
TUNEL assay
To visualize apoptotic cells in situ, brain coronal cryosections were labeled with a TUNEL kit (Lablead, B0013, Beijing, China). Slices were fixed with paraformaldehyde (PFA) for 20 min and cleaned with phosphate-buffered saline (PBS) for 5 min, repeated three times. Continue the IF procedures to stain NeuN. The TUNEL staining process was performed according to the manufacturer’s instructions. The nuclei were labeled with 4’-6-diamidino-2-phenylindole (DAPI). The image was captured using a laser scanning confocal microscope (FV1000 MPE-B; Olympus, Japan). The TUNEL analysis was conducted at least three times with different biological samples.
Cell viability (CCK8) assays
Cells (5.5 × 104/mL) were cultured nightly in 96-well plates (100 µL/well). After reaching 50%–60% confluence, cells received treatments with a conditioned medium or the relevant medicines. After 48 h, each well got 10 µL CCK8 assay (TargetMol) for 1 h at 37°C before being terminated. The optical density was determined at 450 nm via a microplate reader (Multiskan Sky; Thermo Fisher).
Analysis of apoptosis by flow cytometry
Cell apoptosis was evaluated using the Annexin V-FITC/PI Kit (Beyotime, C1062M, Shanghai, China). HT22 cells were incubated with UA (100 µM) for 12 h and exposed to Aβ1–42 (20 µM) for another 48 h. After gathering, cells were resuspended in 195 µL of binding solution. Following adding 5 µL annexin V-FITC and 10 µL PI, cells were incubated for 15 min at room temperature (RT). The percentage of apoptosis was assessed with flow cytometry (Beckman CytoFLEX S, USA) and analyzed with FlowJo.
WB assay
After homogenizing brain tissue and cells in RIPA buffer (SparkJade Biotechnology, China) with 1% proteinase and phosphorylase inhibitor (Topscience, China), the mixture was centrifuged for 15 min at 12,000× rpm at 4°C. Then, the BCA Kit (Beyotime, China) was chosen for measuring the protein content in the supernatant. We performed WB electrophoresis and subsequently transferred the proteins to polyvinylidene difluoride (PVDF) membranes (Millipore, USA). After 1 h of blocking with 5% skim milk, the PVDF membranes were incubated overnight at 4°C with primary antibodies. Then it was incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at RT. The membranes were subsequently cleaned three times with tris-buffered saline with Tween 20 (TBST). Finally, the blots were photographed using the ECL Super Kit on the imaging equipment (US Azure, C300), and the results were quantified with ImageJ (version 2.0).
TEM
TEM was applied to observe the ultrastructure of mitochondria and mitophagosomes. After extracting mice brains, tissue blocks from the hippocampus (2 × 2 × 2 mm) were soaked in 2.5% glutaraldehyde for 2 h at RT before being moved to 4°C for cryopreservation. Following modification, 1 mm3 tissue blocks were dehydrated using a gradient of alcohol (30%, 50%, 70%, 80%, 95%, and 100%) and acetone ahead of being implanted in pure epoxy resin. Then, ultrathin slices (80 nm) were produced using an ultramicrotome (UC7, Leica, Germany) and stained sequentially with 2% uranyl acetate and 2.6% lead citrate. Finally, an electron microscope was utilized to capture ultrastructure pictures of materials.
RNA extraction and real-time quantitative PCR
Total RNA in HT22 cells was obtained with TRIzol reagent (Sangon, Biotech, Shanghai, China), and RNA concentrations were measured using ThermoScientific NanoDrop One. RNA was reverse transcribed into cDNA by the Tiangen Reverse Transcription Kit (Beijing). SYBR Green Master Mix was used to measure the relative mRNA expression levels (Yeason, Shanghai, China). All samples were analyzed in duplicate and adjusted to glyceraldehyde-3-phosphate dehydrogenase. All reactions were performed in triplicate in 0.1 mL 8-tube strips. The 2−ΔΔCT method was utilized for analyzing relative changes in gene expression. The primer sequences are listed in Table 2.
Primer Sequences
Bnip3, BCL2 and adenovirus E1B 19-kDa-interacting protein 3; Dnm1, dynamin-related protein 1; Fundc1, FUN14 domain containing 1; Gapdh, glyceraldehyde-3-phosphate dehydrogenase; Mfn1, mitofusin1; Mfn2, mitofusin 2; Opa1, optic atrophy 1; Ppargc1a, PPARγ coactivator-1α; Nfe2l2, nuclear factor erythroid 2-related factor; Pink1, phosphatase and tensin homolog (PTEN)-induced kinase 1; SQSTM1/p62, protein sequestosome 1.
Intracellular ROS and MtROS measurement
Intracellular ROS generation was measured using the fluorescent probe DCFH-DA (D2215, LABLEAD). HT22 cells were pretreated with UA (100 µM) for 12 h, exposed to Aβ1–42 (20 µM) for another 24 h, and incubated with DCFH-DA at 37°C for 30 min. After washing with PBS, cells were imaged in 3 random regions per sample. As for MtROS, HT22 cells were incubated with 5 μM MitoSOX™ Red Mitochondrial Superoxide Indicator (RM02822, ABclonal, China) according to the manufacturer’s instructions. Finally, we calculated the fluorescent intensity in the images using ImageJ (NIH, Bethesda, MD) and obtained the mean fluorescence intensity through dividing by total cell numbers.
Measurement of MMP (ΔΨm)
JC-1 (Beyotime, C2006, Shanghai, China) was implemented to detect the MMP by the manufacturer’s instructions. Laser confocal microscopy (FV1000 MPE-B, Olympus, Japan) was adopted to view the staining. The quantification of fluorescence values was quantified using ImageJ. Flow cytometry (Beckman CytoFLEX S, USA) was used to determine the fluorescence intensity of 10,000 cells/sample (488 nm excitation and 530 nm emission), which was then analyzed with FlowJo. At least three different biological samples were used to repeat the MMP analysis.
ATP content determination
The ATP content in HT22 cells was measured using a Chemiluminescence Assay Kit (#E-BC-F002, Elabscience, China). First, HT22 cells were pretreated with UA (100 µM) for 12 h and exposed to Aβ1–42 (20 µM) for another 24 h, and cells were lysed in 0.5 mL of lysis buffer. A portion of the lysate was analyzed for protein content using the bicinchoninic acid (BCA). Following centrifugation of the remaining lysate, the supernatant was obtained and combined with luciferin and luciferase to start a chemiluminescent reaction. Luminescence intensity was recorded using a microplate reader (#Varioskan Flash, Thermo, USA). The ATP concentrations in the samples were calculated based on their comparison with a standard curve. The ATP levels were adjusted based on the total protein content measured by the BCA method, and the relative ATP content for each group was calculated accordingly.
IF staining
Cultured cells were seeded onto confocal dishes, given UA (100 µM) for 12 h, and then exposed to Aβ1–42 (20 µM) for an additional 24 h. Then the cells were fixed in 4% PFA at 4°C for 20 min. After washing in PBS, cells were penetrated with 0.3% Triton X-100 and blocked in 5% bovine serum albumin (BSA, Sigma-Aldrich, SRE0096) for 1 h at RT. Cells were treated in primary antibodies at 4°C overnight and then in secondary antibodies conjugated to Alexa Fluor 488 or 568 at RT. for 1 h before being mounted in DAPI mounting solution (D9542, Sigma). Representative IF images were taken (Nikon A1R; Nikon, Japan).
Mouse brains were frozen in 4% PFA at 4°C overnight, dried in 30% sucrose, and implanted into an optimal cutting temperature (O.C.T.) medium. The blocks were sliced at 20 µm and fixed in 4% PFA at RT. for 20 min. The sections were put in antigen retrieval with 10 mM sodium citrate buffer (pH 6.0) (PR30001, Proteintech) in boiling water for 10 min, cooling down to RT., and rinsed by PBS for 5 min, repeated three times. Tissues were penetrated with 0.5% Triton X-100 and blocked in 5% BSA at RT. for 1 h before being incubated in primary antibodies at 4°C overnight, followed by secondary antibodies at RT. for 1 h, then mounted with DAPI mounting solution. Representative fluorescence pictures were taken (FV1000 MPE-B; Olympus, Japan). For Aβ staining, 5×FAD and 5×FAD+UA mice received intraperitoneal methoxy-X04 (Tocris, 4920; 10 mg/kg) 3 h prior to transcardial perfusion with ice-cold PBS. Table 1 showed the primary and secondary antibodies utilized, as well as their dilutions. Each IF experiment was performed at least three times with different biological samples.
Mitophagy detection
The HT22 cells were diluted to 5 × 104/mL and transferred into a 12-well plate at 1 mL per well. The cells were placed in a 5% CO2 incubator at 37°C overnight. After 12 h, 500 µL of culture medium containing the mt-Keima adenovirus (Hanbio Biotechnology Co., Ltd.) was added, and the volume was brought up to 1 mL after 4 h of culture. Following 8 h of infection, the culture medium with the virus was removed and replaced with fresh complete medium. At 36 h post-transfection, HT22 cells were treated with UA and Aβ1–42, and mitophagy was detected by fluorescence microscopy.
siRNA in HT22 cells
The siPINK1 sequences (sense: UCAUCUUGUCCAAUUUCAG; antisense: CUGAAAUUGGACAAGAUGA) were designed and supplied by Tsingke Biotech Co. (Beijing, China). As a control, the company provided negative siRNA (sense: UUCUCCGAACGUGUCACGUTT, antisense: ACGUGACACGUUCGGAGAATT). For transfection, HT22 cells were grown in 6-well plates at a density of 60% overnight attachment. One hour before transfection, fresh media was added, and siRNA (10 µmol) was delivered into the cells using Lipofectamine 2000 transfection reagent (11668019, Invitrogen, CA, USA). All processes were carried out on antibiotic-free media. Following 12 h of transfection, the cells were treated with a conditioned medium or the indicated drugs and harvested for mRNA detection and flow cytometry.
Statistical analyses
All data are presented as mean ± SEM. Statistical analyses were conducted with GraphPad Prism 8.0 software. Differences between the two groups were analyzed by unpaired Student’s t-test and one-way/two-way ANOVA for multiple groups. p < 0.05 was considered statistically significant. An electronic laboratory notebook was not used.
Ethics Approval and Consent to Participate
Ethical approval for the animal study was approved by the Animal Care and Use Committee of Xiamen University (Animal Ethics No. XMULAC20240246). All animal experiments were undertaken in the XMULAC.
Footnotes
Acknowledgments
The experiments were carried out in the Central Clinical Research Core, Xiamen University. The authors thank the Core Facility of Biomedical, Xiamen University for assistance with confocal microscopy (FV1000MPE-B, Olympus). The authors also thank Qiuyang Zheng, Xiang You, and Jingru Huang.
Authors’ Contributions
Q.Z., D.X., B.C., and J.C. conceived and designed research. Q.Z., D.X., B.C., L.Y., J. Chen, and Y.Y. performed experiments and analyzed data. J.C., B.C., M.Z., Q.W., Y.X., T.Y., and H.K. interpreted results of experiments. Q.Z., D.X., and B.C. prepared figures, drafted article, and edited and revised the article. J.C. approved final version of article. All authors approved the final version of the article. J.C. takes responsibility for the integrity of the data.
Author Disclosure Statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported. The authors have nothing to disclose.
Funding Information
This work was supported by the National Natural Science Foundation of China (No.82370895, No.81570772, and No.82260163); Fujian Provincial Industry-University-Research Collaborative Innovation Project Plan, Clinical Application and Industrialization Research on the Screening of Intestinal Bacterial Strains for the Prevention and Treatment of Hyperuricemia (2022Y4007); the Natural Science Foundation of Fujian Province (No.2020J01018); and the Gout Research Foundation (Japan, 2019).
Supplementary Material
Supplementary Figures
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.