
Abstract
Acute altitude hypoxia is a syndrome that manifests at elevations exceeding 2500 m, posing significant health challenges to individuals who travel or work at high altitudes. Uncoupling proteins are integral proteins located within the mitochondrial inner membrane, playing a crucial role in modulating proton leakage across the mitochondrial membrane. This study investigates the potential role of uncoupling protein 4 (Ucp4) overexpression in an intermittent hypobaric hypoxia (IHH) model and its underlying mechanisms in the cerebellar dyskinesia phenotype. An IHH model was developed using a low-pressure hypoxic chamber, exposing mice to 16 h of hypoxia daily for 5 days. Three mouse strains were used: C57BL/6J, Pcp2Cre; Ucp4fl/fl, and Pcp2Cre; Mito-GFP. Behavioral tests, including rotarod, open field, balance beam, and Morris water maze, were conducted. Ucp4-overexpressing virus was administered to cerebellar lobes 4/5. Mitochondrial morphology was assessed via transmission electron microscopy, 3D reconstruction, and network analysis, while function was evaluated through reactive oxygen species, mitochondrial membrane potential (MMP), glutathione/glutathione disulfide ratio, adenosine triphosphate levels, qPCR, and Western blotting. Results showed that IHH induces hypoactivity without affecting spatial cognition. IHH-induced hypoactivity is linked to Ucp4 upregulation and increased mitochondrial fragmentation in Purkinje cells (PCs), though overall mitochondrial dynamics remain balanced. Ucp4 deficiency exacerbates IHH-induced hypoactivity and mitochondrial fragmentation. Conversely, Ucp4 overexpression in PCs significantly alleviates these effects. Mechanistically, Ucp4 protects PCs by stabilizing MMP and regulating oxidative stress, maintaining mitochondrial integrity. This study reveals that Ucp4 protects cerebellar PCs from oxidative stress in IHH, improving motor function and identifying Ucp4 as a potential therapeutic target for intermittent high-altitude syndrome. Antioxid. Redox Signal. 43, 483–508.
Introduction
Acute altitude hypoxia is a syndrome that manifests when individuals ascend to an altitude of 2500 m or higher, with presenting symptoms including cephalalgia, emesis, nausea, and insomnia (West, 2012). Some patients even succumb to high-altitude cerebral edema (Luks and Hackett, 2022). The expansion of human activities in recent decades has led to an increase in the number of people traveling or working at high altitudes, which poses new challenges to human health (Huan et al., 2023). Under hypobaric hypoxia environment, the alveolar oxygen partial pressure decreases (significantly starting at an altitude of 1500 m), resulting in a decrease in blood oxygen saturation and insufficient oxygen supply to tissues (Bussotti and Marchese, 2018). In plateau areas (above 2700 m), the oxygen content in the air decreases by approximately 30%, and the oxygen uptake decreases under the same respiratory efficiency (Koester-Hegmann et al., 2018; Tarver et al., 2025).
Innovation
Our research demonstrates that hypoactivity, a symptom induced by the intermittent-altitude hypoxia (IHH) model, is associated with the overexpression of Ucp4 within the mitochondrial inner membrane of the cerebellum. The conditional knockout of Ucp4 in PCs exacerbates IHH-induced hypoactivity, mitochondrial fragmentation, collapse of the MMP, and oxidative stress. Conversely, the targeted overexpression of Ucp4 in PCs substantially alleviates IHH hypoactivity, mitochondrial fragmentation, and oxidative stress levels, while also promoting the recovery of MMP. Ucp4, a critical molecule for neuroprotection, endows neurons with resistance against oxidative stress caused by intermittent-altitude hypoxia.
The brain is among the body’s most oxygen-intensive organs and is highly susceptible to hypoxia (Chen et al., 2022). The motor coordination function deteriorates above 3000 m, accompanied by manifestations such as tremors and convulsions (Midha et al., 2023; Ando et al., 2020). Acute hypoxia (7000 m) can even lead to the loss of judgment (Burtscher et al., 2021). The cerebellum, a pivotal region influencing motor function, is crucial for the body’s coordination and balance. It achieves this by integrating proprioceptive feedback and motor-related information, enabling the precise execution of motor commands (Koziol et al., 2014). Purkinje cells (PCs) are one of the largest neurons in the cerebellum and are essential for controlling body movements, while their dysfunction and degeneration can lead to dyskinesia (Yamada et al., 2018). Acute hypobaric hypoxia can directly inhibit the discharge frequency of PCs in the cerebellar cortex. Studies have shown that perinatal hypoxemia leads to a reduction of approximately 40% in the frequency of simple spike potentials in PCs and a disorder in the synchronicity of complex spike potentials (Kaur et al., 2005), thereby disrupting the precise regulation of motor coordination and timing control by the cerebellum (Finder et al., 2020). Human experiments have shown that after acute exposure to a simulated hypoxic environment at 3600 m above sea level, the speed of the finger-tapping test decreased by 18% and the accuracy (error rate) increased by 32% in subjects, suggesting impaired cerebellar temporal control (Brochu et al., 2011). In the animal model, the residence time of hypoxic mice in the rotating rod test was shortened to 40% of that of the control group, and gait analysis showed that the coefficient of variation of the hind limb step length increased by 2.1 times (Wu et al., 2021).
Uncoupling proteins (Ucps) are a family of specialized transport proteins located in the inner mitochondrial membrane. Their function is to uncouple the electrochemical gradient produced during adenosine triphosphate (ATP) synthesis by shuttling protons back from the intermembrane space into the matrix (Kim-Han and Dugan, 2005). The Ucp family comprises five known subtypes, with uncoupling protein 2 (Ucp2), uncoupling protein 4 (Ucp4), and uncoupling protein 5 (Ucp5) being particularly widespread within the central nervous system (Kumar et al., 2022). Ucp2 plays a significant role in the central nervous system by mitigating inflammatory responses in neurodegenerative diseases and facilitating synaptic remodeling triggered by hypoxia (Varela et al., 2016). Ucp5 is involved in the regulation of neurotransmission and synaptic plasticity (Andrews et al., 2005). Ucp4 participates in the regulation of neuronal death processes and cellular energy metabolism (Mao et al., 1999). Acute hypoxia significantly increases the expression of Ucp4 in the cerebellum (Wu et al., 2021). The uncoupling activity of Ucp4 not only suppresses oxidative stress but also curtails the production of reactive oxygen species (ROS), thereby conferring a protective effect against neurodegenerative diseases (Andrews et al., 2005). Research has indicated that the deletion of Ucp4 in PCs of healthy mice results in motor retardation (Wang et al., 2024). Nonetheless, the precise function of Ucp4 in motor retardation or hypoactivity induced by hypoxia remains unclear.
The objective of this study was to elucidate the role of Ucp4 in cerebellar PCs in mitigating oxidative stress and associated motor dysfunction in a mouse model of intermittent hypobaric hypoxia (IHH)-induced hypoactivity. To achieve this, we generated two transgenic mouse models: (i) Pcp2Cre; Ucp4fl/fl mice, in which Ucp4 is conditionally knocked out specifically in cerebellar PCs, and (ii) Pcp2Cre; Mito-GFP mice, in which green fluorescence protein (GFP) is selectively expressed on the outer mitochondrial membrane of PCs. The IHH model was established using C57BL/6J, Pcp2Cre; Ucp4fl/fl, and Pcp2Cre; Mito-GFP mice. Motor performance was comprehensively assessed using a battery of behavioral tests, including the rotarod test, open field test (OFT), video tracking, balance beam test, Noldus EthoVision system, and Morris water maze. To investigate the effects of Ucp4 overexpression, a Ucp4-overexpressing viral vector was delivered to the cerebellar lobules 4/5 via stereotactic injection. Mitochondrial morphology was examined using transmission electron microscopy (TEM), three-dimensional reconstruction of mitochondrial networks, and mitochondrial network analysis (MiNA). Mitochondrial function was evaluated by measuring ROS levels, mitochondrial membrane potential (MMP), the glutathione/glutathione disulfide (GSH/GSSG) ratio, and ATP levels, and through molecular analyses using qPCR and Western blotting. The current research demonstrated that Ucp4 in cerebellar PCs plays a crucial role in alleviating motor dysfunction by protecting PCs from oxidative stress in mice exposed to intermittent altitude hypoxia. These results suggest that Ucp4 may represent a critical protective target for the clinical management of intermittent high-altitude syndrome.
Results
IHH induces hypoactivity without affecting spatial cognition
C57BL/6J mice were divided into two groups, each comprising six animals. One group was maintained under standard laboratory conditions and served as the control. The other group was exposed to an IHH regimen, which involved placement in a low-pressure hypoxia chamber simulating an altitude of 5000 m. The IHH group underwent daily exposure for 16 h over a period of 5 consecutive days. Following the exposure period, behavioral assessments were conducted on both groups (Fig. 1A).

The OFT (Fig. 1B–J) showed that IHH significantly damaged the spontaneous movement capacity of mice, with a reduction in movement distance of approximately 45% (Fig. 1C, p < 0.001), a reduction in movement speed of approximately 48.7% (Fig. 1D, p < 0.01), a reduction in activity time of approximately 17.4% (Fig. 1E, p < 0.01), and an increase in rest time of approximately 42.9% (Fig. 1F, p < 0.01). The OFT (Fig. 1B–J) revealed that IHH significantly impaired the spontaneous locomotor activity of mice. Specifically, the movement distance was reduced by approximately 45% (Fig. 1C, p < 0.001), movement speed decreased by approximately 48.7% (Fig. 1D, p < 0.01), and active time was reduced by approximately 17.4% (Fig. 1E, p < 0.01). Conversely, rest time increased by approximately 42.9% (Fig. 1F, p < 0.01). These findings indicate a substantial negative impact of IHH on the spontaneous motor behavior of mice.
To assess the exploratory behavior of mice within the open field, we measured the time and distance spent by the mice in both the central and peripheral zones of the arena. The activity distance and time of mice from the center to the periphery of the open field were simultaneously measured. Analysis revealed that, compared with controls, mice in the IHH group exhibited significant reductions in both movement distance and time from the center of the open field (Fig. 1G, H, p < 0.01). Specifically, the movement distance in the peripheral zone was decreased by approximately 61% (Fig. 1I, p < 0.01), while the dwell time in the periphery increased by approximately 30% (Fig. 1J, p < 0.01). These findings collectively indicate that IHH induces hypoactivity and may be associated with anxiety-like behavioral changes in mice. To further document these behavioral alterations, a behavioral video tracking system (Noldus EthoVision) was employed to record the locomotor activity of mice under normal control (NC) conditions (Supplementary Video S1) and in mice exhibiting IHH-induced movement disorders (Supplementary Video S2).
The results of the rotarod test (Fig. 1K) indicated that IHH did not significantly impair the coordination and balance abilities of mice. This finding was further corroborated by the balance beam test, which also demonstrated no significant effect of IHH on these motor functions (Fig. 1L). In addition, the Morris water maze experiment revealed that IHH did not influence the spatial cognition and memory abilities of mice (Fig. 1M–R). Collectively, these findings confirmed that IHH selectively induces hypoactivity in mice without compromising their coordination and balance, as well as spatial cognition and memory (Fig. 1S).
IHH-induced hypoactivity is associated with upregulation of Ucp4 expression and increased mitochondrial fragmentation
To elucidate the underlying mechanisms of the behavioral changes induced by IHH, we harvested the cerebellar lobules 4/5 (4/5Cb) for subsequent qPCR analysis (Fig. 2A).
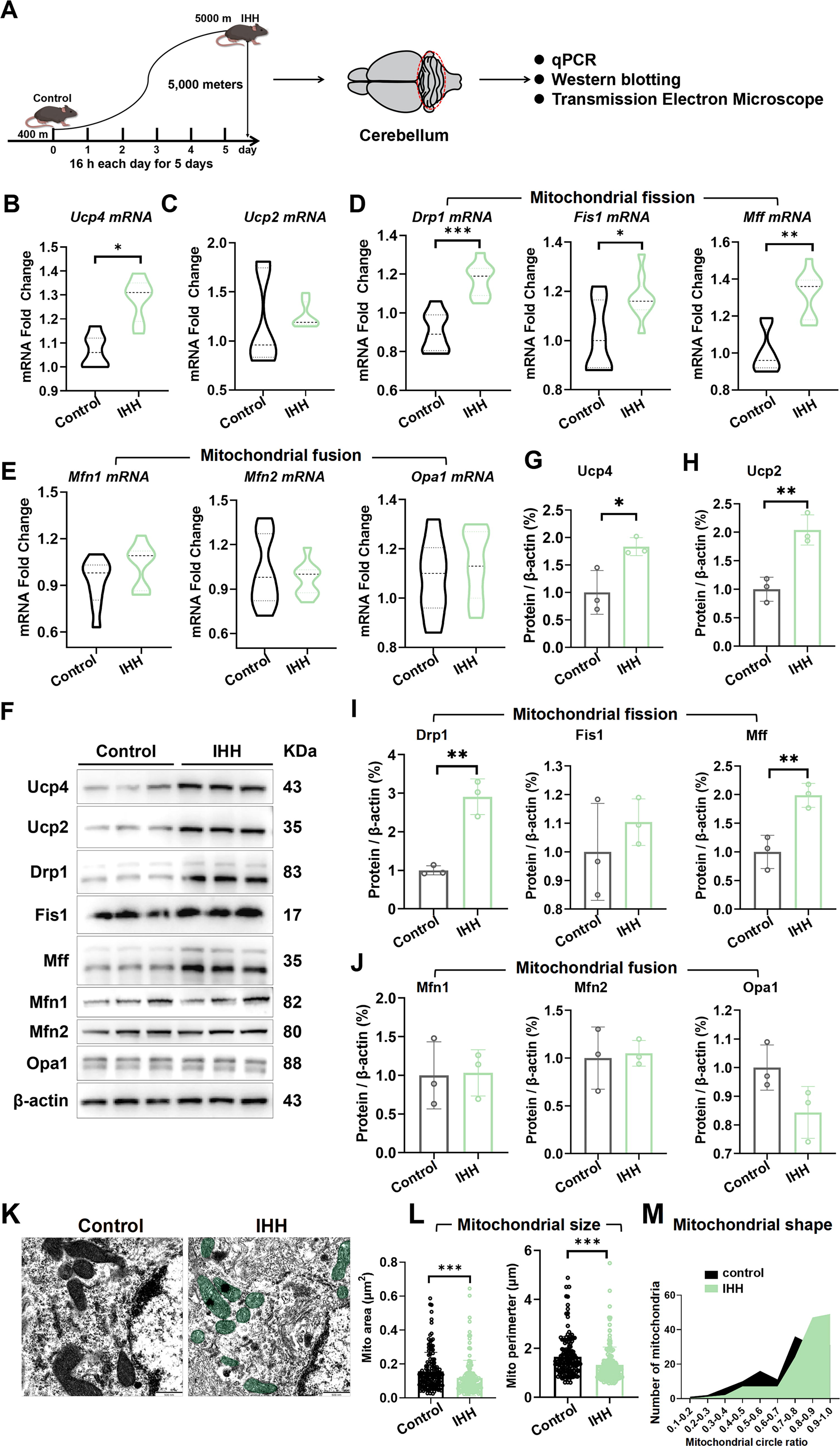
Our findings revealed that IHH significantly increased the expression of Ucp4 by approximately 16.8% (Fig. 2B, p < 0.05), while the expression of Ucp2 remained unchanged (Fig. 2C). In addition, the expression levels of three key mitochondrial fission factors—dynamin-related protein 1 (Drp1), mitochondrial fission protein 1 (Fis1), and mitochondrial fission factor (Mff)—were significantly elevated by approximately 23% (p < 0.001), 14% (p < 0.05), and 24% (p < 0.01), respectively (Fig. 2D). In contrast, the expression levels of three important mitochondrial fusion factors—mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy 1 (Opa1)—did not exhibit significant changes (Fig. 2E). Western blotting analysis further confirmed that the protein levels of Ucp4 (Fig. 2G, p < 0.05), Ucp2 (Fig. 2H, p < 0.01), Drp1 (Fig. 2I, p < 0.01), and Mff (Fig. 2I, p < 0.01) were significantly upregulated following IHH exposure, whereas no significant alterations were observed in the other proteins (Fig. 2I, J). The primary motor cortex (M1) was a critical region in the cerebral cortex responsible for motor control. However, qPCR analysis revealed no significant changes in the expression of mitochondrial-related genes in the M1 area (Supplementary Fig. S1A–E). Collectively, these findings demonstrated that IHH selectively upregulated Ucp4 and mitochondrial fission-related proteins in the cerebellar M1 region, without altering the expression of mitochondrial fusion factors or other mitochondrial genes.
Further analysis using TEM (Fig. 2K) revealed that IHH induced significant mitochondrial fragmentation. The mitochondrial area was significantly reduced from 0.15 ± 0.0027 µm2 to 0.11 ± 0.0024 µm2, corresponding to a decrease of approximately 25% (Fig. 2L, p < 0.001). Similarly, the mitochondrial perimeter was significantly decreased from 1.66 ± 0.0089 µm to 1.32 ± 0.0079 µm, representing a reduction of approximately 20% (Fig. 2L, p < 0.001). In addition, the proportion of “grain”-type mitochondria, which were characterized by a more rounded morphology, significantly increased from 0.7 ± 0.0059 to 0.84 ± 0.0062, corresponding to an increase of approximately 11% (Fig. 2M, p < 0.001). These findings collectively demonstrated that IHH induced mitochondrial fragmentation, as evidenced by the reduced mitochondrial area and perimeter, as well as the increased prevalence of rounded mitochondrial morphology.
The mitochondrial dynamics of PCs induced by IHH remained balanced at an overall level
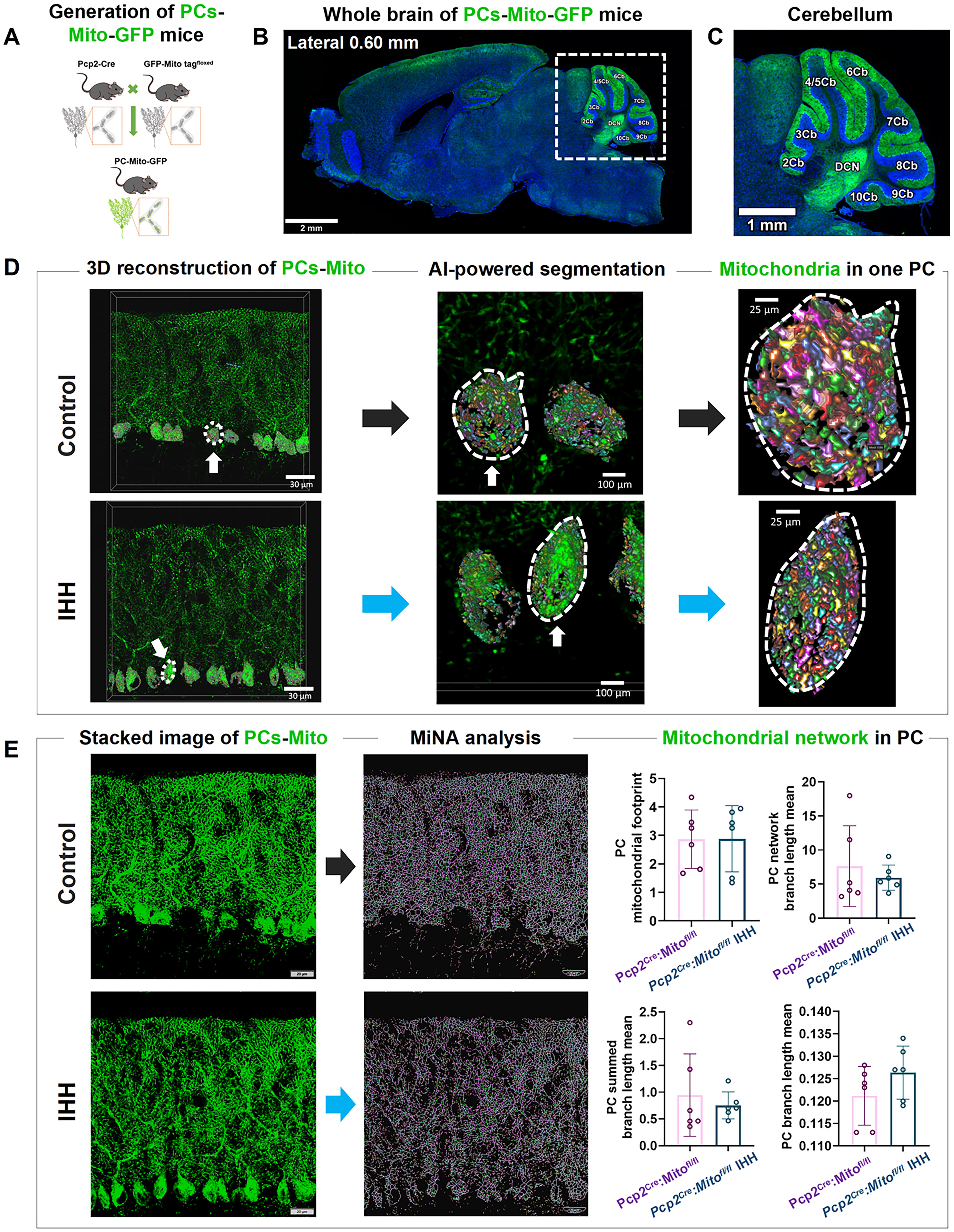
To further evaluate the morphological differences in mitochondria networks across the entire layer of PCs in the IHH model, we used the Pcp2Cre; Mito-GFP transgenic mice24 to conduct a 3D reconstruction of mitochondrial networks and to perform MiNA. In these mice (Fig. 3A), GFP could be specifically expressed on the outer mitochondrial membrane of PCs. Consequently, all mitochondria within the giant neuronal soma, complex dendritic arborization trees, and long projection axons could be readily visualized using a laser scanning confocal microscope. Confocal images (Fig. 3B) confirmed that most mitochondria within the PCs in the cerebellar cortex expressed GFP in the whole brain section. In the enlarged image (Fig. 3C), GFP stain outlined the PC layer clearly throughout the cerebellar lobes ranging from 2 to 10 (Cb2–Cb10). Mitochondrial networks were defined as a continuous matrix lumen. Through the application of artificial intelligence-powered Aivia segmentation and classifiers, there were no significant changes in the mitochondrial networks of the IHH model (Fig. 3D). MiNA analysis further confirmed that there were no significant changes in the four mitochondrial network parameters, including the mitochondrial footprint, network branch length, summed branch length, and mean branch length (Fig. 3E). The findings suggested that the overall mitochondrial dynamics across the entire layer of PCs, induced by IHH, are maintained in equilibrium at the intercellular level.
Ucp4 deficiency exacerbates IHH-induced hypoactivity without affecting spatial cognition
Our team had previously published research detailing the generation of cerebellar PC conditional Ucp4 knockout mice, Pcp2cre; Ucp4fl/fl mice (Ucp4 KO), which presented with Ucp4 deficiency-induced hypoactivity disorder (Wang et al., 2024). Hence Pcp2cre; Ucp4fl/fl mice were further studied in the context of IHH (Ucp4 KO IHH) or sham (Ucp4 KO), with Ucp4fl/fl mice used as the control (Ucp4fl/fl sham and Ucp4fl/fl IHH) (Fig. 4A). The OFT results confirmed that, compared with the Ucp4fl/fl sham group, Ucp4fl/fl IHH mice exhibited significant hypoactivity, characterized by a 50% decrease in movement distance (Fig. 4C, p < 0.001), a 49% reduction in movement speed (Fig. 4D, p < 0.01), and a 21% decrease in activity time (Fig. 4E, p < 0.05). In addition, rest time increased by approximately 40% (Fig. 4F, p < 0.05). The movement distance from the center was significantly reduced by 49% in the IHH group (Fig. 4G, p < 0.05), while central dwell time, peripheral distance, and peripheral dwell time showed no significant differences (Fig. 4H–J, p > 0.05).
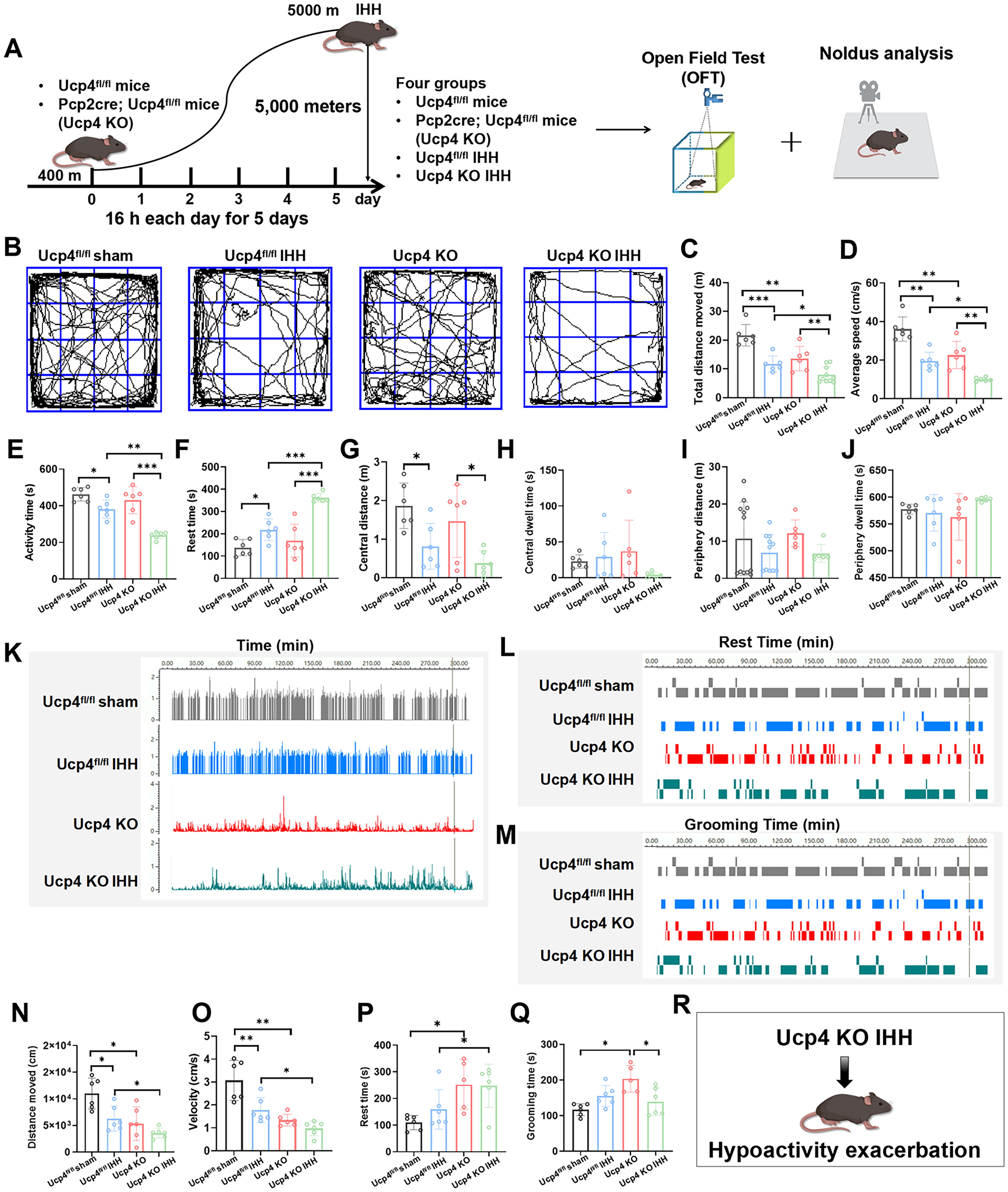
Further analysis revealed that Ucp4 deficiency exacerbated IHH-induced hypoactivity. Compared with the Ucp4fl/fl IHH group, Ucp4 KO IHH mice exhibited a significant worsening of hypoactivity, with a 48% decrease in movement distance (Fig. 4C, p < 0.05), a 49% reduction in movement speed (Fig. 4D, p < 0.05), and a 40% decrease in activity time (Fig. 4E, p < 0.05). Rest time increased by approximately 39% (Fig. 4F, p < 0.001). However, central distance, central dwell time, peripheral distance, and peripheral dwell time did not show significant differences (Fig. 4G–J, p > 0.05).
To identify mice exhibiting movement reluctance, we initially compared resting time (Fig. 4L) and grooming duration (Fig. 4M) among the four experimental groups. Subsequently, we employed the Noldus EthoVision system to assess locomotor capacity by measuring the distance moved (Fig. 4N) and velocity (Fig. 4O). Compared with the Ucp4fl/fl sham group, Ucp4fl/fl IHH mice demonstrated significant hypoactivity, with a 39% reduction in both distance moved (Fig. 4N, p < 0.05) and velocity (Fig. 4O, p < 0.01). Furthermore, IHH exacerbated hypoactivity in Ucp4fl/fl KO IHH mice, resulting in a 40% decrease in both distance moved (Fig. 4N, p < 0.05) and velocity (Fig. 4O, p < 0.05) compared with the Ucp4fl/fl KO group.
The results from the Noldus EthoVision system revealed that, compared with the Ucp4fl/fl sham group, Ucp4fl/fl KO mice exhibited a significant increase in resting time (Fig. 4P, p < 0.05) and grooming duration (Fig. 4Q, p < 0.05), by approximately 31% and 25%, respectively. Furthermore, Ucp4 deficiency exacerbated the increase in resting time in response to IHH, with an approximate increase of 36% (Fig. 4P, p < 0.05), although no significant change was observed in grooming duration (Fig. 4Q, p > 0.05). Subsequently, the Noldus EthoVision system recorded locomotor behavior, capturing Ucp4 deficiency-induced hypoactivity (Supplementary Video S3) and the exacerbation of IHH-induced hypoactivity by Ucp4 deficiency (Supplementary Video S4). These findings collectively demonstrated that Ucp4 deficiency significantly aggravates IHH-induced hypoactivity (Fig. 4R).
The results of the rotarod test indicated that IHH treatment did not significantly impact the coordinated motor ability or passive motor performance in mice, regardless of whether they were wild-type or Ucp4 KO mice (Supplementary Fig. S2A, B). The balance beam test further confirmed that in Ucp4-KO mice, the time spent on the balance beam was increased, but IHH treatment did not further exacerbate this impairment (Supplementary Fig. S2C, D). In addition, the spatial cognition and learning and memory abilities of mice were evaluated using the Morris water maze experiment, which revealed that IHH did not affect these cognitive functions in mice (Supplementary Fig. S2E–J). Collectively, these findings indicated that although IHH exacerbated motor deficits in Ucp4-deficient mice, it did not affect their motor coordination, balance capacity, or spatial cognition.
Ucp4 deficiency exacerbates IHH-induced upregulation of mitochondrial Ucp4 expression and promotes increased mitochondrial fragmentation
qPCR analysis revealed significant alterations in mRNA expression levels across various experimental groups. Compared with the Ucp4fl/fl sham group, IHH treatment significantly upregulated the mRNA levels of Ucp4 and Fis1 by approximately 2.1-fold (Fig. 5A, p < 0.01) and 1.4-fold (Fig. 5C, p < 0.01), respectively. However, no significant changes were observed in the mRNA expression levels of Ucp2, Drp1, Mff, Mfn1, Mfn2, and Opa1 (Fig. 5B–D, p > 0.05).
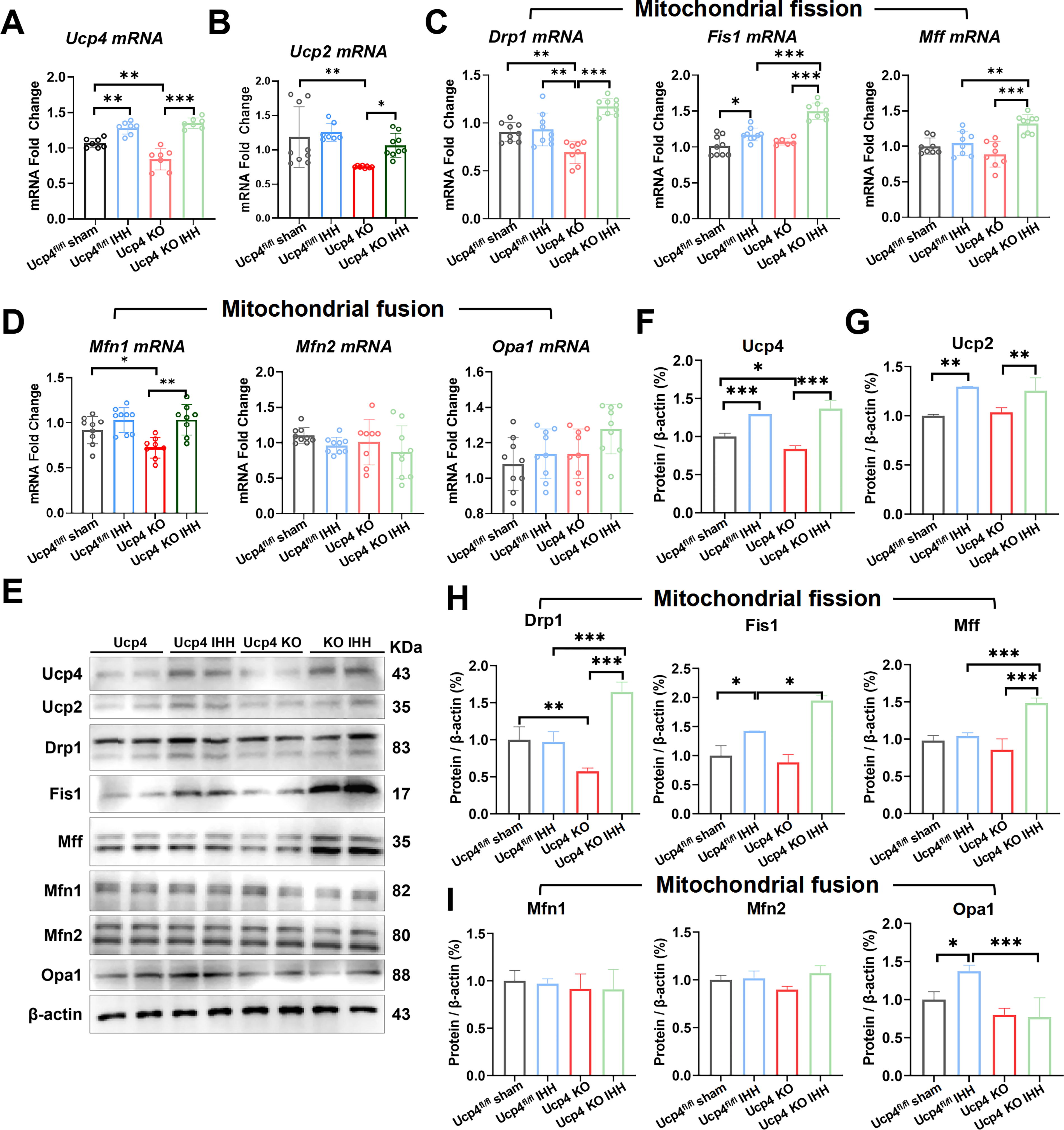
In the Ucp4 KO group, the absence of Ucp4 led to a significant reduction in the mRNA expression of Ucp4 (30%, Fig. 5A, p < 0.01), Ucp2 (39%, Fig. 5B, p < 0.01), Drp1 (19%, Fig. 5C, p < 0.01), and Mfn1 (12%, Fig. 5D, p < 0.05). Conversely, the mRNA expression levels of Fis1, Mff, Mfn2, and Opa1 remained unchanged (Fig. 5C, D, p > 0.05).
When comparing Ucp4fl/fl KO mice with Ucp4fl/fl KO mice subjected to IHH, significant increases were observed in the mRNA levels of Ucp4 (20%, Fig. 5A, p < 0.001), Ucp2 (18%, Fig. 5B, p < 0.05), Drp1 (38%, Fig. 5C, p < 0.001), Fis1 (21%, Fig. 5C, p < 0.001), Mff (28%, Fig. 5C, p < 0.001), and Mfn1 (23%, Fig. 5D, p < 0.01). However, no significant changes were detected in the mRNA expression levels of Mfn2 and Opa1 (Fig. 5D, p > 0.05).
Western blotting analysis further confirmed these findings. Compared with the Ucp4fl/fl sham group, IHH treatment significantly increased the protein levels of Ucp4 (Fig. 5F, p < 0.001), Ucp2 (Fig. 5G, p < 0.01), Fis1 (Fig. 5H, p < 0.05), and Opa1 (Fig. 5I, p < 0.05), while no significant changes were observed in the protein expression levels of Drp1, Mff, Mfn1, and Mfn2 (Fig. 5H, I, p > 0.05).
Similarly, in the Ucp4fl/fl KO group subjected to IHH, significant upregulation was observed in the protein levels of Ucp4 (Fig. 5F, p < 0.001), Ucp2 (Fig. 5G, p < 0.01), Drp1 (Fig. 5H, p < 0.001), Fis1 (Fig. 5H, p < 0.05), Mff (Fig. 5H, p < 0.001), and Opa1 (Fig. 5I, p < 0.001). In contrast, no significant changes were detected in the protein expression levels of Mfn1 and Mfn2 (Fig. 5I, p > 0.05).
Furthermore, the RNAscope technique was employed to validate the Ucp4 mRNA levels in the cerebellar lobules 4/5 (4/5Cb region) across four experimental groups: Ucp4fl/fl sham, Ucp4fl/fl IHH, Ucp4 KO, and Ucp4 KO IHH (Fig. 6A). The results confirmed that the primary source of Ucp4 mRNA expression was the PCs’ soma. Following IHH treatment, Ucp4 mRNA expression was significantly upregulated in the PC soma, granular layer, and molecular layer (Fig. 6B–D, p < 0.001).
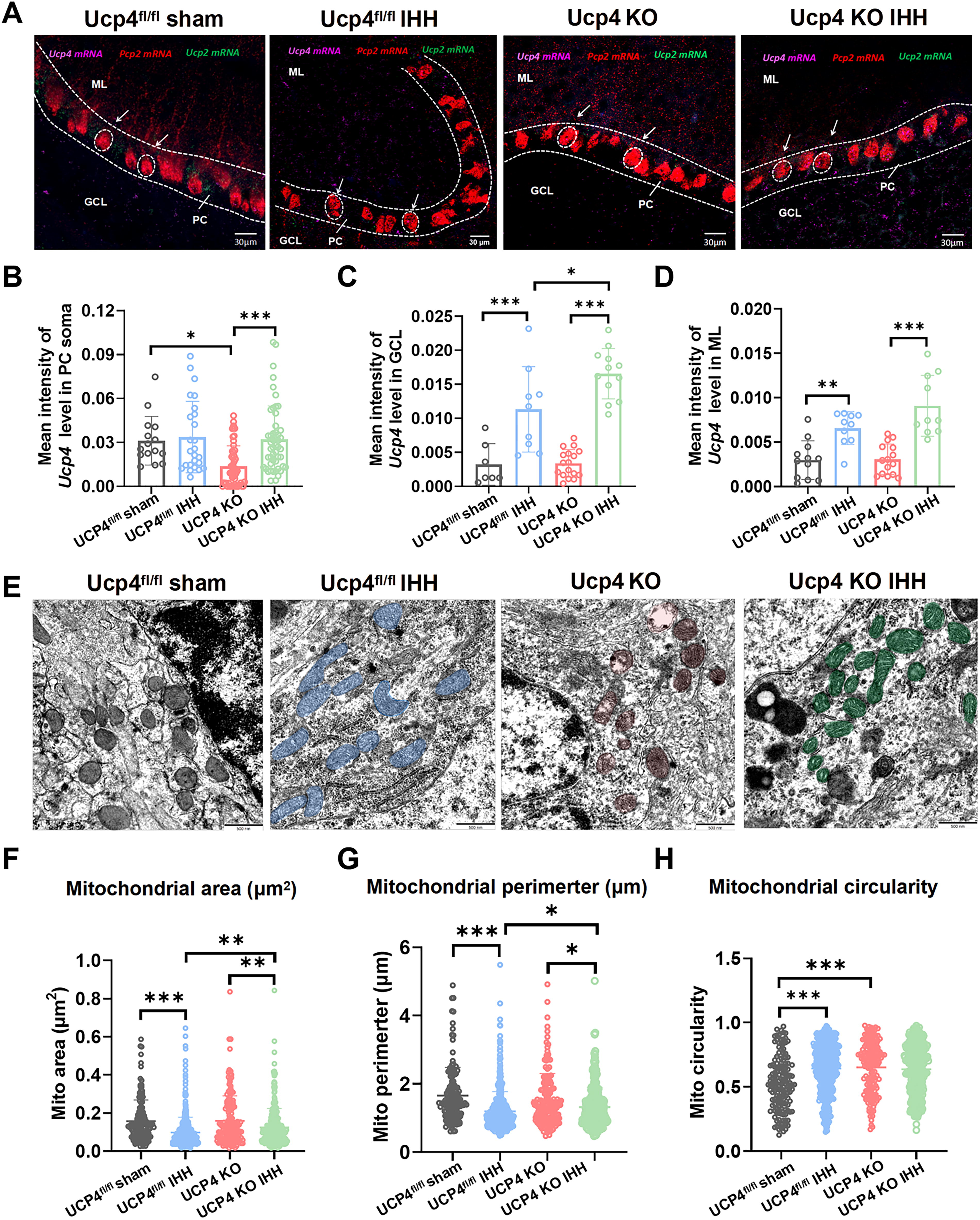
TEM analysis further revealed that, compared with the Ucp4fl/fl sham group, Ucp4fl/fl IHH mice exhibited a marked decrease in mitochondrial size (Fig. 6E). Specifically, the mitochondrial area was significantly reduced from 0.15 ± 0.0093 µm2 to 0.09 ± 0.0037 µm2, corresponding to an approximate decrease of 40% (Fig. 6F, p < 0.001). In addition, the mitochondrial perimeter was significantly diminished from 1.6 ± 0.0685 µm to 1.17 ± 0.022 µm, representing a reduction of approximately 28% (Fig. 6G, p < 0.001). Moreover, the mitochondrial shape became more rounded, as evidenced by an increase in mitochondrial circularity from 0.54 ± 0.0174 to 0.65 ± 0.0028, corresponding to an approximate increase of 20% (Fig. 6H, p < 0.001).
Compared with the Ucp4fl/fl IHH group, Ucp4 KO IHH mice exhibited a marked decrease in mitochondrial size. Specifically, the mitochondrial area was significantly reduced from 0.12 ± 0.0011 µm2 to 0.09 ± 0.0009 µm2, corresponding to an approximate decrease of 20% (Fig. 6F, p < 0.01). In addition, the mitochondrial perimeter was significantly diminished from 1.32 ± 0.0038 µm to 1.21 ± 0.0033 µm, representing a reduction of approximately 8% (Fig. 6G, p < 0.05). However, no significant alteration was observed in mitochondrial shape (Fig. 6H, p > 0.05). These findings indicate that Ucp4 deficiency exacerbates IHH-induced mitochondrial fragmentation.
Specific overexpression of Ucp4 in PCs significantly alleviated hypoactivity in mice subjected to IHH
Subsequently, mice were divided into four experimental groups: NC, Ucp4 OE (Ucp4 overexpression), NC IHH (NC subjected to IHH), and Ucp4 OE IHH (Ucp4 overexpression subjected to IHH) (Fig. 7A). The Ucp4-overexpressing (Ucp4 OE) virus was bilaterally administered into the cerebellar lobules 4/5 (4/5Cb) of C57BL/6J mice, and behavioral testing was conducted after a 4-week interval (Fig. 7B). The successful expression of the Ucp4 virus was confirmed using confocal microscopy (Fig. 7C). The OFT results revealed that, compared with the NC group, Ucp4 OE mice exhibited severe hypoactivity, characterized by a 50% decrease in movement distance (Fig. 7E, p < 0.01), a 32% reduction in movement speed (Fig. 7F, p < 0.01), and a 35% decrease in central distance (Fig. 7I, p < 0.05). However, no significant alterations were observed in activity time, rest time, central dwell time, peripheral distance, or peripheral dwell time (Fig. 7G, H, J–L, p > 0.05).

Compared with the Ucp4 OE group, the Ucp4 OE IHH group exhibited significant improvements in locomotor function, with an approximately 49% increase in movement distance (Fig. 7E, p < 0.01). The Noldus EthoVision system was used to record the locomotor behavior, with the NC treatment group serving as the negative control (Supplementary Video S5). Ucp4 overexpression significantly rescued IHH-induced hypoactivity (Supplementary Video S6). Collectively, these results confirmed that Ucp4 overexpression significantly ameliorateed IHH-induced hypoactivity.
The results of the rotarod test (Fig. 7M) revealed that, compared with the NC group, Ucp4 OE mice exhibited a significant decrease in passive exercise time (Fig. 7N, p < 0.05). Further assessment using the balance beam test indicated that, relative to the NC group, Ucp4 OE mice required significantly more time to traverse the balance beam, with some mice even falling directly (Fig. 7O, p < 0.001). However, no significant differences in balance performance were observed between the NC IHH and Ucp4 OE IHH groups (Fig. 7P, p > 0.05). The water maze experiment demonstrated that neither Ucp4 overexpression nor IHH exposure significantly affected spatial cognitive ability in mice (Fig. 7Q–V, p > 0.05). However, both interventions significantly impaired motor ability and coordination and balance abilities in mice.
Specific overexpression of Ucp4 in PCs significantly alleviates mitochondrial fragmentation and hypoactivity in IHH mice
qPCR analysis revealed significant changes in mRNA expression levels across various experimental groups. Compared with the NC group, overexpression of Ucp4 (Ucp4 OE) significantly upregulated the mRNA levels of Ucp4 (42%, Fig. 8A, p < 0.01), Drp1 (38%, Fig. 8C, p < 0.01), Fis1 (51%, Fig. 8C, p < 0.05), Mff (32%, Fig. 8C, p < 0.01), Mfn1 (35%, Fig. 8D, p < 0.05), and Mfn2 (34%, Fig. 8D, p < 0.01). However, no significant changes were observed in the mRNA expression levels of Ucp2 and Opa1 (Fig. 8B, D, p > 0.05).

Compared with the Ucp4 OE group, IHH treatment further increased Ucp4 mRNA expression by 47% (Fig. 8A, p < 0.01) while reducing the expression of Drp1 (50%, Fig. 8C, p < 0.01) and Fis1 (49%, Fig. 8C, p < 0.05). Conversely, compared with the NC IHH group, Ucp4 overexpression significantly increased Ucp4 mRNA levels by 51% (Fig. 8A, p < 0.001), with no significant changes observed in other genes.
Western blotting analysis further confirmed these findings. Compared with the NC group, Ucp4 overexpression significantly increased the protein levels of Ucp4 (45%, Fig. 8F, p < 0.001), Fis1 (15%, Fig. 8H, p < 0.05), and Mfn2 (35%, Fig. 8I, p < 0.05). Compared with the Ucp4 OE group, IHH treatment increased Ucp4 protein expression by 37% (Fig. 8F, p < 0.001) while reducing Drp1 (39%, Fig. 8H, p < 0.01) and Fis1 (19%, Fig. 8H, p < 0.05) expression. In addition, compared with the NC IHH group, Ucp4 overexpression significantly increased Ucp4 protein levels by 48% (Fig. 8F, p < 0.001) and reduced Drp1 expression by 38% (Fig. 8H, p < 0.01).
TEM analysis (Fig. 8J) confirmed that the Ucp4 OE group exhibited significant mitochondrial fragmentation, characterized by a 50% reduction in mitochondrial unit area (Fig. 8K, p < 0.001) and a 45% decrease in mitochondrial perimeter (Fig. 8L, p < 0.001) compared with the NC group. In addition, mitochondrial rounding was increased by 31.9% (Fig. 8M, p < 0.001) in the Ucp4 OE group relative to the NC sham group. In contrast, the Ucp4 OE IHH group, which was subjected to IHH, exhibited less mitochondrial fragmentation compared to the Ucp4 OE group. This was evidenced by a 30% increase in mitochondrial unit area (Fig. 8K, p < 0.001), a 15% increase in mitochondrial perimeter (Fig. 8L, p < 0.001), and a 20.9% decrease in mitochondrial rounding (Fig. 8M, p < 0.01). These findings demonstrated that overexpression of Ucp4 significantly mitigates mitochondrial fragmentation induced by IHH.
Ucp4 guards against IHH by preserving MMP stability and regulating oxidative stress levels
To identify differentially expressed genes (DEGs) associated with neuronal mitochondria under hypobaric hypoxia conditions, we utilized the Comparative Toxicogenomics Database and the Gene Set Enrichment Analysis (GSEA) database to predict target genes. These predicted genes were then intersected with the mitochondrial gene set from MitoCarta3.0. Following this screening process, we identified 43 DEGs related to mitochondrial function. Functional annotation clustering of these DEGs revealed that they were predominantly associated with specific molecular functions (Fig. 9A), including binding to flavin adenine dinucleotide, transferase activity (particularly acyl group transfer), electron transfer activity, and oxidoreductase activity. These DEGs also exhibited significant enrichment in biological processes (Fig. 9B), with a notable focus on the catabolism of organic acids and fatty acid oxidation pathways. In addition, these genes were linked to specific cellular components (Fig. 9C), such as the mitochondrial inner membrane, oxidoreductase complexes, and components of the mitochondrial respiratory chain. These findings highlighted the critical role of these DEGs in cellular metabolism and energy production, particularly in the context of mitochondrial function and redox reactions. The Gene Ontology (GO) analysis suggested that hypobaric hypoxia induces mitochondrial oxidative stress and dysfunction, thereby affecting key metabolic pathways and mitochondrial integrity.

Ucp4 was essential for protecting mitochondria from oxidative stress, as evidenced by increased ROS levels and a decreased GSH/GSSG ratio, and for maintaining the stability of MMP. Levels of MMP and ROS were measured in control and IHH groups (Fig. 9D). IHH significantly induced MMP collapse (Fig. 9E, p < 0.001), accompanied by a burst of ROS (Fig. 9F, p < 0.001). The GSH/GSSG ratio decreased (Fig. 9G, p < 0.01), indicating impaired mitochondrial antioxidant capacity, but ATP synthesis was unaffected (Fig. 9H, p > 0.05).
MMP and ROS levels were compared across four groups: Ucp4fl/fl sham, Ucp4fl/fl IHH, Ucp4 KO, and Ucp4 KO IHH (Fig. 9I, K). IHH exacerbated IHH-induced MMP collapse (Fig. 9J, p < 0.05) and further increased ROS production (Fig. 9K, p < 0.01). The GSH/GSSG ratio decreased (Fig. 9L, p < 0.05). Ucp4 deficiency significantly aggravated IHH-induced MMP collapse (Fig. 9J, p < 0.001) and ROS burst (Fig. 9K, p < 0.01), with a concomitant decrease in the GSH/GSSG ratio (Fig. 9L, p < 0.05), indicating impaired mitochondrial antioxidant capacity. However, ATP synthesis remained unaffected (Fig. 9M, p > 0.05).
Further comparisons of MMP and ROS levels were made between the NC, Ucp4 overexpression (Ucp4 OE), NC IHH, and Ucp4 OE IHH groups (Fig. 9N). Ucp4 overexpression significantly rescued IHH-induced MMP collapse (Fig. 9O, p < 0.001) and ROS burst (Fig. 9P, p < 0.01). The GSH/GSSG ratio increased (Fig. 9Q, p < 0.05), and ATP synthesis was restored (Fig. 9R, p < 0.01).
These results suggest that the upregulation of Ucp4 mRNA induced by IHH represents a feedback mechanism to counteract oxidative stress and mitochondrial dysfunction. Ucp4 overexpression significantly mitigates IHH-induced mitochondrial damage by restoring MMP and enhancing resistance to oxidative stress.
Discussion
In the present study, we investigated the effects of IHH on hypoactivity and spatial cognition, as well as the underlying mitochondrial dynamics and Ucp4 expression. Our results demonstrated that IHH-induced hypoactivity did not impair spatial cognition. This phenomenon was associated with an upregulation of Ucp4 expression and increased expression levels of three key mitochondrial fission factors, namely, Drp1, Fis1, and Mff, which collectively contributed to increased mitochondrial fragmentation. However, the overall mitochondrial dynamics of PCs remained balanced under IHH conditions.
Furthermore, Ucp4 deficiency exacerbated IHH-induced hypoactivity without affecting spatial cognition. In Ucp4-deficient mice, IHH led to a more pronounced increase in mitochondrial Ucp4 expression and mitochondrial fragmentation compared with wild-type controls. Conversely, specific overexpression of Ucp4 in PCs ameliorated hypoactivity in IHH-exposed mice. This improvement was accompanied by a reduction in mitochondrial fragmentation, indicating that Ucp4 plays a protective role in maintaining mitochondrial integrity. In addition, Ucp4 was found to guard against the detrimental effects of IHH by preserving MMP stability and regulating ROS levels. These findings highlight the critical role of Ucp4 in modulating mitochondrial dynamics and protecting against the neurophysiological consequences of IHH.
The brain is one of the greatest oxygen-consuming organs in the body and is highly susceptible to hypoxia (Chen et al., 2022). Exposure to high altitudes causes a decrease in the oxygenation of both blood and tissues, which in turn triggers an increase in mitochondrial oxidants and a decrease in exercise performance (Ciarlone et al., 2023). The hypoxic environment that is prevalent in high-altitude regions can affect the functioning of cardiovascular, digestive, and nervous systems, consequently affecting cognitive functions such as learning, memory, and visual perception (Pun et al., 2018). Long-term hypobaric hypoxia induced oxidative stress, which was positively correlated with elevation (Joanny et al., 2001). Therefore, the elevated ROS levels caused by exposure to high altitude may have a direct and indirect promoting effect on the progression of cerebellar neuronal pathology.
The main functions of the cerebellum include coordinating movements, maintaining balance, and regulating muscle tone (Benterud et al., 2018). Together, these functional foundations constitute the central role of cerebellar PCs in motor control (Vožeh, 2017). Hypoxia results in the abnormal development of cerebellar neurons and oxidative stress. Lee Y et al. reported that zebrafish experiencing intermittent hypoxia exhibited impaired learning and memory abilities, as well as cerebellar neuron proliferation and replacement (Lee et al., 2018). Clinical studies have found that intermittent hypoxia caused by respiratory control disorders during neonatal development can lead to cerebellar development defects. The white matter and cortex of the cerebellum exhibited elevated levels of oxidative stress and diminished electrical activity in PCs (Leroux et al., 2022; Kundu et al., 2025). Our previous study found that a single hypoxia induced adaptive improvement of motor performance and exploration behavior in mice. Single intermittent hypoxia increased the mRNA expression of Ucp4 in cerebellar cortex, and the knockdown of Ucp4 in PCs caused abnormal mitochondrial division and dysfunction (Wang et al., 2024).
ROS are oxygen-containing active molecules produced during the metabolic processes of aerobic organisms, including superoxide anions (O2 -), hydrogen peroxide (H2O2), and hydroxyl radicals (-OH). (Brieger et al., 2012). The duality of ROS is reflected in its concentration dependence: at low concentrations, it acts as a signaling molecule to regulate physiological functions, while at high concentrations, it leads to oxidative stress and cellular damage. In nerve cells (PC12 and SH-SY5Y), 100 μM of H2O2 induces protective autophagy by activating the AKT/mTOR-HIF-1α/TFEB signaling pathway (Li et al., 2018). Study on the dose–effect relationship and mechanism of ROS promoted neuroprotection (Villalpando-Rodriguez and Gibson, 2021). Low-dose ROS activates antioxidant defense systems (such as SOD, GSH-Px). It enhances the resistance of cells to subsequent oxidative stress through the Nrf2-Keap1 pathway (Feinendegen, 2002). After 6 h of exposure to carbon black nanoparticles, the ROS level increased by 20 times, resulting in mitochondrial calcium overload and membrane potential collapse, and eventually triggering cell apoptosis (Verma et al., 2021). High-altitude cerebral hypoxia promotes ROS production, resulting in neuronal apoptosis, mitochondrial dysfunction, and oxidative stress disorders in mice (Huan et al., 2023). Mitochondria are the main source of ROS in cells (Zhao et al., 2019). Our results also confirmed that mitochondrial ROS significantly increased after IHH treatment. The double-edged sword effect of ROS is jointly determined by dose, spatiotemporal distribution, and cell type. Low-concentration ROS, as an evolutionarily conserved signaling molecule, regulates proliferation, autophagy, and immune defense. High concentrations of ROS cause diseases through oxidative damage and death pathways.
The classical “mild uncoupling” mechanism associated with mitochondrial Ucp represents one of the strategies the body employs to combat oxidative stress. Ucp safeguards neurons against oxidative stress by diminishing the MMP and curbing the production of ROS. This is accomplished through the facilitation of proton leakage, which induces a slight uncoupling of oxidative phosphorylation, albeit at the cost of reduced energy efficiency (Kim-Han and Dugan, 2005; Ulgherait et al., 2020). However, a series of recent studies suggest that neuronal Ucp (Ucp2,4,5) may regulate ROS production by stabilizing MMP, rather than simply decreasing the membrane potential. Ucp1, predominantly expressed in brown adipose tissue, and Ucp3, primarily found in skeletal muscle, mitigate the production of MMP and ROS by facilitating proton leakage (Shabalina et al., 2013; Gustafsson et al., 2004; Hoeks et al., 2006). Ucp2,4,5 show the same uncoupling activity, that is, MMP and ROS decrease synchronously when the expression is upregulated, and MMP and ROS increase synchronously when the expression is downregulated (Mao et al., 1999; Brand and Esteves, 2005; Cho et al., 2016; Crivelli et al., 2024). Paradoxically, in the presence of pathological factors, the uncoupling activity of Ucp2,4,5, predominantly expressed in the brain, seems to be disabled. De Simone R et al. reported that downregulation of microglia Ucp2 in neuroinflammation caused MMP depolarization and increased ROS production (De Simone et al., 2015). A recent report by Geng Z et al. showed that MMP decrease and ROS increase are caused by hypoxia and reperfusion of cardiomyocytes, while MMP levels’ rise toward normal and mitochondrial ATP content also increases after Ucp2 overexpression (Geng et al., 2024). In vivo and in vitro data suggested that Ucp4-mediated neuroprotection against oxidative stress damage may not involve classical uncoupling effects. Chu AC et al. reported that human neuroblastoma exposure to 1-methyl-4-phenylpyridinium (MPP), a mitochondrial complex I inhibitor, induced dysATP production, decreased MMP, and oxidative stress. Under MPP toxicity, Ucp4 overexpression could preserve ATP levels, restore reduced MMP, and inhibit ROS, thereby reducing neuronal death (Chu et al., 2009). Kwok et al. also arrived at a similar conclusion in their study concerning the role of Ucp5 in neuronal MPP toxicity (Kwok et al., 2010). Our previous studies showed that Ucp4 was significantly increased in the cerebellum of a single hypobaric hypoxia simulation, and further knockout of Ucp4 in the cerebellum PC of normal mice caused parallel increases in MMP and ROS (Wu et al., 2021; Wang et al., 2024). Under the pathological condition of intermittent hypoxia, MMP decreased, and ROS increased in cerebellar PCs, and oxidative stress damage was aggravated after Ucp4 was knocked out (lower MMP, higher ROS). The overexpression of Ucp4 in cerebellum MMP decreased and ROS increased (Fig. 9), but the overexpression of Ucp4 in IHH induced in PCs restored MMP to physiological levels and significantly alleviated oxidative stress. These findings were in contrast to the classical “proton leakage” theory of the Ucp family. Consequently, we are introducing the novel hypothesis that Ucp possesses the capability for bidirectional proton transport. In other words, under physiological conditions, it may exhibit a moderate uncoupling effect, while in pathological states, it could contribute to the restoration of MMP and the regulation of metabolic equilibrium, actions that are distinct from its uncoupling function.
Under hypoxic conditions, Ucp4 mitigates cellular oxidative stress through a mechanism involving the uncoupling of oxidative phosphorylation, a reduction in MMP (ΔΨm), and a subsequent decrease in the production of ROS. This adaptive upregulation of Ucp4 represents a cellular response to oxidative stress. Experimental evidence indicates that Ucp4 overexpression is associated with elevated mRNA levels of key mitochondrial fission proteins, including Drp1, Fis1, and Mff. These findings suggest that Ucp4 may directly or indirectly modulate the transcription of these fission-related molecules via specific signaling pathways.
The SIRT1/PGC-1α axis has been identified as a regulator of mitochondrial fission molecules, and this pathway may function independently of Ucp4 (Tian et al., 2019). In the absence of Ucp4, acute hypoxia can activate the expression of Drp1, Fis1, and Mff through alternative stress signaling pathways, such as the SIRT1/PGC-1α pathway. When Ucp4 is overexpressed, mitochondrial fission appears to be predominantly driven by Mff. In addition, the overexpressed Ucp4 may indirectly inhibit the synergistic action of Fis1 by modulating MMP or ROS levels, thereby attenuating the detrimental effects of hypoxia on cellular function. Ucp4 thus protects mitochondrial integrity through uncoupling mechanisms under hypoxic conditions, and its expression level directly influences the regulatory network governing Drp1, Fis1, and Mff (Wu et al., 2021). Conversely, Ucp4 knockout results in the activation of fission molecules via alternative stress pathways during hypoxia. However, Ucp4 overexpression effectively alleviates oxidative stress, thereby inhibiting the propagation of hypoxic signals and preventing further changes in the expression of fission molecules.
Hypoxia induces a decrease in MMP (ΔΨm) and a concomitant surge in ROS production (Li et al., 2019). Elevated ROS levels activate oxidative stress signaling pathways, such as JNK, ERK1/2, or PGC-1α, which in turn drive the phosphorylation of Drp1 and promote mitochondrial fission. While the upregulation of Ucp4 under hypoxic conditions reduces ROS levels through uncoupling, residual ROS levels remain relatively high, potentially stimulating the expression of fission molecules (e.g., Drp1, Fis1, Mff) to facilitate the clearance of damaged mitochondria. Mitochondrial fragmentation under these conditions may serve a dual purpose: maintaining ATP supply through an increase in mitochondrial number (“quantitative” compensation) while simultaneously clearing damaged mitochondria via autophagy.
Notably, overexpression of Ucp4 alone can induce mitochondrial fragmentation, characterized by decreased mitochondrial area, shortened perimeter, and increased circularity. However, this process is associated with a significant upregulation of Drp1, Fis1, and Mff mRNA levels, suggesting a complex regulatory mechanism. This paradox may be explained by the following factors: Overexpression of Ucp4 enhances proton leak, resulting in a sustained reduction in MMP (ΔΨm). The decline in ΔΨm may trigger the fission mechanism, potentially through AMPK-mediated sensing of cellular energy status and subsequent recruitment of Drp1 (Zhou et al., 2022). Furthermore, Ucp4 may directly regulate the transcription of fission molecules by modulating mitochondrial calcium homeostasis or activating the PKA signaling pathway. For instance, the cAMP-PKA pathway may enhance the expression of fission-related genes by phosphorylating transcription factors such as CREB (Yu et al., 2022).
When confronted with oxidative damage, the bidirectional regulatory capacity of Ucp4 on the mitochondrial respiratory chain resembles the “Hormesis effect” as reported by Calabreseci (Calabreseci, 1999). The hormesis effect theory holds that stress can regulate the adaptive responses of organisms and moderately overcompensate, thereby effectively reducing damage. However, generating a higher stress response often backfires and has harmful effects (Calabrese and Fariello, 1988). For instance, low-level oxidative stress may activate NrF2-mediated adaptive responses and regulate the neuroinflammatory cascade of neurodegenerative diseases, while persistent high-level stress can disrupt these defenses, leading to cellular senescence (Calabrese et al., 2023).
The fragmentation caused by overexpression of Ucp4 may be a “preconditioning” mechanism, segmenting mitochondria in advance to cope with future stress (such as ROS burst). When mice overexpressing Ucp4 are subjected to IHH treatment again, mitochondrial area and perimeter increase, and circularity decreases (fewer round mitochondria), indicating enhanced mitochondrial fusion or inhibited fission. Possible mechanisms include: Blockage of oxidative stress signals: Overexpression of Ucp4 has minimized ROS levels, and hypoxia cannot further activate the ROS–Drp1 axis. At this time, mitochondrial fission signals (such as Drp1 phosphorylation) are inhibited, while fusion molecules (such as Mfn1/2, Opa1) may be compensatorily upregulated. The uncoupling effect of Ucp4 reduces ATP synthesis efficiency, but glycolysis is compensatorily enhanced. Chronic hypoxia may trigger metabolic adaptive adjustments, increasing mitochondrial volume (fusion) to enhance the energy output efficiency of individual mitochondria.
UCP may contribute to the mitochondrial antioxidant defense by shuttling fatty acid peroxides from the inner to the outer membrane leaflets. Collectively, neuronal UCP modulates cellular energy transduction, and these mechanisms endow UCP4-upregulated mice with enhanced antioxidant stress resilience (Fig. 10). Ucp4 could serve as a promising therapeutic target for diseases characterized by mitochondrial damage and oxidative stress due to hypoxia.
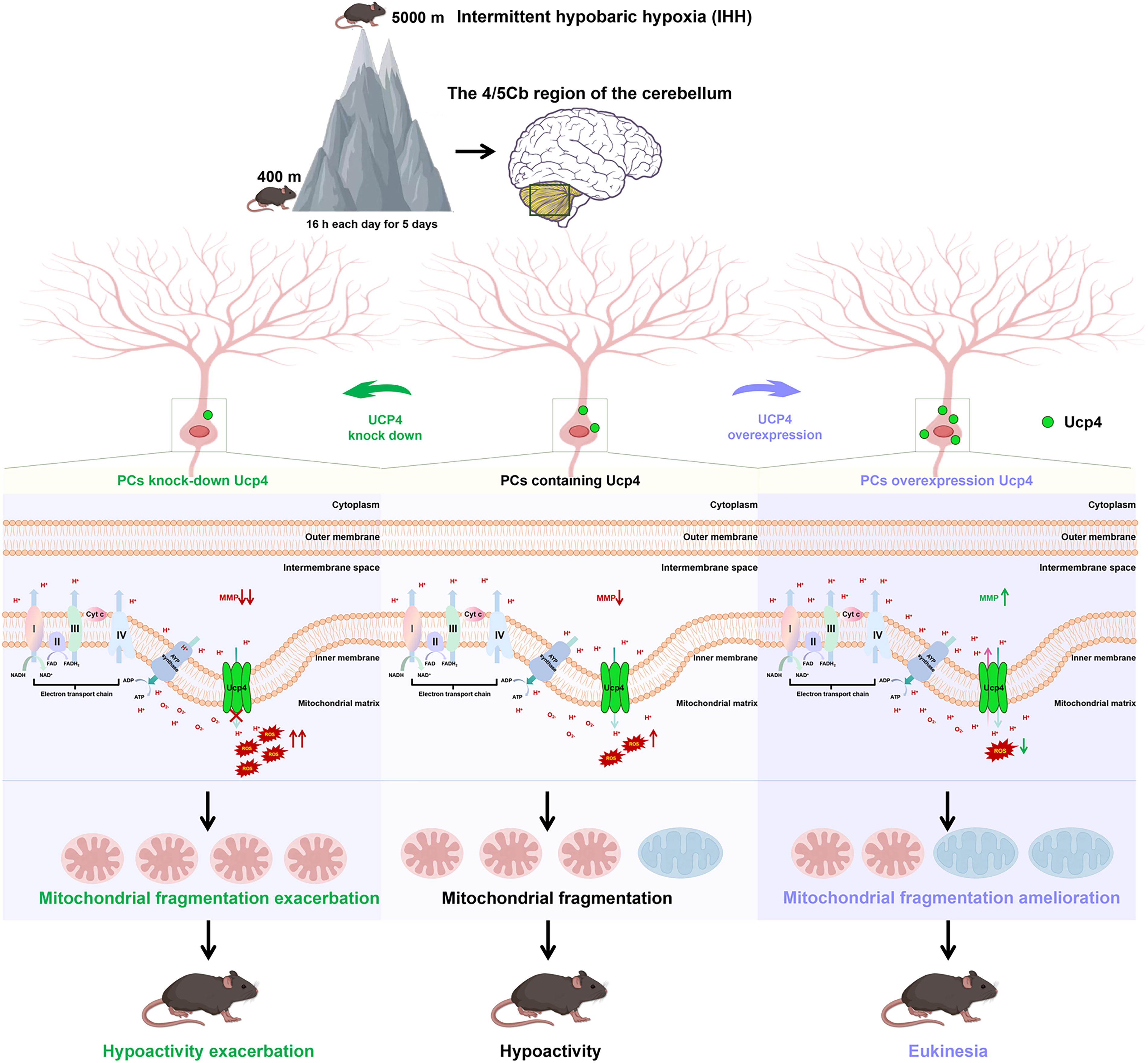
It is important to note that there are some limitations to this study. First, our study focused on the relationship between mRNA expression of Ucp4 and motility, accompanied by changes in MMP and ROS, but other possible mechanisms and signaling pathways were not explored in depth. In addition to the protective effect of mild uncoupling, Ucp is also involved in regulating ion transport, synaptic plasticity, or calcium homeostasis (Andrews et al., 2005), and the role of these mechanisms in oxidative stress damage induced by hypoxia remains unclear.
Our proposed bidirectional proton transport capability of Ucp4 may complement the protective mechanism of hypoxic damage, but further evidence is needed. Furthermore, while the RNA levels of Ucp4 are frequently reported, there is a scarcity of data on protein regulation. Studies have indicated that Ucp4 protein levels do not always correlate with RNA levels, suggesting that post-transcriptional regulatory mechanisms may be at play (Kim-Han and Dugan, 2005). Future research may delve deeper into the post-transcriptional interactions between IHH and Ucp4, as well as investigate how additional related mechanisms influence motor function in mice. Looking ahead, we plan to employ various approaches to develop intermittent-altitude hypoxia models to further validate the correlation between cerebellar Ucp4, oxidative stress, and exercise capacity.
Conclusion
The present study elucidates that hypoactivity induced by the IHH model is closely associated with the overexpression of Ucp4 in the inner mitochondrial membrane of the cerebellum. Conditional knockout of Ucp4 in PCs exacerbates IHH-induced hypoactivity, mitochondrial fission, collapse of the MMP, and oxidative stress. Conversely, targeted overexpression of Ucp4 in PCs significantly alleviates IHH-induced hypoactivity, mitochondrial fission, and oxidative stress levels, while promoting the recovery of the MMP. Mitochondrial Ucp4 exerts neuroprotective effects by safeguarding PCs against oxidative stress-induced damage, thereby improving motor dysfunction. Ucp4 may emerge as a critical protective target in the clinical management of intermittent-altitude hypoxia syndrome.
Materials and Methods
Experimental animal animals
C57BL/6J mice
Adult male mice with an average weight of 20 ± 3 g were obtained from the Laboratory Animal Center of the Air Force Medical University. Mice were kept on a 12:12 h light/dark cycle at 25°C ± 1°C. All animal experimental protocols were approved by the Ethics Committee of the Fourth Military Medical University (IACUC-20220107) and followed the agency’s guidelines on the use of laboratory animals.
Pcp2Cre; Ucp4fl/fl mice
Ucp4fl/fl mice were crossed with Pcp2Cre mice to produce Pcp2Cre; Ucp4fl/fl mice, in which the Ucp4 gene was specifically knocked out in mouse cerebellar PCs. Pcp2cre mice were purchased from Extractive Medicine (Jiangsu, China). The Cre gene was expressed in exon 4 of Pcp2 gene, so that Cre recombinase was specifically expressed in cerebellar PCs. The Pcp2 promoter was combined with Cre recombinase to precisely modulate the target molecule in cerebellar PCs using the cre-loxp system. Ucp4fl/+ mice were purchased from Cyaye Co., Ltd (Guangzhou, China). The Ucp4 gene was located on chromosome 17 of the mouse genome. Using CRISPR/Cas9 gene editing technology, LoxP sites were inserted on either side of Ucp4 exon 3, and flox mice were mated with tissue-specific Cre tool mice to obtain mouse models of Ucp4 gene knockout in specific cell types. Ucp4fl/fl mice were constructed by hybridizing Ucp4fl/+ mice (Table 1).
Summary of the Relevant Information in the Article
Pcp2Cre; Mito-GFP mice
Mito-GFP mice were crossed with Pcp2Cre mice to produce Pcp2Cre; Mito-GFP mice, in which the outer mitochondrial membrane of cerebellar PCs expressed GFP. Rosa26-CAG-LSL-GFP-Mito transgenic mice was constructed by Extract Medicine (Jiangsu, China): A CAG-LBL-GFP-Mito gene fragment was inserted into the mouse Rosa26 site using CRISPR/Cas 9 technology. GFP could be specifically expressed on the outer mitochondrial membrane by crossing with mice carrying the Cre enzyme (Table 1).
IHH model
The intermittent hypobaric hypoxia (IHH) mouse model was established using a hypobaric chamber (Taikang Biotechnology, Shanghai, China. In brief, after placing the mice into the low-pressure oxygen chamber, the height was adjusted to 5000 m to open the vacuum pump, and the gas in the cabin was continuously extracted, resulting in a reduction in the cabin pressure, which was continuously monitored. An IHH model was constructed in the 5000 m hypoxia chamber for 16 h daily for 5 days. During the experiment, the temperature was maintained at 20°C ± 3°C and the humidity was 65% ± 5%.
Behavioral assays
Rotarod test
We performed the rotarod test to analyze the effects of IHH on balance in mice. The rotarod test system was from Shanghai Geely Ang Software Technology Co., Ltd., China. Mice were trained at a constant speed for 3 consecutive days before testing (8 rpm/min on day 1, 11 rpm/min on day 2, and 15 rpm/min on day 3). For testing, mice were placed on the runner device, and rotation started at 8 rpm and reached a maximum speed of 20 rpm/min. The maximum time allowed for each trial was 300 s, and the fall latency of the mice was recorded. Each mouse underwent triplicate measurements.
OFT
To measure the free spontaneous movement of transgenic mice, mice were placed in the center of a 50 × 50 × 50.5 cm opaque square box (DigBehv, Shanghai), before recording their autonomous movements with a camera connected to a computer for 15 min. The movement of the mice was automatically tracked and analyzed by the any-maze software. The following four parameters were analyzed: (i) total distance (m); (ii) average velocity (cm/s); (iii) activity time (s); and (iv) rest time (s); (v) central distance (m); (vi) central dwell time (s); (vii) periphery distance (m); and (viii) periphery dwell time (s).
Behavioral video tracking system
The floor of the PhenoTyper Model 3000 chamber was 30 cm × 30 cm, with a height of 40 cm. The PhenoTyper chamber was equipped with a top unit, including a matrix of infrared LED lights and an infrared CCD camera, with a high-pass filter blocking visible light. The floor of the cage was modified to include a stainless-steel grid (inter-bar separation: 0.9 cm) connected to an electric shock generator (Shock Scrambler ENV-414S; Med Associates, St. Albans, VT). Automated tracking and shock delivery control were performed using EthoVision 3.1 software (Noldus Information Technology, Holland) (Spink et al., 2001). Individual mice were introduced into each chamber and allowed to become accustomed to the apparatus for 60 min. Then, all mice were removed and left undisturbed inside their home cages. Subsequently, the mice were placed individually in the same cages that they had experienced and were tested for 60 min. Behavioral data were collected, and then digital data were analyzed. The following indicators related to motor activity were observed: distance moved (cm), velocity (cm/s), rest time (s), and grooming time (s).
Morris water maze test
During the training stage, the water surface was divided into four equal areas according to the pool markings, and the hidden platform was fixed at the center of the first quadrant, keeping the top of the platform 1–2 cm below the water surface. During each training session, the laboratory mice were randomly placed into the water facing the pool wall from any entry point in the four quadrants. The automatic tracking camera system was adopted to record the swimming trajectory of the laboratory mice and the time it took to reach the platform throughout the process. Each laboratory mouse underwent four training trials, starting from different entry points into the water. If a mouse failed to locate the platform within 60 s, it was guided to the platform and remained there for 20 s before the next trial. The escape latency from all four daily trials was recorded, and the mean value served as the learning ability metric for that day. Throughout the five consecutive days of training, the spatial cognition and memory abilities was assessed by three metrics: (1) total path length to the platform, (2) duration in the target quadrant, and (3) heading angle.
Balance beam test
Prior to formal testing, animals were acclimated to the balance beam apparatus. The training protocol comprised three phases: (i) Environmental habituation: Animals were housed in the testing room for 60 min to minimize novelty stress. (ii) Graduated beam training: Animals were trained to traverse beams of decreasing diameters (initial: 30 mm → final: 10 mm), with five successful traversals required per diameter before rogression. (iii) Pre-test preparation: Animals fasted for 1 h before testing and were transferred to the testing room 30 min pre-procedure. During formal trials, each animal was positioned at the beam start point. Three parameters were recorded: Traversal latency (time to cross 100-cm beam), Fall frequency, Motor coordination (quantified by paw slips, body sway angle, and stride pattern). Each animal underwent five consecutive trials with 15 min inter-trial intervals to prevent fatigue. The mean values served as the final datase.
Stereotaxic AAV viral vector injection
The Ucp4-overexpressing virus, pAAV-CMV-MTS-Ucp4-EGFP-3 x Flag-Wpre (Ucp4 OE), was provided by OBiO (China), and the Ucp4-overexpressing virus was injected into the 4/5 Cb of the mouse cerebellum, with pAAV-CMV-MTS-EGFP-3xFLAG-WPRE used as the negative control (NC). The mice were anesthetized with sodium pentobarbital (100 mg/kg) by intraperitoneal injection and placed in a stereotaxometer (RWD, China). All mice were injected bilaterally with 1 μL of viral vector (2.6 × 1013 viral genome/mL) at a rate of 100 nL/min. Following injection, the needle was left in place for 10 min and then pulled out slowly, to avoid possible leakage of the needle tract. Construct the IHH model, and perform behavioral testing after a 4-week interval.
qRT-PCR
To detect changes in the mRNA levels of mitochondria-associated genes, mice were sacrificed after IHH modeling and subjected to qRT-PCR measurements. Mouse cerebellum tissues were collected and homogenized, and total RNA was measured using TRIzol reagent (Invitrogen, CA) according to the manufacturer’s instructions. The corresponding cDNA was synthesized using the extracted RNA as a template and assayed by qRT-PCR. The genes tested included Ucp4, Ucp2, Drp1, Fis1, Mff, Mfn1, Mfn2, and Opa1. The primer sequences are shown in Table 1. The internal reference was β-actin (Shanghai labor). The reaction conditions were as follows: the program was set to two-step real-time quantification, predenaturation at 95°C, 15 s; denaturing at 95°C, 5 s; and annealing and extension at 60°C, 30 s for a total of 40 cycles. Following amplification, dissolution curve analysis was performed to check for the homogeneity of the products. The relative content was calculated using the 2-△△Ct method and used for statistical analysis.
Western blotting
The cerebellar 4/5 region was dissected and homogenized in lysis buffer (Sigma). Protein samples (30 μg) were separated on SDS–PAGE gel. The blot was blocked and incubated overnight at 4°C with antibodies against Ucp4/Ucp2/Drp1/Fis1/Mff/Mfn1/Mfn2/Opa1/β-actin (antibody information was provided in Table 1). These blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies HRP-Goat antiMouse (1:5000, Cat. #A21010 Abbkine) and HRP-Goat anti-Rabbit (1:5000, Cat. #A21020 Abbkine, CN). Membranes were scanned by the Fusion FX EDGE chemiluminescence imaging, and gray value analysis was conducted using Image J. Each experimental group included three biological replicates.
RNAscope
Mice were anesthetized by intraperitoneal injection of pentobarbital (100 mg/kg). Perfusion: 4% paraformaldehyde. The brain tissue was immersed in a sucrose gradient solution for dehydration. Sections were performed along the sagittal plane. The thickness of the frozen brain tissue sections was 10 (μm), and the frozen brain tissue sections were directly mounted on the precooled adhesive slides. The slides with brain tissue mounted were baked at 37°C for 6 h, and the slides were washed with an appropriate amount of 0.01 M phosphate-buffered saline (PBS) for 5 min to remove the embedding agent. An appropriate volume of RNAscope® hydrogen peroxide was applied to each tissue section and incubated at room temperature for 10 minutes. Sections were then treated with RNAscope® target repair solution, followed by hydrophobic barrier delineation around the tissue perimeter. The slides were treated with RNAscope® protease III, probe hybridization was performed, and AMP signal enhancement treatment was carried out. An appropriate amount of HRP-C1 was dropped onto the slides for fluorescence labeling of the probe channels, DAPI staining was performed, the slides were sealed, and the slides were analyzed using an Olympus confocal fluorescence microscope.
Mitochondrial function detection
Transmission electron microscopy
TEM was used to observe the morphological changes in cerebellar mitochondria. Briefly, mice were perfused with 0.01 M PBS and a 4% formaldehyde solution containing 0.1% glutaraldehyde after anesthesia. The cerebellum was removed and placed into a 4% glutaraldehyde solution for tissue fixation. After fixation, each cerebellum was rinsed twice with 0.1 M PBS, before staining with 1% osmium (TED PELLA, Inc. No.18451) for 2 h. Subsequently, each cerebellum was successively dehydrated with 50%, 70%, and 90% ethanol and 100% acetone, followed by treatment with acetone:embedding agent (Embed812, DDSA, NMA, DMP-30) at a 1:1 ratio for 2 h at room temperature. Finally, the cerebellar tissue was removed from the embedding agent and stored overnight at room temperature. The next day, the tissue was placed on a 60°C special electron microscope plate for 48 h. Next, 70-nm-thick sections were prepared using an ultrathin microtome. Sections were placed on copper grids and stained with lead nitrate and uranium acetate for 10 min. Images were taken with a TEM (JEM1400, Olympus, Japan).
MMP
MMP was assessed in isolated mitochondria using a JC-1 Assay Kit (C2003S, Beyotime, China). The procedure was as follows: First, the cerebellar lobules 4/5 (4/5Cb) were harvested and washed with PBS. Trypsin was added for digestion, and the mixture was agitated every 5 min for a total of 15 min. Subsequently, DMEM supplemented with 10% fetal bovine serum was added to terminate the reaction. After filtration, the sample was centrifuged at 600 g for 5 min to collect the cell pellet. The pellet was resuspended in 0.5 mL of JC-1 staining working solution and mixed thoroughly by inversion. The sample was then incubated at 37°C in a cell culture incubator for 20 min. Following incubation, the cells were centrifuged at 600 g at 4°C for 5 min to pelletize. The supernatant was removed, and the cells were resuspended in JC-1 staining buffer. The sample was then analyzed using flow cytometry.
The flow cytometer was activated and checked for normal operation. The parameter settings were as follows: Two scatter plots were opened. In the first scatter plot, forward scatter was selected as the X-axis and side scatter as the Y-axis to visualize cell size and granularity. In the second scatter plot, the PE (575 ± 26 nm) detection channel was selected as the Y-axis and the FITC (530 ± 30 nm) detection channel as the X-axis to measure the fluorescence intensity of JC-1 in cells. Unstained cell samples (Blank) were used as controls and placed in the flow cytometer sample holder. For cell population selection, a gate was set in the first scatter plot based on cell size and granularity to select the cell population for subsequent analysis.
Fluorescence intensity measurements were conducted sequentially for cells in other sample tubes. The voltage of the fluorescence channels was adjusted to ensure that the cell fluorescence signals were centered in the scatter plot. The MMP of each sample was calculated by comparing the ratio of red fluorescence intensity to green fluorescence intensity (red/green ratio). A decrease in this ratio indicated mitochondrial membrane depolarization. At high MMP level, JC-1 accumulated in the mitochondrial matrix to form polymers (J-aggregates), which emitted red fluorescence. Conversely, under low MMP conditions, JC-1 remained monomeric and exhibited green fluorescence. Flow cytometry was used to measure red–green fluorescence, and the proportion of mitochondrial depolarization was determined by the relative ratio of red to green fluorescence.
ROS
ROS was performed as described in our previous report (Li et al., 2024). ROS levels were quantified in isolated mitochondria using the Reactive Oxygen Species Assay Kit (S0033S, Beyotime, China). Fluorescence intensity was measured by flow cytometry. Each experimental condition included three biological replicates, and each replicate was subjected to three technical replicates to ensure robust data validation.
ATP content
ATP content levels were quantified in isolated mitochondria using the Enhanced ATP Assay Kit (S0027, Beyotime, China). Tissue samples were lysed by adding lysis buffer at a ratio of 100–200 µL per 20 mg of tissue, followed by homogenization using a glass homogenizer or other suitable homogenizing equipment to ensure complete tissue lysis. The homogenate was then subjected to centrifugation at 12,000 g at 4°C for 5 min, and the supernatant was collected for ATP measurement. A standard curve was prepared, and ATP concentration was determined using a luminometer according to the manufacturer’s instructions.
GSH/GSSG ratio
The levels of GSH (reduced glutathione) and GSSG (oxidized glutathione) in isolated mitochondria were quantified using the GSH and GSSG Assay Kit (S0053, Beyotime, China). The assay was based on the reduction of GSSG to GSH by glutathione reductase, followed by the reaction of GSH with the chromogenic substrate DTNB (5,5′-dithiobis-(2-nitrobenzoic acid)) to produce yellow 2-nitro-5-thiobenzoic acid (TNB) and GSSG. The total glutathione (GSSG + GSH) served as a rate-limiting factor for chromogenic development, with TNB formation proportional to total glutathione content. Consequently, the total glutathione concentration was determined by measuring the absorbance at 412 nm (A412). For GSSG quantification, GSH was first eliminated from samples using masking reagents. The remaining GSSG was then measured based on the reaction principle described above. The GSH content was subsequently calculated by subtracting the GSSG content from the total glutathione content (GSSG + GSH). The calculation formula is as follows:
The total GSH concentration was determined by multiplying the GSSG concentration derived from the standard curve by a factor of 2. This adjustment accounted for the stoichiometric conversion wherein one GSSG molecule yields two GSH molecules during reduction. Correspondingly, the measured GSSG content after GSH depletion was also multiplied by 2 to reflect this 1:2 molecular ratio.
Three-dimensional reconstruction of mitochondrial networks
Mitochondrial networks are defined as a continuous matrix lumen. We used our published framework to capture and analyze the neuronal mitochondrial networks using a four-step process composed of 2D and 3D observation (Li et al., 2023), and primary and secondary virtual reality analysis, with the help of artificial intelligence-powered Aivia segmentation and classifiers. In the PCs Mito-GFP mice, GFP could be expressed on the outer mitochondrial membrane specifically on the cerebellar PCs; thus, all mitochondria in the giant neuronal soma, complex dendritic arborization trees, and long projection axons could be easily detected under a laser scanning confocal microscope. The four-step process resolved the complicated neuronal mitochondrial networks into discrete neuronal mitochondrial meshes. We acquired 2D images, which were prepared under confocal microscopy (FV3000, Olympus, Japan). PC-Mito-GFP mice, aged 8 weeks, were perfused transcardially with PBS, followed by 4% fresh formulated paraformaldehyde (Sigma). Tissues were infiltrated in 30% sucrose solution overnight at 4°C. The cerebellum was isolated and frozen in OCT for sagittal sectioning using a cryostat (CM 1950, Leica, Germany) at a thickness of 25 µm. After washing in PBS, the sections were incubated with DAPI (28718–90-3; Solarbio Co., Ltd., China) for 5 min. The brain slides of PC-Mito-GFP mice were used for 3D reconstruction according to a previous report (Li et al., 2023), and at least six slides were taken from each brain tissue.
MiNA
MiNA was performed according to the method described in our previous report (Bai et al., 2023). Briefly, we used the freely available FiJi distribution of the ImageJ platform and amalgamated open-source tools into a simple macro toolset. The brain sections of Pcp2Cre; Mito-GFP mice were used for MiNA analysis under an inverted laser scanning confocal microscope. The number of individual mitochondria was evaluated. Six mice were taken from each group, and at least seven slides were taken from each brain tissue.
Bioinformatics analysis
By employing the keywords “Hypoxia-Ischemia, Brain” and “Consciousness,” we predicted target genes using the Comparative Toxicogenomics Database and GSEA databases and then intersected them with the mitochondrial gene set from MitoCarta3.0. Utilizing the SRplot (Scientific and Research plot tool) platform, we generated a Venn diagram (Yu et al., 2012), which revealed 46 intersecting genes. These intersection targets were then inputted into the STRING database for protein interaction analysis, where we eliminated unbound targets and subsequently identified 43 mitochondrial-related target genes. These genes were designated as our target genes. We further utilized the SRplot platform for GO functional analysis (Luo and Brouwer, 2013; Tang et al., 2023, and presented the outcomes using a Chiplot.
Statistical analysis
Data are expressed as the mean ± standard deviation. All data were first tested for normality and homogeneity of variance. For comparison of two samples, analysis was performed using unpaired two-tailed Student’s t test or paired two-tailed Student’s t test. For comparisons of three groups, differences between groups were assessed using one-way analysis of variance and multiple comparisons between groups using the least significant difference post hoc test. Data that did not meet the tests of normality and homogeneity of variance were analyzed using the Kruskal–Wallis test or Mann–Whitney U test. Statistical analysis was performed using GraphPad Prism software. p Values <0.05 were considered to represent statistical significance.
Electronic laboratory notebook was not used.
Footnotes
Authors’ Contributions
Y.-Y.W. and Y.-L.Y. designed the research plan and interpreted experimental results; F.-F.W., B.-Z.L., R.-Q.W., and Y.-Q.H. were responsible for the collection of clinical samples, sample preparation, the majority of the fundamental experiments, and wrote the article; H.L. conducted the image recognition part of the work; B.-Y.L. and Y.-Z.S. also supported the fundamental experiments; Z.-W.N. supported the bioinformatics analysis; Y.-L.Y. and Y.-Y.W. wrote and edited the article. All authors read and approved the final article.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This work was supported by the National Natural Science Foundation of China (82201627) to F.-F.W., the Medicine Program of Air Force Military Medical University (JJ24JH09, 2024JC003) from Y.-Y.W., the Shaanxi Innovation Capability Support Plan (2023-CX-PT-33) from Y.-Y.W., the Shaanxi Basic Research Program of Natural Sciences (2024JC-ZDXM-60) from Y.-L.Y., the Xi-Jing Hospital Clinical New technology (XJZT24CY15, XJZT25KX01, 2023XJSY27, 2023XJSY27) from Y.-L.Y., and the Shaanxi Basic Research Program of Natural Science (2022JQ-820) from F.-F.W.
Supplementary Material
Supplementary Data
Supplementary Video S1
Supplementary Video S2
Supplementary Video S3
Supplementary Video S4
Supplementary Video S5
Supplementary Video S6
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.