
Abstract
Significance:
Reactive oxygen species (ROS) are a double-edged sword in the context of oncoviruses. The effects of ROS on cells depend on the cellular environment, the stage of the disease, and the specific molecular pathways involved. In general, ROS levels in oncovirus-infected cells are usually increased and produce two distinct outcomes on cancer progression and metastasis through multiple mechanisms. Therefore, identifying the relationship between ROS and tumor viruses at the molecular level is essential for cancer prevention and treatment.
Recent Advances:
ROS play an important role in oncoviral infection and disease progression. The excessive accumulation of ROS induces ferroptosis, which has an important role in tumor therapy and the immune microenvironment, thus providing a theoretical basis for the development of new anticancer treatment strategies.
Critical Issues:
This review summarizes the complex relationship between ROS and oncoviral infection, with the aim of providing a deeper understanding of tumor pathogenesis and new therapeutic strategies.
Future Directions:
The relationship between ROS induced by oncoviral infection and host metabolic pathways, including lipids, lipoproteins, amino acids, and polyamines. Understanding how metabolism is reprogrammed in cancer cells may elucidate the impact of these processes on viral infection and tumor progression and help develop effective treatment strategies. Antioxid. Redox Signal. 43, 528–546.
Introduction
Reactive oxygen species (ROS) encompass oxygen-based free radicals and peroxides that arise as byproducts of cellular oxygen metabolism. This includes the superoxide anion (O2·−), hydrogen peroxide (H2O2), hydroxyl radicals, singlet oxygen, and ozone. The two main sources of ROS are NADPH oxidase (NOX) and the enzyme family of the respiratory electron transport chain (ETC) in the mitochondria (Lambeth and Neish, 2014; Zorov et al., 2014). The NOX family of proteins, which is considered one of the most important sources of ROS production in eukaryotic cells, is a family of heme-containing proteins. The NOX protein family consists of seven members (NOX1-5 and DUOX1-2). NOX is a membrane protein containing a conserved catalytic core of six transmembrane α-helices, with each member sharing a catalytic structural domain and an intracellular dehydrogenase site (Vermot et al., 2021). Its primary function is to transfer electrons from NADPH to oxygen to form ROS. NOX1-3 and NOX5 mainly produce O2·−, whereas NOX4 and DUOX1-2 primarily generate H2O2 (Magnani and Mattevi, 2019). NOX can diffuse through water channel proteins and modulate various cell signaling pathways. NOX proteins are activated by the stimulation of inflammatory mediators and oncoproteins, which suggests that cancer cells can induce increased ROS levels via NOX. The key to mitochondrial ROS production is a single-electron leak. Complexes I, II, and III in the ETC are the major sites of mitochondrial ROS production. Several mitochondrial enzymes and mitochondrial cytochrome P450 (CYP) metabolism also generate ROS (Junco et al., 2024). In tumor cells, mitochondrial dysfunction results in the reaction of electrons with oxygen to generate superoxide (Yang et al., 2016). Lactate, which is a product of glycolysis enriched during the Warburg effect, promotes mitochondrial activity through oxidative phosphorylation, which further increases ROS production (Liberti and Locasale, 2016). ROS are essential mediators of cell signaling and homeostasis and play an important role in the biophysiology and adaptive responses. Examples of signaling pathways disrupted by ROS include extracellular signal-regulated kinase 1/2 (ERK1/2), nuclear factor-κB (NF-κB), activator protein-1 (AP-1), phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt), Ras, Rac, and signal transducer and activator of transcription (STAT) (Averill-Bates, 2024), thereby regulating essential cellular processes including proliferation, differentiation, invasion, and cell death (Quilaqueo-Millaqueo et al., 2024). One possible mechanism is that ROS activate the corresponding receptor or src kinase, which activates the signaling pathway (McCubrey et al., 2006).
ROS levels often correlate with cancer, with different tumor cell types exhibiting higher levels of ROS compared with normal cells. ROS play a dual role in the regulation of both physiological and pathological processes (Cheung and Vousden, 2022). As a second messenger involved in the activation of multiple signaling pathways, ROS can regulate physiological and pathological processes such as cell proliferation, differentiation, metabolic reprogramming, and immune response (Benhar et al., 2002; Cheung and Vousden, 2022). ROS can damage various macromolecules, such as proteins, lipids, and nucleic acids, causing DNA double-strand breaks and damage, which can activate oncogenes or inactivate tumor suppressor genes. This is one mechanism by which ROS promotes tumorigenesis (Poetsch, 2020). In addition, ROS can regulate immune responses at multiple levels. STAT3 is a key transcription factor in ROS-mediated immune pathways (Qin et al., 2025). ROS can promote the phosphorylation and activation of STAT3 through pro-inflammatory cytokines such as interleukin-6 (IL-6). Activated STAT3 recruits immunosuppressive cells, including myeloid-derived suppressor cells, regulatory T cells, and M2-polarized tumor-associated macrophages (TAMs), thereby reducing the function of CD8+ T cells and natural killer (NK) cells and promoting tumor immune evasion (Soler et al., 2023; Thuya et al., 2025). However, high concentrations of ROS can also cause oxidative damage to cellular macromolecules and impair mitochondrial function, thereby triggering cellular stress responses and activating programmed cell death mechanisms such as apoptosis and ferroptosis (Zhang, 2018). Therefore, manipulating ROS levels to regulate downstream signaling pathways may induce tumor cell apoptosis, thus offering a potential cancer treatment strategy.
Oxidative stress occurs when the rate of ROS production exceeds the capacity of the antioxidant system to eliminate them (Hayes et al., 2020). To ensure ROS signaling processes are maintained and oxidative damage is avoided, cells contain a range of antioxidant systems, including superoxide dismutase (SOD), catalase, thioredoxin (TRX), glutathione peroxidase (GPX), glutathione (GSH), and peroxiredoxin. High ROS concentrations oxidize the cysteine residues on Keap1, an E3 ubiquitin ligase, which translocates nuclear factor (erythroid-derived 2)-like 2 (NRF2) to the nucleus and induces the expression of genes containing antioxidant response elements (Menegon et al., 2016).
Cancer caused by infections accounted for 13% of all cancer cases in 2018 (de Martel et al., 2020). Seven human oncoviruses have been identified, including Epstein-Barr virus (EBV), Kaposi’s sarcoma herpes virus (KSHV or human herpesvirus 8), hepatitis B virus (HBV), hepatitis C virus (HCV), human papillomavirus (HPV), human T-cell leukemia virus type 1 (HTLV-1), and Merkel cell polyomavirus (MCPyV) (MacDonald and You, 2017; Morris et al., 1995). Tumorigenesis caused by viral infections usually involves the sustained expression of specific viral gene products that interfere with normal cell processes, such as cell division, apoptotic pathways, and immune evasion (Schiller and Lowy, 2021). In this review, we discuss the mechanisms of altered ROS levels caused by oncoviral infecting cells and their effects on cell proliferation and transformation and introduce new strategies for tumor therapy. Currently, studies on MCPyV remain limited and we will focus on the intricate interactions between six other tumor viruses and ROS.
Oncoviral Infection and ROS
For many oncoviruses, host cells produce ROS through several pathways, such as activating intracellular NOX, interfering with mitochondrial function, and inducing endoplasmic reticulum (ER) stress. Notably, elevated ROS have been shown in a variety of oncoviruses to be able to in turn promote viral replication and facilitate viral infection. Elevated ROS could cause DNA damage and genomic instability, which promotes viral integration, mutation accumulation, and oncogenesis (Gorrini et al., 2013). On the other hand, high ROS levels can affect cell-cycle progression and apoptosis, thereby determining whether the cell survives or undergoes programmed cell death following viral infection (Yang and Lian, 2020). To avoid the cytotoxic effects of ROS, oncoviral infection causes elevation of a range of antioxidant enzymes. This provides multiple strategies for treating cancers caused by viral infections by modulating ROS levels. Notably, certain drugs such as 2-deoxy-D-glucose, dehydroepiandrosterone, and Auranofin have been shown to enhance oxidative stress by inhibiting glycolysis, the pentose phosphate pathway (PPP), and TRX reductase, thereby selectively inducing cancer cell death. However, this strategy has not yet been validated in oncovirus-associated cancers (Glorieux et al., 2024; Li et al., 2015).
EBV and ROS
EBV is a DNA virus classified as human herpesvirus type 4 (Damania et al., 2022). It is highly prevalent and infects approximately 95% of the adult population worldwide. There is a clear link between EBV and malignancies, such as T-cell lymphoma, Burkitt’s lymphoma, Hodgkin’s lymphoma, EBV-associated gastric cancer (EBVaGC), and nasopharyngeal carcinoma (NPC) (Ko, 2015). The EBV life cycle consists of two phases: latency and lytic replication. The establishment of viral latency is a key strategy for EBV to achieve long-term persistence, transmission, immune evasion, and tumorigenesis. EBV latency is associated with six EBV nuclear antigens (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA5) and three latent membrane proteins (LMP1, LMP2A, and LMP2B). In vitro, EBV latent genes such as LMP1, EBNA1, EBNA2, and EBNA3C can cooperatively activate host cell proliferation and antiapoptotic signaling pathways, leading to B-cell immortalization and the formation of lymphoblastoid cell lines (Chen et al., 2016). B-cell immortalization facilitates the establishment of lifelong latent infection (Kenney and Mertz, 2014). EBV infection induces ROS production, activates ROS-related signaling pathways and DNA damage in vivo, which are largely associated with the expression of viral oncogenic proteins (Sun et al., 2015).
EBNA1 is a viral antigen that is consistently expressed in all EBV-associated malignancies as well as throughout both latent and lytic infections. In B cells, EBNA1 can directly or indirectly bind to the promoter region of the NOX2 gene, acting synergistically with B-cell-specific transcription factors, to activate its transcription, significantly increasing ROS levels (Gruhne et al., 2009). EBNA1-induced ROS can activate oncogenes such as Ras and Myc, induce DNA double-strand breaks and genomic instability, thereby promoting B-cell transformation and immortalization (Studstill et al., 2023). In EBVaGC, EBNA1 is involved in the survival of gastric cancer cells by regulating the miR34a-NOX2-ROS signaling axis (Kim et al., 2017). LMP1 results in the formation of the AP-1 complex through the activation of the c-Jun N-terminal kinase (JNK) pathway, which in turn, binds to the promoter region of the p22phox gene (a key regulatory subunit of the NOX complex) to promote its transcription, and NOX-induced ROS overproduction similarly causes DNA damage, thereby facilitating LMP1-mediated malignant transformation of human nasopharyngeal epithelial cells (Sun et al., 2015). LMP1 can also interact with and inhibit Protein kinase RNA-like ER kinase via a conserved transmembrane motif, leading to suppression of NRF2 activity and increased ROS accumulation (He et al., 2021a). EBV-encoded RNA induces IL-10 production in EBV-infected cells by activating retinoic acid-inducible gene I -mediated interferon regulatory factor 3 signaling, and IL-10 induces ROS production (Cerimele et al., 2005; Samanta et al., 2008). Produced ROS results in high methylation of p16INK4a (tumor suppressor gene) and mitogen-activated protein kinase (MAPK) activation, which can promote tumorigenesis (Cerimele et al., 2005). One possible mechanism is the upregulation of DNA methyltransferases by ROS (Arbiser, 2005; Arbiser et al., 2006). Beyond latent genes, EBV replication can impair mitochondrial electron transport, leading to ROS generation (Lassoued et al., 2010), which in turn, induces the expression of BZLF1 to promote EBV lytic and reactivation (Sausen et al., 2021). The possible mechanism is that oxidative stress affects AP-1 signaling pathway and BZLF1 can be functionally or structurally classified as an AP-1 family member. This indicates that EBV replication and ROS production amplify one another to create a feedback cycle. This is the main cause of radioresistance due to oxidative stress (Hu et al., 2020). ROS production also activates TLR signaling, which activates MAPK and JNK, upregulates programmed death-ligand 1, and reduces monocyte survival. Thus, EBV-mediated intracellular ROS production also mediates immunosuppression (Gilardini Montani et al., 2018) (Fig. 1).
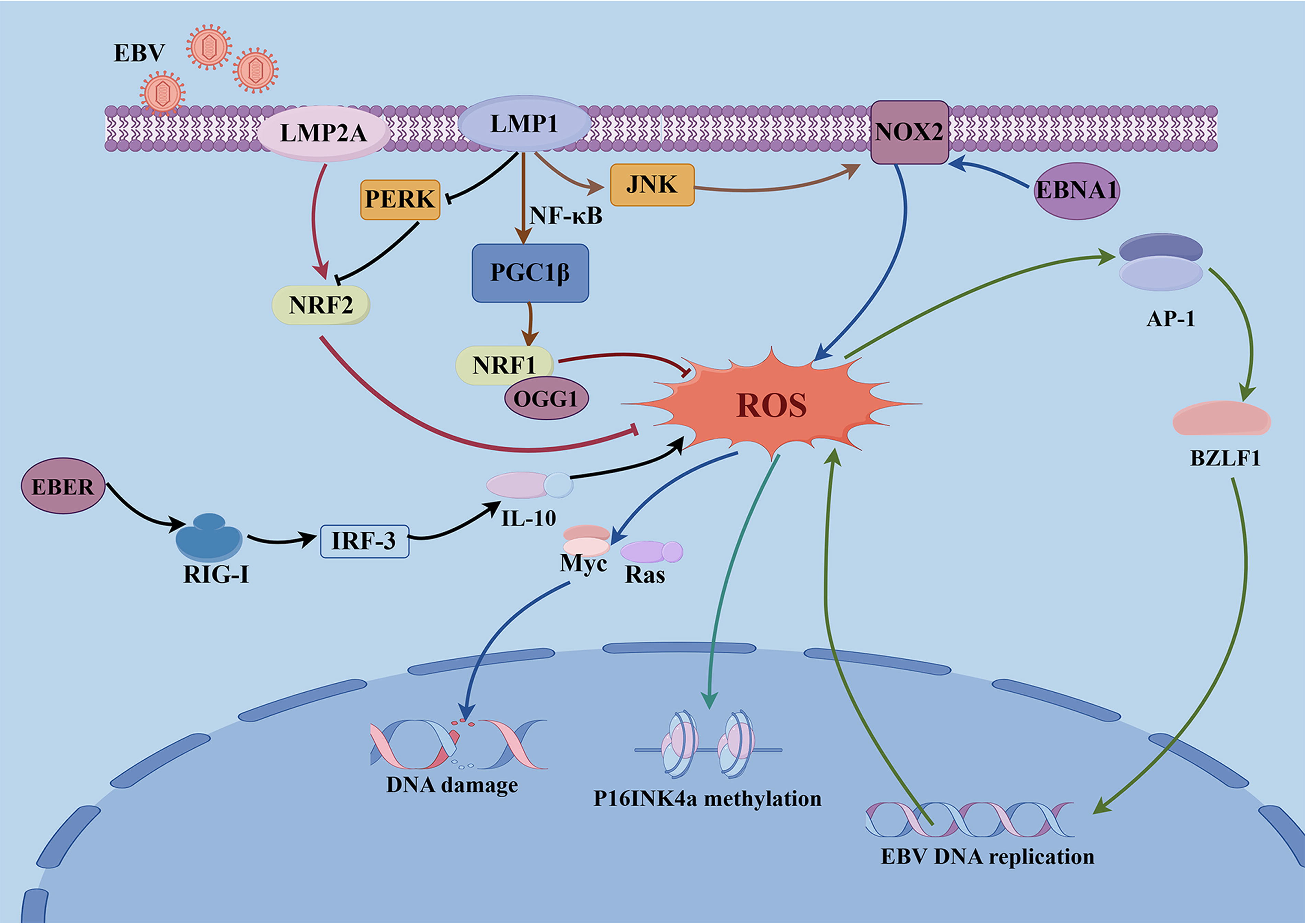
Excess ROS levels can also produce toxic effects in cells. It was reported that ROS could activate caspase-9 through CD70, thereby causing apoptosis in EBV-infected B cells (Park et al., 2010). Additionally, EBV infection can drive the induction of lipid ROS, including EBNA2 strongly stimulating host lipid metabolism (Wang et al., 2019b) and LMP1 upregulating fatty acid synthase (Hulse et al., 2021), rendering newly infected B cells sensitive to ferroptosis. To avoid the deleterious effects of ROS, EBV leads to adaptive responses in host cells by activating a variety of antioxidant defense mechanisms, thereby maintaining persistent infection and enhancing the survival capacity of tumor cells (Galadari et al., 2017). LMP1 can upregulate peroxisome proliferator-activated receptor-γ coactivator-1β (PGC1β) by activating NF-κB. PGC1β, in turn, enhances the expression of NRF1 and the DNA repair enzyme 8-oxoguanine DNA glycosylase, thereby reducing ROS-induced damage to the viral genome and supporting EBV persistence (Feng et al., 2021). Furthermore, LMP1 finely regulates glycolytic metabolism through ROS generation, helping EBV-transformed cells survive and proliferate under harsh metabolic conditions (Yee and Wang, 2025). LMP1 and LMP2A can both induce NRF2 activation in EBV-transformed B cells, NRF2 exerts antioxidant effects through its downstream target heme oxygenase-1 (HO-1) (Yun et al., 2019) (Fig. 1). ROS also activates p62-mediated selective autophagy, which targets ubiquitinated substrates and promotes DNA repair (Wang et al., 2019a). Notably, p62 can also inhibit DNA repair depending on its subcellular localization and state. This dual mechanism of action contributes to the survival of EBV-transformed cells during oxidative stress (Wang et al., 2019a).
EBV and ROS-Targeted Therapeutics
EBV infection not only promotes the generation of ROS, but also activates multiple adaptive mechanisms to cope with oxidative stress, thereby playing a dual role in cell survival, viral latency, and tumorigenesis. Based on this, ROS-related pathways have emerged as important therapeutic targets in EBV-associated malignancies. On the one hand, reducing or neutralizing ROS levels can enhance radiosensitivity and reverse EBV-induced radioresistance; On the other hand, inducing ROS overload, particularly lipid ROS accumulation, can trigger ferroptosis or apoptosis in tumor cells, demonstrating strong antitumor potential.
Studies have demonstrated that LMP1-mediated high oxidative stress promotes EBV reactivation, which in turn enhances radioresistance of NPC cells both in vitro and in vivo (Hu et al., 2020). The use of N-acetylcysteine (NAC) can reverse radioresistance induced by radiotherapy and chemotherapy. In addition, in EBVaGC, targeting the LMP1/PGC1β signaling pathway can disrupt the ROS positive feedback loop, inhibit viral replication, and block tumor progression (Yuan et al., 2023).
Various strategies have been developed to eliminate EBV-associated tumor cells by increasing intracellular ROS levels. For example, cephalosporin antibiotics exhibit highly selective cytotoxicity toward NPC cells by inducing ROS production through HO-1 mediated iron release and the Fenton reaction, while exerting minimal on normal cancer cells (He et al., 2021b, 2024). In addition, natural compounds such as cucurbitacin B and lupeol have been reported to induce ferroptosis in NPC cells by inhibiting GPX4, depleting GSH, and increasing lipid ROS accumulation (Huang et al., 2021; Zhou et al., 2022). In EBVaGC, tremella fuciformis polysaccharides have shown potential therapeutic effects by inhibiting the NRF2/HO-1 pathway and inducing ferroptosis (Kong et al., 2024) (Fig. 2a).

KSHV and ROS
KSHV, also referred to as HHV8, is a double-stranded DNA virus that encodes more than 90 open reading frames (ORFs) (Losay and Damania, 2025). It is closely associated with the development of three malignancies, including Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman disease (Iftode et al., 2020), all of which are characterized and hallmarked by excessive inflammation and oxidative stress. Similar to other herpesviruses such as EBV, the KSHV life cycle consists of latent and lytic phases (Han et al., 2024). Most tumor cells associated with KSHV are latently infected, indicating that latent infection is essential for the initiation and maintenance of KSHV-associated malignancies (Ye et al., 2011; Li et al., 2011). During latency, the viral genome persists stably within host cells and mainly expresses latent-associated genes such as LANA (ORF73), viral Fas-associated death domain protein-like IL-1β-converting enzyme-inhibitory protein (vFLIP) (ORF71), vCyclin (ORF72), kaposin (K12), and a cluster of 12 precursor microRNAs. In the lytic phase, the virus initiates the expression of lytic genes including RTA (ORF50), bZIP (ORFK8), and the viral G-protein-coupled receptor (vGPCR) (Losay and Damania, 2025). Reactivation of the virus from latency not only leads to the release of infectious virions but also often contributes to disease progression.
Multiple oncogenic products encoded by KSHV not only regulate the viral life cycle but also influence tumor cell proliferation, metastasis, and remodeling of the tumor microenvironment by modulating ROS levels, thereby playing a critical role in the initiation and progression of KSHV-associated malignancies (Losay and Damania, 2025). One notable mechanism involves the KSHV-encoded vGPCR, which induces the production of ROS (O2·-) via the Rac1-NOX signaling pathway (Ma et al., 2013). Rac1-activated ROS promote cell proliferation and angiogenesis by regulating the transcription of c-Myc and vascular endothelial growth factor (VEGF) (Ma et al., 2009). In addition, latent viral proteins such as LANA, v-Cyclin, and vFLIP also can induce ROS production (Ma et al., 2013a). ROS can activate the ERK1/2, JNK, and p38 MAPK pathways, which in turn, activate KSHV from latency by increasing the expression of viral lytic proteins, such as RTA, ORF57, and ORF65, thus contributing to KSHV-induced tumorigenesis (Ye et al., 2011; Xie et al., 2008). During the early stages of KSHV infection, ROS effects include changes in the cytoskeleton and membrane structure and amplification of signaling pathways, such as focal adhesion kinase and Rac1, to induce viral entry and replication (Bottero et al., 2013) (Fig. 3). It is worth noting that Incani et al. reported that KSHV infection may exacerbate oxidative stress in patients with type 2 diabetes by increasing ROS production, thereby promoting disease progression and associated complications such as atherosclerosis (Incani et al., 2020).
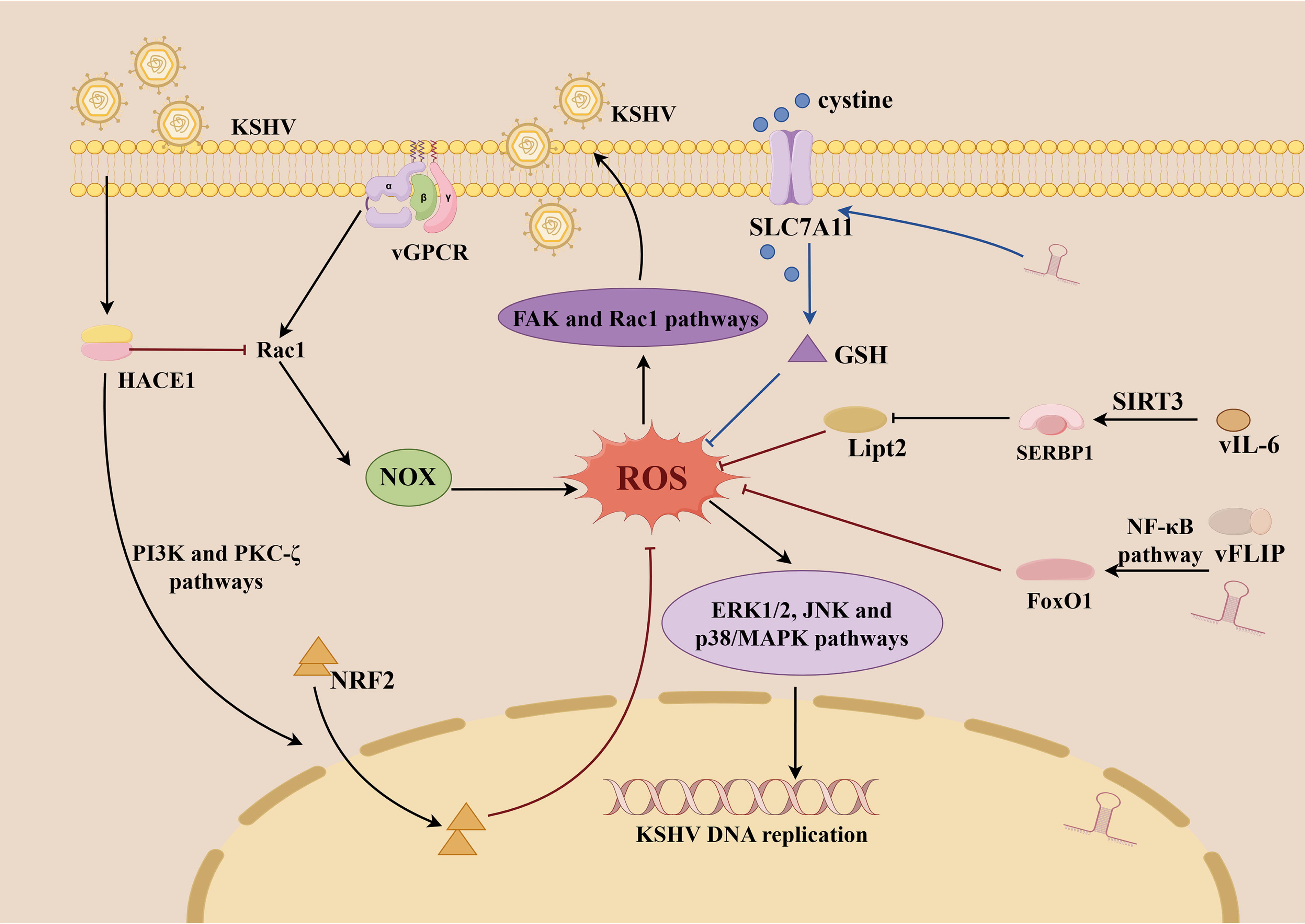
ROS have cytotoxic effects, studies have shown that excessive ROS can induce G1 cell-cycle arrest in KSHV-transformed cells, inhibit their proliferation and colony formation in soft agar, disrupt viral latency, and ultimately lead to cell death (Li and Gao, 2023). Paradoxically, most KS tumor cells harbor latent KSHV infection within a highly inflammatory and oxidative tumor microenvironment, suggesting the existence of mechanisms that enable these cells to withstand oxidative stress-induced cytotoxicity (Gjyshi et al., 2014; Thurau et al., 2009). KSHV-encoded miRNAs and vFLIP upregulate the transcription factor and major antioxidant defender Forkhead box protein O1 (FoxO1) by activating the NF-κB pathway. FoxO1 can directly bind to the promoters of antioxidant genes such as catalase, thereby mitigating the toxic effects of ROS (Li and Gao, 2023) (Fig. 3). SOD prevents O2·− accumulation in endothelial cells and inhibits oxidative stress-induced apoptosis by reducing ROS levels (Thurau et al., 2009), and catalase can reduce JNK and Bcl-2 phosphorylation and inhibits M-CSF-induced monocyte-to-macrophage differentiation, thereby facilitating immune evasion by KSHV (Gilardini Montani et al., 2019). Kaposin A can induce the expression of the host protein HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1, which activates NRF2 nuclear translocation through PI3K and PKC-ζ pathways and promotes the ubiquitin-mediated degradation of Rac1, thereby reducing Rac1-NOX-mediated ROS and mitigating oxidative stress (Kumar et al., 2019). KSHV-encoded miR-K12-11 upregulates the expression of solute carrier family 7 member 11 (SLC7A11), which facilitates cystine transport into cells and promoting GSH, thereby inhibiting ferroptosis and enhancing resistance to oxidative stress (Miao et al., 2025). Additionally, KSHV-encoded viral IL-6 activates the deacetylase sirtuin 3, which deacetylates the mRNA stability factor SERPINE1 mRNA binding protein 1, and promotes the degradation of lipoyltransferase 2 mRNA, thereby inhibiting lipid peroxidation and ferroptosis (Zhou et al., 2024).
KSHV and ROS-Targeted Therapeutics
ROS-targeted therapeutic strategies for KSHV-associated malignancies are gradually demonstrating clinical potential. KSHV infection not only promotes ROS accumulation but also relies on ROS signaling to maintain viral latency, drive cell proliferation, and stimulate angiogenesis. Therefore, either scavenging ROS or inducing ROS overload can exert anti-tumor effects, regulating ROS levels has become a critical approach to suppress the oncogenic mechanisms of the virus.
In the KS animal model, NAC was found to reduce the phosphorylation of STAT3, thereby inhibiting its transcriptional activity. This leads to the downregulation of c-Myc and VEGF expression, suppress cell cycle progression and inhibit angiogenesis, resulting in significant tumor growth suppression (Ma et al., 2013). Similarly, in the PEL mouse model, NAC reduces ROS levels, delays tumor progression, and extends survival (Ye et al., 2011). Natural or semisynthetic antioxidants such as theaflavin and novel curcumin derivatives can rapidly lower intracellular ROS levels, leading to the collapse of ROS-associated stress systems, mitochondrial damage and cell death (Das et al., 2022).
Many therapeutic agents exert their effects by inducing oxidative stress-related cell death. Primaquine specifically targets KSHV-infected PEL cells by inducing ROS accumulation, which activates caspase-3/7, initiates ER stress pathways and upregulates the pro-apoptotic factor CHOP, thereby inducing cell death (Gothland et al., 2022). Silver nanoparticles (nAg) exhibit heightened cytotoxicity against KSHV-infected cells by reactivating lytic replication, a process dependent on ROS induction (Wan et al., 2019). More importantly, nAg can directly disrupt KSHV virions and block primary infection, even at non-cytotoxic concentrations, suggesting that nAg may be a good treatment for KSHV-associated diseases (Fig. 2b).
HBV and ROS
HBV, a member of the hepatophilic DNA virus family, possesses a compact 3.2 kb partially double-stranded, loosely looped DNA genome that exhibits strong tropism for hepatocytes (Xiao et al., 2025). The HBV genome encodes several proteins, such as HBV X protein (HBx), hepatitis B surface antigen (HBsAg), and hepatitis B core antigen (HBcAg) (Naidu and Margeridon, 2025). HBV infection can lead to various liver-related diseases, including liver fibrosis, acute liver failure (ALF), and hepatocellular carcinoma (HCC). A hallmark of HBV infection is hepatocyte injury accompanied by host immune-mediated inflammatory responses (Sayaf et al., 2024). Studies have shown that HBV can generate ROS through multiple pathways, thereby impairing host antiviral immunity and exacerbating inflammatory injury (Choi et al., 2019).
Several HBV-encoded proteins can induce the production of ROS, among which HBx plays a central role in this process (Choi et al., 2019). HBx induces ROS through a two-step process. First, it interferes with the function of the ETC by downregulating the enzymatic activities of mitochondrial complexes I, III, IV, and V, which is a p53-independent mechanism that initiates ROS (O2·−/H2O2) production (Lee et al., 2004). Second, HBx proteins activate p53 through the ataxia telangiectasia mutated-checkpoint kinase 2 pathway to amplify ROS in a p53-dependent manner (Kim et al., 2024). However, ROS can induce the expression of the E3 ligase seven in absentia homolog 1 (Siah-1) in a p53-dependent manner. Siah-1 facilitates the proteasomal degradation of HBx protein, suggesting that H2O2 can act as an inhibitor of HBV replication under certain conditions (Yoon et al., 2023). The exact opposite role of ROS in the regulation of HBV replication has also been reported. The augmenter of liver regeneration (ALR) acts as an antioxidant protein, and HBx can inhibits ALR translation by upregulating miR-181a, exacerbating ROS accumulation and thereby promoting autophagy and viral replication (Mishra et al., 2025). HBx also increases the expression of proteinase-activated receptor 2, which enhances mitochondrial oxidative stress and dysfunction, disrupting the balance between hepatocyte proliferation and apoptosis and ultimately contributing to malignant transformation of normal hepatocytes (Li et al., 2022). HBx may also suppress SLC7A11 expression via enhancer of zeste homolog 2, promoting ferroptosis in hepatocytes during ALF, thereby exacerbating liver damage (Liu et al., 2021). However, in another research, HBx can upregulate SLC7A11 and GPX4 expression to inhibit ferroptosis and promote HCC cell proliferation (Wang et al., 2023). This suggests that during chronic hepatitis, promotion of ferroptosis leads to hepatocyte damage, but during tumor progression, inhibition of ferroptosis may favor cancer cell survival. Additionally, the accumulation of HBsAg and HBcAg in the ER results in ER stress, subsequent unfolded protein response (UPR), and ROS production (Choi et al., 2019). It was demonstrated that serum NOX2 concentrations are significantly higher in patients with HBV-related complications compared with healthy subjects, which suggests that one of the mechanisms involved in elevated ROS levels in cells of HBV-infected patients may be NOX2 production (Xiong et al., 2018); however, the precise mechanism is unclear. In HBV-associated liver fibrosis, tripartite motif 37 is a key pro-fibrotic factor whose transcription depends on ROS-mediated activation of NF-κB (Xie et al., 2020). This activation further reinforces the pivotal role of ROS in the progression of HBV-related disease. ROS upregulate transcription factor SNAIL, which leads to the methylation of the suppressor of cytokine signaling 3 (SOCS3) promoter, whereas the epigenetic silencing of SOCS3 promotes hepatocarcinogenesis. Meanwhile, the epigenetic silencing of SOCS3 negatively feeds back and results in the accumulation of STAT3, which in turn, promotes the development of HCC (Yuan et al., 2016) (Fig. 4).
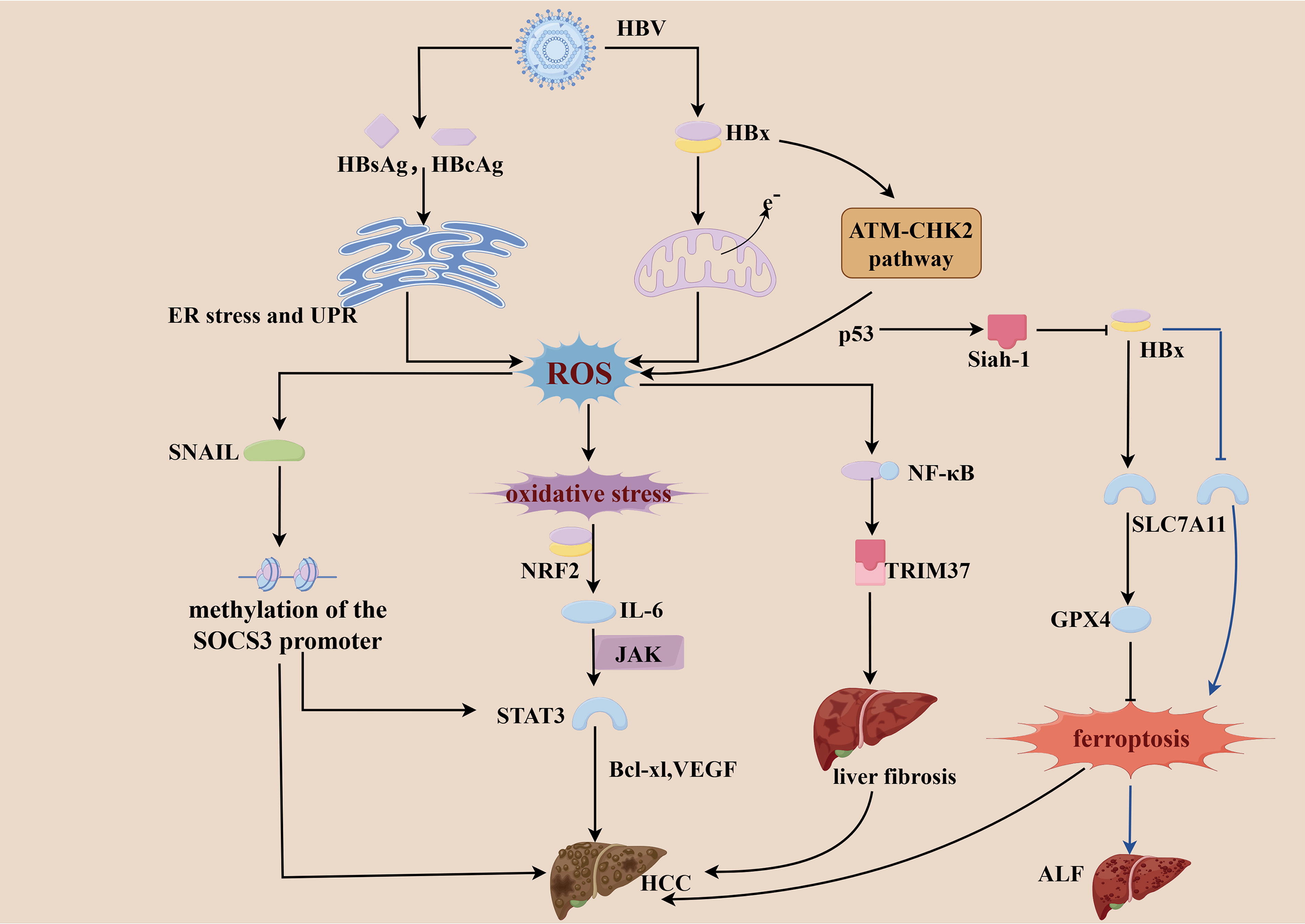
During HBV infection, the interaction between the host immune system and the virus is a central driving force in disease progression (Jabeen et al., 2021). Pro-inflammatory cytokines produced by HBV infection induce ROS production and result in IL-6 overexpression through the activation of multiple redox-sensitive transcription factors (such as NRF2) (Wruck et al., 2011; Yuan et al., 2016). IL-6 phosphorylates STAT3 and activates various genes associated with tumor development, such as Bcl-xl (anti-apoptotic protein) and VEGF (Yu and Jove, 2004) (Fig. 4). In addition, ROS can downregulate the expression of the key immune regulatory protein family with Sequence Similarity 26, Member F, thereby weakening the host’s antiviral immune response (Jabeen et al., 2021). NK cells and T cells promote immune-mediated liver injury by releasing ROS and pro-inflammatory cytokines, which results in necroinflammatory episodes, hepatocyte regeneration/healing, and remodeling of the hepatic microenvironment (Rehermann and Nascimbeni, 2005). Notably, TAMs in the liver also play a critical role in HBV-related HCC, and M1-type TAMs typically suppress tumor growth and enhance immune responses (Tian et al., 2025). HBV infection can promote ferroptosis of M1-type macrophages via up regulating miR-142-3p, affecting the production of GSH and Fe2+, thereby accelerating HCC development (Hu et al., 2022).
HBV infection can induce oxidative stress and trigger cell death; however, HBV also possesses the ability to modulate redox balance to overcome oxidative stress-induced cell death. For instance, HBx promotes hepatocarcinogenesis by counteracting oxidative stress-induced cell death through activation of the high-mobility group at-hook2/stanniocalcin 2 signaling pathway (Huang et al., 2022). Additionally, Activation of the NRF2/ARE antioxidant defense pathway and induction of the Forkhead transcription factor FoxO4 are involved in protection against oxidative stress (Chung, 2011).
HCV and ROS
HCV is a positive-stranded RNA virus, with its genetic material encoding a single multimeric protein. This polyprotein yields 10 distinct structural and nonstructural proteins, which include three structural proteins (core protein, E1, E2), and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Tabata et al., 2020). HCV infection is a global health problem and a major risk factor for ALF, liver fibrosis, liver cirrhosis, and HCC. As early as the mid-1990s, studies indicated that HCV infection induces ROS production and even oxidative stress. Compared with other types of hepatitis, such as hepatitis B, oxidative stress appears to be more prominent in HCV-associated diseases, suggesting it plays a central role in HCV pathogenesis (Muriel, 2009).
HCV-encoded oncogenic proteins can induce ROS generation through various mechanisms, contributing to oxidative stress. Core proteins induce the expression of ER oxidoreductin 1α. The latter increases mitochondrial Ca2+ unidirectional transporter activity by depleting ER Ca2+ stores. This contributes to mitochondrial depolarization, increased ROS (H2O2) production, and the subsequent generation of ER stress and UPR (Ivanov et al., 2015; Wang et al., 2010). HCV core proteins recruit NOX for the transfer of electrons through the membrane in an autocrine transforming growth factor-β (TGF-β)-dependent manner, which results in the generation of O2·− or H2O2 (Boudreau et al., 2009). Co-expression of the HCV core protein with cytochrome P450 2E1 (CYP2E1) increased ROS production, whereas the inhibition of cytochrome enzyme activity attenuated stress (Wen et al., 2004). Similar to the core protein, the NS5A protein accessory domain I of HCV can also induce oxidative stress by activating the transcription and translation of NOX1 and NOX4 through the enhanced expression of TGFβ1. Moreover, the NS5A protein accessory domain I of HCV increases ROS production through CYP2E1 and can further induce ROS production through the TGF-β → COX1 → COX2 → NOX4 cascade (Smirnova et al., 2016). NS5A activates JNK signaling through increased ROS production, which results in decreased phosphorylation and nuclear accumulation of FoxO1, ultimately leading to increased glucose production. This metabolic reprogramming meets the high energy demand of tumor cells and promotes their proliferation and survival (Deng et al., 2011) (Fig. 5).

In chronic HCV infection, immune cells show hyperactivation and increased DNA damage, correlating with elevated risks of chronic liver disease and HCC. NS3 enhances the ability of NOX in phagocytes to induce ROS, and the generated ROS induce apoptosis in T cells and NK cells. This suggests that ROS production catalyzes immune evasion by the virus (Thorén et al., 2004). HCV infection alters mitochondrial regulatory proteins, such as downregulating PGC-1α and mitochondrial transcription factor A, leading to reduced mtDNA content and mitochondrial dysfunction in CD4 T cells. This dysfunction promotes ROS accumulation, disrupts T-cell homeostasis, and causes immune failure (Schank et al., 2021). HCV can also induce excessive topoisomerase 1 cleavage complexes (Top1cc), leading to DNA damage. CD4 T cells, due to high oxygen consumption, are particularly sensitive to Top1-mediated damage, which further promotes mitochondrial ROS production and exacerbates immune dysfunction (Dang et al., 2022). Notably, ROS can feedback to enhance Top1cc formation, creating a vicious cycle of DNA damage. Fibrinogen-like protein 2 (Fgl2) is significantly upregulated in the liver of HCV patients and is strongly induced in macrophages during infection or cytokine stimulation. In mitochondria, Fgl2 inhibits the interaction between heat shock protein 90 and its target protein Akt, suppressing Akt and downstream FoxO1 phosphorylation (Tao et al., 2023). The resulting mitochondrial ROS modulate pro-inflammatory polarization of hepatic macrophages, further promoting chronic inflammation and liver damage.
ER and oxidative stress can lead to cell death. HCV-induced ROS production causes JNK activation, which transcriptionally upregulates pro-apoptotic BH3-only protein (Bim) expression to activate pro-apoptotic multidomain proteins (Bax). This leads to Bcl-2-mediated caspase 3-dependent apoptosis (Deng et al., 2015). In response to stress-generated cell death caused by excess ROS, HCV infection results in the activation of the antioxidant system. The HCV core, E1, E2, NS4B, and NS5A proteins can induce NRF2 activation through protein kinase C (Jhaveri et al., 2011). HCV under oxidative stress also induces the upregulation of 3β-hydroxysterol Δ24-reductase, which prevents apoptosis by blocking p53 acetylation and its interaction with E3 ubiquitin ligase (Tsukiyama-Kohara, 2012) (Fig. 5).
Hepatitis Virus and ROS-Targeted Therapeutics
In chronic HBV and HCV infections and their associated complications, the accumulation of ROS has been confirmed as a key mediator of viral replication, exacerbated inflammation, and hepatocellular injury (Liu et al., 2021; Yasukawa et al., 2003). Therefore, targeting ROS not only helps suppress the activity of hepatitis viruses but may also enhance the efficacy of conventional therapies such as chemotherapy while alleviating liver inflammation and tissue damage.
Various plant extracts and flavonoids have demonstrated potent anti-ROS and antiviral activities. Extracts from Lactuca sativa and luteolin-7-O-glucoside significantly decrease ROS levels and inhibit HBV replication (Cui et al., 2017), as does Pu-erh tea extract (Pei et al., 2011). Synthetic antioxidants such as NAC may enhance hepatic GSH levels, reduce ROS accumulation, and alleviate liver injury caused by HBV and HCV (Wu et al., 2024). To address immune dysregulation in HBV-related liver cirrhosis, three antioxidants, polyphenols, MTA (which directly targets mitochondrial function), and iACAT (which modulates mitochondrial activity indirectly via fatty acid oxidation) have been shown to reduce pyroptosis in mucosal-associated invariant T cells, a unique T-cell subset, thus potentially compensating for their loss in HBV-related cirrhosis (Xu et al., 2025).
Most chemotherapeutic agents induce ROS accumulation, leading to mitochondrial dysfunction, cellular damage, and even viral reactivation. For example, cisplatin enhances HBV replication by activating the ROS/JNK pathway and inhibiting the Akt/mTOR signaling pathway (Chen et al., 2019). Similarly, 5-fluorouracil activates LC3B-II via the ROS-BNIP3 signaling axis, triggering autophagy and promote HBV replication (Yang et al., 2024). Although chemotherapeutic agents can be used in HCC therapy, their side effects require special caution (Yang et al., 2024). In addition, further research has shown that sodium diethyldithiocarbamate, through the inhibition of SOD1, can enhance the therapeutic efficacy of sorafenib in HBV-related HCC. This effect is mediated by modulation of the PI3K/Akt/mTOR pathway and the promotion of ROS-induced cell death (Lee et al., 2024). Berberine (BBR) was previously reported to inhibit HCC cell growth, BBR treatment enhanced HCV replicon-induced ROS production, and ROS inhibition with NAC attenuated cell death (Tai et al., 2020) (Fig. 2c and d).
HPV and ROS
HPV is an envelope-less, double-stranded DNA virus. HPV infection of the skin and mucous membranes is the leading cause of cervical cancer, head and neck squamous cell carcinoma (HNSCC) and other anogenital cancers. Although more than 200 HPV genotypes have been identified, only high-risk HPV types are capable of inducing carcinogenesis (Doorbar et al., 2012). Here, we discuss the interrelationships between ROS and HPV, particularly HPV16 and HPV18. The HPV DNA contains eight openreading frames, categorized into early (E1, E2, E4, E5, E6, and E7) and late (L1 and L2) regions (Baedyananda et al., 2022). E6 and E7 are major promoters of cell transformation. E6 interferes with tumor suppressor proteins such as p53, blocks apoptosis and facilitates viral DNA replication (Hu and Ma, 2018). E7, another major oncoprotein, interacts with the retinoblastoma protein, disrupting cell cycle regulation and promoting malignant transformation (Münger et al., 2004; Tomaic, 2016). The process from HPV infection to cell transformation into cancer involves multiple steps and several viral-associated factors influence this process. ROS are also involved through different mechanisms.
The pathogenesis of cervical cancer is closely associated with oxidative damage caused by persistent infection with oncogenic types of HPV, which facilitates the integration of the HPV genome into the host chromosome. This damage arises from oxidative stress, which promotes tumor progression through metabolic pathways (Preci et al., 2021). HPV E6 and its minor subtype E6* induce oxidative stress by reducing antioxidant protein levels, resulting in excessive ROS production and DNA damage. Of these, E6 suppresses the expression and activity of catalase and GSH (Cruz-Gregorio et al., 2018), while E6* decreases the levels of SOD2 and GPX1/2 (Williams et al., 2014; Williams et al., 2011). Elevated ROS lead to sustained activation of JNK signaling, which in turn recruits transcription factor c-Jun to the promoter region of matrix metalloproteinase-1 (MMP-1), thereby upregulating MMP-1 expression in cancer cells and enhancing their migratory capacity (Naderzadeh et al., 2025). At the same time, ROS accumulation results in oxidative DNA damage, contributing to the formation of double-strand breaks. This activates the ataxia-telangiectasia mutated and Rad3-related kinases, triggering cellular stress responses and increased oxidative stress (Cuadrado et al., 2006) (Fig. 6).

HPV-induced oxidative stress promotes lipid peroxidation and cellular damage, which may result in cell death. For example, HPV tumor cells induce β-oxidation to produce H2O2, which damages mitochondria, leads to mitochondrial uncoupled electron leakage, and generates more ROS, which have cytotoxic effects (Cruz-Gregorio et al., 2021). Reactivation of transcriptional reporter activity acts through the hyperpolarization of mitochondria and the accumulation of ROS to induce necroptosis in cervical cancer cells (Mohanty et al., 2022). HPV-infected cells contain multiple mechanisms to resist cell death. For example, HPV16 often inserts near the c-Myc motif when it integrates into the host cell genome. C-Myc may play an important role in ferroptosis resistance by regulating the miR-142-5p/HOXA5/SLC7A11 signaling axis, which contributes to the progression of cervical squamous cell carcinoma (Chen et al., 2024). Moreover, the presence of HPV16 E7 has been associated with elevated intracellular GSH levels (Shim et al., 2008). Similarly, HPV18 E7 can modulate the expression of catalase, contributing to resistance against oxidative stress-induced cell death (Hochmann et al., 2023). These findings suggest that E7 may exert a protective effect by counteracting oxidative stress-induced cytotoxicity. HPV16 E6 promotes the proliferation of cervical cancer cells by inhibiting the lactosylation modification of glucose-6-phosphate dehydrogenase K45. This activates the PPP and increases NADPH and GSH levels, thereby alleviating intracellular oxidative stress (Meng et al., 2024). E6 also activates the TP53-induced glycolysis and apoptosis regulator (TIGAR) and facilitates its mitochondrial targeting, which reduces glycolysis and enhances NADPH production, ultimately protecting cells from oncogene-induced oxidative genotoxicity (Yapindi et al., 2023). However, it has also been reported that E6 can also suppress p53 protein expression, thereby reducing apoptosis and DNA damage while increasing mutations (Tommasino et al., 2003) (Fig. 6).
HPV and ROS-Targeted Therapeutics
In recent years, an increasing number of studies have revealed that ROS levels not only influence cervical cancer cells proliferation and apoptosis but also play a critical role in modulating treatment sensitivity and drug resistance.
Antioxidants reduce ROS-induced damage and regulate the cellular redox state, demonstrating potential therapeutic value. Epidemiological studies have shown that higher serum albumin levels and increased four dietary intake of antioxidants, vitamins A, B2, E, and folate, are associated with a reduced risk of high-risk HPV infection (Lin et al., 2021). Mentha spicata extract has been shown to modulate oxidative stress by increasing tissue GSH levels in the K14HPV16 transgenic mouse model, although its role in mitigating HPV-induced pathogenesis remains unclear (Jesus et al., 2025). In addition, epigallocatechin gallate and curcumin can activate NRF2 and induce the expression of antioxidant enzymes such as HO-1, thereby contributing to the prevention and treatment of cervical cancer (Jesus et al., 2025; Silva et al., 2018).
Oxidants exert antitumor effects by inducing excessive ROS. A3K2A3 is a new synthetic dibenzylideneacetone that increases ROS levels and reduces the antioxidant defense capacity, thereby significantly upregulating Bax and caspase 9 expression and promoting apoptosis in cervical cancer cells (Zani et al., 2023). Ginsenoside Rb1 is more effective against HNSCC cells by increasing ROS levels and causes DNA damage, while exhibiting reduced toxicity in non-cancerous cells (An et al., 2024). Cisplatin exerts its anticancer effect in HNSCC partly by promoting mitochondrial ROS release. However, in HNSCC, the overexpression of NRF2 enhances the expression of antioxidant proteins such as SOD and GPX, leading to cisplatin resistance (Griso et al., 2022) (Fig. 2e).
HTLV-1 and ROS
HTLV-1 is a retrovirus with single-stranded RNA encoding proteins (Lairmore et al., 2012). It is one etiologic factor that contributes to adult T-cell leukemia/lymphoma. One of the hallmark molecular features of HTLV-1 infection is the induction of oxidative stress mediated by several viral regulatory and accessory proteins, including the HTLV-1 transactivator protein Tax, HTLV-1 basic leucine zipper factor (HBZ), Rex, p12, and p30/p13 (Yaghoubi et al., 2019). In addition to promoting clonal expansion and immortalization of T cells, HTLV-1 infection alters the host immune system, enabling infected cells to evade immune surveillance (Miao et al., 2025).
The Tax protein is considered a major trigger of oxidative stress. It induces ROS production, DNA damage, and reactive cellular senescence (Kinjo et al., 2010). One putative mechanism is that Tax interaction with ubiquitin-specific protease 10 stimulates T cells to produce ROS by inhibiting stress granule formation (Takahashi et al., 2013). HTLV-1 produces a protein of 87 amino acids in length (known as p13), which targets the inner mitochondrial membrane and results in the swelling of the mitochondria. P13 also induces depolarization, with a corresponding increase in respiratory chain activity and an increase in ROS production (Silic-Benussi et al., 2009, 2010). The expression of CD30 in HTLV-1-infected cells promotes non-Tax-dependent ROS production, which are involved in cancer progression by triggering DNA double-strand breaks and chromosomal imbalances via the PI3K pathway (Watanabe et al., 2023) (Fig. 7).

The majority of tumor cells have high ROS levels. Once a certain threshold is surpassed, an increase in ROS may lead to cell senescence and apoptosis (Angsutararux et al., 2015). A large increase in p13-promoted ROS affects the likelihood of mitochondrial permeability transition pore opening, which triggers cell death through energy shortages and the release of pro-apoptotic factors located in the mitochondria (Silic-Benussi et al., 2009, 2010). HTLV-1-infected cells possess multiple mechanisms to counteract ROS-induced cytotoxicity. For example, HTLV-1-infected T-cell lines abnormally express dihydroorotate dehydrogenase, it can support NADPH production through the pyrimidine synthesis pathway. NADPH helps regenerate GSH and maintain redox homeostasis (Miao et al., 2025). Moreover, the viral accessory protein p30II can upregulate p53 expression and promote the transcription of TIGAR. TIGAR reduces ROS levels by activating the PPP, thereby inhibiting oxidative stress and apoptosis. Additionally, the HBZ protein activates the transcription of antioxidant genes through ARE-mediated signaling, leading to the upregulation of heme oxygenase 1. This, in turn, helps infected cells resist ROS-induced cytotoxicity. (Darwiche et al., 2007; Hutchison et al., 2018) (Fig. 7).
HTLV-1 and ROS-Targeted Therapeutics
ATL is an extremely difficult hematological malignancy to treat. Despite continuous advancements in technology, significant progress has been made in understanding the pathogenesis and progression of ATL, and chemotherapy as well as other therapeutic approaches have been widely applied in clinical treatment. Several studies have attempted to enhance the efficacy of antiviral therapy through drug combinations. Many drugs act by inducing ROS production to promote apoptosis. The combination of thymoquinone and doxorubicin induces ROS production and apoptosis through the mitochondrial pathway (Fatfat et al., 2019). N-(4-hydroxyphenyl) retinamide induces the production of ROS as a key mediator of cell-cycle blockade and apoptosis in malignant T cells (Darwiche et al., 2007) (Fig. 2f).
Prospects
Oncovirus infection causes elevation of intracellular ROS through different mechanisms. ROS can affect cell proliferation through signaling pathways and cell survival through apoptotic and immune pathways. Therefore, altering intracellular redox levels is an effective approach for treating diseases associated with oncoviral infection (Bhandarkar et al., 2008; Gothland et al., 2022). Because redox regulation and stress responses involve multiple factors, redox alterations in cancer cells are complex and variable. One approach is the use of antioxidants to target ROS involved in tumorigenesis, progression, and maintenance by enhancing ROS scavenging enzymes or by targeting NOX (Wang and Yi, 2008). However, some experimental data indicate that antioxidants may promote the survival of tumor cells by inhibiting ROS-mediated apoptosis (Bjelakovic et al., 2007). Another is that chemotherapeutic and radiotherapeutic agents, such as oxidants, induce oxidative stress, and ROS-mediated cell damage in cancer (Kim et al., 2016; Yang et al., 2018). Harnessing the toxic effects of ROS against cancer cells may be achieved through two approaches: (1) directly inducing the generation of ROS within neoplastic cells and (2) blocking the antioxidant defense mechanisms of these cells. The antioxidant capacity is enhanced in some advanced tumor cells, a state that may allow cancer cells to repair ROS-induced damage more quickly after radiation, thus causing radiation resistance (Hu et al., 2017). So there is still a lack of conclusive evidence to suggest that this approach is widely applicable to human patients, particularly for long-term applications.
ROS play a role in oncoviruses, and other mechanisms through which the oncoviruses induce oxidative stress remain to be discovered. Notably, the choice of oxidants or antioxidants in combination is an effective means of cancer treatment for oncoviruses that replicate more actively under conditions of high oxidative stress. Therefore, combinations including radiation therapy or standard chemotherapy with drugs that eliminate the antioxidant systems in cancer cells should also be explored in the context of other oncoviral infection (Hu et al., 2017; Yang et al., 2018). In the coming years, attempting to unravel the effects of enhanced ROS production on the altered metabolism of carbohydrates, lipids and lipoproteins, amino acids and polyamines, as well as cytokines and the immune system will be important and challenging in this field. These studies will provide insight into the role of these events in disease progression.
Conclusions
ROS play an important role in the development of relevant tumors caused by oncoviruses, in this review, we discuss how oncoviruses regulate ROS levels and how they architect context-dependent ROS ecosystems to trigger viral dissemination and drive malignant tumors. ROS represent a double-edged sword, they are both a foe and friend in oncoviral infection, and its role depends on the viral type, the stage of infection, and the cellular microenvironment. Thus, therapeutic strategies should focus on precise regulation of ROS homeostasis. After ROS production by viral infection, oncoviruses enhance antioxidant pathway to prevent apoptosis because of oxidative stress caused by excessive intracellular ROS. This is one of the reasons for oncoviruses are resistant to many cancer treatments. Only by deciphering the intricate relationship between viral pathogenesis and redox biology can we transform ROS from enemies to friends in the war against virus-driven cancers.
Footnotes
Acknowledgment
The authors gratefully acknowledge Figdraw for the support of Figures 1–![]() .
.
Authors’ Contributions
R.S., X.Z., and K.S.: Writing—original draft preparation. Y.Z. and B.L.: Writing—review and editing. All the authors have read and agreed to the published version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Natural Science Foundation of Shandong Province (ZR2020MC020 [Y.Z.], ZR2020MH302 [B.L.]) and National Natural Science Foundation of China (32370177 [B.L.]).