
Abstract
Background:
RNA interference (RNAi) therapy has tremendous potential in treating diseases that are characterized by overexpression of genes. However, the biggest challenge to utilize the therapy is to engineer delivery systems that can efficiently transport small interfering RNA (siRNA) to appropriate target sites. Our objective in this study was to develop and evaluate multi-compartmental systems for the oral delivery of siRNA that targets the overexpressed TG2 gene (TG2-siRNA) in the small intestine for the treatment of celiac disease (CD).
Materials and Methods:
Two types of multicompartmental systems were developed and evaluated: (1) a solid-in-solid multicompartmental system featuring “nanoparticle in microsphere oral system (NiMOS)” where type B gelatin nanoparticles containing TG2-siRNA (TG2-NiMOS) were encapsulated within poly(ɛ-caprolactone) (PCL) based microspheres, and (2) a solid-in-liquid multicompartmental system, “Nanoparticle-in-Emulsion (NiE)” consisting of type-B gelatin nanoparticles containing TG2-siRNA encapsulated within safflower oil containing water-in-oil-in-water (W/O/W) multiple emulsion (TG2-NiE).
Results:
Evaluation of the biodistribution and pharmacokinetics (PK) after a single oral dose of siRNA containing multicompartmental systems to C57BL/6 mice showed that TG2-siRNA was delivered to the small intestine (duodenum, jejunum and ileum), and colon with minimal systemic exposure via both TG2-NiE and TG2-NiMOS systems. TG2-siRNA exposure (AUC0–t) in the duodenum, jejunum, ileum and colon was 56.4-, 34.3-, 85.5- and 35.5-fold greater for the TG2-NiMOS formulation, relative to the TG2-NiE formulation.
Conclusion:
The results of this study suggest that TG2-NiMOS formulation was more superior than TG2-NiE formulation in facilitating intestinal delivery of siRNA via the oral route of administration and can be potentially used in the treatment of CD.
Introduction
Celiac disease (CD) is a chronic inflammatory condition of the small intestine triggered by the ingestion of dietary gluten in the susceptible patient population. This autoimmune disorder is further characterized by enteropathy ranging from intraepithelial lymphocytosis to total villous flattening. 1 It is not as uncommon as previously believed, with the global prevalence of the disease being 1.4% based on serological test results in a 2018 study. 2 It is a multifactorial disease with pathogenesis involving a combination of environmental, genetic, and immunological factors. 3
Gluten peptides present in dietary cereals such as wheat, rye, and barley are the known toxins that trigger CD. 4 Glutenin polymers and gliadin monomers are two fractions of gluten that cause the activation of the disease. 5 These peptides have large amounts of prolines (20%) and glutamines (40%) that are resistant to hydrolysis by protease-mediated hydrolysis, making it difficult to be completely degraded and digested in the gastrointestinal tract (GIT).6,7 The accumulation of the gluten peptides activates the innate and adaptive immunity and elicits an immune response leading to the onset of CD in the genetically predisposed population. 8
The major histocompatibility complex class II proteins, located on 6p21, is the main genetic predisposing factor for CD, 1 including alleles encoding HLA-DQ2 and HLA-DQ8. 3 HLA are glycoproteins present on the surface of antigen-presenting cells (APCs). After the uptake of antigens from deamidated gluten peptides, by APC, the HLA-antigen complex is presented to CD4+ T lymphocytes. This elicits an immune response that ultimately leads to the onset of the disease symptoms. 9
In the genetically predisposed population, intestinal tissue transglutaminase-2 (TG2) was found to be directly responsible for catalyzing the deamidation of gliadin peptides, which increased the affinity to bind to the HLA-DQ2 and HLA-DQ8 molecules. 10 Given the role of TG2 in the pathogenesis of the disease, inhibiting the protein production would be a suitable approach to treat CD. This could be achieved by using oral small interfering RNA (siRNA) delivery to silence TG2 gene and subsequent protein production in the small intestine, which would potentially stop the disease from propagating.
The siRNA are 22–24 bases oligonucleotides with an electrically charged backbone and are highly susceptible to degradation in the biological environment. For efficient oligonucleotide delivery to the GIT, there is a critical need to develop formulations that can overcome various barriers, both intra- and intercellularly. Although chemical modifications of siRNA make it less susceptible to degradation and reduce the off-target effects,11,12 the mode of administration dictates its fate till it reaches the targeted tissue or cell. 13 The siRNA must overcome fewer barriers when delivered locally versus systemically. Local delivery of nucleic acid would overcome the drawbacks of systemic administration and has advantages such as lower doses, higher probability of cellular uptake, improved bioavailability, and minimized off-target effects.
Although a number of studies have been carried out pertaining to the local delivery of siRNA, 14 oral delivery still remains an elusive barrier to be overcome. The harsh acidic environment of the stomach and the presence of nucleases in the GIT are some of the barriers the payload needs to overcome to be efficacious. To address this challenge, we have developed hybrid multicompartmental systems in our laboratory, engineered to protect the nucleic acid carrier from the harsh GIT environment, promote delivery of the payload at the target site, and assist in cellular uptake and cytosolic release of the nucleic acid payload. These systems have one or more internal compartment(s), surrounded by one or more external compartment(s) to confer enhanced cargo protection. These systems can be designed to release the payload in response to an external stimulus, for example, pH or enzymes. In addition to this, the compartments can also be modified to target a particular cell type, for example, macrophage specific uptake can be improved with the use of tuftsin-modified hydrophilic nanoparticles. 15 The compartments can have different hydrophilicity, having the ability to incorporate both hydrophilic and hydrophobic drugs in the respective compartment. This hybrid system can also act as a sustained release formulation, releasing the drug over a prolonged period. 15
Our lab at Northeastern University in Boston pioneered in the development of the multicompartmental system for oral delivery of nucleic acids. Three types of multicompartmental systems have been developed and evaluated for distinct applications; they are (1) solid-in-solid,15–19 (2) solid-in-liquid, 20 and (3) liquid-in-liquid 21 multicompartmental systems. We have shown that a solid-in-liquid multicompartmental system, “Nanoparticle-in-Emulsion (NiE)” consisting of type B gelatin nanoparticles (GNPs) containing IL-10 plasmid DNA was encapsulated within safflower oil containing water-in-oil-in-water (W/O/W) multiple emulsion for anti-inflammatory gene therapy in macrophages. 20 Liquid-in-Liquid system was developed as a W/O/W multiple emulsion for DNA vaccine delivery system for immunotherapy of melanoma. 21 For oral delivery of nucleic acids, we have shown a solid-in-solid multicompartmental system featuring “Nanoparticle in Microsphere Oral System (NiMOS)” where type B GNPs containing a nucleic acid payload (pDNA or siRNA) were encapsulated within poly(ɛ-caprolactone) (PCL)-based microspheres to be efficient in promoting efficient intracellular delivery of encapsulated payload at target sites in the GIT.15–19
The focus of this research article is to evaluate multicompartmental delivery technology to enable oral intestinal delivery of TG2-siRNA. Since the small intestine is where CD is initiated and propagated, the intestinal delivery of siRNA that inhibits TG2 production would be an attractive strategy to combat this disease.
Materials and Methods
Preparation and characterization of NiMOS containing TG2-siRNA (TG2-NiMOS)
Formulation and characterization of siRNA containing GNPs has been previously described. 22 The siRNA-containing NiMOS were formulated by using methods adapted from previous studies.16,19 Briefly, PCL with an approximate molecular weight of 14 kDa was obtained from Sigma Aldrich (St. Louis, MO). Five hundred microliters of 2.5 mg/mL suspension of GNPs encapsulating desired siRNA was homogenized with 5 mL of 0.5% (w/v) PCL in dichloromethane at 9000 rpm, using a Silverson lab mixture (Model L4RT-A; Silverson Lab Machines, Bucks, England) for 10 min. The system just cited was further homogenized with 10 mL of 0.1% (w/v) poly vinyl alcohol (PVA) in deionized water at 9000 rpm for another 5 min. The resulting system was magnetically stirred overnight for removal of dichloromethane, by evaporation and hardening of microspheres. The next day, the dispersion was centrifuged at 5000 rpm for 20 min and washed thrice with deionized water for removal of excess PVA, followed by freeze drying and storage at 4°C, until it was used. The siRNA loading in NiMOS was determined with the use of a custom-designed quantitative polymerase chain reaction (qPCR) assay that employed stem-loop primers for TG2-siRNA. 22 The TG2-NiMOS were digested with lipase buffer to degrade PCL matrix, followed by digestion of GNPs in protease buffer protease buffer (0.2 mg/mL) at 37°C for 30 min, and heating in triton buffer, which was then used for the qPCR analysis.
Preparation and characterization of TG2-NiE-containing TG2-siRNA (TG2-NiE)
The TG2-NiE formulation was formulated by a two-step emulsification method similar to a method previously described. 20 The first step consisted of formulation of W/O primary emulsion using an oil soluble surfactant Span® 80. Naked siRNA or siRNA encapsulated in the GNPs was incorporated in the aqueous phase of the primary emulsion to give the final gelatin concentration of 1 mg/mL in the W/O/W multiple emulsion. The appropriate amounts of each ingredient in the optimized formulations are listed in Table 1. A stable primary W/O emulsion dispersion with siRNA-encapsulated nanoparticles was formed by using the Silverson homogenizer L4RT at the speed of 9000 rpm for 15 min. The primary emulsion of dispersion was mixed with an additional aqueous phase consisting of a water-soluble surfactant Tween® 80, and the formulation was prepared by homogenization at 4000 rpm for 4 min. Increasing the speed to greater than 4000 rpm in this step resulted in rupture of the multiple emulsion globules; hence, it was important not to exceed the speed of 4000 rpm for the second homogenization step.
The Composition of the Nanoparticle-in-Emulsion Formulation
W/O/W, water-in-oil-in-water.
Stability of the tri-phasic NiE system was evaluated by using accelerated centrifugation and evaluation of phase separation under different conditions. Multiple emulsion or NiE was centrifuged at up to 4000 rpm for 1 h, and phase separation was evaluated visually and under a light microscope. In addition, we also examined the effect of up to 1000-fold dilution in aqueous media on the leaching and release by fluorescence microscopy of rhodamine-labeled dextran either when administered directly into the W/O primary emulsion or on nanoparticle encapsulation. The siRNA loading in TG2-NiE was determined with use of a custom-designed qPCR assay that employed stem-loop primers for TG2-siRNA; TG2-NiE (1 mL) was first de-stabilized with 100 μL of 5 M NaCl and centrifuged at 20,000 rpm for 30 min. After this step, the GNP palette obtained was treated with 5 mL of protease buffer for 30 min at 37°C for digestion of gelatin and release of encapsulated siRNA. The resulting solution was heated with 1:1 solution of 0.5% triton in 1 × phosphate-buffered saline (PBS) buffer at 95°C for 15 min, which was then used for the qPCR analysis. 22
In vivo biodistribution experiments with TG2-NiMOS and TG2-NiE
All studies were executed in compliance with provisions of the USDA Animal Welfare Act, the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the U.S. Interagency Research Animal Committee Principles for the Utilization and Care of Research.
Male C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). The study design for evaluating pharmacokinetics (PK) and biodistribution of siRNA after oral administration of TG2-NiMOS and TG2-NiE is tabulated in Table 2.
Study Design for Evaluating Pharmacokinetics and Biodistribution of Small Interference RNA After Oral Administration of Nanoparticle-in-Microsphere Oral System
GNP, gelatin nanoparticle; NiE, Nanoparticle-in-Emulsion; NiMOS, nanoparticle-in-microsphere oral system; PBS, phosphate-buffered saline; TG2, transglutaminase-2.
A total of 104 male C57BL/6 mice (20–30 g) were divided into 8 groups. Mice in Group 1 received a single oral dose of TG2-NiMOS formulation at a final siRNA dose of 0.5 mg/kg. Mice in Group 2 received TG2-GNP formulation (TG2-siRNA encapsulated within GNPs) at a final siRNA dose of 0.5 mg/kg. Mice in Group 3 received a single oral dose of TG2-siRNA at 0.5 mg/kg. Mice in Groups 4, 5, and 6 received a single oral dose of blank NiMOS, Blank GNPs, and PBS (formulations without any siRNA). Mice in Group 7 received a single oral dose of TG2-NiE formulation at a final siRNA dose of 0.5 mg/kg. Mice in Group 8 received a single oral dose of blank NiE (formulations without any siRNA). Before each oral dose, mice were fasted overnight to avoid any food interferences with administered formulation. Mice were euthanized at time points specified in Table 2. At each time point, stomach, duodenum, jejunum, ileum, colon, liver, spleen, kidney, as well as blood were collected. Blood was collected in K2EDTA tubes on ice and processed for separation of plasma within 30 min of collection. Tissues were flash frozen in liquid nitrogen immediately after collection and stored at −80°C until they were used for siRNA bioanalysis.
Quantification of TG2-siRNA in plasma and tissue samples
A stem-loop qPCR assay performed by using siRNA-specific primers was developed and qualified for plasma and tissue bioanalysis. 23 Primer and probe sequences used for the stem-loop qPCR assay are shown in Table 3. Plasma samples were diluted 1:1 with 0.5% TritonX-100 buffer in 1 × PBS at 95°C for 15 min, followed by sample processing for qPCR assay. Tissue samples were pulverized at below freezing temperature (cooled with liquid nitrogen) by using a Spex GenoGrinder® (Metuchen, NJ). Tissue powder obtained after the pulverization step was weighed and lysed by using 0.25% TritonX-100 buffer in 1 × PBS by heating at 95°C for 15 min at a final tissue concentration of 100 mg/mL. Tissue lysates were centrifuged at 15,000 rpm for 15 min at 4°C, and supernatants were collected and used for qPCR assay. Both free and encapsulated siRNA was extracted by this method, as the lysates were heated at 95°C. The siRNA concentrations were estimated in ng/mL or ng/g units, with the use of a matrix-matched Ct versus siRNA concentration liner-log calibration curve. The lower limit of quantification for siRNA in plasma and tissue samples was 4 pg/mL and 0.4 ng/g, respectively.
Reagents for the Stem-Loop Quantitative Polymerase Chain Reaction Assay
qPCR, quantitative polymerase chain reaction; siRNA, small interference RNA.
Noncompartmental PK analysis
Noncompartmental PK parameter estimates were calculated by using Phoenix WinNonlin 6.4 software. Parameters such as terminal elimination half-lives (t1/2β), maximum observed concentration (Cmax), time to achieve maximum observed concentration (tmax), and area under the concentration-time curve (AUC) were determined. An R package, tidyverse 24 was used for data analysis, visualization, and generating summary statistics.
Statistical analysis
An unpaired Student's t-test was performed while testing differences in means between two groups. For comparing more than two groups, one-way ANOVA (analysis of variance) with a post hoc (Tukey) test for multiple comparisons was performed. p < 0.05 was regarded as statistically significant.
Results
Formulation characterization and siRNA loading estimation
Gelatin nanoparticles
Formulation characterization for GNPs was previously described. 22
TG2-NiMOS
Particle-size analysis Malvern's Zeta-sizer® showed that the average mean hydrodynamic diameter of TG2-NiMOS was 2.63 ± 0.169 μm. Morphological evaluation using scanning electron microscopy showed that TG2-NiMOS were spherical in shape with an approximate diameter of 2–4 μm, 17 correlating with results obtained from Malvern's Zeta-sizer. The mean ± SD (standard deviation) siRNA loading was estimated to be 55.38% ± 8.13%.
TG2-NiE
Bright-field and fluorescent microscopy images of NiE (Supplementary Fig. S1) showed stable nonleaky multiple emulsion globules of ∼3–5 μm. When the TG2-NiE was centrifuged at up to 4000 rpm for 1 h, there was no phase separation observed under a light microscope. Further, up to 1000-fold dilution of TG2-NiE in aqueous media did not show any leaching or release of rhodamine-labeled GNPs encapsulated in the innermost phase of the W/O/W multiple emulsion. These showed that the TG2-NiE formulations did not phase separate on storage and even after an accelerated centrifugation cycle. The mean ± SD estimate of siRNA loading was ∼65.1% ± 10.5%.
Biodistribution and PK of TG2-siRNA
Concentration-time profiles of TG2-siRNA in plasma and tissues after oral administration via TG2-NiMOS or TG2-NiE are shown in Figure 1. Noncompartmental PK parameter estimates are shown in Table 4. Maximum concentration of TG2-siRNA in plasma and tissues is shown in Figure 2. TG2-siRNA concentrations in the TG2-GNP treated group were below the limit quantitation in plasma and all tissues analyzed.
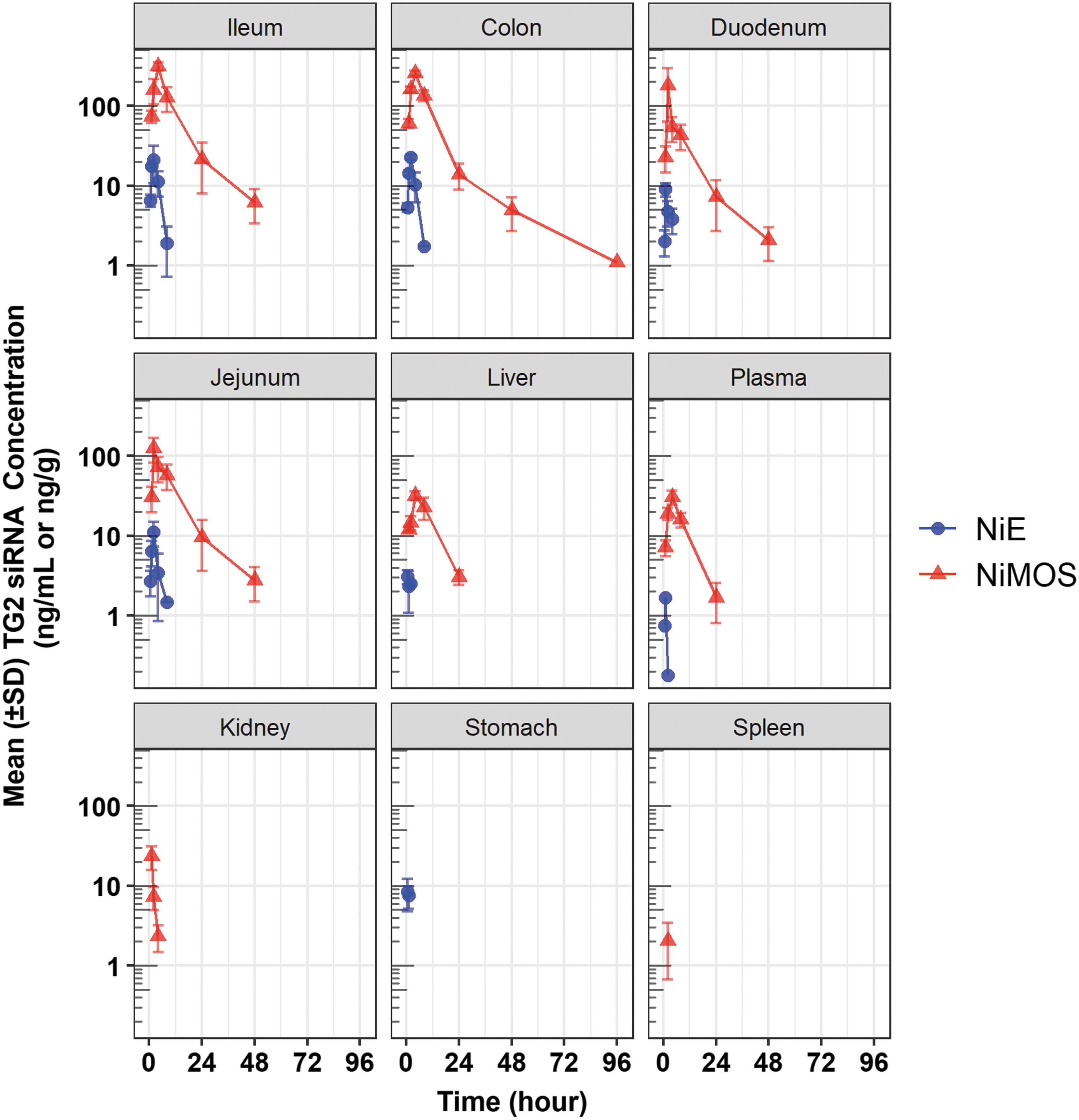
Pharmacokinetic profiles of tissue TG2 siRNA in mouse plasma and tissue samples after oral administration in NiMOS and NiE multicompartmental formulations. The oral siRNA dose in both formulations was 0.5 mg/kg. NiE, nanoparticle-in-emulsion; NiMOS, Nanoparticle-in-Microsphere Oral System; siRNA, small interference RNA; TG2, transglutaminase-2.
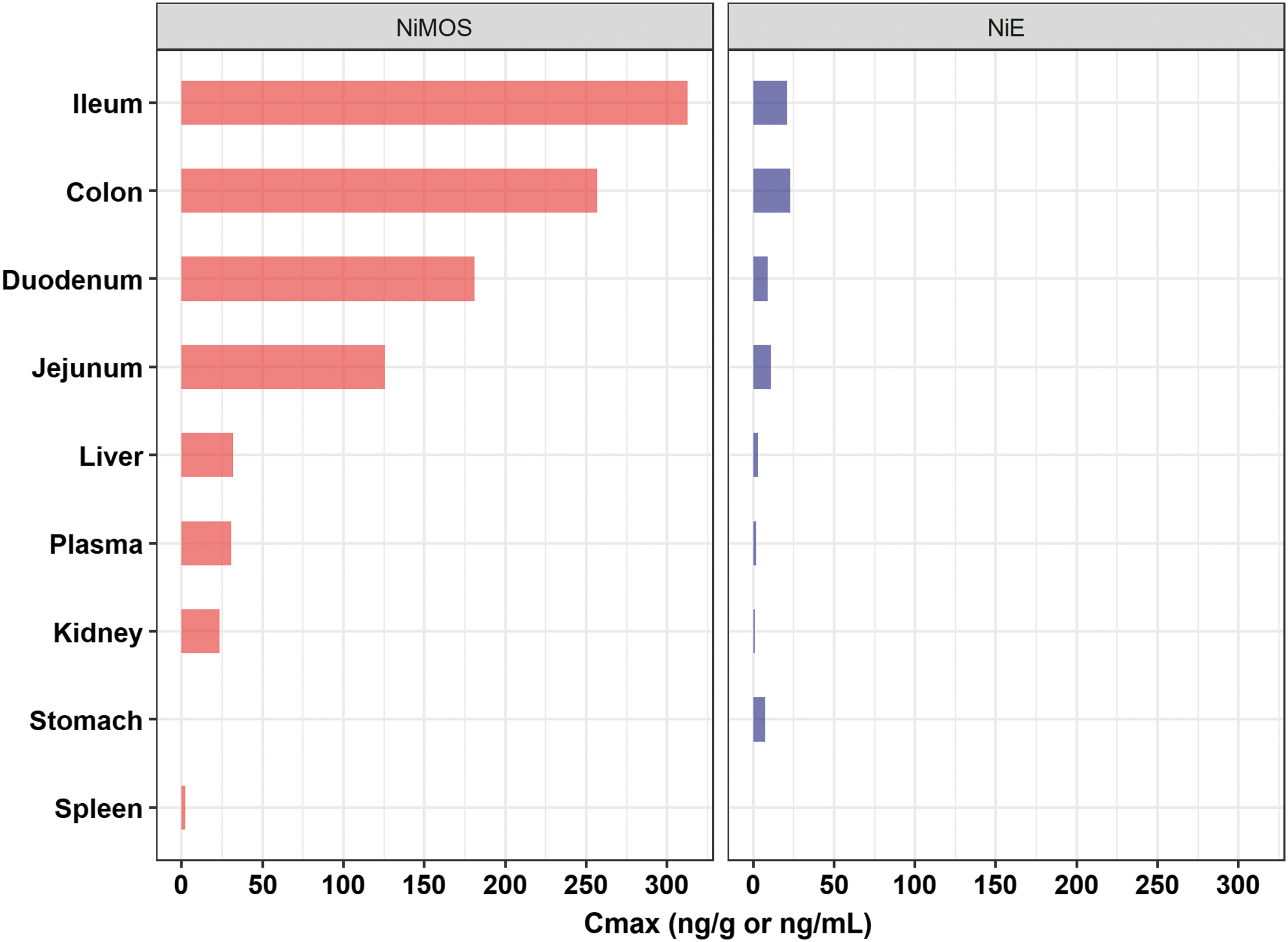
Comparison of the plasma and tissue maximum concentrations (Cmax) values of tissue TG2 siRNA after oral administration in NiMOS and NiE multicompartmental formulations. The oral siRNA dose in both formulations was 0.5 mg/kg.
Pharmacokinetic Parameters of Transglutaminase-2 Gene Silencing Small Interfering RNA in the Plasma and Tissues After a Single Oral Dose of Nanoparticle-in-Microsphere Oral System and Nanoparticle-in-Emulsion at 0.5 mg/kg Small Interfering RNA Dose
NR, not reportable (<3 quantifiable timepoints after tmax).
Biodistribution and PK of TG-siRNA after oral administration through TG2-NiMOS
In the TG2-NiMOS group (Group 1), quantifiable levels of TG2-siRNA were observed in the colon up to 96 h postdose and in the duodenum, jejunum, and ileum, for at least 48 h postdose. The maximum concentrations of TG2-siRNA in the duodenum, jejunum, and ileum were observed between 2 and 4 h postdose, with an apparent elimination half-life value of ∼9.3 h. Between the three portions of the small intestine, the ileum showed maximum TG2-siRNA exposure (area under the curve from 0 to last quantifiable time point [AUC0−t]), followed by the jejunum and duodenum. Exposure (AUC0−t) of TG2-siRNA in the colon was comparable to that observed in the ileum; however, the apparent terminal elimination half was ∼2-fold greater (20 h) than in ileum. Levels of TG2-siRNA quantified from liver, kidney, spleen, and plasma were very less compared with those observed in the GIT. The TG2-siRNA levels in the stomach were below the limit of quantitation at all the study points.
Biodistribution and PK of TG-siRNA after oral administration through TG2-NiE
In the TG2-loaded NiE group (Group 7), quantifiable levels of TG2-siRNA were observed in the jejunum, ileum, and colon up to 8 h and in the duodenum up to 4 h. The maximum concentrations of TG2-siRNA in the duodenum, jejunum, and ileum were observed between 1 and 2 h postdose. Apparent terminal elimination half-lives were not reportable for any of the tested tissues or plasma due to insufficient quantifiable concentration-time data points in the terminal phase (at least three quantifiable data points after Cmax are needed for calculation of half-life). Between the three regions of the small intestine, the ileum showed maximum TG2-siRNA exposure (AUC0−t), followed by the jejunum and duodenum. Exposure (AUC0−t) of TG2-siRNA in the colon was comparable to that observed in the ileum. Levels of TG2-siRNA quantified from the liver, kidney, and plasma were very less compared with those observed in the GIT. The TG2-siRNA levels in the spleen were below quantitation at all the study time points.
Comparison of TG2 siRNA exposure after a single oral dose of TG2-NiMOS or NiE
Between the two multicompartmental formulations, TG2-siRNA exposure was greater in the case of TG2-NiMOS formulation when compared with TG2-NiE formulation (Fig. 2 and Table 4). When tested for statistical significance (where possible), TG2-siRNA concentration values at mean tmax for TG2-NiMOS were significantly greater in the colon, ileum, jejunum, liver and plasma, compared with TG2-NiE. The Cmax and AUC0−t values for TG2-siRNA in duodenum were 20.1- and 56.4-fold greater in the case of TG2-NiMOS when compared with TG2-NiE formulation, respectively. The Cmax (maximum concentration) and AUC0−t values for TG2 siRNA in the jejunum were 11.3- and 34.3-fold greater in the case of TG2-NiMOS when compared with the TG2-NiE formulation, respectively. The Cmax and AUC0−t values for TG2 siRNA in the ileum were 21.0- and 85.5-fold greater in the case of TG2-NiMOS when compared with TG2-NiE formulation, respectively. The Cmax and AUC0−t values for TG2 siRNA in the colon were 11.3- and 35.5-fold greater in the case of TG2-NiMOS when compared with TG2-NiE formulation, respectively. Both formulations led to minimal exposure in plasma and systemic tissues, including the liver, spleen, and kidney (Table 4).
Discussion
Oral route of administration is very challenging for polyelectrolytes such as siRNA due to several extra- and intracellular barriers. Prominent extracellular barriers include low gastric pH that can promote depurination of nucleic acids, and the presence of nucleases in the GIT, both of which can rapidly degrade the administered nucleic acid dose. In addition to these, the cellular internalization could be potentially hindered due to electrostatic charge repulsion between negatively charged nucleic acids, negatively charged mucus surrounding the GI epithelium, and the negatively charged cell surface. On cellular uptake, the nucleic acid cargo must resist degradation via enzymes and low pH in the endo-lysosomal transit compartment before it is released into the cellular cytoplasm. The multicompartmental systems evaluated in this study, TG2-NiE and TG2-NiMOS, were specifically designed to evade these extra- and intracellular barriers to facilitate oral nucleic acid delivery.25,26
Prior in vitro evaluations with GNPs showed that GNPs were capable of efficiently encapsulating siRNA, delivering siRNA to the cellular cytoplasm, and enabling functional RISC loading of delivered siRNA. Further, when tested in an in vitro model of CD, the knockdown of IL-15, either specific or an adjunct with TG2, showed significant downregulation in the expression of proinflammatory cytokines—TNF-α and IFN-γ. 22
Our current objective was to develop an oral siRNA delivery system that is capable of inhibiting expression of target proteins in the small intestine, aimed at developing a potential treatment for patients with CD. When GNPs loaded with TG2-siRNA were administered to mice without being formulated as a multicompartmental system, no local or system siRNA exposure was seen, suggesting that siRNA encapsulation with gelatin particles alone is not enough for enabling siRNA delivery via oral route. To be effective in an in vivo system, the GNPs would need to remain protected in the GIT before reaching target sites in the small intestine. For this to happen, the delivery vehicle must be engineered such that the carrier-associated oligonucleotide load is stable during transit through the degrading physiological environment. Protection against low gastric PH, proteases, and nucleases would be needed for stable GI transit of the carrier system to promote payload accumulation at the intestinal target sites and allow subsequent cytosolic release of the nucleic acid payload.
In this study, we have engineered two distinct multicompartmental systems, where siRNA was first encapsulated within GNPs (to confer protection against nucleases), which was then encapsulated within a solid or liquid outer hydrophobic matrix (to confer protection against low gastric PH, proteases, and nucleases). In case of TG2-NiMOS, 27 the outer hydrophobic matrix was composed of solid PCL microspheres, whereas in the case of TG2-NiE, 20 it was composed of safflower oil-based multiple emulsion.
On in vivo biodistribution and PK evaluations in mice after oral TG2-siRNA administration, we observed that both TG2-NiE and TG2-NiMOS promoted siRNA delivery to the small intestine and colon with minimal systemic exposure. TG2-NiMOS was shown to be more efficient in promoting intestinal siRNA delivery relative to TG2-NiE, possibly due to the greater stability of PCL microspheres during the GI transit. It is noteworthy that no siRNA exposure was seen in the stomach with TG2-NiMOS, suggesting that PCL microspheres remained intact during the transit through the stomach; whereas with TG2-NiE, detectable siRNA levels were seen in the gastric tissue—indicative of gastric degradation of the TG2-NiE system and a possible cause of low siRNA exposure levels across all tissues. Taken together, these data suggest that TG-NiMOS are capable of efficiently delivering siRNA to the small intestine and colon with minimal exposure to systemic tissues after oral administration in mice. The siRNA tissue exposures observed in this study are anticipated to have robust mRNA silencing effect in nonclinical species and humans, based on an established noteworthy relationship between siRNA tissue PK and a subsequent pharmacodynamic effect seen with other systemically delivered siRNA candidates.28,29 Based on encouraging biodistribution and PK results observed in this study, further nonclinical development of the TG2-NiMOS is warranted to fully characterize kinetics of target mRNA silencing and to evaluate subsequent disease-modifying effects in a relevant animal model of CD after oral delivery of TG2-NiMOS.
Conclusions
In the past, we have shown that GNPs can efficiently encapsulate siRNA, deliver it into the cellular cytoplasm, and enable functional RISC loading in Caco-2 cells. They are also efficient in target knockdown in both Caco-2 and murine alveolar macrophage (J774A.1) cell lines. As a next step, to utilize this system for localized delivery into the intestines, GNPs would need to be protected from the physiological conditions of the GIT. This was achieved by encapsulating the nanoparticles in microspheres, as in the case of TG2-NiMOS and in an emulsion, as in the case of NiE. A biodistribution and PK study was performed with TG2-siRNA loaded in NiMOS and NiE by using the corresponding controls. After this study, we can conclude that GNPs are not enough to deliver siRNA to the intestines orally. The results also suggest that the TG2-NiMOS formulation provides a superior protection to the encapsulated siRNA payload, resulting in higher siRNA exposures in different tissues and plasma in C57BL/6 mice after oral administration. Collectively, these data suggest that multicompartmental systems hold promise in enabling local intestinal delivery of siRNA. In our next phase, we will design studies to evaluate siRNA-mediated knockdown of target proteins in the small intestine for the treatment of CD.
Footnotes
Authors' Contributions
A list of contributions from each of the authors is given later. Please note that all the authors have reviewed and consent to the content of this article. Also, this article has not been submitted to any other journal. Original idea: H.Z.A. and M.M.A. Conducting Experiments: H.Z.A. Analysis of Data: H.Z.A. and M.M.A. Writing Article: H.Z.A. and K.S. Article Editing: H.Z.A., K.S., and M.M.A.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This article did not receive funding from federal or private sources.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.