
Abstract
Introduction:
The psychoactive properties of Δ10-THC isomers (trans- and cis-Δ10-THC) are poorly understood. To shed more light on the biological effects of these compounds, we studied in vitro receptor binding of Δ10-THC isomers at cannabinoid CB1 and CB2 receptors.
Materials and Methods:
We first optimized and simplified catalytic synthesis of trans- and cis-Δ10-THC to allow their safe and cheap large-scale synthesis. In our synthesis, BuLi was replaced with KO t Bu, and DMSO/anisole or NEt3/heptane solvent systems were used instead of HMPA/toluene. Single crystal X-ray analysis confirmed the structure of both isomers and the configuration of their chiral centers.
Results:
In the radioligand replacement assay, both isomers showed strong affinity toward the CB1 receptor, with IC50=29.1 nM for the trans isomer and IC50=294.2 nM for the cis counterpart. However, the IC50 values were significantly higher than that of Δ9-THC (2.1 nM), a naturally occurring psychoactive component of cannabis sativa, suggesting a lower affinity of Δ10-THCs toward this receptor. In function assays, in contrast to Δ9-THC, both isomers failed to show any agonist properties at concentrations up to 10 μM suggesting a lack of THC-like psychoactivity for trans- and cis-Δ10-THC.
Conclusions:
Our results established Δ10-THC isomers among antagonists of the CB1 receptor as both cis and trans isomers antagonized CP55,490 with IC50=460 nM for trans and IC50=1040 nM for cis. This functional property has not been previously observed for any other THC isomers.
Introduction
Tetrahydrocannabinol (THC) refers to a variety of structural isomers differing in the position of the unsaturated double bond of the cyclohexene ring (Fig. 1). The primary psychoactive component of cannabis Sativa, Δ9-THC, produced by spontaneous or heat-accelerated decarboxylation of phytochemical Δ9-THCA, has been consumed for medical and recreational purposes for over two millennia. 1 Although some other isomers are also found naturally in the cannabis plant, it is still unclear whether they are produced directly by the plant or due to biosynthesis-independent Δ9-THC/Δ9-THCA rearrangement. However, most THC isomers can be synthesized in the laboratory starting from Δ9-THC. Acid-promoted isomerization yields Δ8-THC, 2 whereas base-promoted one produces a mixture of Δ10-THC diastereomers. 3

Structural isomers of THC differing in the position of the unsaturated double bond. THC, tetrahydrocannabinol.
On the one hand, further acid isomerization of Δ10-THCs affords Δ6a,10a-THC enantiomers. 3 On the other hand, the addition of hydrogen chloride to Δ8-THC followed by stereospecific elimination gives Δ9,11-THC, commonly known as exo-THC. 4 In addition, Δ8-THC can be converted to the Δ7 isomer by a multistep synthesis. 5 Previous studies reported the synthesis of Δ6a,7-THC and Δ7-THC from chemical building blocks (terpene and phenol), 6 but the production of these isomers from Δ9-THC in a multistep chemical reaction can be envisioned as well.
Despite the structural similarity of these isomers, experimental and anecdotal reports on their psychoactivity and health benefits are inconsistent, indicating a large gap of knowledge in understanding the biological properties of less-known THC isomers. The second most well-known isomer, Δ8-THC, is psychoactive and believed to be moderately less potent than Δ9-THC. 7 Experimental information on the psychoactivity of other isomers, mainly obtained by studies conducted in the 70s and 80s, is conflicting and perplexing.8–10 Perhaps, among these isomers, Δ10-THC marks the most ambiguous case. This compound has two chiral centers, totally producing four structural isomers. However, starting from natural enantiopure Δ9-THC [(6aR,10aR)-Δ9-THC], only two diastereomers (6aR,9R)-Δ10-THC and (6aR,9S)-Δ10-THC can be obtained, referred to in this study as trans- and cis-Δ10-THC based on the alignment of hydrogens located on C6a and C9 (Fig. 2).
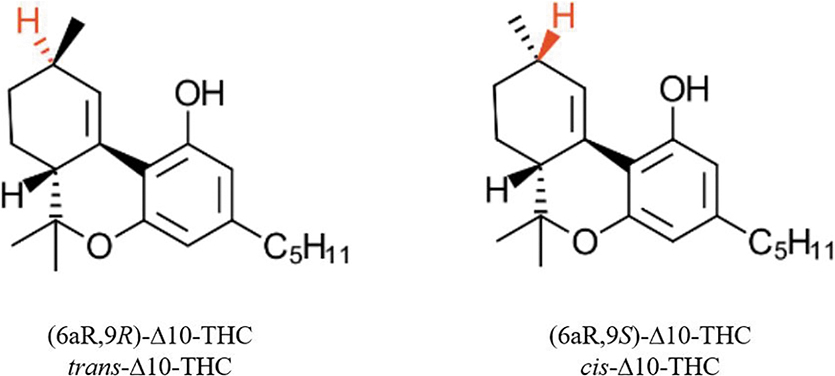
Structures of trans- and cis-Δ10-THC.
Studies on the biological properties of trans- and cis-Δ10-THC are scarce. In a drug discrimination study conducted in 1988 on pigeons trained to discriminate between the presence or absence of Δ9-THC (0.56 mg/kg), cis isomer and its acetate derivative (cis-Δ10-THC-O-acetate) failed to substitute for Δ9-THC even at doses as high as 17.5 mg/kg. 11 The other isomer, trans-Δ10-THC, was not included in this study, but its acetate substitute (trans-Δ10-THC-O-acetate) was found to be ∼26 times less potent than Δ9-THC. 11 Although limited available experimental results suggest a lack of significant cannabimimetic effect for Δ10-THCs, anecdotal reports exclusively claim the psychoactive nature of these compounds. 12 In recent years, Δ10-THC isomers have entered the U.S. cannabis market, partially to circumvent tight regulation on Δ9-THC in some states. 13 Online reviews, individual testimonies, and even some published expert opinions 14 imply notable psychoactivity for Δ10 isomers.
It is believed that Δ10-THC can get individual “high,” although with less potency than Δ9-THC. In some cases, attempts have been made to put their activity into perspective by claiming that they produce sativa-like energizing effects, anecdotal classification with no solid attachment to reliable experimental results. 15
To address some confusion about Δ10-THCs, this study aims to shed light on the chemistry and in vitro cannabinoid receptor binding of these isomers. In this study, we discuss the synthesis of these compounds in detail, and for the first time, report their crystal structure. In vitro affinity and functional properties of Δ10-THC isomers at cannabinoid receptors (CB1 and CB2), recognized as primary receptors involved in the psychoactivity of Δ9- and Δ8-THC, will be explored. The physical, chemical, and biological differences between Δ9- and Δ10-THC isomers will be underlined.
Experimental Method
Synthesis
Method A
To a 50-mL test tube containing a magnetic stir bar and Δ9-THC (0.354 g, 77.9% purity, 0.877 mmol, 1.0 equiv.) under N2 was added solid KO t Bu (0.890 g, 7.94 mmol, 9.0 equiv.). The tube was equipped with a septum and carefully purged again with N2. To the tube was added DMSO (2.0 mL, 26 mmol, 30 equiv.) and anisole (4.0 mL) via a syringe, and the mixture was stirred and heated to 100°C for 1.5 h under N2. The tube was cooled to room temperature (22°C) and quenched with 10% wt/wt aq. citric acid (7.6 mL, 3.9 mmol, 4.5 equiv.) with vigorous stirring for 10 min under N2. Heptane (10 mL) was added, the layers were separated, and the organic layer was washed with 2×10 mL water. The organic layer was concentrated in vacuo to give 0.360 g brown tar. HPLC analysis showed a 90% conversion of Δ9-THC to Δ10-THCs in the ratio trans:cis=6.4.
Method B
To a Schlenk flask containing a magnetic stir bar and Δ9-THC (5.33 g, 92% purity, 15.6 mmol, 1.0 equiv.) was added solid KO t Bu (8.826 g, 78.7 mmol, 5.05 equiv.). The flask was equipped with a reflux condenser and septum and carefully purged with N2. To the flask was added anisole (20 mL) and triethylamine (20 mL, 143 mmol, 9.20 equiv.) by syringe, and the mixture was stirred and heated gradually from 80°C to 125°C over 4 h, adding an additional 30 mL anisole when the mixture began to precipitate solids.
The flask was cooled to room temperature (22°C) and transferred by cannula into a solution of excess citric acid (21 g, 111 mmol, 7.12 equiv.) in water with vigorous stirring for 10–30 min under a blanket of N2. The mixture was diluted with heptane, the layers were separated, and the organic layer was washed with 3×50 mL water. The wet organic layer was evaporated under reduced pressure (65°C, 30 mbar) and concentrated twice from heptane to give a red-brown resin (5 g) that gradually precipitated crystals on standing for several days.
Typical isolation procedure of cis- and trans-Δ10-THC
The isomerization at 110°C of 4.88 g of 90% Δ9-THC provided 4.8 g of brown resin. After standing for several days, crystals could be filtered from the resin and washed with cold heptane to give cis-Δ10-THC. Alternatively, the residue was purified by flash column chromatography on silica gel. Elution with a gradient of 0–10% MTBE in heptane provided the first trans-Δ10-THC as a yellow oil that crystallized on standing after adding a seed crystal (3.7 g, 76% yield). Repeated recrystallization from a 50% solution in pentane at −80°C gave colorless prisms of trans-Δ10-THC. Further chromatographic elution yielded cis-Δ10-THC as colorless needles (0.377 g, 8% yield). Finally, recrystallization by slow evaporation from Et2O/heptane provided colorless prisms suitable for X-ray crystallography.
Crystallography
The crystals obtained by synthesis from DMSO/anisole solvent system were used for single-crystal X-ray analysis. The crystal sample was mounted on a MiTeGen polyimide micromount with a small amount of Paratone N oil. All X-ray measurements were made on a Bruker Kappa Axis Apex2 diffractometer at a temperature of 110 K. The unit cell dimensions were determined from a symmetry constrained fit of 5821 reflections with 5.22° < 2θ < 52.78°. The data collection strategy was a number of ω and ϕ scans, which collected data up to 57.208° (2θ). The frame integration was performed using SAINT. 16 The resulting raw data were scaled and absorption corrected using a multiscan averaging of symmetry equivalent data using SADABS. 17 The structures were solved using a dual space methodology using the SHELXT program. 18
All nonhydrogen atoms were obtained from the initial solution. The hydrogen atoms were introduced at idealized positions and were allowed to refine isotropically. The absolute configuration was assigned in consultation with the sample originator. The structural model was fit to the data using full-matrix least-squares based on F2. The calculated structure factors included corrections for anomalous dispersion from the usual tabulation.
The structure was refined using the SHELXL program from the SHELX suite of crystallographic software. 18 Graphic plots were produced using the Mercury program. 19
Receptor bindings
Binding assay
The pure Δ10-THC isomers synthesized from DMSO/anisole solvent system were used for receptor-binding studies. Radioligand binding at CB1 and CB2 receptors was conducted using the high-throughput CB1-binding filter plate assay applying Human CB1-overexpressing membranes (Merck-Millipore Co., Darmstadt, Germany). 20 In summary, 200 μL prewarmed binding buffer (50 mM Tris-HCl, 5 mM MgCl2, 1 mM CaCl2, 2 mg/mL BSA, pH 7.4), 6 μg/well CB1-overexpressing membranes, and 10 μL DMSO stock solution of tested compounds at different concentrations were added to a polypropylene 96-well plate. The plate was covered with a lid and carefully shaken before incubating for 20 min at 37°C. The radiolabeled ligand, [ 3 H]CP55.940 (PerkinElmer), at the final concentration of 0.8 nM (CB1 receptor) and 0.6 nM (CB2 receptor), was added to each well (except for the blanks) to start the reaction. In the meantime, FC-filter plates were presoaked with 200 μL of 50 mM Tris–HCl containing 0.33% PEI.
Ten minutes before terminating the reaction, PEI was removed by vacuum and filters were washed three times with washing buffer (50 mM Tris–HCl, 500 mM NaCl2, 1 mg/mL, BSA, pH 7.4). After 1 h at 37°C, the reaction solution from the 96-well plate was transferred to the FC-filters, and the filters were rewashed four times with 300 μL of washing buffer. Filters were dried, by placing the plate for 30 min in an incubator at 37°C. Each filter was transferred to the corresponding scintillation vial containing 500 μL of 0.1% Triton in the buffer (50 mM Tris–HCl, 1 mg/mL BSA, pH 7.4). Finally, 3.5 mL of liquid scintillation cocktail (Ultima Gold™ XR) was added, and radioactivity was measured in a scintillation β-counter.
It is noteworthy that the homologous competitive study with cold CP55.490 produced KD=0.8 nM and KD=0.6 nM at CB1 and CB2 receptors, respectively, showing that the radioligand binds with correct affinity. The inhibition curve was fitted for IC50 measurements using GraphPad Prism 6 (GraphPad Software, Inc.), and the Cheng–Prusoff equation 21 was applied to estimate Ki values.
PathHunter® assay
For agonist and antagonist assays, PathHunter CHO-K1 CNR1 (Eurofins DiscoverX, Catalog number: 93-0959C2) and CBR2 (Eurofins DiscoverX, Catalog number: 93-0706C2) β-Arrestin cells were expanded from freezer stocks according to standard procedures provided by Eurofins. The cell line manual was followed for cell growth (including cell culture media and supplementation, the handling and preparation of cells, etc.), test procedure, and signal detection. This manual can be obtained from the Eurofins website free of charge.
In summary, cells were seeded 40,000 cells/well in 96-well plates and incubated at 37°C for the appropriate time before testing. For agonist determination, cells were incubated with samples to induce a response. After preparation of stock DMSO solution at 150 μM concentration, compound intermediate concentrations were achieved by an 11-point series of three-fold compound serial dilutions in a compound dilution buffer, in a separate dilution plate. The concentration of each dilution was prepared at 5×of the final screening concentration. Twenty microliters of samples was added to cells (highest final concentration=10 μM) and incubated at 37°C for 90 min (5% CO2).
For antagonist determination, cells were preincubated with antagonist/compounds followed by agonist challenge at the EC80 concentration. Intermediate dilution of sample stocks was performed to generate 11-point series of three-fold compound serial dilutions in a compound dilution buffer in a separate dilution plate. The concentration of each dilution was prepared at 10×of the final screening concentration.
Ten microliters of samples was added to cells (highest final concentration=10 μM) and incubated at 37°C for 30 min (5% CO2). Ten microliters of (±)-CP 55,940 stock solution was added to the cells to produce a final concentration equal to EC80 (5.04 nM for CB1 and 2.7 nM for CB2). Cells were incubated at 37°C for 90 min. The pure Δ8-THC, Δ9-THC, Δ9-THCA, and CBN for agonist assay were purchased as analytical standards from Sigma, Cerilliant, and Cayman Chemicals.
The assay signal was generated through a single addition of 12.5 (50% v/v) of the PathHunter Detection reagent cocktail, followed by a 1-h incubation at room temperature (24°C). β-Arrestin recruitment was quantified using a Tecan Spark plate reader. Replacement assay was conducted in duplicates, while PathHunter® agonist and antagonist experiments were performed in triplicates. The inhibition curve was fitted for EC50/IC50 measurements using GraphPad Prism 6 (GraphPad Software, Inc.). For the agonist assay, 100% relative activity was normalized to the maximum (±)-CP 55,940 stimulation and 0% relative activity was normalized to compound vehicle control. For the antagonist assay, 100% relative activity was normalized to the maximum rimonabant (CB1) and SR144,528 (CB2) inhibition and 0% relative activity was normalized to compound vehicle control. This study does not involve any human material, neither prospectively nor retrospectively, so IRB does not apply.
Results and Discussion
Synthesis and chemistry
Base-catalyzed isomerization of natural Δ9-THC in aprotic solvents results in a mixture of two diastereomers, cis-Δ10-THC and trans-Δ10-THC. This is thought to occur through the double deprotonation of the phenolic and benzylic positions. Using n-butyllithium (BuLi) in hexamethylphosphoramide (HMPA) at a low temperature produces mainly the kinetic cis-diastereomer. In contrast, the weaker base potassium tert-amylate in refluxing toluene-HMPA provides access to the thermodynamic trans-isomer as the major product. 3 To make the process more scalable, we sought alternative reagents and procedures that would be safer and easier to handle and use solvents permitted as trace impurities in cannabis products by Health Canada.
THC isomers are all very sensitive to autoxidation, especially under basic conditions, so all isomerizations must be conducted under an inert atmosphere. Several strong bases were surveyed (KOH, NaH, LDA, and MgO) but failed to promote any isomerization (Table 1, entries 1–6). KO t Bu in the nonpolar solvent heptane or the protic solvent ethanol also failed. From the literature, it is clear that a highly polar solvent is an essential component of this reaction, 3 as polar solvents can make the base anions more nucleophilic by deaggregating metal–anion clusters in solution.22,23 Replacing the carcinogenic HMPA with DMSO and toluene with anisole, KO t Bu was able to effect efficient isomerization at 100°C to a thermodynamic mixture of Δ10-THC isomers (Table 1, entry 11).

Variation of the reaction temperature was found to affect the trans:cis ratio from 6.4°C at 100°C to 3.8 at 125°C (Table 1, entry 12). This solvent system allows higher temperatures to achieve synthetically useful ratios of trans:cis isomers using the easily handled KO t Bu instead of BuLi. It is noteworthy that high-purity Δ9-THC isolate is not required for this reaction. Typical THC-dominant distillates containing 75–95% Δ9-THC can be applied as starting materials for this isomerization.
Under deprotonating conditions, DMSO has been reported to undergo explosive decomposition at elevated temperatures. 24 A sulfurous smell of isomerization reaction mixtures suggested some DMSO decomposition occurred. Still concerned about the safety of DMSO under these reaction conditions, we looked for a more stable polar solvent. Triethylamine (NEt3) in heptane, both ICH class 3 solvents, proved to be a satisfactory substitute for DMSO-anisole in the KO t Bu-promoted thermodynamic isomerization (Table 1, entry 13). However, solidification over time of concentrated NEt3–Heptane reaction mixtures was a drawback of this system, prompting lower concentrations of reactants. Further improvements to the aqueous workup procedure were made, including a safer reverse quenching under an inert atmosphere and using citric acid instead of HCl to prevent acid-catalyzed isomerization to Δ6a,10a-THC. The BuLi-promoted kinetic isomerization has not yet been attempted in NEt3.
Crystal structure
Cis and trans isomers were found to have dramatically distinct solubility profiles. After concentrating isomerization reaction mixtures, cis-Δ10-THC often precipitates over time from cold concentrated heptane solution, and purification can be conveniently accomplished through recrystallization. Chromatography is required to isolate trans-Δ10-THC, which is reluctant to crystallize and is highly soluble in most solvents. The trans isomer can be recrystallized through cryogenic cooling (−80°C) or low-temperature evaporation (−40°C) of pentane or pentane–hexamethyldisiloxane mixtures. As previously reported, these diastereomerically pure Δ10-THC isomers can serve as starting materials for the facile acid-catalyzed isomerization of pure Δ6a,10a-THC enantiomers. This can be achieved quantitatively using catalytic tosylic acid in toluene. 3
The structure of Δ10 isomers and configurations of their chiral centers were confirmed by single crystal analysis. ORTEP diagrams are shown in Figure 3, and crystal structure data are presented in the Supplementary Data. In the cyclohexene ring of both isomers, the C10-C10a bond length is significantly shorter than other C-C bonds confirming that a double bond is formed between these two carbons. As naturally occurring enantiopure (6aR,10aR)-Δ9-tetrahydrocannabinol was used as starting material, both isomerization products share the same configuration (R) at the C6a center. Configuration of C9 carbon, the new chiral center formed during isomerization, is the main difference between the structure of cis- and trans-Δ10-THC. In cis isomers, the S configuration of C9 allows the alignment of H9 and H6a, while trans-Δ10-THC holds the R configuration at the C9 center.
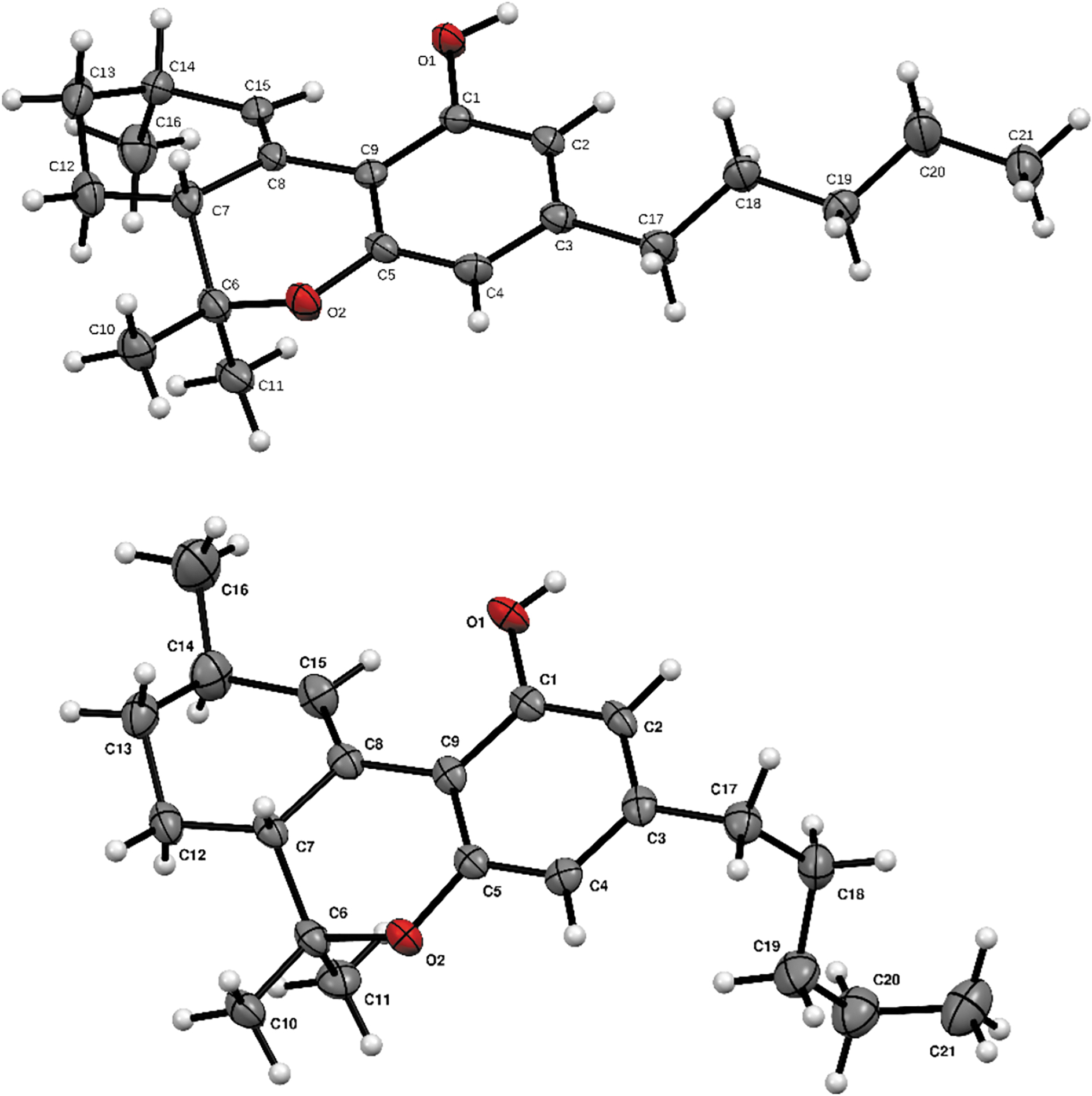
ORTEP drawings of cis- (up) and trans-Δ10-THC (bottom). Ellipsoids are at the 50% probability level, and hydrogen atoms were drawn with arbitrary radii for clarity.
Receptor bindings
Previous structure–activity studies by Reggio et al. showed that the orientation of the C9 substituent plays a vital role in the psychoactivity of THC-like cannabinoids. 25 The orientation of the C9 substitute is, interestingly, the major structural difference between trans-Δ10-THC, cis-Δ10-THC, and Δ9-THC. Our [ 3 H]CP55,940 radioligand displacement assay at the CB1 receptor showed that Δ10 isomers have a lower affinity toward cannabinoid receptors than Δ9-THC (Table 2). In addition, at both CB1 and CB2 receptors, trans-Δ10-THC revealed ∼10-folds lower IC50 and Ki than the cis isomer, revealing its stronger affinity toward the receptors than its cis counterpart. Some selectivity toward the CB1 receptor was observed for Δ9-THC. In contrast, Δ10-THC isomers did not demonstrate any sign of selectivity toward CB1 or CB2, producing similar IC50 and Ki at both receptors. In addition, at the CB2 receptor, Δ9-THC and trans-Δ10-THC displayed comparable displacement activity.
Displacement of [ 3 H]CP55,940 Bound to CB1/CB2
Average of two independent measurements.
Estimated using the Cheng–Prusoff equation.
Despite lower affinity than Δ9-THC, measured IC50 of Δ10 isomers for the displacement of [ 3 H]CP55,940 at CB1 and CB2 were still in the nanomolar range, which encouraged us to further investigate their functional property at these receptors.
Agonist and antagonist assays revealed very unexpected results. Both Δ10 isomers failed to show any agonist property in concentrations up to 10 μM (Table 3, Figs. 4 and 5). Instead, both isomers were capable of antagonizing the agonist effect of CP55,940, a synthetic cannabinoid and full agonist of CB1, with IC50 of 460 (trans) and 1040 nM (cis). Furthermore, the antagonist property at CB1 was found to be approximately a thousand times weaker than rimonabant, an inverse agonist of the cannabinoid CB1 receptor. 26 In addition, although both Δ10 isomers appear to have a significant affinity toward the CB2 receptor, we detected weak agonist and antagonist properties at this receptor (RE <20%) up to 10 μM concentration. However, the observed activity was influenced by the background noise of the test, which prevented us from drawing a solid conclusion about the functional property of isomers at the CB2 receptor.

Agonist mode CB1 (left) and CB2 (right) β-arrestin assay for CP55,940 and Δ10-THC isomers after 90 min incubation time using PathHunter® assay. One hundred percent relative activity was normalized to the maximum (±)-CP 55,940 stimulation and 0% relative activity to compound vehicle control. Error bars represent the standard deviation of three independent measurements.

Antagonist mode CB1 (left) and CB2 (right) β-arrestin assay for synthetic antagonists (rimonabant and SR144528) and Δ10-THC isomers after 30 min incubation time followed by 90 min (±)-CP 55,940 challenge at (5.04 nM for CB1 and 2.7 nM for CB2) using PathHunter assay. One hundred percent relative activity was normalized to the maximum rimonabant (CB1) and SR144,528 (CB2) inhibition and 0% relative activity was normalized to compound vehicle control. Error bars represent the standard deviation of three independent measurements.
EC50 and IC50 Values for Agonist/Antagonist Effects of d10-THC Isomers and Three Synthetic Cannabinoids at CB1 and CB2 Receptors
Values in parentheses are standard deviations of three independent measurements.
RE (%) at 10 μM.
ND, not determined.
It is noteworthy that the β-arrestin pathway, the pathway investigated for agonist and antagonist properties of Δ10 isomers in this study, is one of the multiple downstream pathways that can be activated after ligand binding to the G-protein-coupled receptors. 27 A recent bias analysis of natural and synthetic cannabinoids by Finlay et al. suggested that Δ9-THC may be a biased ligand, with it being less active in the β-arrestin pathway than in cyclic adenosine 3′,5′-monophosphate (cAMP) and PKR-like endoplasmic reticulum kinase pathways. 28
To ensure that our β-arrestin assay is capable of detecting differences in the activity of THC isomers, we compared the CB1 agonist properties of Δ10 isomers with that of Δ9-THC, Δ8-THC, Δ9-THCA, and cannabinol (CBN) at a single 10 μM concentration (Fig. 6). In this test, we observed clear sign of CB1 agonist property for Δ9- and Δ8-THC with relative efficacy (% of CP55,940) of 25.5% and 20.2%, respectively. As the psychotropic property of Δ9- and Δ8-THC is directly associated with their agonist property at the CB1 receptor, these results clearly suggest the lack of cannabinoid-related psychoactivity for trans- and cis-Δ10-THC.

Agonist mode CB1 β-arrestin assay for CP55,940, Δ10-THC isomers, Δ9-THC, Δ8-THC, Δ9-THCA, and CBN at 10 μM concentration after 90 min incubation time using PathHunter assay. One hundred percent relative activity was normalized to the maximum (±)-CP 55,940 stimulation and 0% relative activity to compound vehicle control. Error bars represent the standard deviation of three independent measurements. CBN, cannabinol.
Conclusion
In conclusion, studying the effect of reaction conditions on the catalytic synthesis of Δ10-THC isomers led to the discovery of simplified and safe production of trans- and cis-Δ10-THC using KO t Bu in ICH class III solvents. Characterization with a single crystal structure, which confirmed the chirality of two chiral centers, can help accurate computational analysis for these THC isomers in the future. Both Δ10-THC isomers not only showed lower affinity toward the cannabinoid CB1 receptor compared with Δ9-THC, but also, in contrast to Δ9-THC, failed to demonstrate any agonist property at this receptor. Instead, they established the ability to antagonize the CP55,940 effect at the CB1 receptor with significant potency. In addition, one of the interesting findings of this study is that the antagonist property of isomers appears to be CB1 specific.
Our in vitro results suggest a lack of THC-like psychoactivity for trans- and cis-Δ10-THC. This finding is aligned with the previous drug discrimination study in which cis isomer failed to produce a THC-like effect.
11
Accordingly, three hypotheses can be envisioned to explain reported psychoactivity in anecdotal reports:
Contamination of Δ10-THC samples with psychoactive compounds such as unreacted Δ9-THC and/or other THC isomers, particularly Δ6a,10a-THC, due to improper synthesis and purification methods. Interaction of Δ10-THC isomers with other brain neuropathways, such as interacting with dopamine and serotonin receptors. One of the limits of this study is focusing only on the β-arrestin pathway for assessing agonist and antagonist properties of Δ10-THC isomers. It should be noted that the functional selectivity at the CB1 receptor may allow activation of the receptor without β-arrestin recruitment.
29
Investigating G-protein-dependent signaling pathways, such as cAMP, is required to shed more light on the molecular pharmacology of trans-and cis-Δ10-THC.
Footnotes
Acknowledgments
The authors acknowledge Mahmood Azizpour Fard for helping with crystallography analysis; Torbjörn van der Meulen and Richard Green for providing laboratory space for the synthesis of Δ10-THC isomers; and Dalriada Drug Discovery for performing receptor-binding study.
Author Disclosure Statement
At the time of the study, M.H., D.B., B.G., M.B., and M.B.-M. were employed by Canopy Growth Corporation and had stock options. M.B.-M. served on the Board of Directors for AusCann Group Holdings Limited.
Funding Information
This study was funded by Canopy Growth Corporation.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.