
Abstract
Abstract
We have developed a new method for the isolation of porcine embryonic stem cells (ESCs) from in vivo-derived and in vitro-produced embryos. Here we describe the isolation and characterization of several ESC lines established using this method. Cells from these lines were passaged up to 14 times, during which they were repeatedly cryopreserved. During this time, ESCs maintained their morphology and continued to express Oct 4, Nanog, and SSEA1. These cells formed embryoid bodies in suspension culture, and could be directed to differentiate into various lineages representative of all three germ layers in vitro. When injected into blastocysts these cells localized in the inner cell mass of blastocysts. To examine their pluripotency further, cells were injected into host blastocysts and transferred to recipient animals. Of the six transfers undertaken, one recipient became pregnant and gave birth to a litter of one male and three female piglets. Microsatellite analysis of DNA extracted from the tail tissue of these piglets indicated that two female piglets were chimaeric.
Introduction
We have developed a new method for the isolation of porcine ESCs from in vivo-derived as well as in vitro-produced (IVP) embryos. Here we describe this method together with isolation and characterization of these lines
Material and Methods
In vitro embryo production
Porcine ovaries from slaughtered mature Large White × Landrace sows (white skin color) were collected from a local abattoir and transported to the laboratory in phosphate-buffered saline (PBS) solution, containing 1 × an antibiotic–antimycotic solution (Invitrogen, New Zealand) at between 33 and 37°C. Follicles with a diameter between 3 and 6 mm were aspirated with a 21-gauge needle, through which a constant suction (1/L min) was applied, and follicular contents were pooled in a collection tube. Cumulus–oocyte complexes with at least three uniform layers of compact cumulus cells were recovered from the collected fluid and matured for approximately 40–42 h in groups of 50 in 600 μL of M199 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5 ng/mL insulin, 0.5 mM cysteamine, 0.2 mM Na-pyruvate, 75 mg/mL Penicillin-G, 50 mg/mL Streptomycin sulphate, 10 ng/mL hEGF (all from Sigma, St. Louis, MO, USA), 5 mg/mL FSH (ICP Bio, New Zealand), and 10% sow follicular fluid under mineral oil (Sigma) in an atmosphere of 5% CO2 in air at 38.5°C (Beebe et al., 2007, 2009). Sow follicular fluid was collected from 3–6 mm follicles, stored at −70°C. Follicular fluid was thawed and filter sterilized (Millipore, Bedford, MA, USA; filter pore size 0.22 μm) immediately prior to use.
Mature cumulus oocyte complexes were treated briefly with 0.1% hyaluronidase (Sigma), washed twice in HEPES-buffered NCSU23 + 10% fetal bovine serum (FBS; JRH Biosciences, Lenexa, KA, USA), twice in the TALP-PVA fertilization medium (Bavister, 1989) supplemented with 3 mM calcium lactate and 2 mM caffeine-sodium benzoate (Sigma), then placed into 90-μL drops of fertilization medium (30 oocytes per drop) under mineral oil (Bavister, 1989). IVP embryos (and in vivo-derived embryos, see below) for embryonic stem (ES) cell isolation were fertilized with semen from a Hampshire boar (CH19; black and white skin color; SABOR, Clare, Australia). Host IVP blastocysts for ES cell injection were fertilized with semen from a Landrace boar (CL1219; white skin color; SABOR). Hampshire semen was used to produce the ES cell lines because this breed is not used commercially in Australia. Hence, any Hampshire contribution to chimaeric pigs will have come from this animal. The semen was allowed to come to room temperature (23°C) from its storage temperature (18°C). It was then centrifuged at 1100 × g for 5 min and the supernatant discarded. The pellet was resuspended in 10 mL of Medium 199 (Invitrogen) sperm wash medium containing 1 mg/mL sodium pyruvate, 9 mg/mL calcium-lactate, 0.075 mg/ml penicillin G, 0.05 mg/ml streptomycin sulphate (all from Sigma), and 10% FBS, then centrifuged and resuspended a second time. The sperm was diluted with TALP-PVA medium, and 10 μL of this added to the fertilization drop to give a final concentration of 5 × 105 sperm/mL. The oocytes and sperm were left to coincubate for 6 h in an atmosphere of 5% CO2 in air at 38.5°C, after which any remaining cumulus cells were removed by manual pipetting and the prospective embryos washed and placed into culture drops. The culture medium used was as described previously (Beebe et al., 2007). A modified version of NCSU23 medium containing 0.2 mM pyruvate, 5.7 mM lactic acid, 0.6 mM glucose (all from Sigma, USA) and MEM-nonessential amino acids (Invitrogen) was used for day 1 to 3 of culture, and NCSU23 medium containing 5.6 mM glucose (Sigma) and MEM nonessential and essential amino acids (Invitrogen) was used for day 3 to 6. Culture was conducted in 50-μL droplets under mineral oil (Sigma) in a humidified atmosphere of 5% CO2, 5% O2, and the balance N2 at 38.5°C.
In vivo embryo collection
Experiments were carried out in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (National Health and Medical Research Council, 2004). Late morulae and early blastocysts were collected from mulitparous Large White × Landrace sows (both white skin color). The sows were synchronized by weaning, given 1000 IU of eCG (Folligon, Intervet Australia, NSW, Australia) on the day of weaning, and 750 IU of hCG (Chorulon, Intervet Australia) 80 h later. Both injections were given intramuscularly. The sows were mated by artificial insemination during estrus using semen from a Hampshire boar (CH19, black and white skin color) during standing estrus, which usually commenced 24 h after hCG treatment. The embryos were collected surgically approximately 5 days after the onset of standing estrus. Each uterine horn was flushed with 50 mL of PBS containing 2% inactivated FBS, which was then searched using a dissecting microscope. Recovered embryos were washed in HEPES-buffered NCSU23 (Petters and Wells, 1993), held in 3 mL of the same medium in a 5-mL tube in a portable incubator at 38.5°C until surgery was completed, and all the collected embryos were transported to the laboratory and then cultured overnight as described above.
Establishment of embryonal outgrowths and isolation of porcine ESC lines
Primary outgrowths were established from either day 7 in vitro-produced embryos or day 6 in vivo-produced embryos as described above. After mechanical removal of the zona pellucida using a hand-drawn glass Pasteur pipette, embryos were plated on mitotically inactivated mouse embryonic fibroblast (MEF) feeder layers by gently pressing the whole embryo into the feeder layer using a 30-G needle. The isolation media used consisted of α-MEM medium with ribo- and deoxyribonucleosides supplemented with 10% Serum Replacement (SR), 20 ng/mL bFGF, 20 ng/mL human recombinant EGF, 1 × Insulin–Transferrin–Selenium solution, 55 μM 2-Mercaptoethanol, 1 × MEM nonessential amino acids, 1 × Glutamax (all from Invitrogen) 10 ng/mL human recombinant LIF (Millipore), 10 ng/mL Activin A (R&D Systems, Indianapolis, IN, USA). Cultures were conducted in humidified atmosphere of 5% CO2 and 5% O2. The media was changed every 2–3 days. After 12–15 days, primary outgrowths that had reached 2–5 mm in diameter were mechanically passaged onto fresh feeder layers. This was done by cutting outgrowths into approximately 80–100 micron square pieces using a hand-drawn glass Pasteur pipette, transferring these onto new feeder layers and then gently pressing the pieces into the feeder using a 30-G needle. Colonies were passaged every 10–12 days. Putative ESCs were identified according to morphological criteria described for bovine ESCs (Cibelli et al., 1998, Mitalipova et al., 2001), namely, a polygonal shape, a relatively small (10–15 μm) diameter, a small cytoplasmic/nuclear ratio, a single nucleus with multiple nucleoli, and multiple lipid inclusions in the cytoplasm. The homogeneity of outgrowths was assessed morphologically and by Oct 4 and Nanog coexpression. Cell lines were vitrified at passage 2 and then thawed. Cell lines that survived vitrification and warming and continued to grow were considered viable.
Vitrification and warming of porcine ES cell lines
ESC colonies were cut into smaller pieces as described above. Pieces were vitrified using solid-surface vitrification using the Cryologic Vitrification System (CVM; Cryologic Pty. Ltd., Victoria, Australia). The solutions used to vitrify and warm the embryos were warmed to 39°C prior to use. The ES cell colony pieces were held in 1 mL of the base medium [α-MEM + Glutamax-1 medium (Gibco, Gaithersburg, MD, USA) containing 2.5% vol/vol HEPES buffer solution (Gibco) and 20% FBS. Between 6 and 10 pieces were then washed in fresh base medium, incubated for 1 min in 1 mL of the base medium containing 10% (vol/vol) ethylene glycol (Sigma) and 10% (vol/vol) dimethyl sulphoxide (Sigma), and then washed for about 1 min in 1 mL of the base medium containing 0.4 M sucrose, 20% ethylene glycol, and 20% dimethyl sulphoxide (vitrification medium). The ESC colony pieces were then loaded in an approximately 3 μL droplet of the vitrification medium onto a nylon hook and the droplet quickly touched onto the surface of a metal block that had been cooled by partial immersion in liquid nitrogen. The nylon hooks were then covered with cooled plastic sleeves, plunged into liquid nitrogen, and stored. The ES cell colony pieces were warmed by stirring the vitrified droplet containing the colony pieces in approximately 1 mL of the base medium containing 0.2 M sucrose and holding them in that drop for approximately 1 min. They were then incubated in 1 mL of the base medium containing 0.1 M sucrose for 5 min, the base medium only for 5 min, and a fresh 1 mL of the base medium for a further 5 min. The warmed pieces were plated onto fresh feeder layer.
Karyotype analysis
Colonies of ESCs were treated with 0.05 μg/mL colcemid (Invitrogen) for 5 h at 37°C to arrest the cell cycle. Colonies were peeled of feeder layer and dissociated with TrypLE Express solution (Invitrogen), treated with hypotonic solution (0.075 M KCl) and then fixed in methanol–acetic acid (3:1). The cells were spread onto slides and stained for 30 sec with 5% Giemsa. Karyotype was examined at 1000 × magnification with oil immersion.
Pluripotent marker expression
To examine pluripotent marker expression, ESC colonies were fixed with 4% PFA and analyzed for expressions of Oct-4, Nanog, SSEA-1, and distribution of F-actin. The primary antibodies were rabbit antibody to Nanog (1:300 dilution; Millipore), mouse antibodies to SSEA-1 (1:200 dilution; Millipore), and goat antibody to Oct 4 (1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, porcine primary outgrowths or porcine embryos were incubated for 30 min with secondary antibodies at room temperature. Secondary antibodies were goat rabbit specific antibody conjugated to Texas Red (1:200 dilution; Millipore) and donkey goat-specific antibody conjugated to FITC (1:200 dilution; Santa Cruz) and sheep mouse specific antibodies conjugated to rhodamine (Millipore). For F-actin distribution, fixed colonies were stained with phalloidin conjugated with alexa-488 (Invitrogen) according manufacturer protocol.
In vitro differentiation
The in vitro differentiation potential of the two ESC lines CH19-1 and CH19v8 was examined in terms of their ability to form embryoid bodies in suspension culture and their ability to undergo directed differentiation to cells representative of all three germ layers. For embryoid body formation, colonies of porcine ES cells were cut onto pieces and cultured in suspension in DMEM/Ham F-12 or RPMI media (both from Invitrogen) supplemented with 2% FBS (Hyclone, Logan, UT, USA) and 55 μM 2-mercaptoethanol in ultralow attachment six-well plates (Corning, Corning, NY, USA) for 21–28 days. For directed differentiation assays, porcine ESCs were plated onto fresh feeder layer and cultured for 5–6 days in ESC medium and then differentiated using protocols described previously for ectoderm, mesoderm, and endoderm germ layers.
For ectoderm differentiation the ES medium was replaced with neurobasal medium containing 2% knock-out SR, 1 × N2/B27 supplements and 55 μM 2-mercaptoethanol (all from Invitrogen) and colonies were cultured for another 28–35 days. The morphology of resulted colonies were examined, colonies were fixed with 4% paraformaldehyde and investigated for expression of markers of neuroectodermal differentiation; glial fibrillary acidic protein (GFAP), and β-tubulin III. For GFAP detection colonies were stained with mouse anti-GFAP monoclonal antibodies (Millipore) and donkey antimouse IgG-alkaline phosphatase secondary antibodies (Rockland Immunochemicals, Gilbertsville, PA, USA). For β-tubulin III expression colonies were stained with mouse antitubulin III monoclonal antibodies and antimouse IgG-Alexa 488 secondary antibodies (Invitrogen).
To induce endoderm and mesoderm differentiation porcine ESC colonies were initially cultured for 5 days in porcine ES cell medium in the presence of 100 ng/mL human recombinant Activin A (R&D Systems). For mesoderm lineage differentiation porcine ES cell medium was replaced with Iscove medium containing 2% FBS (Hyclone), 1 μM dexamethasone, 50 μg/mL ascorbic acid, 450 μM thioglycerol (all from Sigma) 1 × ITS and 1 × of penicillin/streptomycin solutions (both from Invitrogen), and colonies were cultured for 28–35 days. At the end of culture period colonies were fixed with 4% PFA and stained with oil red O to detect adipose cells. For endoderm lineage differentiation ES culture medium were replaced with RPMI medium supplemented with 2% SR, 10 μM Nicotine amide, 10 nM exendin 4 (both from Sigma) and 50 ng/mL insulin growth factor II (IGF-II; Invitrogen), and colonies were cultured for another 28–35 days. After culture, colonies were fixed and expression of pancreatic and duodenal homeobox 1 protein (PDX-1), marker of precursors of pancreatic cells, were examined with rabbit anti-PDX1 polyclonal antibodies and goat antirabbit IgG-FITC secondary antibodies (both from Millipore).
Immunofluorescence analysis and confocal microscopy
Oct-4, Nanog, SSEA-1, PDX1 expression, and F-actin distribution were examined using immunofluorescence analysis and confocal microscopy. Porcine ES cell colonies and their derivatives were fixed in 4% paraformaldehyde (Sigma) in PBS (Sigma) for 5–20 min at room temperature. To facilitate antibody entry into cells, 0.3% saponin (Calbiochem, Switzerland) was included in all solutions except for SSEA-1. Cells were then washed twice in PBS for 10 min. Nonspecific antibody binding was blocked using PBS supplemented with 1% BSA (Invitrogen) and 3% normal donkey serum (Jackson Immuno Research Labs, West Grove, PA, USA) or FBS for 30 min at room temperature. Cells were then incubated overnight at 4°C with primary antibodies in the blocking solution. After washing, samples were incubated for 30 min with secondary antibodies at room temperature. After a final wash samples were mounted in Prolong Gold mounting medium with DAPI (Invitrogen) on glass coverslips. Imaging analysis was done using a confocal laser microscope SP5 (Leica, Richmond, IL, USA) using Z-stack sectioning with 0.4-μm sections.
Production of porcine chimaeric blastocysts and chimaeric animals
Colonies from the ESC line CH19-1 were treated for 2–3 min with TrypLE Express to produce single cells or small clumps of cells (four to six cells), which were washed twice in HEPES-buffered NCSU23 containing 10% FBS and then resuspended in 100-μL drops of the same medium. Twenty microlitre drops from these suspensions were then placed under mineral oil on 35-mm Petri dish lids. Day 5 in vitro-produced blastocysts with large blastocoel cavities were used for ES cell microinjection. For chimaeric blastocyst production single porcine ES cells were stained with 10 nM SYTO 64 red fluorescent nucleic acid stain (Invitrogen) and injected into the blastocoel of host blastocysts. On the following day incorporation of injected ES cells into ICM of host blastocysts were examined under fluorescent microscope TS 100 (Nikon, Japan).
To examine the pluripotency of this cell line further, cells at passage 3 were used for blastocyst injection to produce chimaeric pigs. Approximately 12 to 15 cells were injected into day 5 IVP blastocysts using an inverted Diaphot-TMD microscope (Nikon) and hand-made injection pipettes. Blastocysts were cultured overnight and then checked for reexpansion the next day. Estrus was synchronized in gilts that had previously cycled at least three times by feeding 20 mg per pig of altrenogest (Regumate, Hoescht) for 15–18 days and injecting 750 IU of hCG (Chorulon; Intervet, Bendigo, Australia) approximately 96 h after the last day of altrenogest treatment. Surgery was performed 5 or 6 days after the hCG injection. Between 32 and 56 injected blastocysts were transferred into one horn of the uterus near the uterotubal junction via midventral laparotomy performed under general anaesthesia induced with thiopentone sodium and maintained using halothane and oxygen.
Microsatellite analysis
Potential chimaeras were investigated using polymerase chain reaction (PCR)-based microsatellite analysis. Genomic DNA was extracted from semen from the boars CL1219 (white skin color; used to fertilize host blastocysts) and CH19 (black and white skin color; used to fertilize oocytes to produce embryos for ES cell isolation), tails of born piglets, and tail of randomly chosen Landrace piglet using a genomic DNA extraction kit (Qiagen, Chatsworth, CA, USA) according to the manufacturer's protocol. For each PCR, 25 ng of purified porcine genomic DNA was used. The PCR reaction mix had a total volume of 10 μL and consisted of 1 × PCR buffer, 1.5 mM MgCl2; 30 μM of each of dATP, dTTP, dGTP, and 15 μM dCTP (all from Geneworks, Australia) 0.1 μCi α33P-dCTP (S.A. 3600 Ci mM−1; Perkin-Elmer, Australia); 33 ng forward primer; 33 ng reverse primer; 0.5 units of Kappa Taq DNA polymerase (Geneworks). Microsatellite primers used were SW122 (forward primer: TTGTCTTTTTATTTTGCTTTTGG; reverse primer: CAAAAAAGGCAAAAGATTGACA) for the polymorphic loci on chromosome 6. PCR cycling conditions were as follows: 2 min at 98°C for initial denaturation, followed by 30 cycles of 30 sec at 94°C, 30 sec at 62°C, and 30 sec at 72°C. A final elongation step of 5 min at 72°C was included. The reaction products were resolved on a 6% nondenaturing PAGE. The gel was transferred to Whatman 3-mm paper and vacuum dried, and exposed to autoradiographic film (Kodak, Japan).
Results
Isolation and establishment of cell lines
Porcine ESC lines were considered to be established if the majority of cells survived following repeated passaging and cryopreservation and continued to grow without any changes in their morphology, karyotype, and marker expression. Two ES cell lines were established from in vivo-derived porcine embryos (10% efficiency) and four from in vitro-produced embryos (2.5% efficiency). All ES cell lines demonstrated relatively high rates of survival (>90%) after repeated passaging and vitrification. Survival postvitrification for the CH19v8 line is shown in Table 1.
ESC, embryonic stem cell.
ES cell lines were passaged up to 14 times, at the end of which they had not differentiated or begun to senesce. During this time they retained their characteristic morphology, namely, a polygonal shape, a relatively small (10–15 μm) diameter, a small cytoplasmic/nuclear ratio, a single nucleus with multiple nucleoli, multiple lipid inclusions in the cytoplasm (Fig. 1a), and karyotype. These cells had an F-actin pattern similar to that for mouse ES cells (Fléchon, 1997; Slager et al., 1992), namely, circular structures on the periphery of cells where these were in direct contact with each other (Fig. 1b). Cell lines continued to express Oct4 (Fig. 1c and d), Nanog (Fig.1e and f) and SSEA-1 (Fig. 1g and h) also following repeated passaging and cryopreservation.
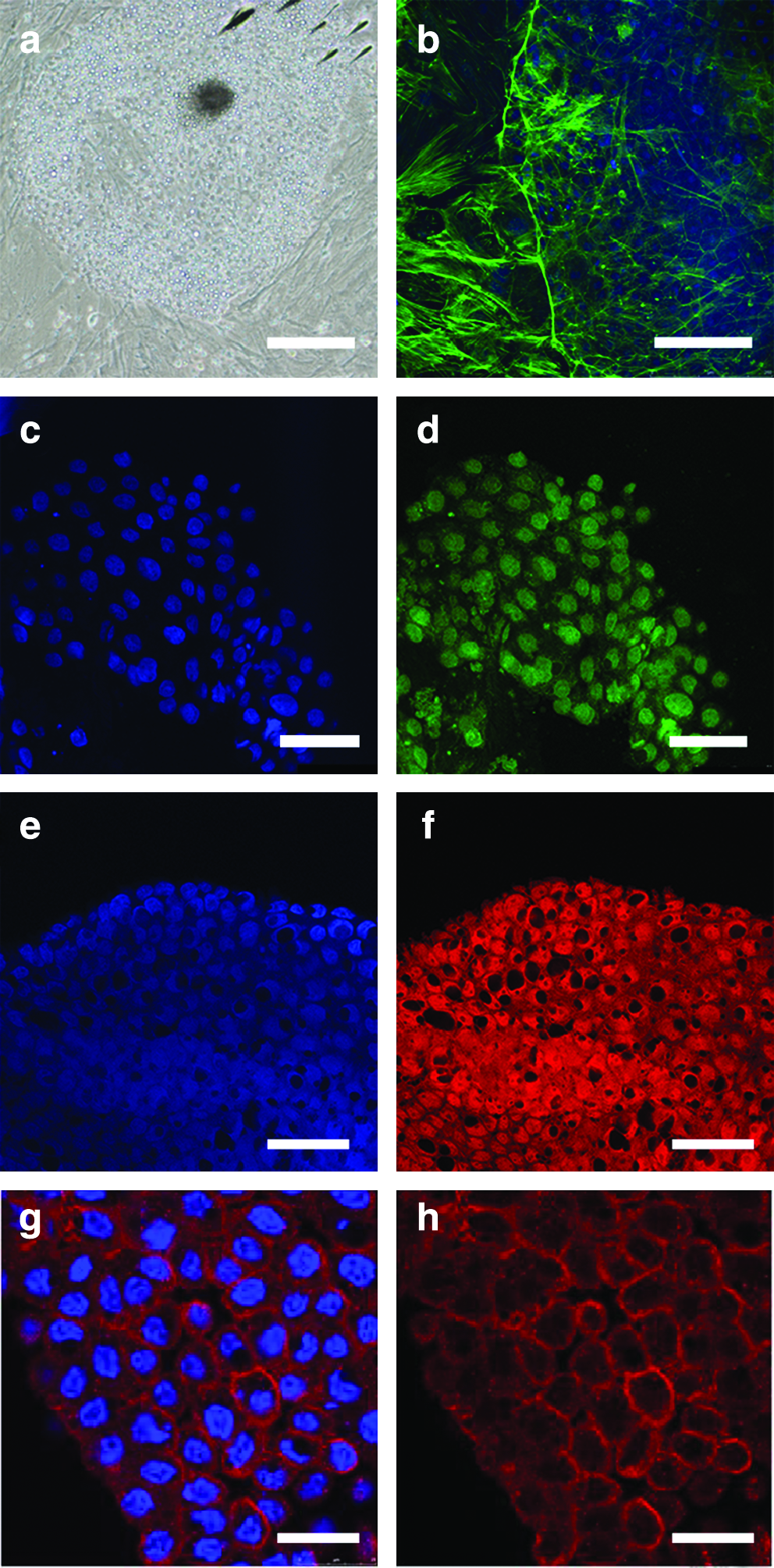
Morphology and pluripotent genes expression in porcine ESCs from line CH19-1. At passage 8 (
In vitro differentiation
ESCs formed embryoid bodies after 3 weeks in suspension culture (Fig. 2a). These had a similar morphology to that described for mice, and consisted of an inner clump of embryonal ectoderm cells surrounded by an outer layer of extraembryonic parietal endoderm cells separated by Reichert's membrane-like structures (Robertson, 1987).
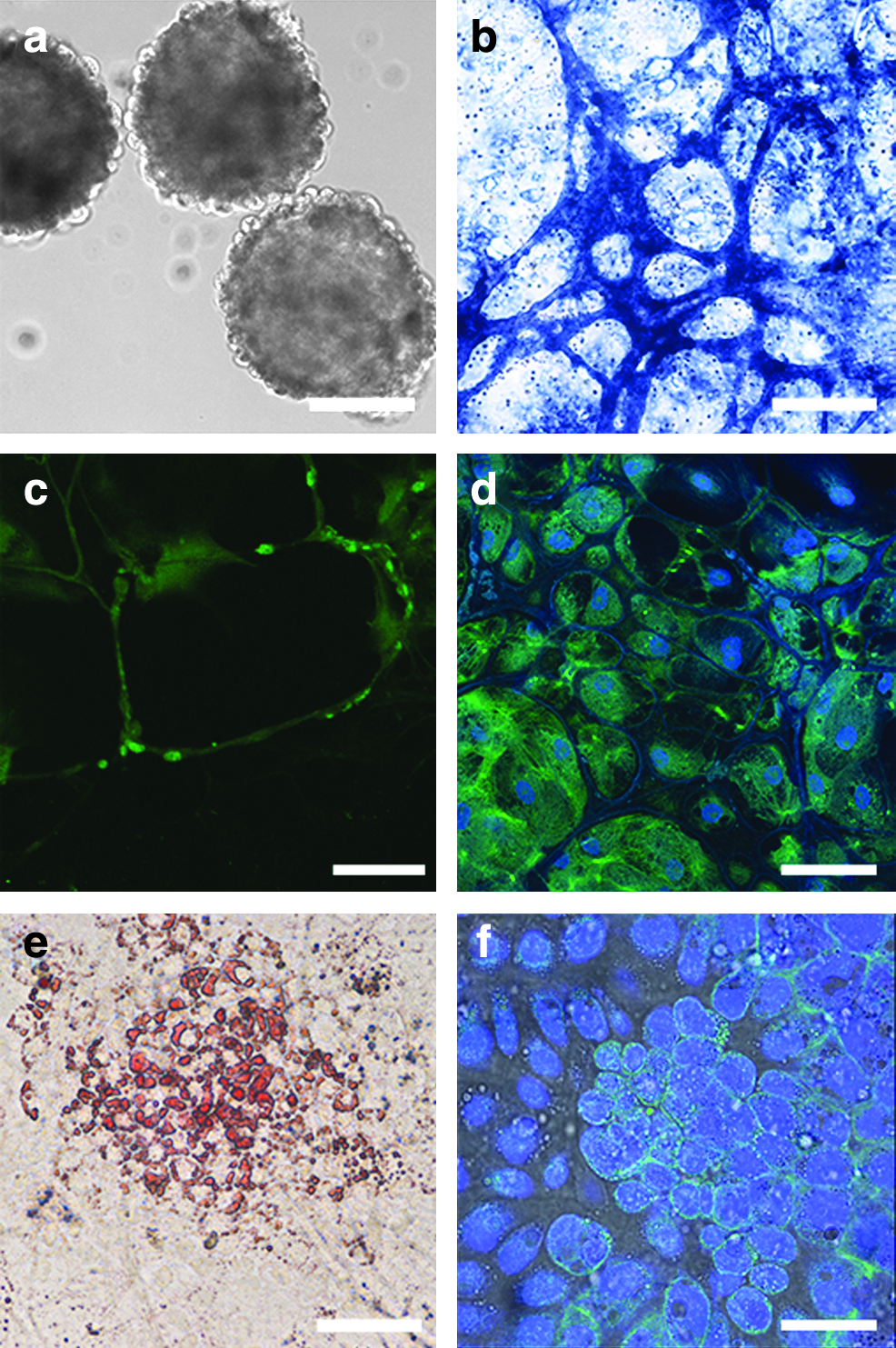
In vitro differentiation of porcine ESCs from line CH19v8. ESCsformed embryoid bodies in suspension culture (
Cells could be differentiated into cell types representative of all three germ layers using established protocols. Culture in Neurobasal medium for 3–4 weeks in the presence of inductors of neuronal differentiation resulted in the formation of multiple neuronal cell types such as glia-like cells (Fig. 2b), neuron-like cells (Fig. 2c), or retinal epithelium-like cells (Fig 2d) (Osakada et al., 2009). Culture of porcine ESCs in Iscove's medium in the presence of dexamethasone, ascorbic acid, and thyoglycerol for 3–4 weeks resulted in formation of muscle cells (not shown) and adipose cells (Fig. 2e). To differentiate ESC to endoderm lineages cells were initially cultured for 5–10 days in induction medium which was RPMI 1640 medium supplemented with high activin A concentration (100 ng/mL) and 2% SR. During this time, cells changed their morphology from ESCs to those characteristic for endoderm-like cells viz epithelial-like morphology with bright cell borders (data not shown) (Notarianni and Flechon, 2001). Subsequent culture in differentiation medium (RPMI 1640 medium with 2% SR, 10 μM Nicotine amide, 10 nM exendin 4, and 50 ng/mL IGF-II) resulted in the development of pdx-1-positive pancreatic islet precursor cells (Fig. 2f).
In vivo developmental potential
Following their injection into the blastocoele cavity, porcine ES cells localized and became incorporated into the ICM (Fig. 3a and b). Of the six embryo transfers undertaken, one recipient became pregnant and gave birth to a litter consisting of one male and three female piglets (born january 22, 2008). All four piglets were healthy at birth and had birth weights within the normal range. One female piglet had a black spot at the base of its tail indicative of skin color chimaerism (Fig. 3c). Microsatellite analysis of DNA extracted from the tail tissue from these piglets confirmed the contribution of the injected ES cells to the tail tissues of this piglet and a second female piglet (Fig. 3d).

In vivo developmental potential of porcine ESCs from line CH19-1. Cells were labeled with Cyto 64 red fluorescent DNA tracer and injected into day 5 in vitro produced blastocysts. Following injection blastocysts collapsed but recovered their shape following overnight culture (
Discussion
The present study was undertaken to determine the pluripotency of putative porcine ESCs lines isolated using a new method developed by us. Previous attempts to isolate porcine ESCs have been largely unsuccessful (reviewed in Vackova et al., 2007). The reasons for this are unclear. In preliminary experiments
Next we determined pluripotent gene expression for these lines. Both lines examined continued to express Oct 4, Nanog, and SSEA-1, following repeated passaging and cryopreservation. Although no definitive panel of pluripotent markers has been described for porcine ESCs (reviewed in Vackova et al., 2007), we have found that Oct 4 and Nanog expression can be used together to determine pluripotency, because, although all cells in porcine blastocysts express Oct 4, only a few cells in the inner cell mass (ICM) express Oct 4 and Nanog together (unpublished results), and in mice these cells are thought to be the cells that give rise to ESCs (Cavalery and Scholler, 2003). In the present study we also examined SSEA-1 expression. SSEA-1 has been shown to be expressed in freshly isolated day 6 to 7 porcine ICMs and in porcine ICMs cultured for 5 days (Wianny et al., 1997). In contrast, epiblasts isolated on day 11 loose SSEA-1 expression after 1–2 days of culture (Wianny et al., 1997). Expression of SSEA-1 has also been demonstrated in the ICM but not in the trophoblast of bovine blastocysts (Vassiliev et al., 2005) and in equine and bovine ES cells (Saito et al., 2004) and in porcine primordial germ cells used to produce chimaeras (Mueller et al., 1999). Together, these finding suggest that Oct 4, Nanog, and SSEA-1 together can be used as evidence of pluripotency.
In vitro we were able to demonstrate that our ES cells could form embryoid bodies in suspension culture similar to those described for mice. Cells from these lines also had a similar F-actin distribution to that of mouse ESCs. Furthermore, we were able to differentiate these into lineages of all three germ layers. In particular, we demonstrated that our cells could be differentiated into several neuroectodermal cell types, and muscle and adipose cell types indicative of mesoderm differentiation. Third, we were able to differentiate these cells to cells that expressed the pancreatic precursor gene, Pdx-1, indicative of definitive endoderm. Although spontaneous differentiation into a variety of different cell types has been reported previously for putative porcine ES cell lines (Keefer et al., 2007), this is the first time to our knowledge that directed differentiation into all three germ layers has been demonstrated. Further development in this area may allow the pig to be used as a large animal model for human ES cells in particular differentiation studies aimed at developing cell-based therapies.
In vivo we demonstrated that these cells localize and become incorporated in the ICM when injected into host blastocysts. In contrast, Chen et al. (1999) reported that their cells become incorporated into both the ICM and trophectoderm layer following their injection. The reason for this difference between their study and our is unclear. We also demonstrated that these cells can be used to produce chimeric pigs. To date, the production of chimaeric pigs from cultured ESC lines is limited to one peer reviewed report (Chen et al., 1999). In the present study, we injected cells at passage three from the IVP embryo cell line into IVP blastocysts and transferred these to recipient animals to produce chimaeric animals. This is the first time to our knowledge that ESCs IVP produced blastocysts have been used to produce chimaeric pigs. Of the six embryo transfers undertaken, one animal became pregnant and gave birth to a litter of three female piglets and one male piglet, all of which appeared healthy at birth and were within the normal birthweight range. One of the female pigs had a black spot at the base of its tail indicative of skin color chimaerism as a result of using Hampshire semen to produce our ESC lines. This breed has a predominantly black skin color with a white band around the middle covering the front legs. This was an unexpected finding because the gene(s) for white skin color are dominant in the pig (Legault, 1998). Rather, we chose to use the Hampshire breed in our study because this is not used commercially in Australia, and as such, is unlikely to be a confounding factor in analyzing chimaerism using microsattelite analysis. Of the four piglets born, the female that had the black spot and a second female piglet were shown to be chimaeric following microsattelite analysis of DNA extracted from a sample of their tail tissue.The extent of the chimaerism in these animals is difficult to determine not only because of white skin being a dominant characteristic but also because extensive microsattelite analysis can only be done by taking representative tissues postmortem. This analysis was essentially a paternity test, and ideally, further studies need to include a second marker of chimaerism such as the presence of an unique DNA sequence obtained, for example, by using a transfected cell line, to confirm this. In the present study we chose to inject cells at passage 3 because this was the stage these cells had grown to at the time the study commenced. Ideally, we also we need to make chimaeras from later passages to demonstrate the stability of these lines. As such, we plan to undertake further studies with these and other cell lines. This will involve the production of additional chimaeric animals using cells from later passages that have been transfected with, for example, a GFP construct to determine their suitability for gene targeting as well as providing an independent assessment of chimaerism.
In conclusion, we have isolated several putative ESC lines from in vivo- derived as well as in vitro-produced embryos using a new method described previously. These cells maintained a morphology characteristic of ESCs following repeated passaging and vitrification, and continued to express Oct 4, Nanong, and SSEA1. These also formed embryoid bodies, and could be differentiated in vitro into various lineages representative of all three germ layers. When injected into blastocysts, cells from early passages localized in the ICM and could be used to produce chimaeric pigs. Further studies are required to fully characterize these cells. In particular, we need to examine the stability of these and other cell lines by making chimaeric pigs using cells from later passages. From a research perspective, we also need to demonstrate the utility of these cells for gene targeting. Finally, we need to demonstrate germline transmission before we can we can state that we have isolated pluripotent ESCs as per the original definition developed for mouse ESCs.
Footnotes
Acknowledgments
This research was supported by a Juvenile Diabetes Research Foundation Project Grant 1-2005-192.
Author Disclosure Statement
The authors indicate no potential conflicts of interest.