
Abstract
Abstract
To understand the epigenetic alterations associated with assisted reproduction technology (ART) and the reprogramming of gene expression that follows somatic cell nuclear transfer (SCNT), we screened a panel of 41 amplicons representing 25 developmentally important genes on 15 different chromosomes (a total of 1079 CpG sites). Methylation analysis was performed on DNA from pools of 80 blastocysts representing three classes of embryos. This revealed a subset of amplicons that distinguish between embryos developing in vivo, produced in vitro, or reconstructed by SCNT. Following SCNT, we observed massive epigenetic reprogramming evidenced by reduced levels of methylation in the resultant embryos. Analysis of data from the 28 most informative amplicons (hotspot loci), representing more than 523 individual CpG sites, we discovered subsets of amplicons with methylation patterns that were unique to each class of embryo and may indicate metastable epialleles. Analysis of eight genes with respect to mRNA expression did not reveal a direct correlation with DNA methylation levels. In conclusion, this approach revealed a subset of amplicons that can be used to evaluate blastocyst quality and reprogramming following SCNT, and can also be employed for the localization of the epigenetic control regions within individual genes and for more general studies of stem cell differentiation.
Introduction
Changes in DNA methylation are observed in the normal restructuring process that follows fertilization. Paternal (sperm) DNA is actively demethylated, while female (oocyte) DNA undergoes passive demethylation as shown in bovine, murine, porcine, rat, and human zygotes (Beaujean et al., 2004; Dean et al., 2001; Mayer et al., 2000; Oswald et al., 2000; Reik et al., 2001; Santos et al., 2002; Xu et al., 2005) before a new wave of CpG methylation affects the entire embryonic genome, the timing of which is species specific (Santos et al., 2002). This switch from genome-wide demethylation to remethylation is correlated with the major onset of transcriptional activity (Niemann and Wrenzycki, 2000). Critical stages of early development such as the timing of the first cell division, activation of the embryonic genome, compaction, blastocyst formation, expansion, and hatching depend on well-orchestrated changes in gene expression which, in part, reflects changes in DNA methylation.
Bovine preimplantation development is important not only because Bos taurus is an agriculturally important species but also because bovine early development is increasingly seen as a model for human development (Niemann and Wrenzycki, 2000; Wrenzycki et al., 2005). Both are uniparous, both have a similar rate of preimplantation development that is slower than murine embryos, and both have a gestation period of 9 months compared to 21 days for the mouse. In vitro production methods for bovine embryos are well advanced due to the availability of large numbers of ovaries from local slaughterhouses. Bovine blastocysts developing in vivo are readily obtained nonsurgically by uterine flushing, and one result of this is a longstanding international trade in bovine embryos (http://www.iets.org/). Somatic cell nuclear transfer (SCNT) and assisted reproductive technologies (ARTs) including in vitro fertilization, intracellular sperm injection (ICSI), cryopreservation, and in vitro embryo culture are associated with changes in DNA methylation and with fetal and placental abnormalities and an increased frequency of rare epigenetic diseases such as Beckwith-Wiedemann syndrome in humans (Dean et al., 2001; DeRycke et al., 2002; Farin et al., 2006; Kang et al., 2001a, 2001b; Niemann et al., 2002; Powell, 2003). Although the relationship between DNA methylation and cancer formation has been extensively studied (Jones and Baylin, 2002; Weber et al., 2005), the methylation profiles of developmentally important genes in preimplantation embryos and changes caused by in vitro manipulation are, to a great extent, unknown.
Here, we screened 41 amplicons (1079 CpG sites) in 25 genes with known functions in embryonic development and the stem cell state, using quantitative bisulfite sequencing, and found a subset of 28 informative amplicons (523 CpG sites) located in 21 genes, which demonstrate specific differences between in vivo bovine embryos (flushed from the uterine horns of donor animals), in vitro produced (IVP) embryos, and embryos produced by SCNT. To evaluate the relationship between DNA methylation at these sites and mRNA transcription, we determined the mRNA levels for a subset of eight of these genes. Results of this study demonstrate for the first time the broad demethylation of the somatic DNA after SCNT and provide clues for improved diagnostics of bovine blastocysts derived from ARTs and in-depth understanding of basic epigenetic events during early preimplantation development.
Materials and Methods
In vitro produced (IVP) bovine embryos
Bovine embryos were produced as described previously (Eckert and Niemann, 1995; Wrenzycki et al. 2001a). Briefly, viable cumulus–oocyte complexes (COCs) derived from abattoir ovaries from Holstein Friesian cows were matured in vitro in groups of 15–20 in 100 μL tissue culture medium (TCM-199) supplemented with 10 IU pregnant mare serum gonadotropin (PMSG) and 5 IU human chorionic gonadotropin (hCG) (Suigonan®, Intervet, Tönisvorst, Germany) and 0.1% bovine serum albumen—fatty acid free (BSA-FAF) (Sigma, A7030, St. Louis, MO, USA) under silicone oil in a humidified atmosphere composed of 5% CO2 in air at 39°C for 24 h. Matured COCs were fertilized in vitro using 1 × 106 sperm/mL frozen/thawed semen from one bull with proven fertility in in vitro fertilization (IVF) over a 19-h period of coincubation under the same temperature and gas conditions used for in vitro maturation. Presumptive zygotes were cultured to the blastocyst stage (day 8 after insemination) in synthetic oviduct fluid (SOF) medium supplemented with BSA-FAF, in a mixture of 5% O2, 90% N2, and 5% CO2 (Air Products, Hattingen, Germany) in modular incubator chambers (ICN Biomedicals, Inc., No. 615300, Aurora, Ohio, USA). For methylation analysis, bovine blastocysts were collected in phosphate buffered saline (PBS) + 0.1% polyvinyl alcohol (PVA) in groups of 5–10 and were stored frozen at −80°C in 0.6 mL siliconized tubes. IVP blastocysts were collected at four time points over a period of 1 year.
In vivo production of bovine embryos
In vivo developing bovine embryos were flushed from the uterine horns of superovulated donor animals (Holstein Friesian cows) that had been artificially inseminated with semen from the same bull used for IVF-based embryo production. Blastocysts were nonsurgically recovered from the donors at days 7 and 8 following artificial insemination (Bungartz and Niemann, 1994). For methylation analysis, bovine blastocysts were collected in PBS + 0.1% PVA in groups of 5–10 and were stored frozen at −80°C in 0.6-mL siliconized tubes. The in vivo blastocysts were collected from 14 different donor animals over a period of 6 months (January to June) to avoid seasonal effects.
Production of somatic cell nuclear transfer-derived embryos
SCNT blastocysts were produced as described previously (Schaetzlein et al., 2004) Briefly, bovine fibroblast cultures were established from ear skin biopsies and cultured in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% fetal calf serum (FCS). The fibroblasts employed for nuclear transfer in these experiments were from passages 4–6. Prior to use, the fibroblasts were cultured in the above medium for 3 days followed by the induction of quiescence (presumptive G0) by serum starvation for 2–3 days (0.5% FCS). For enucleation and nuclear transfer, cytochalasin B (7.5 μg/mL) was added to TCM-air [N-2-Hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-buffered tissue culture medium]. For fusion, 0.285 M mannitol containing 0.1 mM MgSO4 and 0.05% BSA was used. In vitro matured oocytes were enucleated by aspirating the first polar body and the metaphase II plate. The donor cells were pelleted and resuspended in TCM and remained in this medium until insertion. A single cell was sucked into a 30μm (outer diameter) pipette and was then carefully transferred into the perivitelline space of the recipient oocyte. Cell fusion was induced with 1–2 DC pulses of ∼1 kV/cm for 30 μsec each in an Eppendorf fusion machine (Hamburg, Germany). At approximately 27 h after the onset of maturation, the reconstructed embryos were chemically activated by incubation in 5 μM ionomycin (Sigma) in TCM-199 for 5 min followed by a 3–4 h incubation in 2 mM N-6 dimethylaminopurine (6-DMAP) (Sigma) in TCM-199 at 37°C. After three washes, the embryos were cultured, as described above, for 7–8 days until they had reached the blastocyst stage. For methylation analysis, the blastocysts were collected in PBS + 0.1% PVA in 0.6-mL siliconized cups. Groups of 5–10 embryos were stored frozen at −80°C. The SCNT blastocyst collection involved four sessions for the production of male blastocysts and four sessions for the production of female embryos, spread over a period of 7 months.
Preparation of adult fibroblasts for methylation analysis
Primary fibroblasts from the same (male and female) cell lines used as nucleus donors for SCNT were thawed from passages 4–6 and cultured to confluency using DMEM supplemented with 10% FCS at 37°C and 5% CO2 in air. Cells were recovered by incubation with 0.05% Trypsin/0.02% ethylenediaminetetraacetic acid (EDTA) in PBS and centrifugation at 160 × g. The cell pellet was then frozen at −80°C for subsequent DNA isolation.
Genomic DNA preparation
Genomic DNA was prepared from embryos, peripheral blood mononuclear cells (PBMCs), and fibroblasts using the DNeasy tissue kit (Qiagen, Hilden, Germany) following the protocol for cultured animal cells. DNA was extracted from pools of 80 blastocysts from the three groups (in vivo, IVP, and SCNT), each yielding approximately 70 ng DNA. The SCNT (cloned) blastocysts and the IVP blastocysts were produced over a total of four to six sessions and pooled to minimize variance. All blastocysts were at the same stage of development showing moderate expansion of the blastocoel. To exclude sex-specific methylation patterns, the pool of SCNT blastocysts was composed of equal numbers of male (n = 40) and female (n = 40) embryos. The in vivo embryos were pooled from a total of 20 separate flushing sessions. The DNeasy eluate was precipitated with 0.1 volumes of sodium acetate and 2.2 volumes of ethanol, and genomic DNA was resuspended in 100 μL TE buffer.
Sodium bisulfite conversion, PCR amplification and sequence analysis
Sodium bisulfite treatment of genomic DNA was performed according to Olek et al. (1996), with minor modifications. The method has been successfully employed for methylation studies of somatic cells and methylation profiling of human chromosomes and yields reliable and highly reproducible data (Baron et al., 2006; Eckhardt et al., 2006). Purified genomic DNA was treated with sodium bisulfite, resulting in the conversion of nonmethylated cytosine to uracil. In subsequent PCR amplification, uracil was replicated as thymine. Methylated cytosines were protected from sodium bisulfite conversion and remained as cytosines. Thus, detection of a “C” in the following sequencing reaction indicated methylation at this (CpG−) site, whereas detection of a “T” indicated that there had been no methylation at that cytosine. A sample of 50 to 70 ng of purified genomic DNA was dissolved in 40 μL water followed by the addition of 85 μL of “bisulfite mix” and 15 μL of “DNA Protect Buffer.” For bisulfite conversion, three denaturation cycles were used. The first denaturation cycle was 99°C for 5 min followed by an incubation at 60°C for 25 min; the second denaturation step was 99°C for 5 min followed by an incubation at 60°C for 85 min; and the third denaturation step was 99°C for 5 min followed by incubation at 60°C for 175 min. Bisulfite converted DNA was purified using EpiTect (Qiagen) spin columns according to the suppliers recommendations. PCRs were performed on MJ Research thermocyclers (Waltham, MA, USA) in a final volume of 25 μL containing 1 × PCR Buffer, 1 U Taq DNA polymerase (Qiagen), 200 μM dNTPs, 12.5 pmol each of forward and reverse primers, and 1.5–3 μL of bisulfite-treated genomic DNA. The amplification conditions were 95°C for 15 min and 40 cycles of 95°C for 1 min, 55°C for 45 sec and 72°C for 1 min and a final extension step of 10 min at 72°C. PCR products were purified using ExoSAP-IT (USB Corp., Cleveland, OH, USA), and sequenced applying PCR primers and ABI Big Dye Terminator v1.1 cycle sequencing chemistry (Applied Biosystems, Foster City, CA, USA) followed by capillary electrophoresis on an ABI 3100 genetic analyzer. ABI-files were analyzed using ESME, a program that normalizes sequence traces, corrects for incomplete bisulfite conversion, and allows for quantification of methylation levels at individual CpG positions (Lewin et al., 2004). (See example in Supplementary data.) Colored “heat maps” (in which yellow represents no methylation and blue indicates full methylation) summarize the methylation status of all amplicons and all samples as characteristic “barcodes.” Blue represents full methylation and yellow represents unmethylated. Analysis of individual amplicons is shown in more traditional format in low (<25%) methylation is represented as an unfilled circle; intermediate methylation (25–75%) is represented as a partially filled circle and high methylation (>75%) is represented as a filled circle.
Genes and primer sequences
Amplicons were designed for a panel of 25 genes known to play a critical role in preimplantation development. Two amplicons each covering a different CpG rich region were designed for each gene. These genes cover a wide range of physiological functions during embryonic development including: intercellular communication, trophoblast function, DNA methylation, epigenetic regulation, imprinting, maternal expression, translation, glucose transport, growth factor signalling, maintenance of pluripotency, differentiation and telomere regulation, thus providing significant insight into the physiological status of the embryos derived from different production methods. A detailed description of the functions of all genes analyzed in this study is found in Supplementary Table 1 “Genes Selected for Methylation Analysis.” The same primers were used for both PCR of bisulfite-treated genomic DNA and for the sequencing reactions. Strand specificity and orientation: Primer pairs “p” and “o” produced amplicons based the +1 strand, whereas primers “r” and “q” produced amplicons originating from the −1 strand. Thus, primer designations “p” and “r” indicate forward and primer designations “o” and “q” denote reverse orientation. For bisulfite-specific primer sequences see Supplementary Table 3.
Isolation of mRNA
Poly(A)+ RNA was isolated using a Dynabeads® mRNA Direct Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instruction with some modifications. Briefly, samples consisting of four to eight blastocysts (developing in vivo, produced in vitro, or produced by somatic cell nuclear transfer) were placed in 40 μL of lysis-binding buffer [100 mM Tris–HCl. pH 8.0, 500 mM LiCl, 10 mM EDTA, 1% lithium dodecyl sulfate (LiDS), 5 mM dithiothreitol (DTT)] and incubated at room temperature for 10 min. Prewashed Dynabeads® Oligo d(T)25 (5 μL) were pipetted into the lysate. After incubation for 5 min at room temperature on a shaker to allow binding the poly(A)+ RNA to the beads, the beads and mRNA were separated using a Dynal MPC-E-1 magnetic separator, the remaining lysate containing DNA was stored at −20°C. These DNA containing lysates were subsequently combined into pools representing 80 blastocysts prior to DNA isolation for methylation analysis as described above.
The beads with the poly(A)+ RNA were briefly washed once in washing buffer A (10 mM Tris-HCL. pH 8.0, 0.15 mM LiCl, 1 mM EDTA, 0.1% LiDS) and twice with washing buffer B (10 mM Tris-HCl. pH 8.0, 0.15 mM LiCl, 1 mM EDTA) by resuspension followed by magnetic separation. The poly(A)+ RNA was eluted from the beads by incubation in 4 μL of sterile water at 65°C for 3 min and stored at −80°C.
Reverse transcription (RT)
RT was performed in a reaction mixture consisting of 2 μL of 10 × RT buffer (Invitrogen), 2 μL of 50 mM MgCl2 (Invitrogen), 2 μL of 10 mM dNTP solution (Amersham Biosciences, Piscataway, NJ, USA), 1 μL (20 Units) of RNAsin® (Applied Biosystems), 1 μL (50 Units) of murine leukemia virus (MuLV) reverse transcriptase (Applied Biosystems), 1 μL of hexamer primers (50 μM) (Applied Biosystems), the entire 4 μL mRNA sample, 0.5 pg/blastocyst of rabbit globin mRNA to control for RT efficiency, and sufficient water to bring the volume to 20 μL.
These samples were incubated at 25°C for 10 min for primer annealing and then incubated at 42°C for 1 h. Finally, the samples were heated to 95°C for 5 min. The cDNA was diluted to a concentration of 0.1 blastocyst equivalent/μL, 2μL of this RT reaction product were used for real-time PCR amplification.
Real-Time PCR
Real-Time PCR was performed in 96-well Optical Reaction Plates (Applied Biosystems). The 20 μL PCR reaction mixture in each well included 10 μL of 2 × Power SYBR_Green PCR Master Mix (Applied Biosystems), 0.8 μL each of the forward and reverse primers (5 μ M), 2 μL of cDNA (0.2 blastocyst equivalents), and 6.4 μL dH2O to bring the reaction volume to 20 μ L.
The PCR reaction was carried out in an ABI 7500 Fast Real-Time System (Applied Biosystems) using the following program, 10 min at 95°C for nucleic acid denaturation and activation of the Taq Polymerase, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min and, finally, a slow heating cycle to obtain a dissociation curve for the products.
In each run, a cDNA dilution standard prepared from pooled blastocysts was included to give a standard curve for each gene. These standard curves were used for calculation of the relative concentration of each target gene normalised to the signal from the globin mRNA that had been included introduced into the mRNA samples as a control. Quantification was performed with the Sequence Detection Software 1.3.1 by fitting the threshold cycle (Ct) values to the standard curves. Supplementary Table 4 shows the primer sequences used for the Realtime PCR based expression analysis.
Results
The quantitative bisulfite sequencing method used in this study has been shown to be highly reproducible for the analysis of methylation differences in a variety of cell types including chondrocytes and T cells and in the analysis of various types of tumours (Floess et al., 2007; Rapko et al., 2007). Here, we used pools of 80 blastocysts in each of the three groups to provide robust results. A total of 25 genes were examined by the quantitative bisulfite sequencing of 41 amplicons. The results are summarized in Figure 1 as a barcode where heavy methylation is indicated by blue, medium methylation is indicated by green, and light methylation is indicated by yellow. Column A shows the dramatic difference in methylation between differentiated cells (PBMSs) and embryos developing in vivo. Column B demonstrates the reprogramming that takes place after transferring fibroblasts into enucleated oocytes. Column C shows the differences in methylation distinguishing the three classes of embryo. Gene-specific methylation patterns showing the methylation level at each CpG site in the 41 amplicons are presented in Supplementary Figure 1.
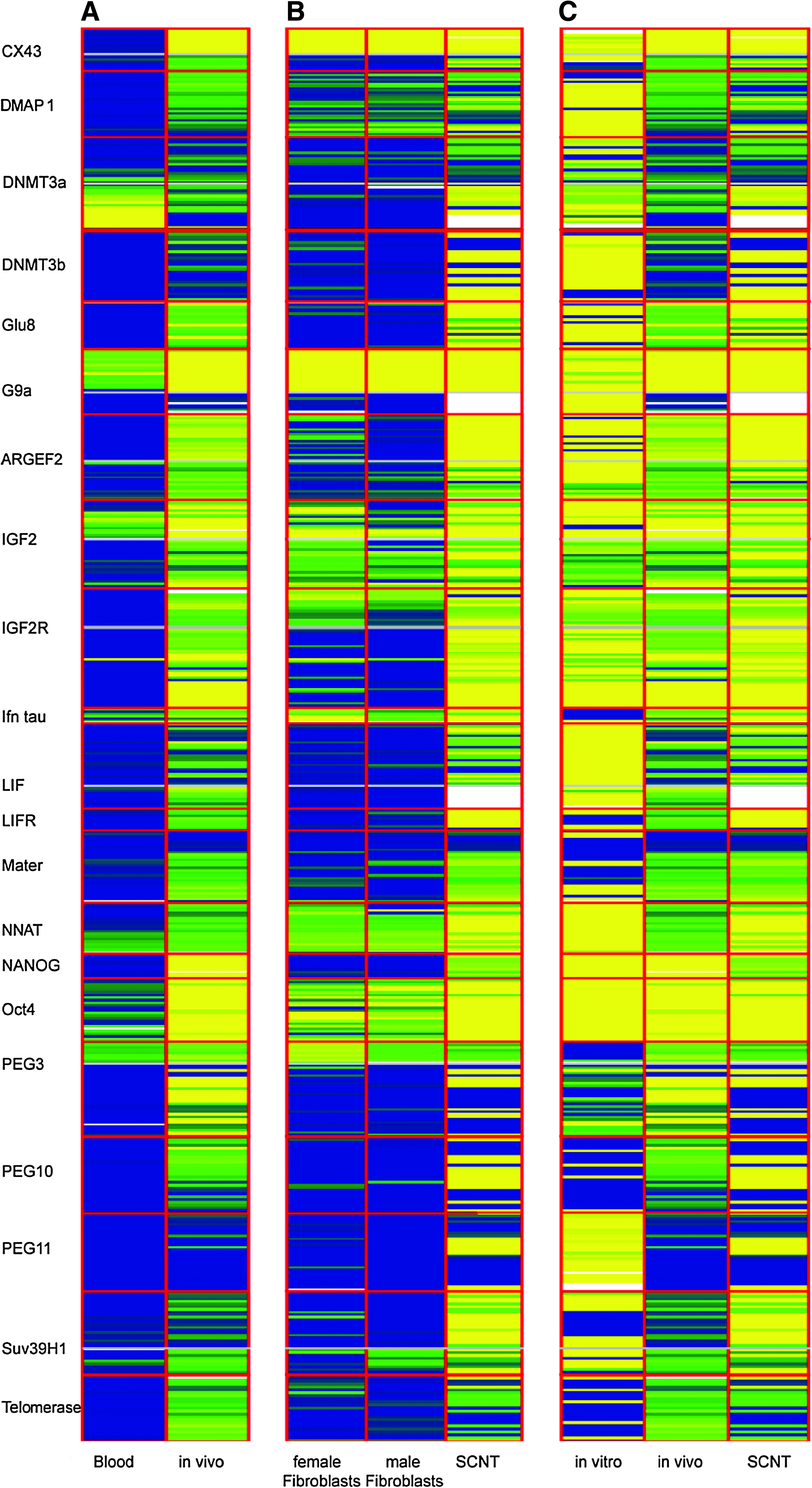
Differences in methylation for 21 genes that play important roles in early mammalian development. DNA was derived from peripheral blood mononuclear cells (PBMCs), a female and a male line of primary bovine fibroblasts, bovine embryos developing in vivo, in vitro produced (IVP) embryos, and embryos produced by somatic cell nuclear transfer (SCNT). Analyzed genes are separated by red lines with each row representing the methylation status of a single CpG. When genes are represented by two amplicons, these are separated by a gray line. Methylation of single CpGs is visualized by a color code: yellow (low methylation), green (medium methylation), and blue (high methylation), white: no CpG information. Differentiated somatic cells are more heavily methylated (blue) while the embryonic samples are less methylated (yellow). Columns A shows a comparison of PBMC (blood) and in vivo embryos. Column B shows the comparison of SCNT blastocysts with the fibroblasts from which they were produced. Column C shows the comparison of the three types of embryos.
The 41 amplicons in this study could be classified into five groups, typical examples of which are shown in Figure 2. The “Nanog” group of amplicons would be useful for studies of the reprogramming of differentiated cells to an embryo-like phenotype but did not distinguish between the three types of embryo. This group included ARGEF2 (both amplicons), GLU8 (2nd amplicon), IGF2R, NANOG, OCT4 (first amplicon), PEG3 (second amplicon), and TERT (second amplicon). The “NNAT” group of amplicons, which included DNMT3B, NNAT, PEG11, and SUV39H1 (both amplicons), distinguished the two in vitro handled blastocyst samples (IVP, SCNT) from the other samples. The “IFN tau” group of amplicons, which distinguished IVP embryos, included DMAP1 (second amplicon), IFN tau, LIF (first amplicon), PEG11, and SUV39H1 (both amplicons). The “LIFR” group of amplicons [ARGEF2 (first amplicon), DNMT3B, GLU8 (first amplicon), LIFR, NANOG, PEG11, and SUV39H1 (both amplicons)] had a methylation pattern that distinguished SCNT produced embryos from other groups. There were 11 amplicons in the “GLU3” group that showed little or no methylation and were thus not very useful for discrimination between the three types of embryos. This group included: CX43 (first amplicon), DMAP1 (first amplicon), EIF2C1, G9A (first amplicon), GLU3 (both amplicons), MASH2 (both amplicons), OCT4 (both amplicons), TERT (first amplicon), and ZAR1 (both amplicons).
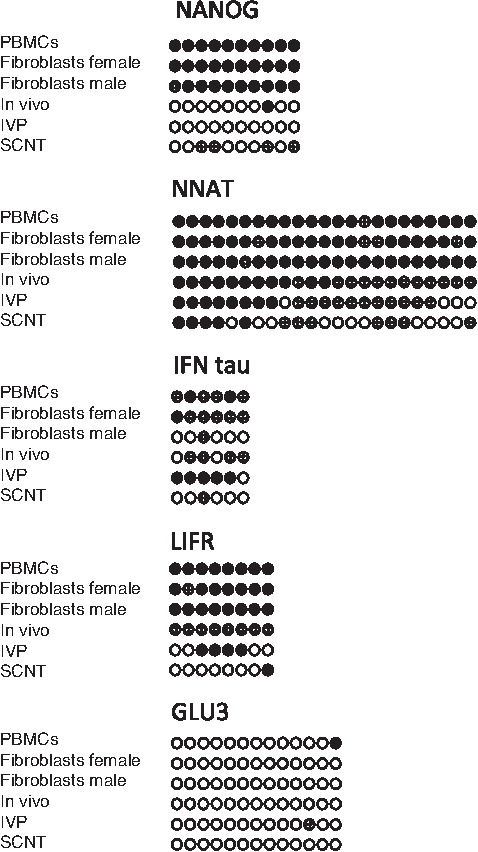
Examples of differential methylation. This is a subset of the patterns included in Supplemenary Figure 1. Nanog is representative of the group of eight amplicons which distinguish somatic from embryonic cells. NNAT is an example from the group of five amplicons characteristic of embryos produced under experimental conditions, that is, IVP and SCNT blastocysts. IFN tau is typical for the six amplicons specific to IVP embryos. LIFR is typical of the eight amplicons that distinguish SCNT blastocysts. GLU3 is an example of the 13 amplicons that showed consistently low methylation and were thus noninformative for embryo evaluation.
The list of the genes analyzed giving their GO functions, their ENSEMBL IDs, their chromosomal location, etc., is presented in Supplementary Table 1. The amplicon length, orientation, localization of the first CpG, total number of CpGs and full BLAST-compatible sequences are listed in Supplementary Table 2. Supplementary Table 3 shows the sequences of all the primers used in this study: these primer sequences are not suitable for a BLAST search in a bovine genome database because they are modified to function after bisulfite treatment. We used 41 out of the 42 pre-selected amplicons. Amplicon no. 27 (MATER) did not yield reliable results.
Real-time RT-PCR data for a selected panel of eight genes is shown in Figure 3. All values were normalized relative to the level found in in vivo embryos that showed the lowest levels of expression in all cases. With the exception of the amplicon in PEG3, there was correlation between methylation levels and expression levels of the respective genes.
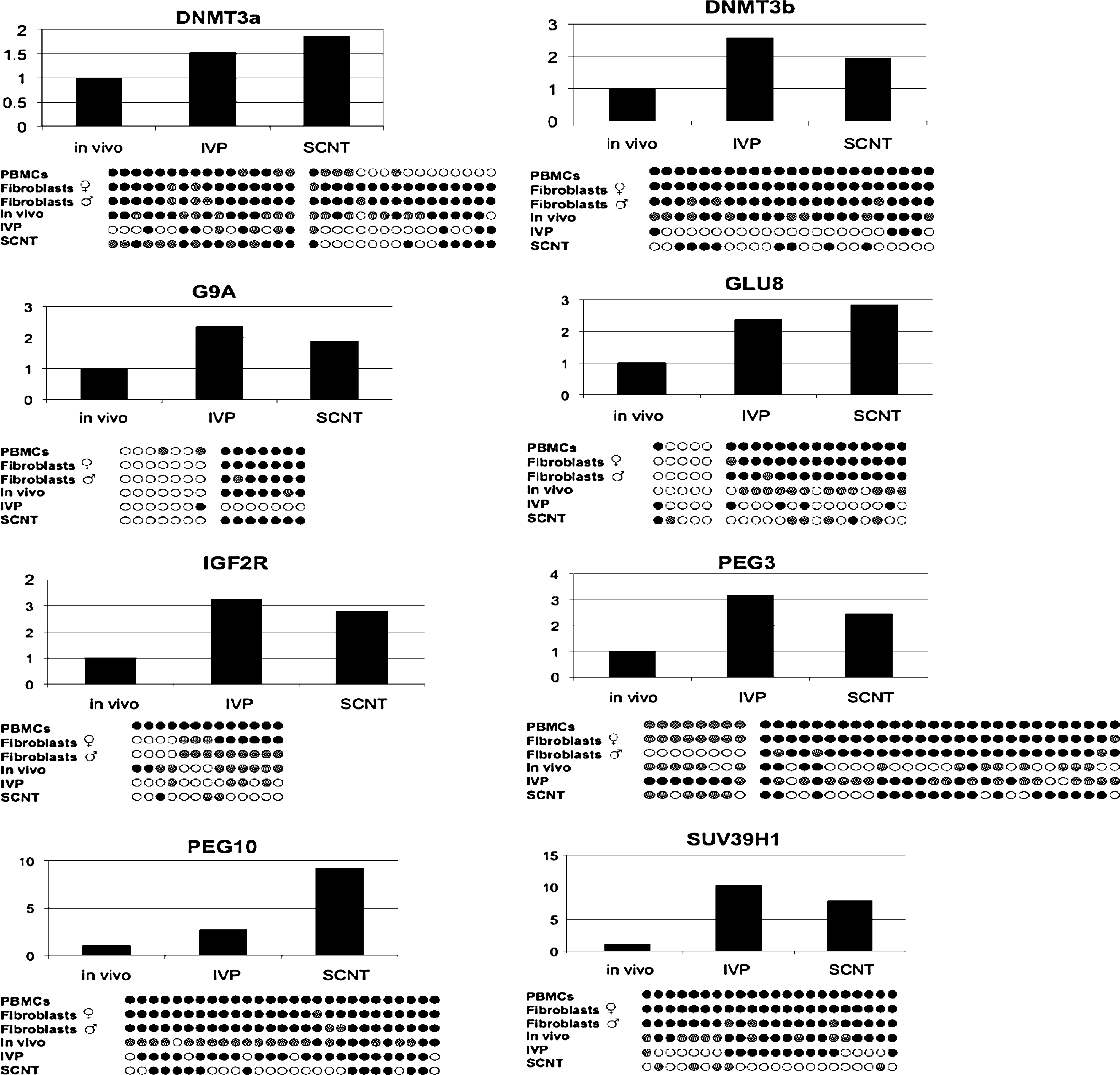
The bars represent gene expression. For the in vivo embryos, we used four pools, each containing four to eight embryos. For the in vitro embryos we used five pools, each containing five embryos. For the SCNT embryos, we used four pools, each containing four to eight embryos. Four pools of male and four pools of female SCNT embryos were analyzed separately for gene expression. The bar for SCNT embryos represents the mathematical mean of male and female embryos for comparison with methylation results for the SCNT group. Expression levels were normalized by setting the level for in vivo embryos to 1.0 and making all other levels in relation to this standard. The DNA methylation pattern for each gene is shown below each expression chart.
Discussion
The goal of this work was to discover informative amplicons that (1) distinguish in vivo developed embryos from differentiated cells, (2) demonstrate epigenetic reprogramming related to SCNT, and (3) differentiate between different types of embryo. We screened a panel of CpG-rich DNA regions in genes critical for embryonic development to assess DNA methylation in bovine blastocysts derived from three different systems, in vivo, in vitro, or somatic cloning. The methylation patterns in 28 of 41 amplicons discriminated between differentiated somatic cells and embryonic cells. This subset of informative methylation patterns, each representing the population mean of 80 blastocysts, can also distinguish between the three classes of embryos. The quality of embryos exposed to in vitro culture can be judged by comparison to the pattern obtained from embryos developing in vivo. This provides an important tool for the improvement of in vitro culture conditions leading ultimately to a reduction of the developmental abnormalities associated with ART. We demonstrate for the first time that heavily methylated fibroblast donor cells are broadly demethylated following nuclear transfer and are pushed toward the methylation profile typical of embryos developing in vivo.
DNA methylation is possibly the best studied epigenetic modification. Environmental factors have been shown to alter gene expression through chromatin changes in: (1) the promoter regions of some housekeeping genes; (2) the transposable elements adjacent to genes with metastable epialleles; and (3) the regulatory elements of imprinted genes (Jirtle and Skinner, 2007). Metastable epialleles are defined as loci that are particularly sensitive to variable, reversible, and often cyclic epigenetic modification to produce different phenotypes from genetically identical cells. At the time of writing, only a few epigenetic hot spots containing genes with metastable epialleles have been identified. The most prominent example is the mouse Avy (viable yellow agouti) gene in which allelic expression is correlated with the epigenetic status of a transposon located near the promoter (Duhl et al., 1994; Waterland and Jirtle, 2003). We propose that some of the genes identified in the present study, which show different DNA methylation profiles in the three types of bovine embryo, have metastable epialleles. This has to be substantiated in future studies. The number of genes with metastable epialleles is thought to be species dependent and related to the insertion pattern of transposable elements (Duhl et al., 1994). The bovine genome is rich in transposable elements but little direct evidence has been found regarding their influence on gene expression (Adelson, 2008). This is also the case for the human genome (Hurst and Werren, 2001) In both species, the exposure of embryos to in vitro culture conditions has been shown to produce immediate effects on gene expression and on subsequent fetal development. (Powell, 2003; Wrenzycki et al., 2005).
Studies of early mammalian development suggest that an early consequence of exposing embryos to extracorporeal culture is aberrant DNA methylation and associated changes in the placental phenotype. The development of mammalian embryos subjected to in vitro culture is associated with an increased frequency of abnormalities in the fetuses and neonates. These abnormalities are thought to be the result of aberrant epigenetic control (DeRycke et al., 2003; Niemann et al., 2002; Niemann and Wrenzycki, 2000; Powell, 2003; Wrenzycki et al., 2005). There is growing evidence that environmental conditions during early development can cause a predisposition to cardiovascular disease and diabetes later in life. Epigenetic modifications and DNA methylation in particular are the most plausible mechanism (Dolinoy et al., 2007). The effects of environmental factors on epigenetic gene regulation can even persist into the next generation (Anyway et al., 2005).
It is apparent from this study that the DNA methylation at some loci is affected by in vitro culture or SCNT. Genes with various physiological functions were included in this analysis and were affected by epigenetic marks supporting the hypothesis that epigenetic changes due to environmental factors are essentially stochastic in nature but biased toward metastable epialleles. Studies of the differentiation of murine and human embryonic stem cell lines also suggest that there are methylation “hotspots,” which are tightly associated with differentiation (Bibikova et al., 2006; Kremenskoy et al., 2003; Shiota et al., 2002). Studies with human embryonic stem cells support the view that these cells have a unique epigenetic status that allows them to rapidly differentiate into a number of derivative cell types depending on the reception of appropriate signals (Lee et al., 2006; Shen et al., 2006; Skottmann et al., 2006). In some cases, the tumorigenic potential of transplanted ES cells seems to be related to the loss of methylation imprinting at critical hotspots (Holm et al., 2005). Our results are consistent with these observations, and demonstrate epigenetic plasticity and the vulnerability of early embryos to environmental factors.
Imprinting is an expression control mechanism by which one allele, either the maternal or the paternal allele, is silenced by the covalent addition of methyl groups to the cytosine residues of CpG dinucleotides (Constancia et al., 2004). Imprinted genes are more sensitive to subsequent epigenetic alterations because of only one active allele (Dolinoy et al., 2007). This results in dramatic effects specifically when tumor suppressor genes are involved. The majority of imprinted genes are important in fetal and placental growth and differentiation, making them primary suspects when developmental aberrations appear following SCNT or in vitro embryo production (IVP). In this study, we included PEG3, PEG10, PEG11, IGF2, and IGF2R, all of which are imprinted in normal bovine embryos. In all cases, the DNA methylation patterns of amplicons located in these genes differed among the three different classes of embryos, suggesting that these regions are sensitive to environmental factors. It is known that somatic cloning perturbs the expression of imprinted genes, such as H19, Xist, and IGF2 in bovine development (Curchoe et al., 2005; Dindot et al., 2004; Zhang et al., 2004)
SCNT involves reprogramming of the donor cell nucleus by erasure of the CpG methylation pattern, which maintains a differentiated cell phenotype and establishment of a new expression profile involving the 10,000 to 12,000 genes required for embryonic and fetal development. It now appears that DNA demethylation is the limiting factor in SCNT cloning efficiency, and conversely, that partial retention of CpG methylation is responsible for what has been called “persistent cellular memory” in lines of embryonic stem cells derived by somatic cell cloning (Eilertsen et al., 2007). SCNT reprogramming is similar to, but less efficient than, the dramatic wave of demethylation that occurs after fertilization (Bourc'his et al., 2001; Dean et al., 2003). In the context of genomic reprogramming after nuclear transfer, efficient demethylation is probably the key to improving efficiency (Hiiragi and Solter, 2005). On a population basis, it is possible to determine threshold levels of global DNA methylation that are compatible with normal development, but it is probably more important to achieve the correct pattern of DNA methylation in specific regions. It has been recently proposed that DNA demethylation of the mouse germ cells involves DNA repair enzymes (Hajkova et al., 2008), and it will be interesting to evaluate this potential improvement to the efficiency of SCNT using the panel of amplicons described in this study. SCNT is now a routine method for producing transgenic animals and reestablishing endangered species. It has great potential for the production of patient-specific embryonic stem cells for human medicine (Trounson, 2006), but it is also a critical technology for studying epigenetic gene regulation.
Extended in vitro culture of mammalian embryos results in aberrations in mRNA expression of both imprinted and nonimprinted genes, which is correlated with significant placental and fetal overgrowth (Lazzari et al., 2002; Young et al., 2001). The effects of somatic cloning on mRNA expression patterns have been most thoroughly analyzed in bovine morula and blastocyst stages. Numerous genes have been identified that are expressed at different levels in cloned embryos than in their in vivo derived counterparts (Lazzari et al., 2002; Smith et al., 2005; Trounson, 2006; Wrenzycki et al., 2001b, 2002, 2005; Young et al., 2001). The effects of DNA methylation on gene expression are complex and an integrated study of DNA methylation, RNA polymerase II occupancy, and the chromatin state of 16,000 promoter elements in human ovaries, somatic and germ line cells revealed that the level of DNA methylation and its silencing potential were correlated with a subset of target sequences in promoter regions and with the molecular function of the gene product (Weber et al., 2007). Strong promoters can have unmethylated CpG islands but remain inactive, whereas weak promoters seem to be more sensitive to differences in methylation (Weber et al., 2007). Dynamic, cyclic, and pulsatile DNA methylation patterns have recently been shown in which DNA methyltransferases play a dual role in the cycle of methylation and demethylation (Kangaspeska et al., 2008; Métivier et al., 2008). The eight amplicons in the present study, where RT-PCR was performed in parallel to methylation analysis, do not appear to represent primary control regions as transcription was not obviously related to the observed pattern, or the mean level, of DNA methylation.
In conclusion, the quantitative bisulfite sequencing shown here as methylation barcode is a powerful system for monitoring epigenetic changes in early embryos. This analysis revealed a set of amplicons that give diagnostic differences between in vivo developing embryos and their IVP and SCNT counterparts and demonstrate epigenetic reprogramming, which is the basis for cloning by SCNT. We propose that amplicons where DNA methylation patterns distinguish between the three types of embryos indicate candidate metastable epialleles that link gene expression and the environment. These are informative sites where methylation analysis can be employed for quality assessment of IVP and SCNT embryos, potentially leading to early diagnosis and/or prevention of the developmental aberrations related to assisted reproduction technologies. It is expected that this system will also be an important tool for monitoring stem cells in culture.
Footnotes
Acknowledgments
The authors thank Klaus-Gerd Hadeler for the collection of in vivo embryos from superovulated donor cows of the Institute's experimental herds and Cornelia Krüger at Epiontis for her expert handling of DNA samples.
Author Contributions
H.N., conceptualization, writing of article; J.W.C., conceptualization, writing of article; D.H., preparation of donor cells, DNA extraction, expression analysis; A.L.H., production of cloned and in vitro blastocysts; E.L., production of cloned and in vitro blastocysts; S.O., conceptualization, writing of arrticle; G.W., bisulfite analyses, editing of article.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.