
Abstract
Abstract
Human embryonic stem cells (hESCs) offer an inexhaustible supply of human somatic cell types through their ability to self-renew while retaining pluripotency. As such, hESC-derived cell types are important for applications ranging from in vitro modeling to therapeutic use. However, for their full potential to be realized, both the growth of the undifferentiated cells and their derivatives must be performed in defined culture conditions. Many research groups maintain hESCs using mouse embryonic fibroblasts (MEF) and MEF conditioned medium (CM). The use of murine systems to support hESCs has been imperative in developing hESC technology; however, they suffer from some major limitations including lack of definition, xenobiotic nature, batch-to-batch variation, and labor-intensive production. Therefore, hESC culture definition is essential if hESC lines, and their derivatives are to be quality assured and manufactured to GMP. We have initiated the process of standardizing hESC tissue culture and have employed two serum-free media: mTeSR (MT) and Stem Pro (SP). hESCs were maintained in a pluripotent state, for over 30 passages using MT and SP. Additionally, we present evidence that hESCs maintained in MT and SP generate equivalent levels of human hepatic endoderm as observed with CM. This data suggests that MT and SP are effective replacements for MEF–CM in hESC culture, contributing to the standardization of hESC in vitro models and ultimately their application.
Introduction
In the last decade, significant progress has been made in developing efficient and functional somatic cell models from hESCs, thus providing useful tools for in vitro and in vivo modeling (Basma et al., 2009; Hay et al., 2008). Although these studies are very promising and demonstrate the potential of hESCs, they rely on undefined components that complicate large-scale manufacture. As the use of stem cells in diagnostic and clinical applications progresses, it is imperative that cell culture systems and tissue culture models are standardized. Consequently, the full potential of hESC can only be realized when defined cell culture and differentiation conditions are established. In this way, quality-assured hESC-derived products can be manufactured in large scale and to GMP if necessary.
Initially, the most common method of culturing hESCs was on mouse embryonic fibroblasts (MEFs) in the presence of serum replacement media. In these cultures the MEF feeder cells supplied both essential factors and supported cell attachment required for the successful maintenance of pluripotent hESCs (Thomson et al., 1998). More recently, feeder free conditions have been developed allowing hESCs to be cultured on a matrigel basement membrane in the presence of MEF-conditioned media (CM) (Xu et al., 2001). Although this system improved hESC culture conditions and scalability, feeder-free CM systems still suffered most of the limitations associated with the previous coculture approach, including a lack of definition, presence of animal components, batch-to-batch variability, and the labor-intensive nature of MEF CM production.
To address these issues we have employed more defined serum-free commercially available media, mTeSR (MT) and StemPro (SP) (Ludwig et al., 2006; Wang et al., 2007). We successfully transitioned hESCs, which were previously maintained feeder free on matrigel in the presence of CM, to MT and SP. Moreover, the transitioned hESC lines were capable of being directly differentiated to human hepatic endoderm (HE) with equal efficiency to hESCs cultured in MEF–CM (Fletcher et al., 2008; Hay et al., 2008). This data demonstrates that one major hurdle in hESC manufacture has been successfully addressed. This, in turn, will help reduce culture variability by providing uniformity between hESC lines, allowing the creation of generic directed differentiation models (Mikkola et al., 2006).
Materials and Methods
hESC culture
H1 hESCs were grown and propagated in feeder free conditions on Matrigel (BD Biosciences, San Jose, CA, USA)-coated plastic in the presence of CM, before being transitioned in to MT or SP. The cells were split at a 1:3 ratio and allowed to settle overnight. They were maintained in a 80:20 ratio of CM (R&D Systems, Minneapolis, IN, USA) (Hay et al., 2008) to MT (Stem Cell Technologies, Vancouver, Canada) (Ludwig et al., 2006), or SP (Invitrogen, Carlsbad, CA, USA) (Mallon et al., 2006) followed by 60:40, 20:80, and finally 100% over a 14-day period. Subsequently, transitioned hESC lines were maintained as per the manufacturers instructions. hESCs were cultured for 30 passages in CM, MT, or SP on matrigel-coated plastic with daily media change and passaged using collagenase (Invitrogen) and physical detachment (Hay et al., 2008).
Embroid body formation
EB cellular aggregation was initiated and characterized as previously described (Fletcher et al., 2006).
Fluorescent-activated cell sorting
Flow cytometry was performed as previously described (Fletcher et al., 2008).
Immunohistochemistry
hESCs cultured in CM, MT, and SP were fixed in 4% PFA and stained as described by (Fletcher et al., 2008). hESC derived HE was fixed in ice-cold methanol as described (Hay et al., 2008). Immunostaining was recorded using a Leica DMIRB using × 10 magnifications to analyze cell morphology and × 20 magnification to analyze immunostaining (Oct 4; Santa Cruz Biotechnologies, Santa Cruz, CA, USA). EB staining was carried out using the following antibodies, α-SMA, AFP, and β-Tub III, the phase contrast images were taken at × 20 and the immunostaining at × 40. HE derived from hESCs cultured in CM, MT, and SP were stained with a variety of hepatic markers such as albumin, AFP, HNF4a, E cadherin, and Cyp3A4; all photos in Figure 4 are at × 40. To calculate the number of hepatic cells within the population, albumin-positive cells were counted at × 20 magnifications in four fields of view, with a total for 2000 dapi positive cells. This was supported by further cell counting, based on morphology alone, using phase contrast images; 20 fields of view from each well (n = 3). An IgG antibody was used as a negative control. Alexa Fluor 488 (Invitrogen–Molecular Probes, Eugene, OR, CA; 1:400) was used for all secondary staining. Antibody details are shown in Supplementary Table 1.
RNA extraction and RT PCR
RNA extraction and RT PCR was carried out as previously described (Hay et al., 2007, 2008). PCR primers are shown in Supplementary Table 2.
Directed differentiation into HE
hESCs cultured in CM, MT, and SP were directly differentiated into HE as previously described, specifically employing the Activin A/Wnt 3a molecules (Hay et al., 2008).
Cyp p450 assay
hESC-derived HE was differentiated as above and at day 17 was cultured in 1 mL of L15 media (Sigma-Aldrich, St. Louis, MO, USA) (Hay et al., 2008) with the Cyp 1A2 luciferin (1:50) and Cyp 3A4 luciferin (1:40); for details please refer to the pGLO manual found on www.promega.com (Promega, Madison, WI, USA). The supernatant was collected for Cyp 1A2 (18 h postaddition) and Cyp 3A4 (4 h postaddition). The supernatant was then assayed as detailed as per manufacturer's instructions. The samples were measured in triplicate on a luminometer and units of activity are quoted as RLU/mg protein.
Ureagenesis assay
HE was cultured in phosphate-buffered saline (PBS) (GIBCO, Grand Island, NY, USA) containing ammonium chloride (Sigma-Aldrich) and the urea produced was determined as before (Filippi et al., 2004). Samples were assayed in triplicate and measured in mmol/mg/h and normalized to per mg of cellular protein.
Statistical analysis
One-way analysis of variance (ANOVA) was used to calculate the significance between the data obtained from urea and CYP P450 assays comparing the various medias, and the asterisk defines the level of significance where *p < 0.05, **p < 0.01, and ***p < 0.001. (Graphpad Prism 5 Software was used for all calculations.)
Results
Culture and characterization of hESCs maintained in CM, MT, and SP
With a view to further defining hESC culture conditions, hESCs maintained in CM were gradually transitioned to MT and SP over a 14-day period. hESCs were maintained in CM or the serum-free media for a further 30 passages before characterizing hESC identity and pluripotency. These attributes were assessed using an established panel of stem cell markers (Hay et al., 2008) and the ability to form embryoid bodies exhibiting all three germ layers (Fletcher et al., 2008). hESCs maintained in all three media were positive for embryonic stem cell markers Oct4 (Fig. 1A, a–c), and Nanog (Fig. 1B) and possessed hESC morphology (Fig. 1A, d–f). hESCs grown in CM formed compact colonies with little spontaneous differentiation (Fig. 1A, d, arrow), whereas hESCs maintained in MT formed highly defined tightly packed dome-like colonies with no observable spontaneous cellular differentiation (Fig. 1A, e, arrow). In contrast, hESCs cultured in SP resulted in loosely associated colonies with spiky edges when compared to the other hESC media (Fig. 1A, f, arrow). Following the observation of differences in cell morphology we decided to characterize hESC surface expression using FACS. Our panel of markers was composed of an established set of hESC surface proteins; SSEA 3, 4, Tra 1-60, and 1-81 (Fig. 1C). There was no significant difference observed in the cell surface expression of these markers between the different hESC maintenance media, except for Tra 1-60 expression in hESCs maintained in MT. Expression levels in hESCs cultured in MT were significantly higher when compared to SP and CM. However, this did not affect either the ability of MT stem cell populations to generate all three germ layers, or Oct 4 and NANOG expression (Fig. 2). This data demonstrate that we were able to maintain hESC populations using more defined serum-free cell culture media, thereby making it possible to scale up hESC numbers for our subsequent analysis.
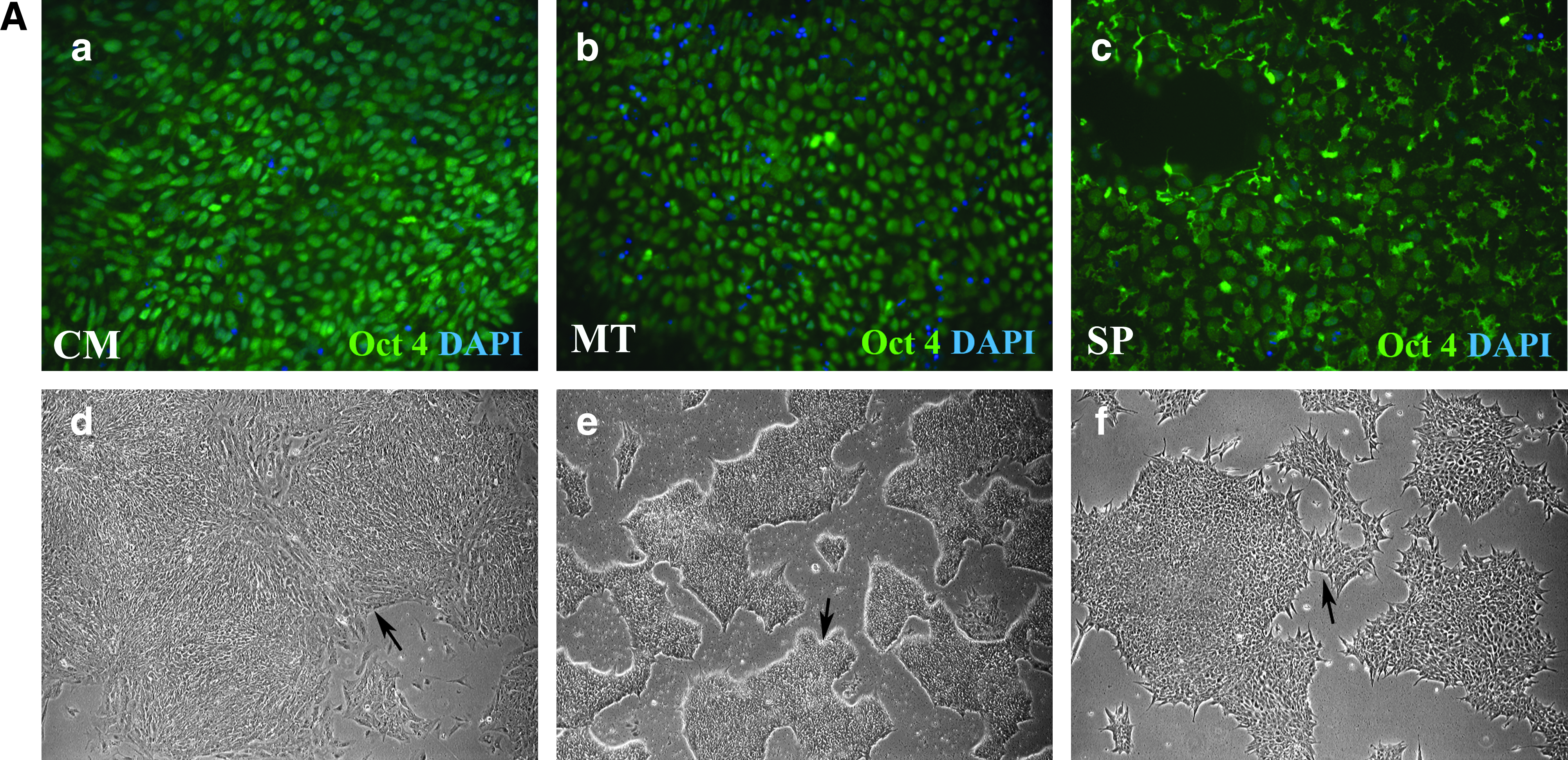
Characterization of hESCs cultured in CM, MT, and SP. (

Investigation of pluripotency in hESCs cultured in CM, MT, and SP. (
Investigating pluripotency of hESCs cultured in CM, MT, and SP
EB formation is a standardized method for measuring hESC pluripotency. Consequently, hESCs were lifted and placed into a suspension suitable for promoting cell aggregation. After 7 days the EBs generated were well defined and vacuolated (Fig. 2A), suggesting efficient EB formation. The EBs were replated on gelatin-coated wells and were allowed to spontaneously differentiate over a 14-day period. The resulting cell types were fixed and stained with specific antibodies representing individual germ layers. Endodermal commitment was measured using alpha-feto protein (AFP), mesodermal specification was measured with α-SMA (smooth muscle actin), and developing ectoderm was detected using β-Tubulin III (Fig. 2B). This data demonstrate that the hESCs cultured in MT and SP remain pluripotent for over 30 passages, comparable to hESCs maintained in CM.
Directed differentiation and characterization of hESC-derived hepatic endoderm
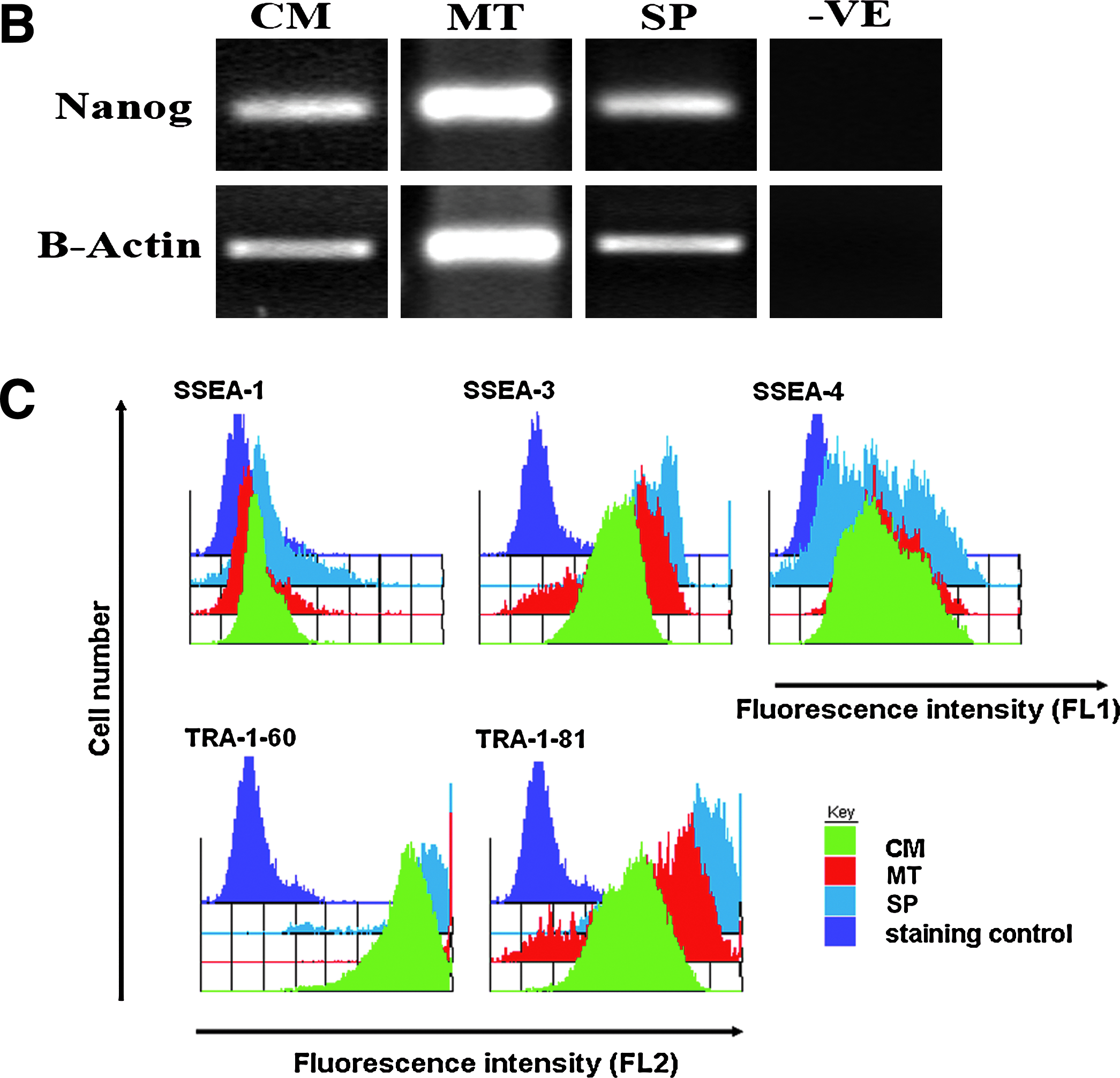
Efficiently directing hESC differentiation to the appropriate cell type is essential if stem cell-derived products are to be manufactured cost effectively. hESCs maintained in the different media were scaled up to the desired quantity and hepatic differentiation was driven using Activin A/Wnt3a stimulation (Hay et al., 2008). At day 17 of differentiation the purity of hESC-derived HE was quantified using an adult liver marker, albumin, and an IgG antibody was used as a negative control. hESC-derived HE purity was determined by the percentage of albumin-positive cells in differentiating cultures and estimated using four random fields of view with ∼500 cells per field of view. This data was further supported by estimating the percentage of HE generated in 20 random fields of view per well using phase contrast microscopy (n = 3). We did not observe any significant differences regarding HE generation, in vitro, between each of the different media (Fig. 3A). The CM control formed 96.5% (±1.5) HE, hESCs cultured in MT resulted in 98.6% HE (±0.38) and hESCs cultured in SP generated 94% (±2.5) HE. To further characterize our in vitro-derived HE we assessed the repertoire of hepatic gene expression by PCR (see Supplementary Table 2 for conditions). HE derived from hESCs cultured in various defined media expressed a number of hepatic transcripts (Fletcher et al., 2008; Hay et al., 2007, 2008), which was comparable among the three different media (Fig. 3B). In addition to PCR, we confirmed hepatic gene expression by immunostaining fixed hESC-derived HE monolayers for a number of proteins involved in hepatic maintenance and function. hESC-derived HE maintained in CM, MT, and SP were fixed on day 17, and the expected patterns were observed when stained with antibodies to albumin (ALB), alpha-feto protein (AFP), cytochrome p450 3A4 (Cyp 3A4), E-Cadherin, and hepatocyte nuclear factor 4α (HNF4 α) (Fig. 4). Immunostaining was controlled using an IgG negative control. Figure 4A displays HE derived from hESCs cultured in CM, Figure 4B displays HE derived from hESCs cultured in MT, and Figure 4C depicts HE derived hESCs cultured in SP. The data presented in Figures 3 and 4 demonstrate that hESCs cultured in CM and the serum-free media (MT and SP) are not only pluripotent stem cells but can also undergo efficient directed differentiated to human HE, in vitro.

Characterization of HE derived from hESC cultured in CM, MT and SP. (
Immunocytochemical Analysis of HE formed from hESCs cultured in CM, MT, and SP, in vitro. (
Characterizing hESC-derived HE function in vitro
The production of functional HE from hESCs is essential for a number of downstream applications ranging from in vitro models to the bioartificial liver and cell-based therapies. Following hepatic differentiation, we focussed on characterizing hepatic function that would have direct application to the generation of predictive toxicology tools and extra-corporeal support devices. As such, we investigated the two most relevant liver functions: cytochrome p450s expression (CYP3A4 and CYP1A2), involved in prescription drug metabolism, and urea production, a vital function carried out by the liver. Both CYP3A4 and 1A2 activity were observed in hESC-derived HE from cells maintained in CM, MT, and SP (Fig. 5). Maximal CYP3A4 activity was detected in hESCs maintained in MT that was threefold greater than SP-derived HE, and twofold higher than in CM-derived HE (both significant); (p < 0.0002, ANOVA) (Fig. 5A). CYP1A2 activity was similar between hESCs maintained in MT and SP, and was significantly greater that that observed for CM; p < 0.0001 (Fig. 5B). Urease activity was measured using an “in-house” assay (Filippi et al., 2004). Urea production was similar between MT and SP maintained cells that was greater than CM maintained hESCs; (p < 0.015) (Fig. 5C). Taken together, this data demonstrates that the introduction of a more defined cell culture technology not only permits scalable hESC-derived HE, but also improves cell function when compared to CM-derived HE.
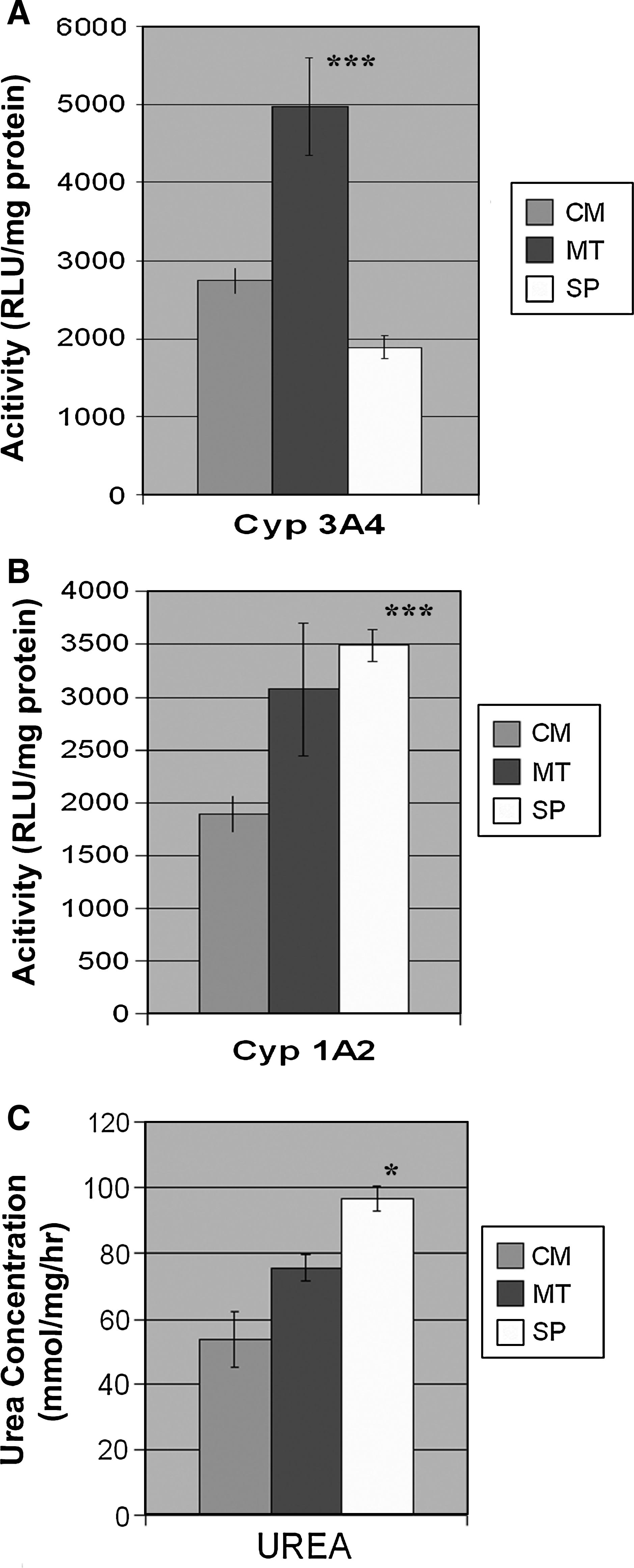
Functional Analysis of HE derived from hESCs cultured in CM, MT, and SP. hESC-derived HE cytochrome p450 function, Cyp3A4, and Cyp1A2 were assessed using the pGlo systems; (n = 3) (Promega). (
Discussion
The ability to manufacture hESCs and their derivatives for large-scale production requires the standardization of cell culture systems and routine tissue culture models. Therefore, as stem cells move closer to diagnostic and clinical applications, further efforts are required to define culture models so that they can be translated more efficiently to quality assurance required for good manufacturing process (GMP). These studies demonstrate success in overcoming one of the major issues that face us in hESC culture, improving culture medium definition.
We demonstrate that serum free media, MT and SP, are suitable replacements for CM supporting hESC self-renewal, pluripotency, and directed differentiation of hESCs to HE. Furthermore, HE function was mainly improved in response to MT and SP in comparison to CM. Morphologically, we also observed differences with the presence of more tightly packed 3D hESC colonies in cells maintained using MT, whilst hESCs cultured in SP or CM exhibited a more ‘‘flat’’ or 2D appearance. The difference in colony structure in MT cultures could be attributed to the elevated levels of Tra 1-60 detected.
The next stage in defining hESC culture conditions will be the identification of suitable extracellular matrices (ECMs) which sustain pluripotent and karyotypically stable cell populations. Of note, a recently identified xeno-free and fully defined ECM, CellStart, (Invitrogen) has shown promise in defining hESC culture and differentiation (Swistowski et al., 2009).
In conclusion, we demonstrate that the growth of hESCs in a more defined culture environment did not compromise hESC self-renewal, pluripotency, or directed differentiation, and in some cases improved HE function. These studies are both important and part of an incremental process that seeks to define hESC cell culture that permits scale-up and the creation of reliable in vitro models. In the future, it will be interesting to see if it is possible to derive other functional somatic cell types from hESCs with equal efficiency using serum free media.
Footnotes
Acknowledgments
We thank Professor Roland Wolf for kindly providing the antibody to Cyp3A4. Ms Zara Hannoun was supported by a MRC PhD studentship. Dr. David Hay was supported by a RCUK fellowship. Dr. Claire Medine, Mr. Sebastian Greenhough, and Ms. Ruchi Sharma were supported by a grant awarded by the UK Stem Cell Foundation and Scottish Enterprise.
Author Disclosure Statement
The authors declare no conflicts of interest.
The first two authors contributed equally to this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.