
Abstract
Abstract
Animal cloning by nuclear transfer (NT) has made the production of transgenic animals using genetically modified donor cells possible and ensures the presence of the gene construct in the offspring. The identification of transgene insertion sites in donor cells before cloning may avoid the production of animals that carry undesirable characteristics due to positional effects. This article compares blastocyst development and competence to establish pregnancies of bovine cloned embryos reconstructed with lentivirus-mediated transgenic fibroblasts containing either random integration of a transgene (random integration group) or nuclear transfer derived transgenic fibroblasts with known transgene insertion sites submitted to recloning (recloned group). In the random integration group, eGFP-expressing bovine fetal fibroblasts were selected by fluorescence activated cell sorting (FACS) and used as nuclei donor cells for NT. In the recloned group, a fibroblast cell line derived from a transgenic cloned fetus was characterized regarding transgene insertion and submitted to recloning. The recloned group had higher blastocyst production (25.38 vs. 14.42%) and higher percentage of 30-day pregnancies (14.29 vs. 2.56%) when compared to the random integration group. Relative eGFP expression analysis in fibroblasts derived from each cloned embryo revealed more homogeneous expression in the recloned group. In conclusion, the use of cell lines recovered from transgenic fetuses after identification of the transgene integration site allowed for the production of cells and fetuses with stable transgene expression, and recloning may improve transgenic animal yields.
Introduction
In the last decades, many techniques have been used in the production of genetically modified animals. Three of them have become more popular due to their technical simplicity and positive outcome; these are pronuclear microinjection of exogenous DNA into zygotes, injection of genetically modified embryonic stem cells (ES) into blastocysts and, more recently, retrovirus-mediated gene transfer (Gordon and Ruddle, 1981; Gossler et al., 1986; Hammer et al., 1985; Hofmann et al., 2003, 2004). Injection of ES cells into the blastocoel, the most popular technique in laboratory animal research, allows for the introduction of targeted genetic modification by transgenesis of ES cells (for review, see Misra and Duncan, 2002). However, ES cells are not yet well characterized in farm species (Keefer et al., 2007; Prelle et al., 2002), limiting the use of this methodology in production.
In the techniques most commonly applied to farm animals, the transgene site integration in the host genome is reported as being random (Clark et al., 2000). Random integration of exogenous DNA into the genome is problematic because position effect variegation can profoundly affect transgene expression, leading to unpredictable transgene expression, including the disruption of endogenous genes (Beard et al., 2006; Rulicke and Hubscher, 2000; Soriano et al., 1987; Williams et al., 2008). Often, transgene expression is highly variable even among cell lines transformed with the same construct. Phenotype effects may be crucial to guarantee the welfare of transgenic animals (Dunn et al., 2005; Lisauskas et al., 2007; Rulicke and Hubscher, 2000), and therefore, identification of copy number and chromosomal location at a particular chromosomal site is highly desirable for the production of animal bioreactors.
To provide more stable transgene expression in animals, somatic cell nuclear transfer (SCNT) has become the method of choice in the production of genetically modified farm animals. The SCNT technique, together with efficient donor cell genetic modification by lentivirus-mediated exogenous gene transduction enables animal modification without mosaicism, compared to other often applied techniques, including the well-characterized pronuclear microinjection (reviewed by Park, 2007).
Although the efficiency of SCNT is still lower than desired, probably due to abnormalities in early development leading to low developmental rates (Arnold et al., 2008; Miglino et al., 2007), this methodology provides the opportunity to use a well-established, modified, and selected cell line before embryo reconstruction (reviewed by Wolf et al., 1998). The use of transgenic cell lineages already proven to be successfully reprogrammed by nuclear transfer may indicate that transgene integration is not deleterious in those particular lineages, resulting in a good source of donor cells that can be used to produce a homogeneous bioreactor herd. In this study we tested the hypothesis that recloning of transgenic somatic cells that have already been characterized and successfully reprogrammed, such as cloned fetal cells, may improve the rate of production of SCNT transgenic bovine animals.
Materials and Methods
Isolation, genetic modification, and culture of primary fetal fibroblasts
A primary culture of bovine fetal fibroblasts was obtained from a 55-day male Nelore (Bos indicus) fetus [primary nontransgenic fibroblasts (PNTF)]. After removal of the organs and head, the tissue was washed in phosphate buffer saline (PBS), minced thoroughly with small scissors and placed on plastic Petri dishes filled with Dulbecco's modified Eagle Medium (DMEM; Gibco BRL, Gaithersburg, MD) supplemented with 20% fetal bovine serum (FBS; Gibco BRL) and antibiotics.
Transduction of replicative-defective lentiviruses was used for the introduction of the reporter gene, enhanced Green Fluorescent Protein (eGFP), into the fibroblast genome. The lentivirus contained the eGFP coding region driven by the ubiquitin promoter (FUGW) (Lois et al., 2002). Briefly, media culture was collected 48 h after lipofection (Lipofectamine, Invitrogen, Carlsbad, CA; following manufacturer's guidelines) of HEK 293 cells with pFUGW, the HIV-1 packaging vector Δ8.9 and the VSVG envelope glycoprotein, filtered through 0.45-μM filters (Millipore, Bedford, MA), concentrated, titrated, aliquoted, and kept at −80°C until use.
Twenty-four hours after plating of fetal fibroblasts in the second passage in wells of a 96-well dish, lentivirus diluted in fresh culture medium supplemented with 8 μg/mL of hexadimethrine bromide (Polybrene, Sigma, St. Louis, MO) was added to cells with normal visual morphology (viral concentration adjusted to one viral particle per cell). Two to 4 h after incubation, the culture medium was replaced with regular medium. Transduction was repeated after 24 h. When the cells were confluent, the cell culture was subcultured progressively into a 24-well dish, a 35-mm dish, and finally to a 60-mm dish, when cell quantity was enough to allow fluorescence analysis by flow cytometry (FACSAria, FACSDiva software; BD, San Jose, CA). Positive cells were sorted and in vitro cultured until approximately 80% of confluence in two 60-mm dishes. The positive cells [primary transgenic fibroblasts (PTF)] containing presumptive random transgene copy numbers and site insertions were used as nuclei donors for the production of the random integrated fibroblast clones at 30 days of pregnancy. One eGFP-expressing embryo on day 30 of pregnancy was recovered by hysterectomy, characterized for the integration site and used as a fibroblast donor for recloning.
Throughout the experiments, unless stated otherwise, cellular cultures were maintained in DMEM medium supplemented with 10% FBS and antibiotics and cultured at 38.5°C, 5% CO2 in air, and saturated humidity. Passages were conducted when cells were confluent or when necessary using 0.25% trypsin (Gibco BRL).
Nuclear transfer
The nuclear transfer procedures were performed as described previously (Miranda et al., 2009). Briefly, donor cells were prepared with serum starvation (DMEM + 0.5% FBS) for 48 h before each round of micromanipulation. Bovine oocytes aspirated from ovarian antral follicles (3–6 mm in diameter) in slaughterhouse-derived ovaries were selected by morphology (de Loos et al., 1989) and matured in vitro. After 18 h, oocytes were denuded and selected for the extrusion of the first polar body. Oocytes were enucleated by micromanipulation and the donor cell was placed into the perivitelline space of each enucleated oocyte (Fig. 1). SCNT couplets were then electrically fused, chemically activated, and cultured for 7 days. Fusion rates were evaluated during the SCNT procedure, cleavage rates were evaluated on the third day of culture, and blastocyst rates on the seventh day. Parthenogenetic embryos were activated and in vitro cultured in the same conditions and used as controls for oocyte quality, activation procedure, in vitro culture, and eGFP fluorescence (Fig. 1, Supplemental Data; data are available online at www.liebertonline.com/cell).
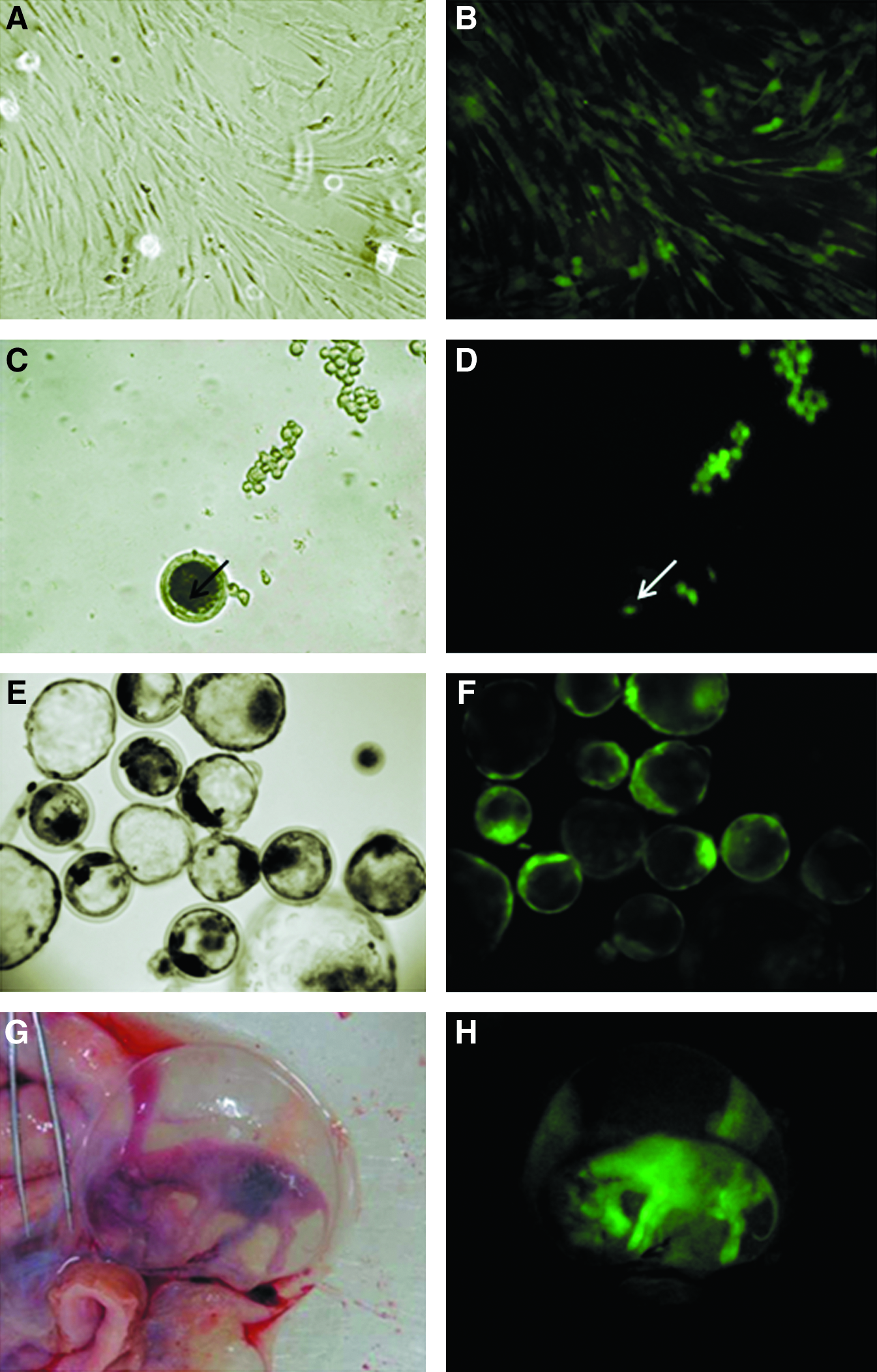
(
In the nuclear transfer procedure, embryos were reconstructed to evaluate the developmental potential of cloned embryos using PTFs as the nuclei donor, called the random integration group. Three 30d conceptuses (clone 1, clone 2, and clone 3) were collected and primary transgenic fibroblasts were isolated in vitro and cultured (PTF1, PTF2, and PTF3). For the recloned group, one fibroblast lineage isolated from the selected transgenic embryo (clone 3, PTF3) was used as the nuclear donor. Cell lines were also derived from resultant recloned conceptuses at 30 (PTF3.1), 60 (PTF3.2, PTF3.3, PTF3.4), or 90 days of gestation (PTF3.5 and PTF3.6).
Embryo transfer and pregnancy monitoring
One or two embryos at the blastocyst stage on day 7 after reconstruction were placed in 0.25-mL straws containing buffered SOF medium and transferred nonsurgically to synchronized recipients. Pregnancy diagnoses were performed by transrectal ultrasonography (Aloka SSD-900, Aloka Co., Switzerland) on day 30 and the embryos were recovered throughout the first trimester of pregnancy (days 30, 60, and 90) by hysterectomy to confirm eGFP gene presence and expression.
Detection of transgene presence and expression
Fluorescence detection
eGFP expression in fibroblast cells, blastocysts, and embryonic tissues was analyzed using a fluorescence microscope (Axioplan 2, Carl Zeiss, Thornwood, NY). In addition, eGFP expression of conceptuses was evaluated with GFsP-5 (BLS Ltd, Ontario, Canada) equipment, which permits macroscopic detection of GFP fluorescence (Fig. 1). Flow cytometric analysis was also performed with fibroblast samples (FACsAria; BD Biosystems). Briefly, harvested single cells were resuspended in media culture, stained with propidium iodide (10 μg/mL) for cell viability discrimination, and data were analyzed based on the examination of eGFP fluorescence on 50,000 cells per sample using FACsDiva software (BD Biosystems).
Extraction of nucleic acid and PCR analysis
Genomic DNA extraction was performed on different fetal fibroblast cultures to identify cell lineages containing the transgene using the NaCl protocol (Sambrook and Russell, 2001). Briefly, cells were incubated with a Proteinase K solution (0.53 μg/mL, Gibco BRL) and 20% SDS for 3 h at 55°C for digestion of protein and lipids. DNA was washed in a 5 M NaCl solution, precipitated with ethanol and eluted in ultrapure water. Total RNA from the same cells was extracted using a commercial kit (RNeasy kit, Qiagen, Chatsworth, CA), and eluted in ultrapure water to identify cell lines expressing the transgene. DNA and RNA quantity and quality were analyzed by spectrophotometry (Nanodrop, Thermo Scientific, Worchester, MA).
The presence of the eGFP gene was determined by polymerase chain reaction (PCR) using eGFP-specific primers and probe, as described by Joshi et al. (2008).
eGFP mRNA expression in fibroblasts was evaluated by qPCR using the same primers and conditions described previously (Joshi et al., 2008), using the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as reference gene. Briefly, reverse transcriptase reactions were performed using random oligo primers [Pd(N)6, Amersham, Arlington Heights, IL] as recommended by the supplier.
Ligation-mediated PCR (LM-PCR)
For identification of the integration site of the transgene in the fibroblast line used for the recloning, genomic DNA was extracted as described above and processed and analyzed similar to as described by Wu et al. (2003). Briefly, 2 μg were digested with the MseI restriction enzyme for 8 h at 37°C. MseI is responsible for widespread genome DNA digestion by cutting at TTAA sequences. Digested DNA was linked to an adaptor sequence that binds to digested DNA ends. The adaptor sequence was obtained by incubation of two sequences called here as Linker forward and Linker reverse at 95°C for 3 min and cooled 1°C per min until 4°C. Sequences are presented in Table 1. A nested PCR was performed to amplify sequences containing genomic DNA adjacent to viral long terminal repeat sequences (LTRs), with annealing at 55°C for 30 sec, for 35 cycles. Amplicons were cloned in competent bacteria using the P-GEM-T Easy Vector System kit (Promega, Madison, WI) as described by supplier. Bacterial clones (n = 36) were sequenced in triplicate for both forward and reverse sequencing primers (M13 primers) in an ABI 377 DNA sequencer (Applied Biosystems, Bedford, MA) and the products obtained (n = 192) were analyzed using Chromas software (Technelysium Pty Ltd). Genomic sequences were only considered to be integration sites when they contained lentiviral specific sequences immediately followed by bovine genome sequences (n = 13, from three different bacterial clones), whereas the remaining sequencing products did not have enough nucleotides or were considered unspecific and were therefore discarded. Finally, nucleotide sequences containing both lentiviral and bovine genome counterparts were analyzed by alignment using BLAST (Basic Local Alignment Search Tool) (Altschul et al., 1990), providing the specific location of transgene in a chromosome.
PCR analysis was conducted to confirm the transgene integration in the genomic DNA of transgenic conceptuses. For that, the forward primer used for second nested PCR, which anneals to the LTR sequence of lentivirus was used together with a primer designed in the bovine DNA genome sequencing, amplifying a 146-bp sequence. Reference gene 18S was used for DNA amplification control, resulting in a 63bp amplicon (Fig. 2, Supplementary Data). Primer sequences for LM-PCR, DNA sequencing, transgene integration confirmation, DNA and cDNA analysis are presented in Table 1.
Statistical analysis
The random integration group consisted of 11 replicates and the recloned group consisted of four replicates. Data on embryo reconstruction and development, and flow cytometric analysis were submitted to analysis of variance (ANOVA) and means were compared by Tukey's test. Quantification of the eGFP mRNA was evaluated by semiquantitative PCR using the 2−ΔΔCt method (Livak and Schmittgen, 2001), based on duplicates from two independent replicates. Both target and reference gene PCRs were equally efficient. The frequency of 30-day pregnancies was tested by Fisher's exact test. The 5% level of significance was used for all experiments.
Results
In the nuclear transfer procedure, 884 embryos were reconstructed using PTFs as nuclei donors. Three embryos were recovered; however, the first conceptus (clone 1, from which PTF1 was derived) did not express the transgene, whereas the second (clone 2, from which PTF2 was derived) and third (clone 3, from which PTF3 was derived) were obtained from a gemelar pregnancy. Of these, clone 2 presented signs of degeneration; however, clone 3 was apparently healthy (Fig. 3, Supplementary Data). One embryo after 30 days of pregnancy was then used to establish a fibroblast lineage (PTF3), characterized with regard to transgene insertion and used as a donor for nuclei to reconstruct 329 embryos in the recloned group (Table 2).
Different superscripts within columns indicate difference (p < 0.05).
Nrc, number of reconstructed couplets; Nfc, number of fused couplets; Nc, number of cleaved embryos; Cleav rate, cleavage rate; Nb, number of blastocysts; Blast rate, blastocyst rate.
The sequenced products from LM-PCR of PTF3 resulted in the identification of a 250-bp segment, approximately, of the genome corresponding to a nontranslated site on chromosome 14 (NW_001493203.1; from nucleotide 882780 to 882705). No other sequenced product matched the criteria to be considered to be from a genuine integration event, as described before. It was therefore assumed that the identified integration site would not compromise developmental competence or normal phenotype, and this fetus-derived cell line was submitted to a second round of nuclear transfer (recloning).
Amplification by PCR was used to confirm the identical transgene integration site in the genomic DNA of transgenic conceptuses clone 2, clone 3, and reclones 3.1, 3.2, 3.3, 3.4, 3.5, and 3.6; hence, because no amplification occurred in PTFs, it showed that the integration site in these cells is a rare event compared to the other random integration sites, and that in clone 1 the integration is not located in the same region of the genome (Fig. 2, Supplementary Data).
Embryo production procedures resulted in no difference in fusion (61.73 and 58.20% for random insertion and recloned groups, respectively) or cleavage rates (68.34 vs. 68.47%). In contrast, the blastocyst rate was higher in the recloned group (25.38%) compared to the random insertion group (14.42%; Table 2).
The competence to establish pregnancies was evaluated in both groups by means of transfer to synchronized surrogate cows, as described before. The recloned group had a higher number of successful 30-day pregnancies, as demonstrated by the percentages of positive results shown in Table 3.
Different superscripts within columns indicate difference (p < 0.05).
ET, number of embryos transferred; No. fetuses, number recovered from 30, 60, or 90 days of pregnancy; No. 30 day preg, number of 30-day pregnancies; 30 day preg rate, pregnancy rate at 30 days (number of pregnancies in relation to number of embryos transferred).
The presence of the transgenic construct was confirmed by PCR in cell lineages derived from cloned fetuses; eGFP expression was confirmed by exposure of pregnant uteri to fluorescence analysis using a GFsP-5 device and fluorescence microscopy (Fig. 1). In addition, PTFs derived from each cloned and recloned embryo were evaluated with regard to the abundance of eGFP transcripts by qPCR (Fig. 2). Conceptuses-derived fibroblasts showed no difference in eGFP expression between groups; however, fibroblasts derived from recloned conceptuses had more homogeneous eGFP expression [coefficient of variance (CV) = 22.2%] when compared to the random integration group (CV = 93.1%). Quantification of eGFP fluorescence by flow cytometry is in agreement with mRNA expression data. No difference was observed between groups; however, fibroblasts derived from recloned conceptuses presented more homogeneous eGFP fluorescence mean (Table 1, Supplemental Data).
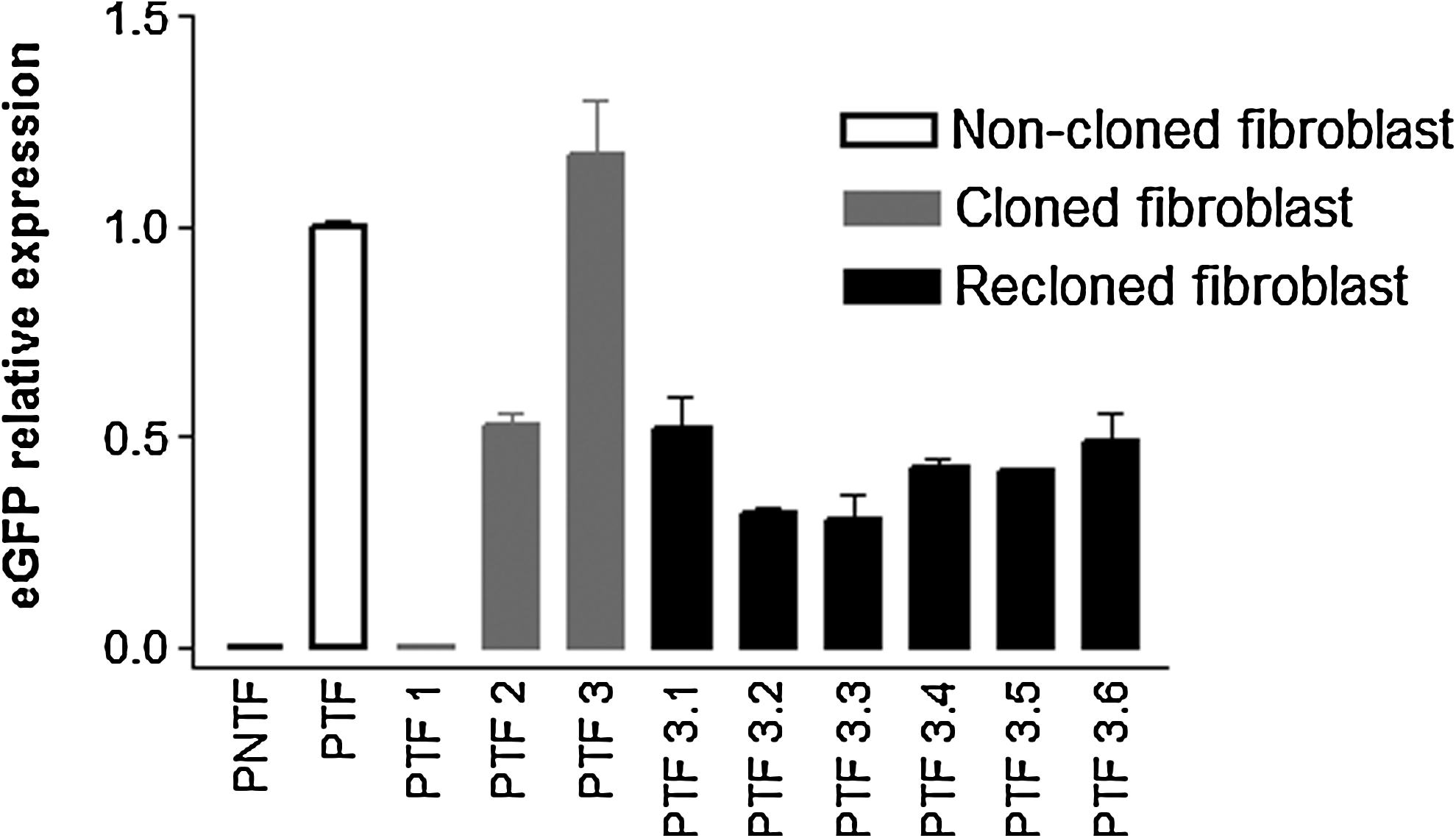
eGFP relative expression. PNTF, primary nontransgenic fibroblasts; PTF, primary transgenic fibroblasts; PTF1, primary fibroblasts derived from clone 1; PTF2, primary fibroblasts derived from clone 2; PTF3, primary fibroblasts derived from clone 3; PTF3.1, primary fibroblasts derived from reclone 1; PTF3.2, primary fibroblasts derived from reclone 2; PTF3.3, primary fibroblasts derived from reclone 3; PTF3.4, primary fibroblasts derived from reclone 4; PTF3.5, primary fibroblasts derived from reclone 5; PTF3.6, primary fibroblasts derived from reclone 6. Error bars indicate standard deviation.
Discussion
The production of transgenic animals is hampered by several problems of the genetic manipulation procedure itself or its consequences. The invasiveness of some techniques and mosaicism are two of most common drawbacks. Nuclear transfer procedures are now widely used to overcome these problems (Clark et al., 2000; Laible and Alonso-Gonzalez, 2009; Wolf et al., 2000). Its major contribution relies on its capacity to properly select genetically modified donor cells before embryo reconstruction. The resulting animals are therefore assumed to contain the desired genetic modification in 100% of the cells. Phenotypic characteristics, however, are dependent on transgene transcription of mRNA and its translation into functional proteins. Such aspects are deeply influenced by genome elements nearby transgene site integration, besides epigenetic mechanism of regulation (Baup et al., 2009).
To identify the transgene integration site, a number of PCR-based methods, often referred to as chromosome walking techniques, have been developed to isolate DNA fragments adjacent to known sequences. One of these methods is ligation-mediated PCR (Bryda et al., 2006; Rosenthal and Jones, 1990). In this study, we speculated that the combination of the cell selection with an apparently innocuous transgene integration site and the developmental ability through the first 30 days of pregnancy, together with recloning by nuclear transfer, would prevent the production of undesired phenotypes such as developmentally disadvantaged individuals. Analysis of blastocyst development, pregnancy rates and transgene expression in cultured fibroblasts revealed that this is an efficient method to produce a homogenous transgenic herd.
In this study, eight pregnancies with nine bovine transgenic conceptuses were successfully established through genetic transfer by lentiviruses in fibroblasts followed by nuclear transfer. Part of the bovine embryos was reconstructed with transduced donor cells and selected for eGFP expression by flow cytometry (random integration group). An eGFP-expressing 30-day-old fetus harboring, presumably, one unique site insertion in a nontranscribed region of chromosome 14 was used to derive a fibroblast cell culture used for recloning (recloned group). To our knowledge, there have been no previous reports that identified the transgene integration site prior to recloning. All fetuses had the transgene stably incorporated to the genome. One 30-day-old fetus derived from the random integration group did not express eGFP (clone 1); however, PCR amplification revealed the presence of the transgene. Further studies on the epigenetic regulation of transgene expression to clarify this event are ongoing.
The other pregnancy derived from SCNT of random integration group cells provided a gemelar pregnancy. Both conceptuses expressed eGFP; however, as described before, one clone appeared apparently healthy, whereas the other appeared apparently in process of degeneration. Therefore, the healthy one was used for transgene site integration analysis and recloning. After confirmation of transgene integration position, and determination that the integration site is a rare event in PTFs, it is clear that both embryos are probably derived from one unique blastocyst.
Overall, the embryo production and pregnancy rates seen in this study are in agreement with those described in the literature. The developmental competence of blastocysts commonly varies from 7 to 30% of SCNT embryos (Cho et al., 2004; Lee et al., 2007; Roh et al., 2000). Success rates in pregnancy establishment range from 6 to 48% (Arat et al., 2002; Gong et al., 2004; Iguma et al., 2005; Zakhartchenko et al., 2001). It is noteworthy that comparisons between the results of transgenic production require extra attention because different methodologies and species are used with different objectives.
In this study, we show a nearly 76% increase in blastocyst rate and a 5.58-fold increase in pregnancy rate in the recloned group compared with the random integration group. Together, these data indicate a 10.45-fold increase in the cloning efficiency, approximately. Indeed, Kuroiwa et al. (2002) and Fujimura et al. (2008), among others, reported a higher rate of blastocyst production, pregnancy establishment, and offspring when recloning was used. In addition to the advantage of recloning in terms of efficiency of development, homogenous expression of the transgene was observed in the cells derived from the recloned conceptus, which supports the use of recloning in the production of a homogeneous bioreactor herd.
The establishment of a cell line with the intended genetic modification after cloning allows for verification of the transgene pattern of expression and integration site into the genome. However, better results seen after recloning of this lineage cannot be attributed solely to the transgene insertion locus. Recloning of fetal cells per se is probably beneficial to SCNT because it enables the use of cell cultures with low population doublings, avoiding the accumulation of aneuploidy. Hence, cells that were previously reprogrammed are somehow more easily reprogrammed, or are more prone to be reprogrammed.
The production of transgenic animals with stable transgene expression and predictable desired phenotypes is extremely important for successful genetic engineering projects. The characterization of transgene site integration together with the selection of potentially reprogrammable cells may avoid the transfer of embryos with undesirable transgene site integration, resulting in events such as disruption of genes controlling embryonic, fetal, and perinatal development (Yamauchi et al., 2007).
This study speculated that the transgene insertion site in this cell lineage would not hamper embryonic or fetal development. This assumption is in agreement with the results of this study, in which a higher blastocyst development rate and a higher number of pregnancies were achieved through recloning of this lineage.
In conclusion, the present study shows that replication-defective lentiviruses used together with nuclear transfer procedures were efficient to maintain viable embryo development in vitro and in vivo and allowed for stable transgene expression. Transgene integration site identification in the donor cell lineage followed by recloning showed an increase in the efficiency of nuclear transfer, in vitro development of transgenic embryos, and in vivo establishment of transgenic pregnancies. Finally, recloning and cell selection procedures were discussed as a possible approach to increase pregnancy efficiency and herd homogeneity after NT of transgenic somatic cells because the use of a primary transgenic cell lineage has already proven to be successfully reprogrammed may indicate that transgene integration is not deleterious in that specific cell lineage, resulting in a good source of donor cells to be used for the production of a homogeneous bioreactor herd.
Footnotes
Acknowledgments
The authors acknowledge Dr. Virgínia Picanço-Castro for LM-PCR assistance, Patrícia V. B. Palma for flow cytometry assistance, Ms. D.V.M. José Rodrigo V. Pimentel and D.V.M. Paulo Fantinato for embryo transfer and hysterectomy procedures, Prof. Luiz L. Coutinho and Ms. Nirlei S. Silva for DNA sequencing, and Prof. Mariz Vainzof for GFsP-5 equipment. This work was supported by the Foundation for the Support of Research of the State of São Paulo (FAPESP), Brazil, and National Counsel of Technological and Scientific Development (CNPQ), Brazil.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.